

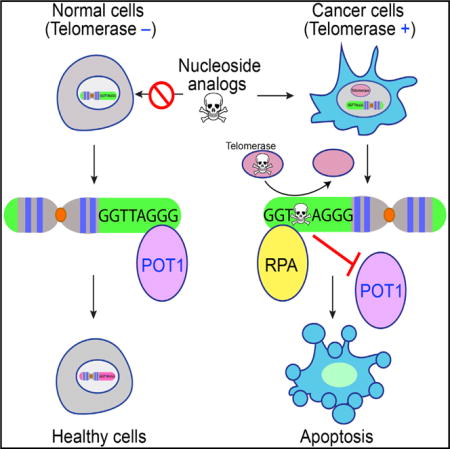
SUMMARY
Telomerase, the end-replication enzyme, is reactivated in malignant cancers to drive cellular immortality. While this distinction makes telomerase an attractive target for anti-cancer therapies, most approaches for inhibiting its activity have been clinically ineffective. As opposed to inhibiting telomerase, we use its activity to selectively promote cytotoxicity in cancer cells. We show that several nucleotide analogs, including 5-fluoro-2′-deoxyuridine (5-FdU) triphosphate, are effectively incorporated by telomerase into a telomere DNA product. Administration of 5-FdU results in an increased number of telomere-induced foci, impedes binding of telomere proteins, activates the ATR-related DNA-damage response, and promotes cell death in a telomerase-dependent manner. Collectively, our data indicate that telomerase activity can be exploited as a putative anti-cancer strategy.
Graphical abstract
In Brief: Telomerase is an attractive target for anti-cancer therapies. Zeng et al. show that several nucleotide analogs, including 5-fluoro-2′-deoxyuridine (5-FdU), are effectively incorporated by telomerase to induce dysfunctional telomeres that activate the ATR-related DNA-damage response, resulting in cancer cell death in a telomerase-dependent manner.
INTRODUCTION
One distinguishing characteristic of cancer cells over healthy somatic tissue is the reactivation of telomerase, the chromosome end-replication enzyme (Blackburn, 1994; Nandakumar and Cech, 2013). Because of the “end replication problem” the ends of chromosomes, called telomeres, of healthy somatic cells shorten with each cell division (de Lange, 2009). This phenomenon limits the number of cell divisions that may occur before the telomeres become so short that a state of replicative senescence is induced (Hayflick, 2000). Through reactivation of telomerase, the majority of cancer cells are capable of maintaining telomere length to undergo an infinite number of divisions (Shay and Bacchetti, 1997). Whereas it is undetectable in most healthy cells, telomerase is abundant in 85%–90% of all metastatic tumors (Shay and Bacchetti, 1997).
Because of its upregulation in cancer cells, telomerase is a prime chemotherapeutic target. Inhibitors of telomerase activity have entered clinical development for treating various types of human cancers, including multiple myeloma and breast, non-small cell lung, and pancreatic cancers, with several advancing to phase III trials (Xu and Goldkorn, 2016). The primary short-coming of telomerase inhibitors, however, is that even after destroying telomerase activity, the cancer cells must go through multiple rounds of DNA replication before telomere attrition results in replicative senescence. This delay can allow cancer cells to develop other mechanisms of survival, such as alternative lengthening of telomeres (ALT), to overcome the effects of telomere shortening caused by telomerase inhibition (Hu et al., 2016; Queisser et al., 2013). To address this problem, we devised an alternative strategy for selectively targeting telomerase-positive cancer cells. As opposed to inhibition, we exploit the increased telomerase activity to incorporate small-molecule nucleotide analogs into telomeres with the goal to induce genomic instability and rapid cell death selectively in telomerase-positive cancer cells. We used direct telomerase extension assays and identified 5-fluoro-2′-deoxyuridine triphosphate (5-FdUTP) as an efficient and effective compound for telomerase-mediated misincorporation into telomere DNA products. We demonstrate that nucleotides with 5-fluoro-2′-deoxyuridine (5-FdU) substituted into the telomere DNA sequence inhibit binding of essential telomere end-binding complexes. In cell lines, administration of 5-FdU induced telomeric DNA-damage responses and subsequent cell death in a manner that was dependent on the presence of active telomerase. These findings reveal an unconventional mechanism of action for the anti-cancer agent 5-FdU. Our study also provides valuable insights for the development of non-native nucleotide analogs that can be incorporated into telomeres of human cancer cells to selectively target a potentially wide range of cancers.
RESULTS
In Vitro Screening of Non-native Nucleotide Analogs for Incorporation by Telomerase
We first conducted an in vitro telomerase extension assay to identify potential nucleotide analogs that could serve as effective substrates for telomerase-mediated synthesis of telomere DNA. Previous work investigating nucleotide analogs and telomerase activity has identified a number of purine analogs that behave as inhibitors (Tárkányi and Aradi, 2008). Therefore, our initial investigation focused on pyrimidine derivatives, which were modified within the nucleobase using a range of moieties that varied in chemical properties (e.g., size variation, hydrophobicity, π-electron density). The individual nucleotide analog triphosphates were substituted into a direct telomerase extension reaction in place of the native nucleotide triphosphate. Screening of the chemically diverse pyrimidine analogs identified several compounds that were successfully and effectively incorporated by telomerase into a telomere single-stranded DNA (ssDNA) product (Figures 1A and 1B). Cumulatively, these data indicate that a diverse collection of non-native pyrimidine analogs serve as effective substrates for telomerase-mediated synthesis of telomere DNA.
Figure 1. Multiple Pyrimidine Analogs Are Successfully Incorporated by Telomerase.

(A) A series of non-native pyrimidine analogs were tested for incorporation into telomere ssDNA by telomerase using an in vitro direct extension assay. The structures and electron density surface potentials of the analogs are indicated. Electronegative regions are indicated in red, neutral regions are indicated in green, and the electropositive regions are indicated in blue.
(B) A telomerase extension assay reveals extended products. LC, loading control. Numbers on the left display the number of hexamer repeats added by telomerase. Numbers at the bottom of telomerase assay correlate with the compounds that were used in place of dTTP in the telomerase extension assay, as labeled in (A).
(C) Extended products were analyzed to quantify the telomerase activity and repeat addition processivity relative to that of native dTTP. Data are represented as mean ± SEM (n = 3).
While many of the tested substrates could be incorporated by telomerase, the degree of telomere extension varied for each compound. Telomerase is a multicomponent ribonucleoprotein enzyme that is minimally composed of a noncoding telomerase RNA component (TR or TERC) that serves as a template for the catalytic telomerase reverse transcriptase (TERT) subunit (Nandakumar and Cech, 2013). Telomerase exhibits repeat addition processivity (RAP) where multiple hexamer repeats are added to a single bound substrate before enzyme dissociation (Greider, 1991). RAP requires melting of the RNA template-DNA product hybrid so that repositioning of the template can be translocated to re-anneal with the 3′ end of the ssDNA product, and the enzyme is prepared for nucleotide addition of the next six nucleotides (Lai et al., 2001). As such, direct interactions between nucleotide analogs and the telomerase active site might be altered to preferentially incorporate or discriminate against specific chemical moieties of the analogs. Additionally, RAP might be indirectly altered due to the interactions formed between telomere DNA and the telomerase RNA template, thereby affecting single-stranded DNA binding and positioning, annealing, and/or dissociation events (Wu et al., 2017). To better understand how each nucleotide analog affected telomerase interactions and incorporation, we quantified telomerase activity for all pyrimidine analogs tested as well as relative RAP for those compounds that were incorporated into at least five hexameric repeats of the telomere ssDNA product (Figure 1C). These data identified 5-hydroxymethyl-2′-deoxyuridine triphosphate (5-OH-methyl-dUTP) and 5-FdUTP as particularly promising substrates for telomerase-mediated incorporation. Total activity for these two compounds was significantly higher than that of the other analogs tested, with relative levels of approximately half of that determined for assays conducted with native deoxythymidine triphosphate (dTTP) as a substrate (Figure 1C). Moreover, RAP for these two compounds was determined to be similar to (~80% for 5-OH-methyl-dUTP), or even higher than (~120% for 5-FdUTP), control experiments using native dTTP as a substrate for telomerase-mediated telomere extension (Figure 1C). In addition, telomerase RAP using 5-hydroxy-2′-deoxyuridine triphosphate (5-OH-dUTP) as a substrate in place of dTTP was approximately 70% of that using native dTTP as a substrate. All other compounds examined revealed telomerase RAP that was less than half of the dTTP control measurements (Figure 1C). Together, these data further indicate that pyrimidine nucleotide analogs, and particularly those that are modified at the 5-position of the nucleobase, serve as efficient substrates for telomerase-catalyzed nucleotide addition activity and RAP.
5-FdU Induces Cell Death in Telomerase-Positive Human Cancer Cells
After identifying nucleotide analogs that were successfully incorporated by telomerase, we sought to determine whether any of them might induce the death of cancer cells in a telomerase-dependent manner. To this end, we treated cells with the respective cell-permeable nucleoside counterparts and determined the induced cytotoxicity using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. For these experiments we focused on pyrimidine analogs that were incorporated by telomerase as nucleotide triphosphates and included 5-hydroxymethyl-2′-deoxyuridine (5-OH-methyl-dU), 2′-deoxypseudouridine (pseudo-dU), and 5-hydroxy-2′-deoxyuridine (5-OH-dU). Treatment of telomerase-positive HCT 116 colon cancer cells with these nucleoside analogs, at concentrations up to 5 μM, did not result in significant cytotoxic effects (Figure 2A). In contrast, 5-fluoro-2′-deoxyuridine (5-FdU), which was most efficiently incorporated as a nucleotide in the telomerase extension assay, induced cell death in HCT 116 cells at low doses (Figure 2A).
Figure 2. 5-FdU-Induced Cell Death Is Enhanced with Telomerase Activity.

(A) HCT 116 cells were exposed to varying concentrations of five non-native nucleoside analogs as indicated for 24 hr, followed by incubation in drug-free medium for an additional 72 hr. MTT assays were performed to assess the cytotoxic effects of each compound on the cells.
(B) The cytotoxicity studies for 5-FdU treatment were expanded to include telomerase-positive HCT 116, MIA-PaCa2, and A549 cells, or telomerase-negative WI-38 and U2OS cells.
(C) In contrast to 5-FdU, 5-EdU induced cell death in both telomerase-positive and telomerase-negative cell lines.
Data are represented as mean ± SEM (n = 3).
We next expanded our analysis to include additional cell lines. As with HCT 116 cells, 5-FdU treatment induced cell death of telomerase-positive pancreatic cancer MIA-PaCa2 and lung adenocarcinoma A549 cells in a dose-dependent manner (Figure 2B). However, telomerase-negative osteosarcoma U2OS and human lung primary fibroblast WI-38 cells were significantly less responsive to 5-FdU-induced cytotoxicity (Figure 2B), indicating that 5-FdU sensitivity correlated with telomerase activity. To explore whether similar effects were encountered with another nucleoside analog, we treated the five different cell lines with 5-ethynyl-2′-deoxyuridine (5-EdU). While 5-EdU is generally recognized by the replication complex to promote DNA damage and cytotoxicity in dividing cells (Ligasová et al., 2015), its nucleotide counterpart does not serve as an efficient substrate for telomerase-mediated extension (Figure 1). Indeed, administration of 5-EdU induced cell death in all treated cells, regardless of their telomerase status (Figure 2C). Finally, the ectopic expression of TERT and TR is sufficient for generating detectable telomerase activity and extended telomeres in U2OS cells (Tilman et al., 2009). Therefore, we further sought to define the relationship between telomerase activity and 5-FdU-induced cytotoxicity by generating telomerase-active U2OS cells and analyzing the response to 5-FdU treatment. These data revealed that the introduction of telomerase activity significantly sensitized U2OS cells to 5-FdU administration (Figure S1). All together, these results show that, while tolerated in telomerase-negative cells, low dosing of 5-FdU treatment promotes cytotoxicity in a diverse set of cancer cell lines that possess detectable levels of telomerase activity.
siRNA-Mediated Silencing of TERT Expression Reduces 5-FdU-Induced Apoptotic Cell Death
We reasoned that, mechanistically, incorporation of 5-FdUTP by telomerase into the telomeres of cancer cells might interfere with the highly sequence-specific recognition and binding of telomere proteins, thereby inducing cell death through activation of DNA-damage checkpoints (O’Sullivan and Karlseder, 2010). To test this possibility, we first monitored poly(ADP-ribose) polymerase (PARP) cleavage, a marker of apoptosis (Oliver et al., 1998), to determine whether 5-FdU treatment activates apoptotic cell death. Additionally, 5-FdU was administered in combination with small interfering RNA (siRNA)-mediated silencing of TERT to identify whether any adverse effects occur in a telomerase-dependent manner.
Silencing of TERT with siRNA resulted in at least a 90%–95% reduction in the expression levels of TERT mRNA when compared to the parallel control siRNA-transfected cells (Figure 3A). Whereas 5-FdU treatment induced a substantial increase in PARP (Asp214) cleavage in control siRNA-transfected cells, siRNA-mediated silencing of TERT expression significantly reduced those effects (Figures 3B and 3C). To further validate these findings, we performed annexin V and propidium iodide staining in conjunction with flow cytometry to determine the relative percentage of cells that remain viable versus those that become apoptotic or necrotic (Vermes et al., 1995) in response to 5-FdU treatment. In this experiment, 67% of the control siRNA-transfected HCT 116 cells underwent apoptosis in response to 3 μM 5-FdU treatment. Consistent with the afore-mentioned results, TERT knockdown decreased 5-FdU-induced apoptosis of treated cells to only 33% (Figure 3D). These data indicate that 5-FdU treatment results in apoptotic cell death in a manner that is largely dependent on functional telomerase.
Figure 3. siRNA-Mediated Silencing of TERT Expression Reduces 5-FdU-Induced Apoptosis.

(A) HCT 116 cells transfected with TERT siRNA results in robust TERT knockdown efficiency compared to control siRNA transfection as revealed by real-time PCR. 5-FdU-treated HCT 116 cells results in apoptosis, as indicated by PARP (Asp214) cleavage.
(B) PARP cleavage is dramatically reduced in response to 5-FdU treatment when TERT is diminished using siRNA.
(C) The expression levels of cleaved PARP in telomerase-positive or telomerase-knockdown HCT 116 were quantified and normalized to tubulin. Bars represent the mean ± SEM (n = 4). p values were calculated using Student’s t test.
(D) Annexin V and propidium iodide staining were performed to determine the percentage of apoptotic cells 3 days after the control- or TERT-siRNA transfected cells 24 hr post-5-FdU (3 μM) treatment.
Replacement of Thymidine with 5-FdU Reduces the Binding Affinity of POT1-TPP1 to Telomere DNA
Telomere DNA is recognized and protected by a set of specialized proteins that collectively form the shelterin complex (de Lange, 2005; O’Sullivan and Karlseder, 2010). Of the shelterin proteins, the protection of telomeres 1 (POT1) protein binds exclusively to the ssDNA overhang that is synthesized by telomerase (Baumann and Cech, 2001). The minimum ssDNA sequence necessary to bind a single POT1 protein with high affinity is 5′-TTAGGGTTAG-3′ (Lei et al., 2004), and the inclusion of its natural binding partner, another shelterin protein called TPP1, increases the affinity of the POT1-TPP1 heterodimer for telomere DNA by an order of magnitude over that of POT1 alone (Wang et al., 2007; Xin et al., 2007). Individual changes to the telomere sequence, particularly substitution of the interior thymidine nucleotides, have been shown to significantly diminish POT1-binding potential (Lei et al., 2004). The binding and selectivity for telomere DNA sequence is even more pronounced when TPP1 is coupled with POT1, as evidenced by a decrease of more than a 100-fold in affinity of the POT1-TPP1 heterodimer for telomere DNA where an individual thymidine base is substituted with ribouridine (Nandakumar et al., 2010). Due to this extreme preference of POT1-TPP1 for telomere sequence, we reasoned that the substitution of thymidine with the structurally similar 5-FdU nucleotide analog would adversely affect the binding of POT1-TPP1 protein to a ssDNA substrate. To address this possibility, we quantitatively characterized the binding of purified, recombinant POT1-TPP1 protein to a series of ssDNA substrates using equilibrium-binding conditions. Each substrate included 5-FdU substituted for the individual thymidine nucleotides that reside within a dodecamer substrate exhibiting telomere sequence.
The individual substitution of the second thymidine with 5-FdU in the telomere dodecameric sequence reduced POT1-TPP1 binding affinity by about four times when compared to that of the native telomere sequence (Figure 4). This finding is consistent with prior studies identifying a primary role for this particular nucleotide in binding of POT1 or POT1-TPP1 to telomere ssDNA (Lei et al., 2004). The individual substitution of 5-FdU for thymidine at other positions within the dodecameric substrate revealed only marginal to no significant changes in POT1-TPP1 binding affinity (Figure 4). However, the substitution of all four thymidines in a telomeric oligonucleotide (5′-GGTTAGGGTTAG-3′) with 5-FdU resulted in a 27-fold decrease in the binding affinity of POT1-TPP1 to the DNA (Figure 4). These findings quantitatively demonstrate that 5-FdU impairs binding of POT1-TPP1 protein to telomere DNA and that the incorporation of multiple 5-FdUs into a single POT1-TPP1 recognition sequence exhibits a synergistic effect that far exceeds the additive properties of multiple individual nucleotide changes.
Figure 4. Replacement of Thymidine with 5-FdUTP Reduces the Binding of POT1-TPP1 to Telomere DNA Sequence.

(A) A representative gel shift used to determine the binding of POT1-TPP1 protein to ssDNA under equilibrium binding conditions. The lower band represents free, radiolabeled ssDNA, and the upper band indicates ssDNA-protein complex.
(B) Gel shift data were quantified and fitted to calculate dissociation constants (KDs) of POT1-TPP1 for several substrates with native thymidine replaced with 5-FdUTP. Data are represented as mean ± SEM (n = 3).
(C) A table summarizing the results indicates that 5-FdUTP exhibits differing positional effects on POT1-TPP1 binding affinity for ssDNA substrates.
5-FdU Treatment Induces Translocation of RPA to Telomeres in Human Cancer Cells
The fidelity of DNA-damage response and repair pathways requires signal transduction events that are coordinated by a group of specialized proteins (Matsuoka et al., 2007). Central to the DNA-damage response pathway are the protein kinases ATM (ataxia-telangiectasia mutated) and ATR (ataxia-telangiectasia mutated and Rad3-related), which respond to double-stranded-DNA- and ssDNA-associated repair, respectively (Matsuoka et al., 2007). Upon detecting DNA damage, ATM and ATR phosphorylate a specific set of proteins to signal an extensive cellular DNA-damage response. The conditional deletion of POT1 from mouse and human cells compromises telomere integrity and induces DNA-damage response (Denchi and de Lange, 2007; Guo et al., 2007; O’Sullivan and Karlseder, 2010). Mechanistically, the binding of POT1 to telomere ssDNA inhibits the localization of replication protein A (RPA), a ssDNA-damage sensor, at telomeres. As such, the loss of POT1 protein results in the illicit accumulation of RPA at the telomere, which persistently activates ATR signaling and activation of DNA-damage checkpoints to ultimately result in cellcycle arrest, senescence, or apoptosis (Flynn et al., 2012; O’Sullivan and Karlseder, 2010).
Having determined that 5-FdUTP is efficiently incorporated by telomerase into telomere ssDNA, 5-FdU incorporation impairs POT1-TPP1 binding to telomere ssDNA and that 5-FdU treatment-induced apoptosis is enhanced with active telomerase, we next asked whether 5-FdU treatment impaired telomere protection and integrity in live cells. Independent of its interactions with telomere ssDNA, POT1 localizes to telomeres through protein-protein interactions orchestrated with other shelterin proteins (de Lange, 2005). These protein-protein interactions are sufficient for recruitment of POT1 to the telomere, even when its DNA-binding domain is ablated (Loayza and De Lange, 2003). Therefore, to determine whether 5-FdU treatment disrupts POT1 binding in cells, we monitored the accumulation of RPA at telomeres by combining immunofluorescence with fluorescent in situ hybridization (IF-FISH) to visualize 5-FdU-induced telomeric DNA damage. Whereas no RPA foci were detected in untreated cells (Figure 5A), IF-FISH studies revealed that treatment of cells with 5-FdU resulted in multiple RPA foci formation, with many of those foci localizing at telomeres (Figure 5).
Figure 5. 5-FdU Treatment Activates the Translocation of RPA to Telomeres.
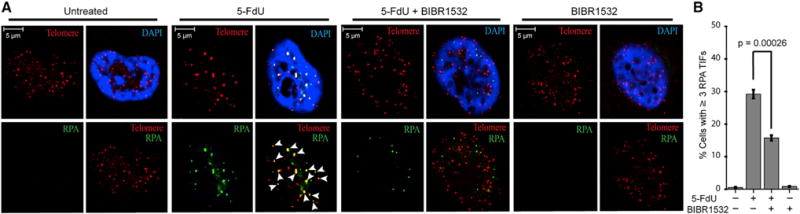
(A) HCT 116 cells treated with 3 μM 5-FdU results in RPA foci accumulating at the telomeres. The pretreatment of cells with 10 μM BIBR1532 significantly reduces the number of RPA foci accumulating at telomeres after 5-FdU treatment (p = 0.00026). RPA foci are indicated in green, and telomeres are indicated in red. The nuclei were counterstained with DAPI (blue).
(B) The percentage of cells with more than three RPA telomere-induced foci was quantitated for each experimental condition. Bars represent the mean ± SEM (n = 3).
BIBR1532 (2-[(E)-3-naphtalen-2-yl-but-2-enoylamino]-benzoic acid) is a potent and selective non-nucleosidic, non-competitive, small-molecule inhibitor of telomerase activity. We used this drug to determine whether telomerase inhibition via co-treatment with BIBR1532 would alter the effects of 5-FdU-induced telomere DNA damage in telomerase-positive cancer cells. While treatment of BIBR1532 alone had no effect on the formation of RPA foci, BIBR1532 co-treatment significantly decreased 5-FdU-induced co-localization of RPA foci with telomeres (Figure 5). These data reveal that administration of 5-FdU results in aberrant RPA accumulation at the telomere in a manner that is dependent on telomerase activity.
Activation of RPA, Chk1, and p53 by 5-FdU-Induced Telomeric DNA Damage
The RPA-mediated recruitment of ATR is characterized by phosphorylation of serine 33 (S33) of the RPA32 subunit (Cai et al., 2009). Additionally, the Chk1 protein functions as the key downstream effector kinase of the ssDNA-damage response and is selectively phosphorylated and activated by ATR (Cai et al., 2009). Finally, ATR activation leads to downstream signaling of effector molecules, including the p53 tumor suppressor (Tibbetts et al., 1999). Consistent with this DNA-damage signaling pathway, 5-FdU treatment of HCT 116 cells resulted in phosphorylation of RPA32 (S33) and Chk1 (S317), as well as p53 activation (Figure 6). Moreover, siRNA-mediated silencing of TERT expression significantly reduced 5-FdU-induced signaling for all of the aforementioned events to approximately half of that observed with active telomerase. These findings indicate that RPA, Chk1, and p53 are all involved in 5-FdU-induced telomeric DNA-damage response signaling.
Figure 6. siRNA-Mediated Silencing of TERT Expression Decreases 5-FdU-Induced Activation of ATR and Signaling of Downstream Targets.

(A–D) The treatment of HCT 116 with 5-FdU results in the activation of p53 (A and B) and phosphorylation of Chk1 (Ser317) (A and C) and RPA32 (S33) (A and D). Expression levels were quantified using the ratio of phospho-RPA (S33), phospho-Chk1 (Ser317), and p53 to actin following drug treatments. Bars represent the mean ± SEM (n = 3). p values were calculated using Student’s t test.
DISCUSSION
While undetectable in most somatic tissues, telomerase is reactivated in ~90% of human cancers (Kim et al., 1994). As such, direct telomerase inhibitors have been extensively explored for treating a diverse set of cancers, including those of the breast, prostate, pancreas, lung, and blood (Mocellin et al., 2013). Unfortunately, the primary shortcoming associated with this methodology reflects the inherent biology of abnormally lengthened telomeres, which requires malignant cells to undergo multiple rounds of cell division before telomere attrition results in replicative senescence. This delay allows cancer cells to develop other mechanisms such as ALT to elongate telomeres and overcome the effects of telomere shortening caused by telomerase inhibition (Hu et al., 2016; Queisser et al., 2013). Our data support an alternate strategy of using telomerase activity in cancer cells as a “Trojan horse” to selectively deliver toxic, non-native nucleotides to induce telomere dysfunction and more immediate cell death in cancer cells. Our initial studies have identified the fluorinated pyrimidine analog, 5-FdUTP, as an effective substrate for telomerase-mediated extension of ssDNA. Intriguingly, our data demonstrate that 5-FdU treatment induces substantial cell death within 3 days and in a telomerase-dependent manner. Together, these findings suggest that telomerase mediates the misincorporation of 5-FdU into telomeres and leads to the rapid killing of human cancer cells at clinically feasible concentrations of the drug.
Nucleoside analogs constitute a large class of antimetabolite agents used for treating viral infections and many cancers, including acute myeloid leukemia and solid tumors (Galmarini et al., 2001). One advantage of using nucleoside analogs is the ability of these small-molecule agents to be actively transported into cancer cells via equilibrative and concentrative transporters that are upregulated, due to a higher demand on nutrient uptake, in rapidly dividing cancer cells. 5-FdU is a Food and Drug Administration (FDA)-approved nucleoside analog that has been used primarily for the treatment of metastatic carcinomas of the colon and liver. The mechanism of action of 5-FdU has traditionally been credited to being similar to that of the more studied antimetabolite, 5-fluorouracil (5-FU) (Longley et al., 2003). The cytotoxicity of 5-FU has been attributed to its conversion to 5-fluorodeoxyuridine monophosphate, which blocks thymidylate synthase to deplete cellular nucleotide pools of deoxythymidine, thereby impairing the ability of cells to properly replicate DNA (Longley et al., 2003). However, previous studies have recently expanded the mechanism of action and cytotoxicity of 5-FU to include its incorporation into RNA products (Silverstein et al., 2011). Subsequent studies have similarly shown that 5-FdU is phosphorylated to serve as a direct substrate for DNA misincorporation, thereby exacerbating its cytotoxic effects (Yan et al., 2016). Our data support this latter role of 5-FdU in mediating carcinoma cytotoxicity, as we demonstrate that the low-fidelity telomerase reverse-transcriptase readily misincorporates non-native nucleotide analogs, including 5-FdU, into its telomere DNA products. Moreover, our data indicate that the cytotoxicity of 5-FdU is largely telomerase dependent and that its incorporation into telomere DNA results in a rapid and overwhelming DNA-damage response that is followed by cell death.
We show that the knockdown of TERT using siRNA or inhibition of telomerase activity with BIBR1532 significantly reduces the cytotoxicity of 5-FdU. In these cases, the remaining cytotoxic effects are likely due to inhibition of thymidylate synthase and/or misincorporation of 5-FdU or deoxyuridine (a by-product of thymidylate synthase inhibition) into genomic DNA. Indeed, both uracil and 5-FU moieties are detected in genomic DNA after treating cells with 5-FdU (Yan et al., 2016). These dual effects are likely recapitulated at the telomere upon treatment of 5-FdU. Specifically, 5-FdUTP gets incorporated directly by telomerase, while thymidylate synthase is concomitantly inhibited to produce higher levels of deoxyuridine. We determined that dUTP functions as an effective substrate for telomerase-mediated extension of telomere ssDNA, albeit with less efficiency than that of dTTP or 5-FdUTP (Figure S2). Also, similar to our results showing that incorporation of 5-FdU into telomere DNA impairs binding of the POT1-TPP1 heterodimer, prior studies have revealed that replacement of a single deoxythymidine with deoxyadenosine or a ribouridine in a telomeric sequence significantly reduces POT- and POT1-TPP1 affinity for the DNA template (Lei et al., 2004; Nandakumar et al., 2010). Therefore, while our data reveal a clear dependence on telomerase for the killing effects of 5-FdU, a more thorough investigation is required to indisputably attribute the adverse consequences to misincorporation of 5-FdU or deoxyuridine into telomere ssDNA.
Whereas the substitution of the second thymidine with 5-FdU in a dodecamer ssDNA sequence has the most pronounced effects on POT1-TPP1 binding, our data reveal that the incorporation of multiple 5-FdU nucleotides into telomere DNA exhibits synergistic effects that further impair POT1-TPP1 binding to a ssDNA substrate. When considering that multiple POT1-TPP1 proteins coat physiological lengths of telomere ssDNA in a semi-cooperative manner (Taylor et al., 2011), it is likely that the disruption of a single protein-binding event exhibits even greater effects for POT1-TPP1 to properly coat and protect telomere ssDNA in the cell.
In addition to evidence showing that 5-FdU impairs binding of POT1-TPP1 to a DNA substrate, our cellular assays support a similar scenario in response to treatment. Specifically, the number of RPA foci that colocalize at telomeres in response to 5-FdU treatment increases in a manner that is dependent on telomerase activity. The concentration of cellular RPA is estimated to be nearly 3 million-fold higher than that of POT1 and TPP1 proteins (Flynn et al., 2012). Despite this extreme difference in cellular protein concentrations, POT1-TPP1 outcompetes RPA for binding of DNA exclusively with telomere sequence (Baumann and Price, 2010). Unlike POT1-TPP1, which shows an extreme preference for telomere ssDNA sequence, RPA recognizes ssDNA indiscriminately and with little or no sequence specificity (Dickey et al., 2013). Our data strongly suggest that telomerase-mediated incorporation of 5-FdU into the telomere ssDNA of cancer cells compromises POT1-TPP1 binding, so that it is outcompeted by the much more abundant and less selective RPA protein. The illicit binding of RPA at telomeres then signals a DNA-damage response cascade that is indicative of the ssDNA repair pathway activated by ATR kinase and is characterized by Chk1 phosphorylation and p53 activation to eventually culminate in apoptotic cell death (Figure 7).
Figure 7. Proposed Model of 5-FdU-Induced Telomere Dysfunction.
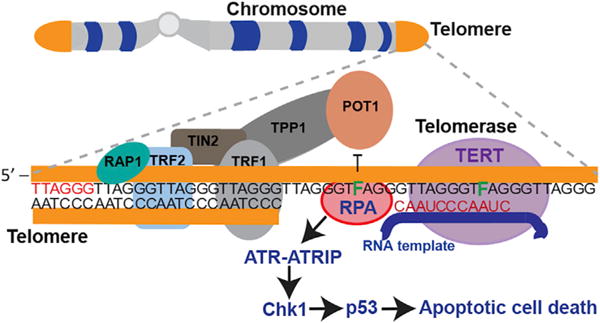
After administration, 5-FdU is intracellularly phosphorylated to 5-FdUTP to serve as a viable substrate for telomerase incorporation into telomere DNA. Once incorporated, 5-FdUTP impairs binding of POT1 protein, which results in the activation and translocation of RPA to the deprotected telomeric ssDNA. RPA-coated, single-stranded regions of telomere ssDNA activates the ATR DNA-damage response at sites of telomeric DNA damage, as evidenced by Chk1 phosphorylation, p53 activation, and apoptotic cell death.
Collectively, our data provide the potential for developing an alternative chemotherapeutic strategy that could improve or complement existing therapies. For example, our findings expand on the mechanisms of action of the anti-cancer drug 5-FdU to include its specific incorporation by telomerase into telomeres in human cancers. More specifically, our data demonstrate that pyrimidines with modifications at the 5-position of the nucleobase can be efficiently incorporated by telomerase into telomeres, whereas modifications at other positions of the thymidine nucleobase tend to limit telomerase-mediated incorporation. Collectively, these findings support a notion of developing additional non-native nucleotides that are selectively incorporated by telomerase over replicative and repair DNA polymerases as an effective strategy for targeting telomerase in cancer cells.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK293T cells; human lung fibroblast WI-38 (VA13) cells; and human cancer cell lines, including human colorectal cancer HCT 116, pancreatic cancer MIA-PaCa2, lung adenocarcinoma A549, and osteosarcoma U2OS cells, were purchased from the American Type Culture Collection (ATCC, Manassas. VA). The cells were maintained in DMEM (Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Norcross, GA, USA), at 37°C in an atmosphere of 5% carbon dioxide.
POT1-TPP1 Expression and Purification
Human full-length POT1 and TPP1 were co-expressed in Spodoptera frugiperda 9 (Sf9) insect cells, using a recombinant baculovirus expression system as previously described (Mullins et al., 2016). Briefly, Sf9 cells expressing full-length POT1 and N-terminal 6 His-tagged TPP1 (89-334) were lysed in a lysis buffer containing 25 mM HEPES (pH 8.0), 300 mM NaCl, 5 mM DTT, 5 mM benzamidine, 1 mM PMSF, and 1 complete ULTRA protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN, USA). The cell lysates were then sonicated and incubated with 2 U/mL of RQ1 DNase (Promega, Madison, WI, USA) for 30 min before pelleting cellular debris through ultracentrifugation at 36,000 rpm for 45 min at 4°C. Following ultracentrifugation, supernatant was applied to a gravity filtration column with buffer-washed nickel-nitrilotriacetic acid (Ni-NTA) beads (QIAGEN). Bead binding was performed at 4°C and then rinsed with a protein buffer containing 25 mM HEPES (pH 8.0) and 300 mM NaCl. The protein-bound beads were subsequently washed with a protein buffer containing 10 mM imidazole before the proteins were eluted with a protein buffer containing 100 mM imidazole. After the affinity purification, POT1-TPP1 was then purified with an AKTA high-performance liquid chromatography (HPLC) system and size-exclusion chromatography using a Superdex 200 HiLoad 16/60 chromatography column (GE Healthcare). Protein fractions were pooled, concentrated with a Millipore Amicon Ultra 10K centrifugal column, and flash-frozen in 20-μL aliquots to be stored at −80°C.
Electrophoretic Mobility Shift Assays
The oligonucleotides with the replacement of each thymidine or all four thymidines in the telomeric oligonucleotide (GGTTAGGGTTAG) for 5-FdUTP were obtained from Trilink Biotechnologies (San Diego, CA, USA). Electrophoretic mobility shift assays (EMSAs) were performed to quantitatively compare the ability of POT1-TPP1 to bind hT12 and 5FdU-modified analogs. All ssDNA substrates were 5′-radiolabeled using ATP[γ-32P] (PerkinElmer; 6000 Ci/mmol; 10 mCi/mL) and T4 Polynucleotide Kinase (New England BioLabs). Protein-DNA binding reactions were performed in buffer containing 60 mM HEPES (pH 8.0), 75 mM NaCl, 5 mM DTT, 5 μg/mL BSA, 1.2 μg/mL tRNA, and 6.25% glycerol. Reactions were performed using 50 pM 32P-labeled DNA and variable concentrations of recombinant POT1-TPP1 protein ranging from 0 to 500 nM. Binding reactions were incubated for 15 min at room temperature before 8 μL of the reaction was loaded onto a 10% Tris-borate non-denaturing gel (Invitrogen). Gels were run at 120 V for 70 min, dried, and scanned using a Typhoon FLA 9500 biomolecular imager (GE Healthcare), and densitometry was performed using ImageQuant TL 1D v8.1 software (GE Healthcare). The Hill equation was used to fit and obtain an equilibrium dissociation constant (KD) for each oligo. Fitted curves were calculated for each trial, and the reported values represent the average, with calculated SEs, derived from three independent experiments for every condition.
Direct Telomerase Incorporation Assay
A telomerase extension assay was performed to identify non-native nucleotide analogs that can be incorporated into the telomeric DNA, as described previously (Mullins et al., 2016). Briefly, human (h)TERT and hTR were co-transfected into HEK293T cells. 48 hr after the transfection, 2 μL of the 293T cell lysate overexpressing hTERT and hTR was then incubated with a 16-μL reaction mixture containing 35 mM Tris-HCl (pH 8.0), 0.7 mM MgCl2, 1.8 mM β-mercaptoethanol, 0.7 mM spermidine, 35 mM NaCl, 100 μM dTTP, 100 μM deoxy-ATP (dATP), 2.9 μM deoxy-GTP (dGTP), 2 μL [a-32P]-dGTP (10 μCi/μL, 3,000 Ci/mmol; PerkinElmer), and 1 μM 18-nt substrate d(GGGTTA)3 primer. The telomerase reaction was carried out for 30 min at 30°C using non-native nucleotide analogs in place of the corresponding native dTTP pools. Reactions were quenched by adding 100 μL of a quenching buffer containing 3.6 M NH4OAc, 300 μM glycogen, and 400 μM EDTA. A 5′-32P-labeled hT18 primer (GGGTTA)3 was used as a loading control. The resulting ssDNA products were ethanol-precipitated and then analyzed on a 12% polyacrylamide/7 M urea/1X Tris-borate-EDTA (TBE) denaturing gel. The gels were dried and exposed overnight to phosphorimager plates, which were imaged on a Typhoon FLA 9500 biomolecular imager (GE Healthcare), and densitometry was performed using ImageQuant TL 1D v8.1 software to quantitate the telomerase assay products.
Quantification of in vitro telomerase assay products was performed as described previously (Mullins et al., 2016). Briefly, relative intensities for each hexamer repeat were calculated and normalized against the loading control for each lane. Total activity was presented as total lane counts by summing the relative intensity of all normalized bands within a lane, after the correction of incorporated Gs. Repeat addition processivity was calculated by first correcting for the number of radiolabeled Gs incorporated within each hexamer repeat and calculating the fraction left behind by dividing the sum of intensities for each round of extension (1 — n) by the entire sum of intensities for the total lane count. The ln(1 — fraction left behind) was plotted against the repeat number of telomerase extension, and the slope was fitted to the linear portion of those data, which was represented as repeat round numbers 5–20 in the telomerase assay. Repeat addition processivity was defined as −0.693 per slope.
Drug Treatments
Cells were seeded ~17–18 hr prior to drug treatments and then exposed to varying concentrations of non-native nucleoside analogs as indicated or treated with 3 μM 5-FdU (Sigma-Aldrich, St. Louis, MO, USA) for 24 hr, after which cells were incubated in drug-free medium for up to 3 days. To assess the effects of inhibition of telomerase on 5-FdU-induced cell death, cells were pre-treated with 10 μM BIBR1532 (Selleckchem, Houston, TX, USA) for 4 hr and then co-treated with 3 μM 5-FdU and 10 μM BIBR1532 for an additional 24 hr, followed by incubation in drug-free medium for up to 3 days.
Stable Expression of hTERT and hTR in U2OS Cells
The pBABE-puro hTERT+hTR plasmid used for the coexpression of untagged human hTERT and hTR was obtained from Addgene (Cambridge, MA, USA) (plasmid #27665) (Wong and Collins, 2006). Stable expression of hTERT and hTR into U2OS cells was performed using retroviral infection. Briefly, GP2-293 cells were seeded at 1.3 × 106 cells per dish, in 60-mm dishes, in DMEM containing 10% FBS. 18–20 hr later, the cells were then transfected with 8 μg pBABE-puro hTERT+hTR and pVSV-G plasmids at a ratio of 3:1 using polyethylenimine (PEI) (Polysciences, Warrington, PA, USA). At 48, 72, and 82 hr after the transfection, the virus-enriched medium was collected and centrifuged at 800 × g for 5 min to infect U2OS cells grown in 60-mm dishes. Three rounds of retrovirus infections were performed on 2 sequential days in the presence of 4.0 μg/mL hexadimethrine bromide (Sigma). 24 hr after the third infection, the cells were replated into 100-mm dishes and allowed to grow for additional 4 days before they were used for subsequent experiments.
Telomeric Repeat Amplification Protocol Assay
For quantitative analysis of telomerase activity in U2OS cells stably expressing TERT and TR, the telomeric repeat amplification protocol (TRAP) assay was used as described previously (Herbert et al., 1999). Briefly, cells were harvested followed by resuspension to 1,000 cells per microliter in ice-cold NP-40 lysis buffer (10 mM Tris-HCl [pH 8.0], 1 mM MgCl2, 1 mM EDTA, 1% NP-40, 0.25 mM sodium deoxycholate, 10% glycerol, 150 mM NaCl, 5 mM β-mercaptoethanol, 0.1 mM AEBSF [4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride]). Following a 30-min incubation on ice, cell lysates were spun at 16,000 × g for 20 min at 4°C. The total protein concentration of cell lysates was determined using the Pierce Coomassie Protein Assay Kit (Thermo Fisher Scientific). The TRAP assay was performed as follows: 1 μg of protein lysate was assayed in a 25-μL reaction mixture supplemented with 1× iQTM SYBR Green Supermix (Bio-Rad), 1 mM EGTA, 100 ng of ACX primer (5′-GCGCGGCTTACCCTTACCCTTACCCTAACC-3′), and 100 ng of TS primer (5′-AATCCGTCGAGCAGAGTT-3′). Reactions were prepared in triplicate in a 96-well PCR plate and then placed at 30°C, protected from light for 30 min to allow for telomerase-mediated extension. The reaction mixture was heated to 95°C for 10 min and then amplified for 40 cycles at 95°C for 15 s and 60°C for 60 s. Using the included telomerase-positive sample dilution series, a standard curve of Ct value versus log10 protein concentration was generated and used to determine the relative telomerase activity (RTA) of each test sample as well as telomerase-positive sample. Values are reported as percentages of RTA of telomerase-positive sample ± SD.
siRNA-Mediated Silencing of TERT Expression
The siGENOME Non-Targeting siRNA Pool (catalog #D-001206-14-05) and the SMARTpool TERT siRNA oligos (catalog #M-003547-02-0005) were chemically synthesized by Dharmacon (Lafayette, CO, USA). siRNAs were introduced into HCT 116 cells using Lipofectamine RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol. Briefly, 1.8 × 105 cells per well were plated and cultured in 12-well plates for 19 hr. Then 6 μL Lipofectamine RNAiMAX or 6 μL 20 μsiRNAs was diluted into 100 μL Opti-MEM and incubated for 5 min at room temperature. The diluted siRNA duplex and Lipofectamine RNAiMAX were then mixed and incubated for 20 min at room temperature. Following the formation of the siRNA-RNAiMAX complex, the mixture was then added to the cells containing 800 μL Opti-MEM, and the cells were cultured under normal conditions. To supplement the culture medium with 10% FBS, 110 μL FBS was added to the cells 5 hr post-transfection. 24 hr after the transfection, the cells were split from 1 well of 12-well plates into 2×60-mm dishes. The siRNA-transfected cells were allowed to adhere for 18 hr and then were treated with 5-FdU as described earlier. Although studies exist that report on the localization and immunoprecipitation of TERT using commercially available antibodies (Abreu et al., 2010), the low abundance of the protein combined with the lack of a good antibody have frustrated efforts to quantitatively and reliably assess levels of TERT using western blotting procedures (Xi et al., 2015). As such, real-time qPCRs were performed to determine TERT knockdown efficiency.
Real-Time qPCR
48 hr after TERT siRNA transfection as detailed earlier, total RNA was isolated from the cells using the TRIzol reagent (Invitrogen) and reverse transcription of RNA (1.5 μg) was conducted to synthesize first-strand cDNA with a high-capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s protocol. After first-strand cDNA synthesis, real-time qPCR was then performed using a FastStart Universal SYBR Green Master (ROX) mixture (Roche) on a StepOnePlus Real-Time PCR System (Applied Biosystems) with the following amplification condition: an initial 10-min incubation at 95°C followed by 40 cycles of denaturation for 15 s at 95°C, annealing for 30 s at 60°C, and extension for 30 s at 72°C. The primers used for the PCR amplifications are as follows: TERT gene (forward, 5′-CGGAAGAGTGTCTGGAGCAA-3′; reverse, 5′-GGATGAAGCGGAGTCTGGA-3′) and GAPDH gene (forward, 5′-GCTCAGACACCATGGGGAAGGT-3′; reverse, 5′-GTGGTGCAGGAGGCATTGCTGA-3′). Relative quantitation of TERT mRNAs was performed using the comparative ΔΔCt method, and the expression levels of TERT mRNA are normalized to those of the endogenous control GAPDH.
MTT Assay
Cellular death in the presence or absence of experimental agents was determined using the MTT (Sigma-Aldrich, St. Louis, MO, USA) assay. Briefly, rapidly growing cells were seeded into 96-well plates at 2,500 cells per well using a multichannel pipette. After cells were treated with the drugs as described earlier, 20 μL MTT prepared at 5 mg/mL in PBS was added to the cells with 100 μL complete culture medium containing 10% FBS. After a 3-hr incubation at 37°C, the MTT-containing medium was removed from each well, and 150 μL MTT solvent (4 mM HCl, 0.1%. NP-40 in isopropanol) was added to each well to solubilize the MTT-formazan product. After thorough mixing with a multichannel pipette, absorbance at 570 nm was measured with a VersaMax Tunable Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
Clonogenic Survival Assay
Cell survival following treatment with 5-FdU was determined with a standard colony-forming assay. Briefly, U2OS cells stably expressing hTERT + hTR were plated onto 60-mm dishes and allowed to adhere for 17 hr. The cells were then treated with 5-FdU (0–7.5 μM) for 24 hr and subsequently incubated in fresh medium for 14 days for colony formation. The colonies were stained with 0.5% crystal violet in methanol/acetic acid (3:1), and those with >50 cells were counted.
IF-FISH
Cells were grown on coverslips coated with poly-L-lysine (Sigma-Aldrich). After the drug treatments, conducted as described earlier, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and then stained with mouse monoclonal antibodies against RPA (Calbiochem, Billerica, MA, USA) and fluorescent-tagged secondary antibodies for 16 hr at 4°C. After the antibody staining, the cells were fixed with 4% paraformaldehyde; washed with PBS; dehydrated in an ethanol series of 70%, 95%, and 100%; and air dried. The dehydrated cells were then hybridized with a Cy5-conjugated telomere probe TelC-Cy5 (#F1003; PNA Bio, Thousand Oaks, CA, USA) in a buffer containing 200 nM TelC-Cy5, 70% formamide, 1% blocking reagent (Roche, Indianapolis, IN, USA), and 10 mM Tris (pH 7.2). Confocal images were acquired with a laser-scanning microscope (LSM 510 Meta; Carl Zeiss, Peabody, MA, USA). Percentages of cells with ≥3 DNA-damage-induced RPA foci that colocalize at the telomere were quantified. Each experiment was performed in triplicate, with each dataset consisting of at least 100 cells.
Annexin V and Propidium Iodide Staining
Annexin V and propidium iodide staining (eBioscience, San Diego, CA, USA) was performed to measure apoptosis, according to the manufacturer’s protocol. Briefly, 48 hr after TERT siRNA transfection, HCT 116 cells were treated with 3 μM 5-FdU for 24 hr and subsequently incubated in drug-free medium for 3 days. The cells were then harvested by trypsinization along with initial culture medium to ensure inclusion of detached cells. After washing once with PBS and 1× binding buffer, the cells were resuspended in 100 μL 1×binding buffer at 4 × 106 cells per milliliter. 5 μL APC-conjugated Annexin V (eBioscience) was added to the cell suspension and incubated at room temperature for 15 min. Following the annexin V staining, the cells were washed once with 1× binding buffer and resuspended in 200 μL 1× binding buffer. 5 μL Propidium Iodide Staining Solution (eBioscience) was then added to the cell suspension, and the samples were stored at 4°C in the dark and analyzed by flow cytometry (BD LSR II; BD Biosciences, San Jose, CA, USA) within 4 hr.
Western Blotting
SDS-PAGE and immunoblot analyses were carried out using standard procedures. Briefly, cells were harvested, lysed in RIPA buffer and Laemmli Sample Buffer, and then heated at 95°C for 10 min. Equal amounts of protein from each sample were electrophoresed through SDS-polyacrylamide and transferred to nitrocellulose membranes. Immunoreactive proteins were detected using SuperSignal West Pico Substrate (Thermo Fisher Scientific, Waltham, MA, USA). The primary antibodies used for immunoblot analyses included rabbit polyclonal antibodies against phosphor-RPA32 (S33) (Bethyl Laboratories, Mongomery, TX,USA), phospho-Ch1(Ser317), phospho-Chk2 (Thr68), p53, cleaved PARP (Asp214) (Cell Signaling Technology, Danvers, MA, USA), and mouse monoclonal antibodies against β-actin and tubulin (Sigma-Aldrich, St. Louis, MO, USA).
Supplementary Material
Highlights.
Several nucleotide analogs, including 5-FdU, are effective substrates for telomerase
5-FdU induces apoptotic cell death in a telomerase-dependent manner
5-FdUTP reduces the binding affinity of POT1-TPP1 to telomere DNA
5-FdU-induced telomeric DNA damage results in activation of RPA, Chk1, and p53
Acknowledgments
The authors would like to thank all members of the Taylor lab for helpful comments and suggestions related to the investigation. We additionally thank E. Jankowsky and W. Schiemann for comments on the manuscript. This work was supported by the NIH (DP2 CA186571 to D.J.T.) and the American Cancer Society (RSG-13-211-01-DMC to D.J.T.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes two figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.020.
AUTHOR CONTRIBUTIONS
X.Z., J.Z., A.J.B., and D.J.T. planned the experiments. M.X. performed the EMSA experiments, D.B. and W.H.-S. performed the telomerase extension assays, and T.L.W. performed the TRAP assay. X.Z. performed the remaining experiments. X.Z. and D.J.T. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, Lingner J, Terns MP. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol. 2010;30:2971–2982. doi: 10.1128/MCB.00240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- Baumann P, Price C. Pot1 and telomere maintenance. FEBS Lett. 2010;584:3779–3784. doi: 10.1016/j.febslet.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH. Telomeres: no end in sight. Cell. 1994;77:621–623. doi: 10.1016/0092-8674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Cai Z, Chehab NH, Pavletich NP. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol Cell. 2009;35:818–829. doi: 10.1016/j.molcel.2009.09.007. [DOI] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- de Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- Dickey TH, Altschuler SE, Wuttke DS. Single-stranded DNA-binding proteins: multiple domains for multiple functions. Structure. 2013;21:1074–1084. doi: 10.1016/j.str.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Chang S, Zou L. RPA and POT1: friends or foes at telomeres? Cell Cycle. 2012;11:652–657. doi: 10.4161/cc.11.4.19061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia. 2001;15:875–890. doi: 10.1038/sj.leu.2402114. [DOI] [PubMed] [Google Scholar]
- Greider CW. Telomerase is processive. Mol Cell Biol. 1991;11:4572–4580. doi: 10.1128/mcb.11.9.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Deng Y, Lin Y, Cosme-Blanco W, Chan S, He H, Yuan G, Brown EJ, Chang S. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26:4709–4719. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L. The illusion of cell immortality. Br J Cancer. 2000;83:841–846. doi: 10.1054/bjoc.2000.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert B, Pitts AE, Baker SI, Hamilton SE, Wright WE, Shay JW, Corey DR. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc Natl Acad Sci USA. 1999;96:14276–14281. doi: 10.1073/pnas.96.25.14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Shi G, Zhang L, Li F, Jiang Y, Jiang S, Ma W, Zhao Y, Songyang Z, Huang J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci Rep. 2016;6:32280. doi: 10.1038/srep32280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Lai CK, Mitchell JR, Collins K. RNA binding domain of telomerase reverse transcriptase. Mol Cell Biol. 2001;21:990–1000. doi: 10.1128/MCB.21.4.990-1000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004;11:1223–1229. doi: 10.1038/nsmb867. [DOI] [PubMed] [Google Scholar]
- Ligasová A, Strunin D, Friedecký D, Adam T, Koberna K. A fatal combination: a thymidylate synthase inhibitor with DNA damaging activity. PLoS ONE. 2015;10:e0117459. doi: 10.1371/journal.pone.0117459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza D, De Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;423:1013–1018. doi: 10.1038/nature01688. [DOI] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Mocellin S, Pooley KA, Nitti D. Telomerase and the search for the end of cancer. Trends Mol Med. 2013;19:125–133. doi: 10.1016/j.molmed.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Mullins MR, Rajavel M, Hernandez-Sanchez W, de la Fuente M, Biendarra SM, Harris ME, Taylor DJ. POT1-TPP1 binding and unfolding of telomere DNA discriminates against structural polymorphism. J Mol Biol. 2016;428:2695–2708. doi: 10.1016/j.jmb.2016.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar J, Cech TR. Finding the end: recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol. 2013;14:69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar J, Podell ER, Cech TR. How telomeric protein POT1 avoids RNA to achieve specificity for single-stranded DNA. Proc Natl Acad Sci USA. 2010;107:651–656. doi: 10.1073/pnas.0911099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11:171–181. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- Queisser A, Heeg S, Thaler M, von Werder A, Opitz OG. Inhibition of telomerase induces alternative lengthening of telomeres during human esophageal carcinogenesis. Cancer Genet. 2013;206:374–386. doi: 10.1016/j.cancergen.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Silverstein RA, González de Valdivia E, Visa N. The incorporation of 5-fluorouracil into RNA affects the ribonucleolytic activity of the exosome subunit Rrp6. Mol Cancer Res. 2011;9:332–340. doi: 10.1158/1541-7786.MCR-10-0084. [DOI] [PubMed] [Google Scholar]
- Tárkányi I, Aradi J. Pharmacological intervention strategies for affecting telomerase activity: future prospects to treat cancer and degenerative disease. Biochimie. 2008;90:156–172. doi: 10.1016/j.biochi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Podell ER, Taatjes DJ, Cech TR. Multiple POT1-TPP1 proteins coat and compact long telomeric single-stranded DNA. J Mol Biol. 2011;410:10–17. doi: 10.1016/j.jmb.2011.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilman G, Loriot A, Van Beneden A, Arnoult N, Londoño-Vallejo JA, De Smet C, Decottignies A. Subtelomeric DNA hypomethylation is not required for telomeric sister chromatid exchanges in ALT cells. Oncogene. 2009;28:1682–1693. doi: 10.1038/onc.2009.23. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, Lei M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–510. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- Wong JM, Collins K. Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita. Genes Dev. 2006;20:2848–2858. doi: 10.1101/gad.1476206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RA, Upton HE, Vogan JM, Collins K. Telomerase mechanism of telomere synthesis. Annu Rev Biochem. 2017;86:439–460. doi: 10.1146/annurev-biochem-061516-045019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi L, Schmidt JC, Zaug AJ, Ascarrunz DR, Cech TR. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol. 2015;16:231. doi: 10.1186/s13059-015-0791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, O’Connor MS, Songyang Z. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature. 2007;445:559–562. doi: 10.1038/nature05469. [DOI] [PubMed] [Google Scholar]
- Xu Y, Goldkorn A. Telomere and telomerase therapeutics in cancer. Genes (Basel) 2016;7:E22. doi: 10.3390/genes7060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Han X, Qing Y, Condie AG, Gorityala S, Yang S, Xu Y, Zhang Y, Gerson SL. Inhibition of uracil DNA glycosylase sensitizes cancer cells to 5-fluorodeoxyuridine through replication fork collapse-induced DNA damage. Oncotarget. 2016;7:59299–59313. doi: 10.18632/oncotarget.11151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.