

 The 4-acylaminopyrazolo[3,4-d]pyrimidine 9a was identified as a highly selective σ1R antagonist, showing as well substantial antinociceptive properties.
The 4-acylaminopyrazolo[3,4-d]pyrimidine 9a was identified as a highly selective σ1R antagonist, showing as well substantial antinociceptive properties.
Abstract
The synthesis of a new series of 4-acylaminopyrazolo[3,4-d]pyrimidines active on the sigma-1 receptor (σ1R) is reported. Compounds were efficiently prepared using a two to three step process starting from commercially available 1H-pyrazolo[3,4-d]pyrimidin-4-amine. A SAR study shows that the σ1R requires the presence of relatively highly lipophilic substituents at opposite sides of the central scaffold, while selectivity versus the σ2R can be improved by shortening the distance of the basic nitrogen to it. Compound 9a was among the most active and selective in vitro derivatives and exhibited potent antinociceptive properties in several pain models in mice, indicating its antagonistic behaviour.
Introduction
The σ receptor (σR) was initially recognized as a new addition to the opioid receptor family, but later it has been shown to be a unique molecular chaperone, which modulates the function of different proteins when they become challenged by disease-related stress or mutations.1 The σR presents at least two different subtypes, σ1R and σ2R, which show distinct functions and are related to different potential therapeutic indications. The σ1R was cloned from guinea pig liver in the late nineties2 and this discovery paved the way for the search for selective σ1R ligands. Since the receptor is widely distributed in peripheral organs and in different areas of the central nervous system (CNS), which are involved in memory, emotion, sensory and motor function control, σ1R ligands were mainly studied in the past for treating different CNS-related pathologies such as schizophrenia and depression.3 The σ2R has remained more elusive and is often associated with proliferation, synaptogenesis and cell plasticity. Ligands acting on the σ2R have been proposed as biomarkers for tumour cell proliferation and show pro-apoptotic properties, thus suggesting a potential role in cancer imaging and treatment.4
More recently, studies on the σ1R knock-out (σ1R-KO) mouse and several σ1R ligands have been key to establishing the role of the σ1R in opioid analgesia modulation and in pain control, when central sensitization occurs, as is the case with several neuropathic pain states.5 A growing body of evidence supports the role of the σ1R in pro-nociception, acting through the up-regulation of sodium and calcium channels as well as NMDA receptor function, all of them linked to pain facilitation and amplification of nociceptive messages. Another important factor is the localization of the σ1R in key areas involved in pain control, such as the superficial layers of the dorsal horn, the periaqueductal grey matter, the locus coeruleus and the rostroventral medulla.6
The reduced levels of hyperalgesia observed in several models, both in σ1R-KO mice and in rodents treated with σ1R ligands, have helped to build up substantial evidence of the role of σ1R antagonism in the treatment of pain.6,7 However, the determination of the functionality of σ1R ligands is not obvious, since the protein is mainly located intracellularly and it exerts a modulatory action by regulating the action of different proteins. Probably, this is why some σ1R ligands do not show the classical linear dose–response curves and the assay conditions and readouts used markedly influence the outcome, and some compounds identified as agonists using one readout act as antagonists using a different one. For these reasons, in vivo tests are frequently used to determine the functional nature of σ1R ligands.8
Good antinociception in rodents has been shown by several types of σ1R antagonists, such as those developed by the group of A. Marrazzo around compound (+)-MR200 (ref. 9) and several series of spirocyclic derivatives studied by the group of B. Wünsch.10 Our group has been involved in the search for σ1R antagonists for several years and developed the highly selective σ1R antagonist E-52862 (1, Fig. 1 and Table 1)11 up to phase II clinical trials for the treatment of different pain conditions.12,13
Fig. 1. General structure of pyrazolo[3,4-d]pyrimidines I and reference compound 1.

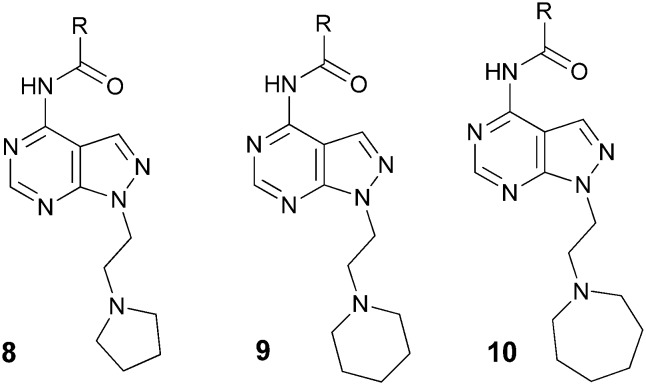
Table 1. Amide derivatives 8–10.
| ||||
| Comp | R | K i σ1 a (h, nM) | HLM c Clint (μl min–1 mg–1 prot.) | clog P d |
| 1 | 17 | |||
| 8a | Cyclopropyl | 32 731 | — | 1.0 |
| 8b | Cyclobutyl | 716 | — | 1.3 |
| 8c | Cyclopentyl | 116 | — | 1.9 |
| 8d | Cyclohexyl | 98 | 13.9 | 2.4 |
| 9a | tert-Butyl | 51 | 2.4 | 2.2 |
| 9b | Adamantyl | 25 | 5.0 | 3.6 |
| 9c | Noradamantyl | 55 | 6.2 | 3.0 |
| 9d | 2,4-Dichlorophenyl | 292 | — | 3.0 |
| 10a | tert-Butyl | 9 | 3.2 | 2.7 |
| 10b | Adamantyl | 9 | 11.7 | 4.2 |
| 10c | Noradamantyl | 15 | 18.0 | 3.6 |
| 10d | Cyclopentyl | 16 | 7.9 | 3.0 |
| 10e | Cyclohexyl | 18 | 51.7 | 3.5 |
| 10f | 4,4-Difluorocyclohexyl | 55 b | 12.2 | 3.9 |
| 10g | Tetrahydropyran-4-yl | 514 | — | 1.1 |
| 10h | 2,2,3,3-Tetramethylcyclopropan-1-yl | 11 b | — | 4.7 |
| 10i | 2,4-Dichlorophenyl | 58 b | — | 3.6 |
| 10j | 4-Fluorophenyl | 156 b | — | 3.1 |
aBinding affinity (Ki) to human σ1R in transfected HEK-293 membranes using [3H]-(+)-pentazocine as radioligand. Each value is the mean of two determinations.
bThe compound also shows binding affinity to the σ2R in guinea pig brain membranes using [3H]-di-o-tolylguanidine as radioligand. Each value is the mean of two determinations. The % inhibition at 1 μM is 81% (10f), 79% (10h), 75% (10i), and 57% (10j).
cIntrinsic clearance in human liver microsomes as a measure of metabolic stability.
dclog P calculated using ChemDraw Ultra 10.0.3.
As a back-up program in the development of 1, we undertook several approaches in order to identify novel scaffolds binding to the σ1R and showing acceptable drug-like properties. A high throughput screening (HTS) campaign over our internal library led to the identification of an interesting new framework,14 while a scaffold hopping strategy toward 1 provided a series of potent triazole derivatives.15 As a different approach, we report here the synthesis and structure–activity relationship (SAR) studies of a new series of pyrazolo[3,4-d]pyrimidines16 (I, Fig. 1), which were designed on the basis of Glennon's σ1R pharmacophore17 and taking into account the information generated in previous SAR studies.
Results and discussion
Chemistry
The synthesis of the final compounds was quite straightforward, since it involved a two to three step process starting from commercially available 1H-pyrazolo[3,4-d]pyrimidin-4-amine 2. As indicated in Scheme 1, the reaction of 2 with an alkylating agent 3 in the presence of a strong base provided the N1-sustituted pyrazoles 4.18 Alternatively, compounds 4 were obtained in two steps by reaction of 2 with a dihalo derivative 5 to afford 6, followed by substitution with an amine 7 under standard conditions.
Scheme 1. Reagents and conditions: (a) NaH, DMF, rt, 16 h; (b) K2CO3, NaI, DMF, rt, 16 h; (c) R3COCl, pyridine, TEA, DMAP, 130 °C, MW; (d) R3NCX, acetonitrile or toluene, 120–150 °C, MW.

Both routes provided the N1-sustituted pyrazoles 4 as major isomers. N2-regioisomers were observed in the crude reaction mixture (1–30% by MS), but were isolated in very low yields or not isolated at all. This result could be attributed to lower solubility. The characterization of regioisomers was effected over the pyrrolidinylethyl derivatives shown in Fig. 2. The minor isomer, 16, which was isolated in 1% yield, showed a NOE effect between the pyrazole proton at position 8 and the methylene protons linked to N9, while the major isomer 4a, isolated in 40% yield, did not show any NOE effect. The identity of 16 was confirmed by the presence of an NH2 signal in the 1H NMR spectrum in CDCl3, thus discarding 17. The amino substituted isomer analogues to 17 were not detected in any case.
Fig. 2. Potential regioisomeric composition in the alkylation of 2.

The last step of the synthesis involved the derivatization of 4 with suitable reagents. Thus, amides 8–12 were obtained with acyl chlorides in pyridine under microwave heating, while reaction with alkyl isocyanates or alkyl isothiocyanates gave ureas 13 and 14 and thioureas 15, respectively.
The compounds were isolated as hydrochloride, maleate or citrate salts, prepared by standard methods from the corresponding bases, which were usually obtained as oils.
SAR
A new series of pyrazolo[3,4-d]pyrimidines I16 was designed taking into account Glennon's σ1R pharmacophore,17 which consists of a positive ionisable group (i.e., a basic amino group) and two opposite hydrophobic regions at a distance of 2.5–3.9 Å and 6–10 Å (Fig. 4A). This elegant and simple model is nowadays still a good reference for designing new σ1R ligands. Only very recently the σ1R structure has been solved by X-ray diffraction,19 and therefore the rational design of σ1R modulators has up to now mostly relied on ligand-based approaches, as recently summarized.20
Fig. 4. (A) Main features of the Laggner σ1R pharmacophore in comparison to the distances described by the Glennon σ1R pharmacophore. (B) 3D superposition of 18 with Laggner σ1R pharmacophoric features. (C) 3D superposition of 9a and 18.

It was hypothesized that the pyrazolo[3,4-d]pyrimidine scaffold could place the primary hydrophobic group and the nitrogen atom in an adequate spatial disposition. Additionally, previous reports on bicyclic frameworks, such as 18 (ref. 21) and 19 (ref. 22) (Fig. 3), suggested that this disposition could be tolerated and that the ideal distance between the nitrogen atom and the central bicycle could be of two to three methylene groups. As shown in Fig. 4B, compound 18 fitted well Glennon's pharmacophore (white lines) and also matched three (HYD1, 2, and 4) out of the four hydrophobic features of the Laggner23 pharmacophore (Fig. 4A), a widely accepted 3D model of σ1R receptor ligands. Compound 9a (Fig. 4C) nicely displayed key features, such as the positive ionisable group and the hydrophobic groups matching HYD1, 2, and 4. However, a potential drawback was the presence of polar atoms in the bicyclic scaffold, which could be detrimental in view of the reported lipophilic nature of the σ1R.8
Fig. 3. Compounds 18 (ref. 21) and 19 (ref. 22).

All the compounds synthesized were evaluated in a primary σ1R binding assay using [3H]-(+)-pentazocine24 as radioligand (Tables 1–3). Because the two σ receptors are pharmacologically differentiated and selectivity among them is desired, the binding affinities to the σ2R were also measured, using [3H]-di-o-tolylguanidine25 as radioligand. Since, in general, the compounds showed low affinity for the σ2R, only the values for the σ1R appear in Tables 1–3, and σ2R binding data for those compounds showing an affinity higher than 50% at 1 μM are indicated as notes.
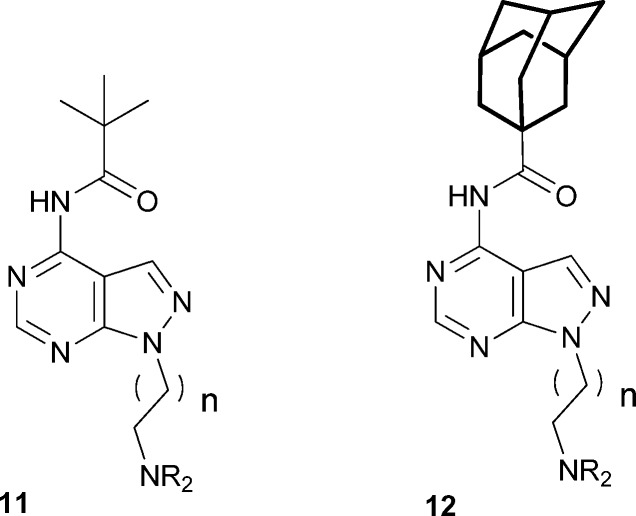
Table 2. Amide derivatives 11–12.
| |||||
| Comp | n | NR2 | K i σ1 a (h, nM) | HLM c Clint (μl min–1 mg–1 prot.) | clog P d |
| 11a | 1 | 4-tert-Butyl-piperidin-1-yl | 12 | 8.6 | 3.9 |
| 11b | 1 | Diisopropylamino | 399 | — | 2.8 |
| 11c | 1 | Morpholin-4-yl | >1000 | — | 1.0 |
| 11d | 1 | Homomorpholin-4-yl | 184 | — | 0.9 |
| 11e | 1 | 4-Methylpiperazin-1-yl | 42 923 | — | 0.1 |
| 11f | 1 | 4-Methylhomopiperazin-1-yl | 813 | — | 0.6 |
| 11g | 2 | Piperidin-1-yl | 45 | 1.1 | 2.5 |
| 11h | 2 | Homopiperidin-1-yl | 33 | 0.2 | 3.1 |
| 11i | 3 | Homopiperidin-1-yl | 62 b | — | 3.2 |
| 11j | 3 | 4-tert-Butyl-piperidin-1-yl | 946 b | — | 4.3 |
| 12a | 1 | Diisopropylamino | 99 | 31.2 | 4.3 |
| 12b | 1 | 4-Isopropylpyperazin-1-yl | 573 | — | 2.3 |
| 12c | 1 | Morpholin-4-yl | 774 | — | 2.4 |
aBinding affinity (Ki) to human σ1R in transfected HEK-293 membranes using [3H]-(+)-pentazocine as radioligand. Each value is the mean of two determinations.
bThe compound also shows binding affinity to the σ2R in guinea pig brain membranes using [3H]-di-o-tolylguanidine as radioligand. Each value is the mean of two determinations. The % inhibition at 1 μM is 89% (11i) and 77% (11j).
cIntrinsic clearance in human liver microsomes as a measure of metabolic stability.
dclog P calculated using ChemDraw Ultra 10.0.3.
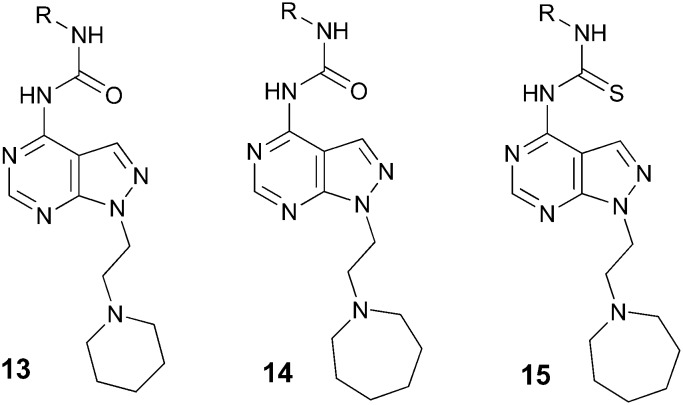
Table 3. Urea and thiourea derivatives 13–15.
| ||||
| Comp | R | K i σ1 a (h, nM) | HLM c Clint (μl min–1 mg–1 prot.) | clog P d |
| 13a | Ethyl | 371 | — | 1.6 |
| 13b | Propyl | 85 | 5.7 | 2.3 |
| 13c | Cyclopentyl | 192 | — | 2.6 |
| 13d | Cyclohexyl | 818 | — | 3.1 |
| 13e | Adamantyl | 181 | — | 3.7 |
| 13f | 2,4-Dichlorophenyl | 141 | — | 3.9 |
| 14a | Ethyl | 79 | 2.8 | 4.2 |
| 14b | Propyl | 24 | 22.2 | 3.1 |
| 14c | Isopropyl | 23 | 31 | 2.5 |
| 14d | Butyl | 29 | 60.3 | 3.2 |
| 14e | Cyclopropyl | 100 | — | 2.2 |
| 14f | Cyclopentyl | 21 | 112.5 | 3.1 |
| 14g | Cyclohexyl | 119 | — | 3.7 |
| 14h | tert-Butyl | 41 | 63.9 | 2.9 |
| 15a | Ethyl | 9 b | — | 2.0 |
| 15b | Propyl | 7 b | — | 2.5 |
| 15c | Butyl | 13 b | — | 3.0 |
| 15d | Cyclopropyl | 22 b | — | 2.0 |
aBinding affinity (Ki) to human σ1R in transfected HEK-293 membranes using [3H]-(+)-pentazocine as radioligand. Each value is the mean of two determinations.
bThe compound also shows binding affinity to the σ2R in guinea pig brain membranes using [3H]-di-o-tolylguanidine as radioligand. Each value is the mean of two determinations. The % inhibition at 1 μM is: 61% (14d), 68% (15a), 82% (15b), 74% (15c), and 75% (15d).
cIntrinsic clearance in human liver microsomes as a measure of metabolic stability.
dclog P calculated using ChemDraw Ultra 10.0.3.
For those compounds showing σ1R Ki values below 50 nM and displaying no binding to the σ2R at 1 μM, clearance in human liver microsomes26 was evaluated to ensure a suitable half-life.
Taking into account the SAR study of compound 1,11 the amines piperidine, azepane and pyrrolidine were chosen as initial surrogates for the secondary hydrophobic group. As indicated in Table 1, several acylated derivatives (8–10) were prepared. The most active compounds exhibited nanomolar affinity for the σ1R. In the pyrrolidinyl series (8) it was shown that a certain degree of lipophilicity in the acyl substituent was needed, since the activity increased in the order cyclopropyl (8a), cyclobutyl (8b), cyclopentyl (8c), and cyclohexyl (8d). Regarding the amine counterpart, activity also improved when the lipophilicity increased, since the azepanyl derivatives 10a–c showed increased affinity in relation to the piperidinylamides 9a–c, while 10d and 10e were more active than their pyrrolidinyl counterparts 8c and 8d. As expected, the ethoxy derivative 20 (Fig. 5) was completely devoid of activity, confirming that the amino group was a requirement for activity. The best activities were obtained with highly lipophilic and bulky acyl substituents, such as tert-butyl (9a, 10a), adamantyl (9b, 10b) and noradamantyl (9c, 10c), but the tert-butyl derivatives were somewhat superior in terms of clearance.
Fig. 5. Compounds 4b and 20versus9a.

The introduction of four methyl groups in the cyclopropyl ring substituent provided the highly active derivative (10h), with less selectivity for the σ2R. The substituted aryl derivatives 9d and 10i showed diminished activity, as did the more polar tetrahydropyranyl derivative 10g, while the fluoro derivative 10j, with intermediate polarity, showed intermediate affinity. Compound 10f, although improving the metabolic stability of 10e, was less stable than 10a.
Two of the best acyl substituents, tert-butyl and adamantyl, were chosen to study the variation of the amine group NR1R2 (Table 2). Introducing a tert-butyl group in position 4 of the piperidine ring (11a) improved the affinity for but impaired microsomal stability vs.9a, probably due to the lipophilicity increase. The acyclic di-isopropylamino derivatives showed reduced affinity both in the tert-butyl (11b) and the adamantyl (12a) series. Although 12a was profiled further, it was discarded because of its low metabolic stability. The 4-morpholinyl (11c and 12c) and homomorpholinyl (11d) derivatives were substantially less active than their piperidinyl counterparts (9a, 9b and 10a, respectively). The methylpiperazinyl derivative 11e displayed submicromolar affinity, which was not sufficiently improved by the homopiperazinyl (11f) and isopropyl (12b) analogues. Overall, these results show that, as in the case of the acyl substituents, introducing heteroatoms in the carbocyclic rings in this part of the molecule is detrimental, probably due to the increase in polarity. In fact, all the compounds with clog P below 1.6 showed Ki values above 1000 nM, indicating that a minimum overall lipophilicity is required by the σ1R.
Elongation of the distance between the basic nitrogen and the pyrazolopyrimidine central scaffold to three (11g, h) or four (11i) methylene groups retained activity, while further elongation (11j) was detrimental. However, selectivity for the σ1R vs. the σ2R decreased in the case of 11i and 11j, a result consistent with that obtained in the SAR study of 1.11
The piperidinyl and azepanyl ethyl derivatives were selected to study the introduction of urea and thiourea groups to replace the acyl moiety (Table 3). The piperidinyl ureas 13 were less active than their azepanyl counterparts 14, which showed interesting potencies but were either less selective (14d) or showed lower metabolic stabilities (14b–h). Regarding the thioureas 15a–d, all of them were not selective for the σ2R.
One of the best compounds, 9a, was profiled further and confirmed to be fairly selective, since in addition to its low affinity for the σ2R (Ki > 10 000 nM), it failed to show any significant affinity (inhibition at 10 μM < 50%) in a panel of 65 receptors.27 Moreover it did not bind significantly to recombinant human cytochrome P450 (rhCYP) isoforms (1A2, 2C9, 2C19, 2D6, and 3A4).28 Furthermore, it had good permeability in Caco-2 cells29 (Papp = 390 nm s–1) and an efflux ratio of 0.9, indicating that it was not a P-gp substrate. Its metabolic stability in rodent liver microsomes was high (mouse Clint = 6.6; rat Clint = 8.9 μl min–1 mg–1 prot.) like in humans (Clint = 2.4 μl min–1 mg–1 prot.). This good in vitro ADME profile was complemented by a low blockage of the human ether-a-go-go-related gene (hERG) potassium channel,30 an interesting result taking into account that it has been described that the pharmacophoric requirements of the σ1R are reported to be similar to those of the hERG channel.31
The in vivo activity of 9a was measured in three different pain models in mice (Table 4). In all of them 9a displayed potent antinociceptive properties, indicating that it acts as an antagonist of the σ1R.11 Compound 9a was active in both phases of the formalin test, where the intraplantar (i.pl.) injection of formalin elicits an early phase (phase I) and a delayed phase (phase II) of pain characterized by paw licking, biting and other behaviours. Phase I is predominantly caused by direct activation of C-fibers, whereas phase II is a result of the combination of an inflammatory reaction in the peripheral tissue and functional changes in the spinal cord, thus involving both peripheral and central sensitization.32 Compound 9a exerted a dose-dependent analgesic effect on both phase I and phase II, with similar efficacy and potency in both phases.
Table 4. In vivo activity of 9a in comparison to 1.
| Formalin
a
|
Capsaicin
b
|
PSNL
c
|
|||||||||||||
| 40 mg kg–1 |
80 mg kg–1 |
20 mg kg–1 (%) |
40 mg kg–1 (%) |
80 mg kg–1 (%) |
|||||||||||
| Phase I (%) | Phase II (%)° | Phase I (%) | Phase II (%) | 40 mg kg–1 (%) | 80 mg kg–1 (%) | VF | PT | CP | VF | PT | CP | VF | PT | CP | |
| 1 | 50 ± 8 | 50 ± 13 | 79 ± 6 | 84 ± 8 | 52 ± 10 d | 69 ± 9 e | 23 ± 21 f | 39 ± 15 f | — | 72 ± 29 d | 80 ± 40 d | — | 98 ± 10 e | 109 ± 38 e | — |
| 9a | 45 ± 12 | 45 ± 8 | 85 ± 6 | 81 ± 7 | 19 ± 3 | 49 ± 7 | 32 ± 7 | 65 ± 22 | 83 ± 9 | 57 ± 6 | 84 ± 26 | 85 ± 9 | 102 ± 15 | 122 ± 17 | 93 ± 6 |
aPercentage of antinociception in the formalin test evaluated as the licking–biting time in drug-treated animals vs. vehicle.
bPercentage reduction of mechanical hypersensitivity in the capsaicin test evaluated as the latency time to the paw withdrawal response to upward pressure by von Frey filament stimulation in drug-treated animals vs. vehicle, with 50 s cut-off.
cPercentage reduction of mechanical (VF: von Frey test) and thermal (CP: cold plate test) hypersensitivity and thermal hyperalgesia (PT: plantar test) induced by partial sciatic nerve ligation, as described in the Experimental section.
dDose: 16 mg kg–1.
eDose: 32 mg kg–1.
fDose: 64 mg kg–1.
It also showed a moderate, but significant, antiallodynic effect in the mouse model of mechanical hypersensitivity induced by i.pl. capsaicin, which produces increased sensitivity to pain, resulting from central sensitization.33 This confirms previous studies, which indicate that the σ1R is essential for the development of capsaicin-induced mechanical hypersensitivity.12
Compound 9a was also active in the partial sciatic nerve ligation (PSNL) model in mice, representative of neuropathic pain.34 This model of peripheral nerve injury leads to a pain syndrome which is characterized by pain appearance in response to normally innocuous stimuli (allodynia) and exaggerated pain feeling in response to noxious stimuli (hyperalgesia). Hyperalgesia to noxious thermal stimuli and allodynia to cold and mechanical stimuli were used as outcome measures of neuropathic pain by using the plantar, cold plate and von Frey test, respectively. Compound 9a dose-dependently inhibited both mechanical and thermal hyperalgesia, achieving complete suppression at 80 mg kg–1. In thermal allodynia (cold plate test), it exhibited a significant effect at three different doses. Post-treatment values on day 14 (1 day after treatments) were not significantly different from the pre-treatment post-surgery values on day 10, indicating that the effect of drug treatment was reversible (data not shown). Altogether, compound 9a exhibited significant antinociceptive effects, which were comparable in efficacy and potency with those displayed by the reference compound 1, demonstrating its antagonistic nature and reinforcing the role of σ1R antagonists in the treatment of pain.
Evaluation of the physicochemical properties of compound 9a (Fig. 5) indicated that it is a highly soluble compound (thermodynamic solubility > 2 g mL–1), with an experimental log P (2.6) in accordance with the calculated value (clog P 2.2), which together with its low molecular weight makes it fully compliant with Lipinski's rule. However, compound 9a (and several additional analogues) showed an indication of suboptimal chemical stability at pH 2, since it slowly converted to the de-acylated amine 4b, which was poorly active against the σ1R (Ki 18 μM). Thus, after treatment with 0.01 M HCl, a decrease in area of the parent compound 9a over time was observed, which was accompanied by a corresponding increase in 4b (1.5% after 1 h, 3.4% after 2 h, 5.3% after 3 h). Although these results did not preclude a potential development, we decided to look for more stable alternatives, which are disclosed in the following paper.
The low binding affinity of primary amine 4b for the σ1R was not surprising since, in accordance with the pharmacophoric features of the σ1R ligands and the results shown in Table 1, a lipophilic substituent linked to the amino group was needed for activity. Hence, it can be concluded that the relatively high polarity of scaffold I is acceptable as long as lipophilic substituents are present in NR1R2 and R3. Indeed, a more lipophilic moiety in NR1R2 was needed in this case in order to achieve high affinity, compared to the series related to 1.11 Whereas in the pyrazole series the morpholine group showed a similar potency to more lipophilic substituents, in this family it was detrimental. It seems that the increased polarity of the central scaffold has to be somehow compensated for by the increased lipophilicity in side chain moieties.
Conclusions
The synthesis and pharmacological activity of a new series of pyrazolo[3,4-d]pyrimidines, designed according to known pharmacophoric features of the σ1R, is reported. The most active compounds exhibited nanomolar affinity for the σ1R and generally good selectivity for the σ2R. The σ1R required the presence of relatively highly lipophilic substituents at opposite sides of the central scaffold and selectivity was improved by shortening the distance of the basic nitrogen to it. Compound 9a was among the most interesting derivatives and exhibited potent antinociceptive properties in several pain models in mice, indicative of its antagonistic behaviour over the σ1R. However, a suboptimal stability of 9a at pH 2 led us to explore alternative substituents in the 4-position of the pyrazolopyrimidine scaffold that would overcome this potential drawback. The results of this new approach are reported in the following paper.
Experimental
Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. Microwave assisted reactions were conducted using an Explorer Synthesizer from CEM. Flash chromatography was performed with a forced flow of the indicated solvent system on SDS silica gel Chromagel 60 ACC-(230-400 mesh) or on a CombiFlash Companion system with Redisep Rf disposable columns. 1H NMR spectra were recorded on an Agilent UNITY 300 MHz spectrometer (fitted with a 5 mm H/F/X ATB probe) or an Agilent Mercury 400 MHz spectrometer (fitted with a 5 mm ID/PFG probe) with 2 H lock in deuterated solvents. Chemical shifts (δ) are in parts per million. Commercially available reagents and solvents (HPLC grade) were used without further purification for all the analytical tests. Analytical HPLC–MS analyses were performed on a Waters 2795-MS ZQ system using reverse phase XBridge C18 columns (4.6 × 50 mm, 2.5 μm), gradient 2–95% B (A = 10 mM ammonium bicarbonate, B = acetonitrile) over 4 min, injection volume: 5 μL, flow: 1.5 mL min–1 (method A) or gradient 2–95% B (A = 10 mM ammonium bicarbonate, B = acetonitrile) over 3.7 min, injection volume 5 μL, flow: 2.0 mL min–1 (method B). PDA spectra were recorded at 220–310 nm using a Waters 2996 PDA detector. Mass spectra were obtained over the range m/z 100–800 at a sampling rate of 0.3 scans per second using a Waters ZQ system. Data were integrated and reported using Water Masslynx software; all compounds displayed purity higher than 95% as determined by this method. Accurate mass measurements were carried out using an Agilent 6540 UHD Accurate-Mass QTOF system and obtained by electron spray ionization (ESI) in positive mode.
1-[2-(Pyrrolidinyl-1-yl)ethyl]-1H-pyrazolo[3,4-d]pyrimidin-4-amine (4a, n = 1, NR1R2 = pyrrolidine)
NaH (60% in mineral oil, 312 mg, 7.8 mol) was added under an argon atmosphere to a solution of 4-amino-1H-pyrazolo[3,4-d]pyrimidine (2, 1.0 g, 7.4 mmol) in anhydrous DMF (30 mL) at 0 °C. The mixture was left to reach room temperature for 1 h, and then, after cooling to 0 °C again, a solution of 1-(2-chloroethyl)pyrrolidine (3, n = 1, NR1R2 = pyrrolidine) (0.989 g, 7.4 mmol) in anhydrous DMF (10 mL) was added dropwise. The mixture was stirred at room temperature for 18 h and then quenched with H2O. The mixture was concentrated in vacuum and partitioned between EtOAc and H2O. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated to dryness to yield a crude residue that was purified by flash chromatography (SiO2, EtOAc/MeOH gradient from 9 : 1 to 7 : 3) to give 4a as a solid (0.687 g, 40% yield) together with the N2-regioisomer 2-(2-(piperidin-1-yl)ethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine, 16 (13 mg, 1% yield). 4a: 1H NMR (400 MHz, CDCl3) δ 8.39 (1H, s, 9-H), 7.92 (1H, s, 2-H), 5.47 (2H, br s, 10-H), 4.59 (2H, t, J = 7.0 Hz, 11-H), 3.07 (2H, t, J = 7.0 Hz, 12-H), 2.69–2.60 (4H, m, 14-H, 17-H), 1.82–1.71 (4H, m, 15-H, 16-H). HPLC (method B) Rt 1.58 min, 98% purity. MS (ESI) m/z: [M + H]+ 233.2; 16: 1H NMR (300 MHz, CDCl3) δ 8.47 (1H, s, 8-H), 8.08 (1H, s, 2-H), 5.45 (2H, br s, 7-H), 4.53 (2H, t, J = 6.3 Hz, 11-H), 3.13 (2H, t, J = 6.3 Hz, 12-H), 2.70–2.51 (4H, m, 14-H, 17-H), 1.89–1.73 (4H, m, 15-H, 16-H). HPLC (method B) Rt 1.47 min, 93.6% purity, MS (ESI) m/z: 233.2.
1-[2-(Piperidin-1-yl)ethyl]-1H-pyrazolo[3,4-d]pyrimidin-4-amine (4b, n = 1, NR1R2 = piperidine)
Using a similar procedure to that previously described, 4b was obtained as a solid (1.63 g, 45% yield). 1H NMR (400 MHz, CDCl3) δ 8.38 (1H, s, 8-H), 7.94 (1H, s, 2-H), 5.55 (2H, br s, 7-H), 4.62 (2H, t, J = 7.1 Hz, 11-H), 3.12–2.91 (2H, m, 12-H), 2.66–2.51 (4H, m, 14-H, 18-H), 1.67–1.61 (4H, m, 15-H, 17-H), 1.47–1.42 (2H, m, 16-H). HPLC (method A) Rt 2.29 min, 96% purity. MS (ESI) m/z: [M + H]+ 247.2.
1-[2-(1,4-Oxazepan-4-yl)ethyl]-1H-pyrazolo[3,4-d]pyrimidin-4-amine (4c, n = 1, NR1R2 = homomorpholine)
Compound 2 (1.0 g, 7.4 mmol) was added in portions to a suspension of NaH (60% in mineral oil, 320 mg, 8.0 mmol) in anhydrous DMF (15 mL) under an argon atmosphere. After stirring for 2 h at room temperature, a solution of 1-bromo-2-chloroethane (5, n = 1, X = Br, Y = Cl) (0.68 mL, 8.14 mmol) in anhydrous DMF (2 mL) was added dropwise. The reaction was stirred at room temperature for 18 h, cooled to 0 °C and quenched with H2O. The mixture was concentrated in vacuum, treated with EtOAc and filtered. The solution was concentrated and the crude product thus obtained purified by flash chromatography (SiO2, gradient from 100% EtOAc to EtOAc/MeOH 9 : 1) to yield 1-(2-chloroethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (6, n = 1, Y = Cl) as a white solid (0.82 g, 56% yield). A mixture of the previous derivative (0.250 g, 1.26 mmol), 1,4-oxazepane hydrochloride (0.313 g, 2.27 mmol), K2CO3 (0.524 g, 3.79 mmol) and a catalytic amount of NaI was dissolved in anhydrous DMF (10 mL) and the mixture stirred at 95 °C for 16 h under argon atmosphere. The mixture was concentrated under reduced pressure, treated with EtOAc and filtered. The solution was evaporated to dryness and the residue was treated with petroleum ether and decanted to give a crude product that was purified by flash chromatography (SiO2, EtOAc/MeOH 9 : 1) to yield 4c (0.08 g, 24% yield). 1H NMR (300 MHz, CD3OD) δ 8.19 (1H, s, 8-H), 8.09 (1H, s, 2-H), 4.47 (2H, t, J = 6.6 Hz, 11-H), 3.67 (2H, t, J = 5.9 Hz, 18-H), 3.64–3.56 (2H, m, 16-H), 3.05 (2H, t, J = 6.6 Hz, 12-H), 2.82–2.70 (4H, m, 14, 19-H), 1.79 (2H, p, J = 5.9 Hz, 15-H). HPLC (method B) Rt 1.51 min, 95.6% purity, MS (ESI) m/z: 263.1.
N-{1-[2-(Piperidin-1-yl)ethyl]-1H-pyrazolo[3,4-d]pyrimidin-4-yl}pivalamide maleate (9a)
Compound 4b (0.50 g, 2.03 mmol), pivaloyl chloride (0.44 g, 3.66 mmol), triethylamine (1.13 mL, 8.12 mmol) and a catalytic amount of DMAP in anhydrous pyridine (5 mL) were heated in a CEM microwave reactor at 130 °C for 20 min. The mixture was concentrated under reduced pressure and partitioned between EtOAc and 10% aqueous NaOH solution. The combined organic layers were washed with H2O, dried and evaporated to dryness. The oily residue (0.5 g, 74% yield) was dissolved in iPrOH (3 mL) and cooled to 0 °C, and a solution of maleic acid (0.18 g in 2 mL of iPrOH) was added dropwise. After stirring for 30 min at room temperature, the solvent was removed under vacuum without heating and the residue was washed with Et2O to give 9a as a white solid (0.48 g, 71% yield).1H NMR (400 MHz, DMSO-d6) δ 10.77 (1H, s, 9-H), 8.72 (1H, s, 2-H), 8.47 (1H, s, 8-H), 6.03 (2H, s, 26-H, 27-H, maleate), 4.80 (2H, t, J = 6.3 Hz, 17-H), 3.58–3.53 (2H, m, 18-H), 3.45–3.20 (4H, m, 20-H, 24-H), 1.70–1.65 (4H, m, 21-H, 23-H), 1.54–1.49 (2H, m, 22-H), 1.31 (9H, s, 14-H, 15-H, 16-H). 13C NMR (101 MHz, DMSO-d6) δ 177.76 (C-10), 167.10 (C-25, C-28, maleate), 154.63 (C-4), 154.54 (C-2), 152.83 (C-6), 136.88 (C-8), 135.67 (C-26, C-27, maleate), 103.99 (C-5), 54.36 (C-18), 52.49 (C-20, C-24), 41.41 (C-17), 39.77 (C-12), 26.55 (C-14, C-15, C-16), 22.68 (C-21, C-23), 21.32 (C-22). The assignation has been done according to the numbering in Fig. 5. HPLC (method B) Rt 2.80 min, 97% purity. HRMS (ESI) m/z: [M + H]+ calculated for C17H26N6O 331.2241, found 331.2239.
1-Cyclopentyl-3-{1-[2-(piperidin-1-yl)ethyl]-1H-pyrazolo[3,4-d]pyrimidin-4-yl}urea hydrochloride (13c)
A mixture of 4b (0.050 g, 0.20 mmol) and cyclopentyl isocyanate (0.034 g, 0.31 mmol) in acetonitrile (2 mL) was heated in a CEM microwave reactor for 1 h at 120 °C. The mixture was concentrated under reduced pressure, treated with acetonitrile and filtered. The crude product was dissolved in anhydrous dichloromethane (1.5 mL) and 4 M dioxane–HCl solution (1 mL) was added at 0 °C. After stirring for 15 min the solid was filtered and washed with dioxane to give 13c (55 mg, 70% yield). 1H NMR (400 MHz, CD3OD) δ 8.68 (1H, s, 16-H), 8.68 (1H, s, 2-H), 4.98 (2H, t, J = 6.0 Hz, 19-H), 4.20 (1H, p, J = 6.3 Hz, 11-H), 3.77 (2H, t, J = 6.0 Hz, 20-H), 3.72 (2H, d, J = 12.1 Hz, 22-H, 26-H), 3.07 (2H, td, J = 12.3, 3.1 Hz, 22-H, 26-H), 2.12–1.44 (14H, m, 12-H, 13-H, 14-H, 15-H, 23-H, 24-H, 25-H). 13C NMR (101 MHz, CD3OD) δ 163.15 (C-8), 154.07 (C-2), 153.96 (C-4), 148.45 (C-6), 136.91 (C-16), 102.48 (C-5), 56.12 (C-20), 54.70 (C-22, C-26), 53.47 (C-19), 43.05 (C-11), 33.72 (C-12, C-15), 24.53 (C-13, C-14), 24.03 (C-23, C-25), 22.52 (C-24). HPLC (method A) Rt 3.56 min, 100% purity. HRMS (ESI) m/z: [M + H]+ calculated for C18H27N7O: 358.235, found 358.2354.
1-{1-[2-(Azepan-1-yl)ethyl]-1H-pyrazolo[3,4-d]pyrimidin-4-yl}-3-butylthiourea maleate (15c)
A mixture of 1-(2-(azepan-1-yl)ethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (0.10 g, 0.38 mmol) and butyl thioisocyanate (0.13 g, 1.15 mmol) in anhydrous toluene (4 mL) was heated in a CEM microwave reactor at 150 °C for 1 h. The mixture was concentrated under reduced pressure, purified by flash chromatography (SiO2, EtOAc), treated with acetonitrile and filtered to afford the free base as a white solid (22 mg, 15% yield). The previous solid was dissolved in MeOH (0.5 mL) and to this mixture a solution of maleic acid (7.5 mg, 0.06 mmol) in MeOH (0.5 mL) was added at 0 °C dropwise. After stirring for 15 min at room temperature, the solid was filtered, and the residue washed with Et2O to give 15c (26 mg, 90% yield). 1H NMR (400 MHz, CD3OD, 400 MHz) δ 8.62 (1H, s, 2-H), 8.52 (1H, s, 15-H), 6.22 (2H, s, 28-H, 29-H), 4.88 (2H, t, J = 5.9 Hz, 18-H), 3.79 (2H, t, J = 5.9 Hz, 19-H), 3.75 (2H, t, J = 7.1 Hz, 11-H), 3.49–3.44 (4H, m, 21-H, 26-H), 1.95–1.90 (4H, m, 22-H, 25-H), 1.80–1.68 (6H, m, 12-H, 23-H, 24-H), 1.56–1.42 (2H, m, 13-H), 1.01 (3H, t, J = 7.4 Hz, 14-H). 13C NMR (101 MHz, CD3OD) δ 181.54 (C-8), 170.72 (C-27, C-30), 155.25 (C-2), 155.14 (C-4), 154.24 (C-6), 136.62 (C-15), 134.57 (C-28, C-29), 102.85 (C-5), 56.69 (C-19), 56.27 (C-21, C-26), 46.37 (C-18), 43.19 (C-11), 31.51 (C-12), 27.50 (C-22, C-25), 24.60 (C-23, C-24), 21.34 (C-13), 14.12 (C-14). HPLC (method B) Rt 3.70 min, 100% purity. HRMS (ESI) m/z: [M + H]+ calculated for C18H29N7S: 376.2278, found 376.2276.
Determination of physicochemical properties
clog P was calculated using ChemDraw Ultra 10.0.3. Solubility was measured as thermodynamic solubility from the solid compound in phosphate buffer at pH 7.4 by HPLC. Log P was determined by using a pH metric technique35 in a GlpKa Sirius Analytical instrument.
Stability determination
A solution of compound 9a in DMSO (10 mM, 25 μL) was treated with 0.01 M HCl and the mixture was maintained at 37 °C for different time periods (1, 2 and 3 h), after which the mixture was left at room temperature for 5 min and the content monitored by HPLC. It was shown that the content of 9a decreased with time (95.5% at 1 h, 93.6% at 2 h, 91.7% at 3 h), while the content of compound 4b increased with time (1.5% at 1 h, 3.4% at 2 h, 5.3% at 3 h).
In vitro assays
Protocols for σ1 and σ2 receptor binding, human liver microsome stability and hERG inhibition tests are provided in the ESI.†
In vivo studies
Descriptions of the formalin, capsaicin and PSNL models are provided in the ESI.†
Abbreviations
- ADMET
Absorption, distribution, metabolism, excretion and toxicity
- Clint
Intrinsic clearance
- clog P
Calculated logarithm of the octanol/water partition coefficient
- CNS
Central nervous system
- rhCYP
Recombinant human cytochrome P450
- 3D
Three-dimensional
- hERG
Human ether-a-go-go-related gene
- HLM
Human liver microsomes
- HRMS
High resolution mass spectrometry
- HTS
High throughput screening
- NOE
Nuclear Overhauser effect
- Papp
Apparent permeability coefficient
- PSNL
Partial sciatic nerve ligation
- Rt
Retention time
- SAR
Structure–activity relationship
- σR, σ1R, σ2R
Sigma, sigma-1 and sigma-2 receptor, respectively
Supplementary Material
Acknowledgments
We thank Adriana Port, Raquel Enrech, Inés Álvarez, Pilar Pérez, Enrique Hernández, Eva Ayet, Ariadna Balada, Raquel Fernández-Reinoso, Mª José Pretel, Enrique Portillo, Beatriz de la Puente and Daniel Zamanillo, for their expert contribution to analytical, in vitro and in vivo studies, Joan Andreu Morató, Monica Carro, and Edmundo Ortega for their excellent technical assistance and Carlos Pérez and Eduardo Villarroel for their contribution to compound management.
We acknowledge the support from Centro Desarrollo Tecnológico e Industrial (CDTI, PROJECT IDI-20110577).
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available: Analytical and characterization data for all the compounds. Experimental details for in vitro and in vivo testing. See DOI: 10.1039/c7md00077d
References
- Hayashi T., Su T. P. Cell. 2007;131:596. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Hanner M., Moebius F. F., Flandorfer A., Knaus H. G., Striessnig J., Kempner E., Glossmann H. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8072. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Zamanillo D., Portillo-Salido E., Vela J. M. and Romero L., in Therapeutic Targets. Modulation, Inhibition and Activation, ed. J. M. Botana and M. Loza, John Wiley & Sons, Inc., Hoboken, NJ, 2012, pp. 225–278. [Google Scholar]; (b) Maurice T., Su T. P. Pharmacol. Ther. 2009;124:195. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K. W., Bowen W. D. Cancer Res. 2002;1:313. [PubMed] [Google Scholar]
- Drews E., Zimmer A. Pain. 2009;145:269. doi: 10.1016/j.pain.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Zamanillo D., Romero L., Merlos M., Vela J. M. Eur. J. Pharmacol. 2013;716:78. doi: 10.1016/j.ejphar.2013.01.068. [DOI] [PubMed] [Google Scholar]
- Vela J. M., Merlos M., Almansa C. Expert Opin. Invest. Drugs. 2015;24:883. doi: 10.1517/13543784.2015.1048334. [DOI] [PubMed] [Google Scholar]
- Almansa C., Vela J. M. Future Med. Chem. 2014;6:1179. doi: 10.4155/fmc.14.54. [DOI] [PubMed] [Google Scholar]
- Marrazzo A., Cobos E. J., Parenti C., Aricò G., Marrazzo G., Ronsisvalle S., Pasquinucci L., Prezzavento O., Colabufo N. A., Contino M., González L. G., Scoto G. M., Ronsisvalle G. J. Med. Chem. 2011;54:3669. doi: 10.1021/jm200144j. [DOI] [PubMed] [Google Scholar]
- Wünsch B. Curr. Pharm. Des. 2012;18:930. doi: 10.2174/138161212799436548. [DOI] [PubMed] [Google Scholar]
- Díaz J. L., Cuberes R., Berrocal J., Contijoch M., Christmann U., Fernández A., Port A., Holenz J., Buschmann H., Laggner C., Serafini M. T., Burgueño J., Zamanillo D., Merlos M., Vela J. M., Almansa C. J. Med. Chem. 2012;55:8211. doi: 10.1021/jm3007323. [DOI] [PubMed] [Google Scholar]
- Romero L., Zamanillo D., Nadal X., Sánchez-Arroyos R., Rivera-Arconada I., Dordal A., Montero A., Muro A., Bura A., Segalés C., Laloya M., Hernández E., Portillo-Salido E., Escriche M., Codony X., Encina G., Burgueño J., Merlos M., Baeyens J. M., Giraldo J., López-García J. A., Maldonado R., Plata-Salamán C. R., Vela J. M. Br. J. Pharmacol. 2012;166:2289. doi: 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abadias M., Escriche M., Vaqué A., Sust M., Encina G. Br. J. Clin. Pharmacol. 2013;75:103. doi: 10.1111/j.1365-2125.2012.04333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz J. L., Christmann U., Fernández A., Luengo M., Bordas M., Enrech R., Carro M., Pascual R., Burgueño J., Merlos M., Benet-Buchholz J., Cerón-Bertran J., Ramírez J., Reinoso R., Fernández de Henestrosa A., Vela J. M., Almansa C. J. Med. Chem. 2013;56:3656. doi: 10.1021/jm400181k. [DOI] [PubMed] [Google Scholar]
- Díaz J. L., Christmann U., Fernández A., Torrens A., Port A., Pascual R., Alvarez I., Burgueño J., Montero A., Balada A., Vela J. M., Almansa C. J. Med. Chem. 2015;58:2441. doi: 10.1021/jm501920g. [DOI] [PubMed] [Google Scholar]
- (a) Cuberes R., Corbera J., Díaz J. L. and Almansa C., WO2013010950, PCT Int. Appl., 2013.; (b) Díaz J. L., Corbera J., Cuberes R., Contijoch M., Almansa C., Montero A. and Dordal A., Part of this SAR study presented as a poster in the XXIII International Symposium on Medicinal Chemistry, Lisbon, 2014. [Google Scholar]
- Glennon R. A., Ablordeppey S. A., Ismaiel A. M., El-Ashmawy M. B., Fischer J. B., Howie K. B. J. Med. Chem. 1994;37:1214. doi: 10.1021/jm00034a020. [DOI] [PubMed] [Google Scholar]
- Rao T. S., Ojwang J. O., Marshall H. B., Revankar G. R. J. Heterocycl. Chem. 1997;34:257. [Google Scholar]
- Schmidt H. R., Zheng S., Gurpinar E., Koehl A., Manglik A., Kruse A. C. Nature. 2016;532:527. doi: 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune S., Pricl S., Wünsch B. J. Med. Chem. 2013;56:9809. doi: 10.1021/jm400660u. [DOI] [PubMed] [Google Scholar]
- Díaz J. L., Zamanillo D., Corbera J., Baeyens J. M., Maldonado R., Pericàs M. A., Vela J. M., Torrens A. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:172. doi: 10.2174/1871524910909030172. [DOI] [PubMed] [Google Scholar]
- Schläger T., Schepmann D., Lehmkuhl K., Holenz J., Vela J. M., Buschmann H., Wünsch B. J. Med. Chem. 2011;54:6704. doi: 10.1021/jm200585k. [DOI] [PubMed] [Google Scholar]
- Laggner C., Schieferer C., Fiechtner B., Poles G., Hoffmann R. D., Glossmann H., Langer T., Moebius E. F. J. Med. Chem. 2005;48:4754. doi: 10.1021/jm049073+. [DOI] [PubMed] [Google Scholar]
- DeHaven-Hudkins D. L., Fleissner L. C., Ford-Rice F. Y. Eur. J. Pharmacol. 1992;227:371–378. doi: 10.1016/0922-4106(92)90153-m. [DOI] [PubMed] [Google Scholar]
- Ronsisvalle G., Marrazzo A., Prezzavento O., Cagnotto A., Mennini T., Parenti C., Scoto G. M. Pure Appl. Chem. 2001;73:1499. [Google Scholar]
- (a) Obach R. S., Baxter J. G., Liston T. E., Silber B. M., Jones B. C., MacIntyre F., Rance D. J., Wastall P. J. Pharmacol. Exp. Ther. 1997;283:46. [PubMed] [Google Scholar]; (b) Obach R. S. J. Pharmacol. Exp. Ther. 1999;27:1350. [Google Scholar]
- Compound 9a did to show any significant activity at 10 μM in the standard Ricerca panel of 65 receptors, ion channels and enzymes
- Stresser D. M., in Optimization in Drug Discovery. In vitro methods, ed. Z. Yan and G. W. Caldwell, Humana Press, Totowa, New Jersey, 2004, pp. 215–230. [Google Scholar]
- Hu M., Ling J., Lin H. and Chen J., in Use of Caco-2 cell monolayers to study drug absorption and metabolism, Optimization in Drug Discovery. In vitro methods, ed. Z. Yan and G. W. Caldwell, Humana Press, Totowa, New Jersey, 2004, pp. 19–36. [Google Scholar]
- Diaz G. J., Daniell K., Leitza S. T., Martin R. L., Su Z., McDermott J. S., Cox J. B. F., Gintant G. A. J. Pharmacol. Toxicol. Methods. 2004;50:187. doi: 10.1016/j.vascn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Corbera J., Vaño D., Martínez D., Vela J. M., Zamanillo D., Dordal A., Andreu F., Hernandez E., Perez R., Escriche M., Salgado L., Yeste S., Serafini M. T., Pascual R., Alegre J., Calvet C., Cano N., Carro M., Buschmann H., Holenz J. ChemMedChem. 2006;1:140. doi: 10.1002/cmdc.200500034. [DOI] [PubMed] [Google Scholar]
- Rosland J. H., Tjolsen A., Maehle B., Hole K. Pain. 1990;42:235. doi: 10.1016/0304-3959(90)91167-H. [DOI] [PubMed] [Google Scholar]
- Entrena J. M., Cobos E. J., Nieto F. R., Cendán C. M., Gris G., Del Pozo E., Zamanillo D., Baeyens J. M. Pain. 2009;143:252. doi: 10.1016/j.pain.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Malmberg A. B., Basbaum A. I. Pain. 1998;76:215. doi: 10.1016/s0304-3959(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Box K. J., Comer E. A. Curr. Drug Metab. 2008;9:869. doi: 10.2174/138920008786485155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.