

 An aryl ether series of potent and selective Nav1.3 inhibitors is described, starting from a series of diphenymethyl amides.
An aryl ether series of potent and selective Nav1.3 inhibitors is described, starting from a series of diphenymethyl amides.
Abstract
In this article, we describe the discovery of an aryl ether series of potent and selective Nav1.3 inhibitors. Based on structural analogy to a similar series of compounds we have previously shown bind to the domain IV voltage sensor region of Nav channels, we propose this series binds in the same location. We describe the development of this series from a published starting point, highlighting key selectivity and potency data, and several studies designed to validate Nav1.3 as a target for pain.
Voltage-gated sodium channels (Nav) comprise a family of nine transmembrane proteins (Nav1.1–Nav1.9) that span cellular membranes.1 The main pore-forming α-subunit has a 6-transmembrane topology of approximately 260 kDa size which assembles as a pseudo-tetramer into four distinct domains each of which contribute pore-forming helices and a spatially separated voltage sensor region to allow the channel to respond to voltage changes across the cell membrane.2 The sequence homology of mammalian Nav subtypes is very high, particularly in the pore region, making subtype selectivity of ligand interactions challenging. Nav channels selectively conduct sodium ions across cell membranes to create action potentials and nerve impulses in electrically excitable cells. These action potentials in turn elicit a plethora of physiological effects to, for example, control muscle contraction, cardiac function and neurological processing. Nav channel modulators have therefore been targeted as potential treatments for diseases as diverse as chronic pain, epilepsy and cardiac arrhythmias leading to a number of successful drug launches.3Fig. 1 shows some selected Nav channel drugs 1–7. All of these drugs show weak and non-selective activity across the Nav family which has limited their utility due to central and/or cardiovascular adverse events.4 Significant research has been dedicated to the identification of subtype selective inhibitors of Nav channels, as potentially safer alternatives to these older, non-selective examples.
Fig. 1. Selected Nav channel ligands.

There has been considerable work carried out to characterise and modify various animal toxins and other natural products which have been shown to engage Nav channels,5 but despite some recent advances, no advanced clinical candidates from this approach have as yet been described. There have been significant efforts to obtain protein crystal structures of bacterial sodium channels6 and carry out modelling studies7 to elucidate ligand binding sites and modes of modulation. In a notable recent publication,8 a bacterial Nav channel was engineered to contain features of the human Nav1.7 voltage-sensor region and a crystal structure obtained of this chimera bound to an inhibitor to elucidate the drivers of subtype selectivity within this region of the protein. Homology models using this structure to explore subtype selectivity of other Nav channels, such as Nav1.3, are anticipated.
For several years now, we have pursued medicinal chemistry approaches to identify potent and selective inhibitors of several Nav channels to aid in the elucidation of the role of individual channels in pain transmission.9 As part of this larger effort, we have identified a series of aryl sulphonamides that show potent and for the most part selective inhibition of the Nav1.3 channel. This paper describes our efforts to successfully improve the physicochemical and pharmacokinetic properties of this series while retaining excellent Nav1.3 potency and broader Nav subtype selectivity.
There are very few reports of potent Nav1.3 inhibitors, and those reports that have emerged tend to describe poorly selective Nav blockers that also carry Nav1.3 activity such as lacosamide 810 (Fig. 2). There have been some more recent reports of aryl sulphonamides from Vertex 911 and Icagen 1012 that have greater Nav1.3 potency and it has been the latter series that we have focussed our efforts on.
Fig. 2. Selected literature Nav1.3 ligands.

Identifying Nav1.3 pharmacological tools
In a previous disclosure, we described the identification of a diphenylmethyl amide 11 (Fig. 3) of this aryl sulphonamide series13 which showed good potency at Nav1.3 and some selectivity for this channel over other Nav subtypes. Compound 11 showed excellent potency at human and rat Nav1.3 with good selectivity against all other human subtypes tested with the exception of Nav1.1 which mirrored Nav1.3 activity. Interestingly, while 11 was very weak at human Nav1.7, it showed significantly greater potency at the rat orthologue. This compound showed poor passive permeability in RRCK and moderate efflux in an MDR1 cell line, and was of modest aqueous solubility, features that we sought to address in subsequent analogues.
Fig. 3. Screening lead compound.

Compound 11 was shown to bind in the voltage-sensor region of voltage-gated sodium channels, based on chimeric channels of Nav1.3 domains combined with varying homologous domains of either Nav1.7 or Nav1.5. These studies suggested domain 4 to be the interaction site for this compound, a region of the protein that we believed would support subtype-selective Nav1.3 channel blockers with the appropriate optimisation, as we had observed large reductions in potency when the S2–S4 regions within domain 4 were mutated away from Nav1.3 sequences to those of Nav1.5 or Nav1.7.13 This region also showed the greatest similarity in amino acid sequence between Nav1.3 and Nav1.1 channels,13 explaining the equivalent potency of 11 at these two channel subtypes.
In developing this lead further, analogues of 11 were prepared to explore peripheral and core template functionality and their effect on Nav1.3 channel potency (Table 1) and subtype selectivity. Compounds were tested for Nav1.3 inhibitory activity in a HEK cell-based electrophysiology PatchXpress platform.
Table 1. SAR exploration around compound 11.
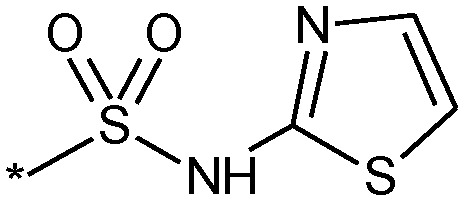
| |||
| Compound | R | clog P (log D) | Nav1.3 IC50 (nM) |
| 11 |
|
3.8 (3.3) | 17 |
| 12 |
|
3.5 (2.5) | 147 |
| 13 |
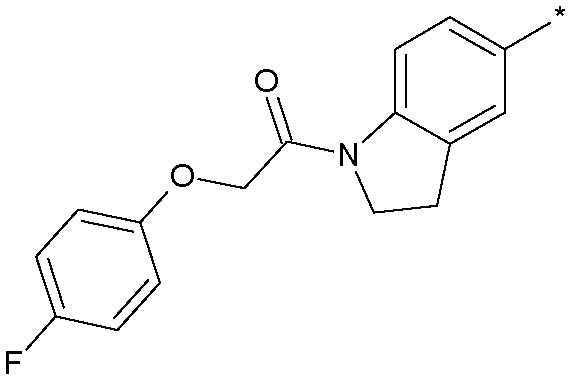
|
2.6 (1.8) | 2506 |
| 14 |
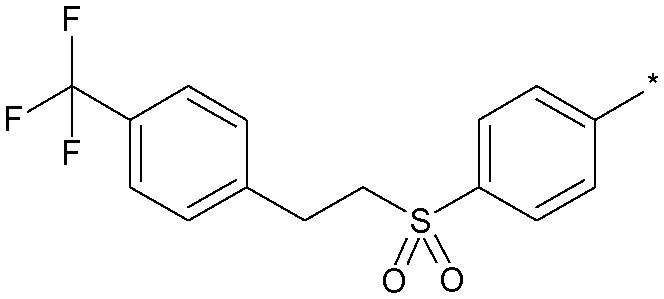
|
3.7 (ND) | 155 |
| 15 |
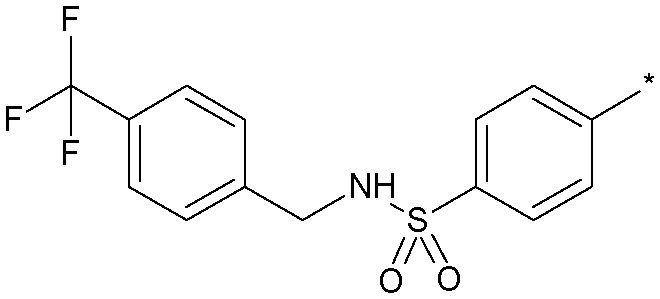
|
3.2 (ND) | 160 |
| 16 |
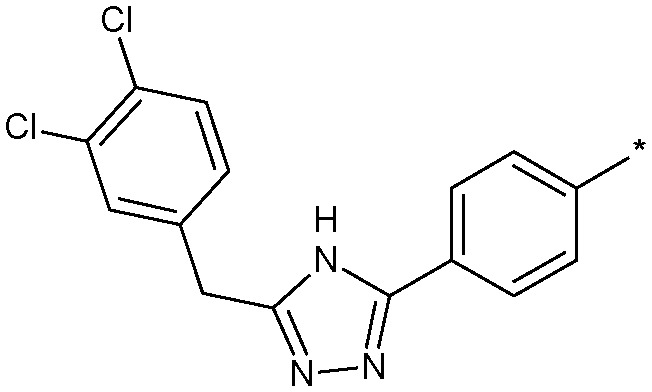
|
4.1 (ND) | 45 |
| 17 |
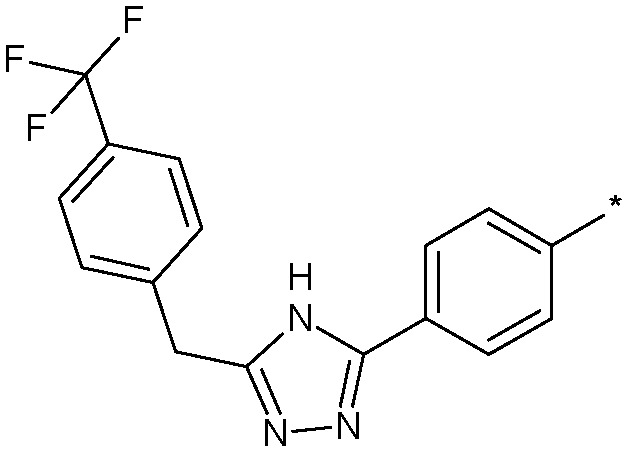
|
3.6 (ND) | 124 |
| 18 |
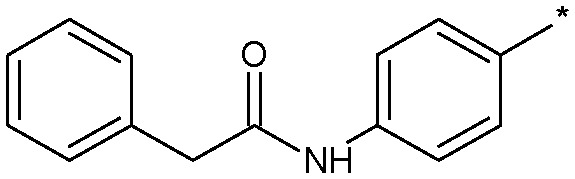
|
2.9 (ND) | 3790 |
| 19 |
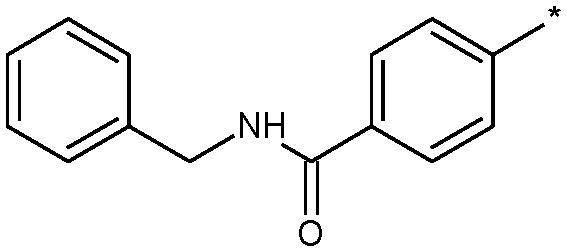
|
2.6 (ND) | 4450 |
| 20 |
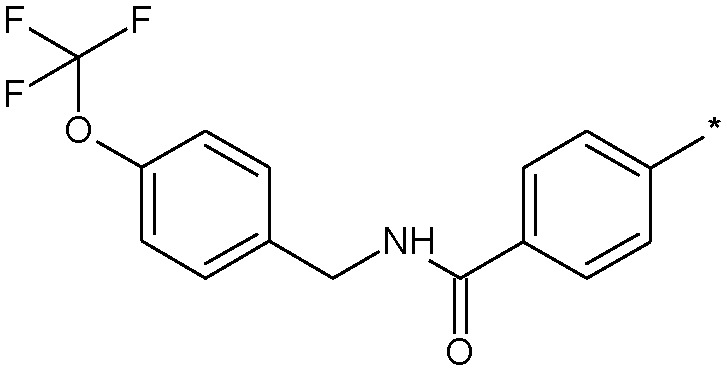
|
4.0 (2.6) | 197 |
| 21 |
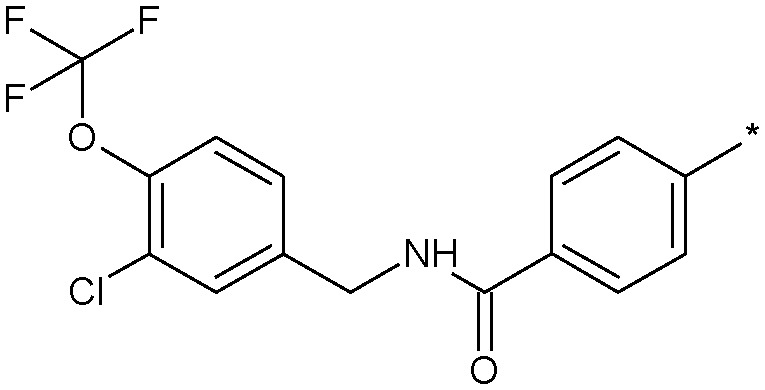
|
4.3 (3.1) | 20 |
| 22 |
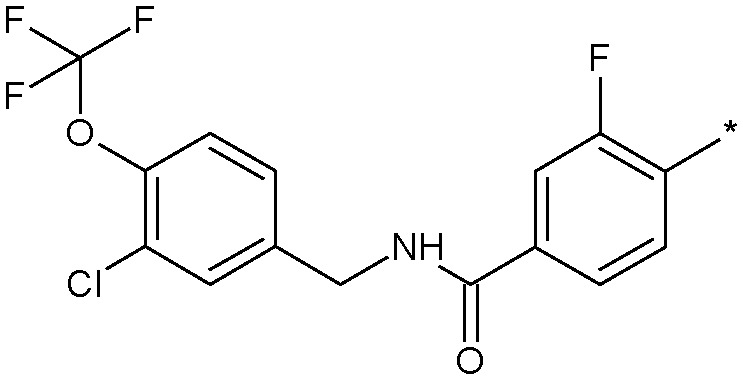
|
4.5 (3.3) | 11 |
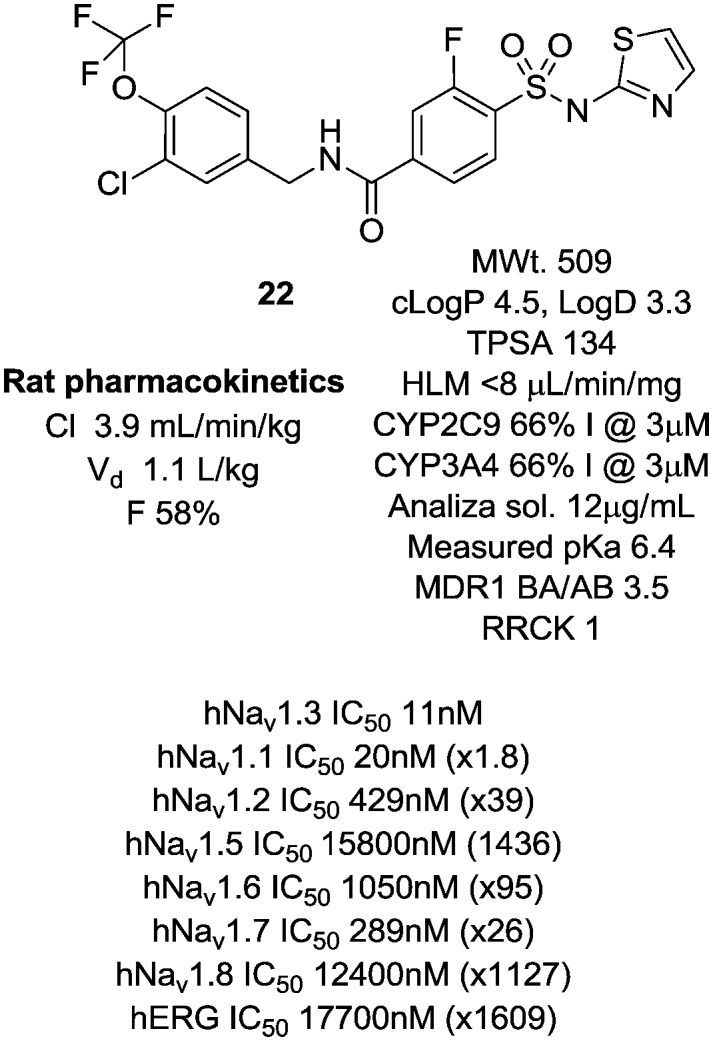
Simply excising one of the phenyl rings (18) led to a sharp drop in potency. Similarly, constraining the aniline function into a ring system such as 13 concomitant with excising one of the phenyl rings also produced a significant loss in activity. Inverting the amide group in 19 was essentially equipotent with 18, which was an important observation as we were concerned that amidolysis of the amide linkage in 18 would produce sulfathiazole which is a known toxin.14 It was the addition of lipophilic substituents, for example the trifluoromethyl group in 12, which gave significant boosts in potency and 12 was now only some 10-fold less active than the starting 11. Other amide replacements such as the sulfone 14 and the sulphonamide 15 were of similar potency to 12, as were the amide isosteric triazoles 16 and 17. It was clear at this stage that several truncated amides or their isosteres could afford potent Nav1.3 activity provided they retained a lipophilic aryl substituent. We chose the synthetically most accessible amide linkage, in the non-anilinic reversed configuration, to further optimise. A trifluoromethoxy group in 20 was similarly potent to the trifluoromethyl congener 12 and further optimisation of the functionality on both phenyl rings led to the chloro derivative 21 and the fluoro-chloro 22 which had excellent potency at Nav1.3, equivalent to the starting point 11. Further profiling (Fig. 4) confirmed its potency at both human and rat Nav1.3 and that 22 retained good selectivity over all subtypes except Nav1.1, at which it was equipotent.
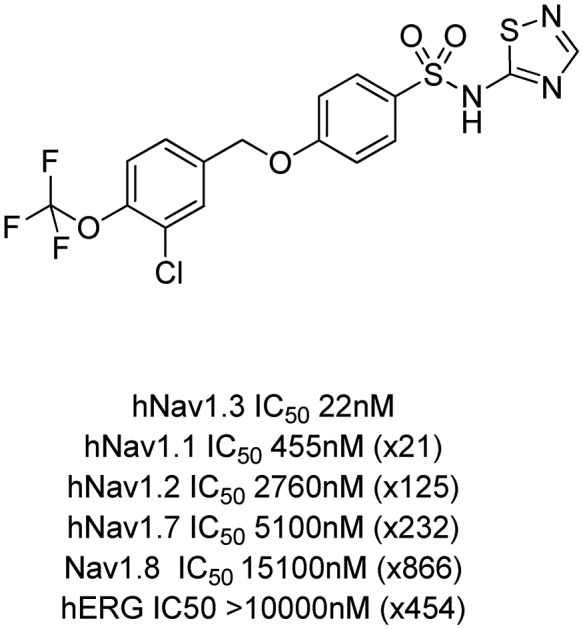
Fig. 4. Early Nav1.3 tool compound.
Evaluating Nav1.3 as a pain target
Despite the equivalent levels of Nav1.1 activity, 22 was an excellent early tool compound to further explore Nav1.3 pharmacology and in vivo efficacy, but as 22 was profiled more extensively, it became apparent that it had some liabilities as a lead due to low aqueous solubility, low passive membrane permeability and inhibition of both CYP3A4 and CYP2C9 isoforms in high throughput inhibition assays. In common with similar chemotypes developed as selective Nav1.7 blockers,1522 was highly bound to plasma proteins in all species tested (rat and human ppb 99.8% as measured by equilibrium dialysis, dog ppb >99.8%). Nonetheless, 22 did allow us to probe Nav1.3 as a drug target more fully at this stage.
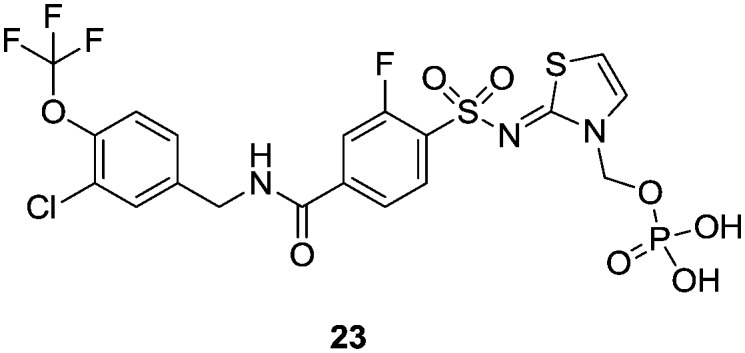
An oral 100 mg kg–1 dose of 22 produced exposure levels in the rat of approximately 12 fold the Nav1.3 IC50, at which concentrations a drop in heart rate, an increase in blood pressure and a decrease in core body temperature were observed. A similar finding was seen at a 500 mg kg–1 dose, which resulted in similar exposure levels, a result of solubility-limited absorption. These findings raised some concern that blockade of Nav1.3 could produce a cardiac safety liability. It was not believed that the findings were confounded by the inherent activity of 22 at the primarily brain-expressed Nav1.1 channel and it became important to check if this observation was purely a rat-specific effect. A follow-up study was conducted in cynomolgus monkey, in which we wished to target in excess of the 12 fold IC50 levels achieved in the rat study. In order to do this with 22, a phosphate prodrug was designed with significantly enhanced solubility (Fig. 5).16 We demonstrated first that 23 was rapidly and completely converted to the parent 22 upon oral dosing to the rat and an equivalent dose of 23 resulted in a significantly higher exposure of parent 22 than dosing 22 directly.
Fig. 5. Phosphate prodrug of 22.
Cynomolgus monkeys were then dosed with 30 mg kg–123 which resulted in a plasma exposure of 22 in excess of some 62 fold the in vitro IC50. This was accompanied by no clinical signs, no changes in heart rate and mean, diastolic or systolic blood pressure, no effects on any ECG parameter and no changes in left ventricular pressure parameters. Our conclusion from this study was that the original observations were likely rat-specific and allowed us to proceed with more confidence in the safety of Nav1.3 as a target.
Based on previous reports of an upregulation of Nav1.3 channels in the DRG and their potential involvement in pain pathogenesis,17 we investigated the ability of 22 to alter neuronal excitability using ex vivo skin nerve recording and furthermore evaluated the effects of 23in vivo, using spinal cord recording in models of peripheral nerve injury. Exogenous administration to the skin-tibial nerve with 22 at 0.01, 0.03, 0.1, 0.3 and 1 μM had no effect on heat evoked afferent firing or ongoing nerve activity. Intravenous administration of 23 produced significant reductions of the noxious pinch evoked activity (3 mg kg–1: difference –759, 95% CI: –1393 to –126, p = 0.02; 10 mg kg–1: difference –788, 95% CI: –1421 to –154, p = 0.02), where exposures up to 15× IC50 of 22 were achieved. In the same study, pregabalin, an α2δ ligand licensed for the treatment of neuropathic pain robustly inhibited pinch evoked activity (9 mg kg–1: difference –711, 95% CI: –1345 to –76, p = 0.03; 18 mg kg–1 difference –1354, 95% CI: –1989 to –719, p = 0.0003). Notably, the magnitude of pregabalin effects were greater compared to that seen with 23. Responses to innocuous brush, mechanical punctate stimuli and heat evoked activity were not significantly modulated by 23. This was in contrast to pregabalin, where clear inhibitions were seen against these parameters. These data indicated that in this study, Nav1.3 blockade did not drive a strong inhibitory response against the majority of the endpoints measured. However, Nav1.3 gene expression is known to be significantly upregulated in a number of nerve injury models of pain including SNT.18–20 Furthermore, data from the Waxman group has shown profound effects of Nav1.3 gene knockdown on several other evoked endpoints in different pain models.18 The increased availability of Nav1.3 could contribute to the pain hypersensitivity in these models.
Gene expression levels of Nav1.3 in rat DRGs following spinal nerve transection (SNT) were measured in-house. Ipsilateral versus contralateral Ct values were compared using a paired T-test. The house keeping gene beta actin was not statistically different between ipsi- and contralateral samples. However both Nav1.3 and ATF3 genes were significantly different at the 5% level when ipsi- and contralateral levels were compared (see Table 2).
Table 2. Changes in gene expression levels of Nav1.3 in rat DRG.
| Ct value change | Lower 95% CI | Upper 95% CI | Std error | P value | |
| Nav1.3 | 3.325 | 1.906 | 4.744 | 0.552 | 0.0018 |
| ATF3 | 4.629 | 2.076 | 7.181 | 0.993 | 0.0055 |
| β-Actin | –0.348 | –0.876 | 0.180 | 0.205 | 0.1508 |
Ipsilateral Ct values were lower for both Nav1.3 and ATF3 indicative of higher gene expression. In line with previous studies,18 Nav1.3 and ATF3 genes were upregulated in ipsilateral DRG following SNT.
The fact that Nav1.3 blockade did not drive a strong inhibitory response for the majority of the endpoints measured in either the ex vivo skin nerve or in vivo spinal nerve recordings, despite showing an increase in Nav1.3 gene expression in the neuronal cell bodies, is in contrast to reports in the literature, which have shown effects of Nav1.3 knockdown on behavioural and electrophysiological endpoints in different pain models. One could speculate that this could be due to differences in the experimental approach i.e. genetic modification to reduce expression versus a pharmacological inhibition, or that the increase in Nav1.3 gene expression was not recapitulated at the protein level in our model. Finally, it could be that a higher multiple of the primary pharmacology is required to drive efficacy against the endpoints measured (maximum of 15× IC50 plasma free drug levels achieved in our in vivo studies, approximately 200 nM; the free drug levels in DRG or spinal cord were not measured and could be markedly less). Interestingly it is worth noting that Nassar et al. showed using Nav1.3 null mutant mice, that Nav1.3 is neither necessary nor sufficient for the development of nerve-injury related pain.21 Further study is required to elucidate these points.
Identifying a non-amino thiazole tool compound
We were concerned that the amino thiazole group could represent a toxicity liability and were very keen at this early stage to identify alternate groups that would avoid a potential structural alert in the amino thiazole group.22 The analogous 1,2,4-thiadiazole 24 demonstrated very similar pharmacology and physicochemical properties but interestingly showed very low oral bioavailability, perhaps due to this more acidic headgroup being more susceptible to transporter-mediated clearance and having a higher overall topological polar surface area (TPSA) and H-bond count (Fig. 6).
Fig. 6. Non-amino thiazole version of 22.
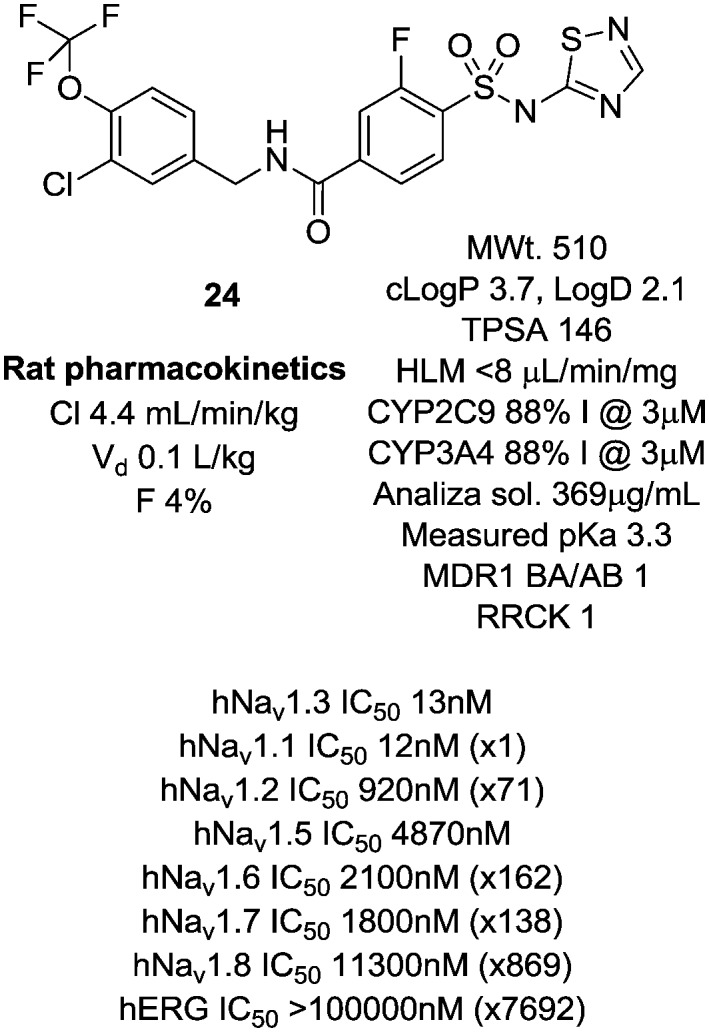
We began to further explore structure activity relationships in this chemotype to specifically improve the physicochemical and pharmacokinetic properties of the series in a non-thiazole containing headgroup and identify an improved compound that retained the favourable potency and selectivity characteristics of 22. In particular, the areas we wished to explore in more depth were variations to the linker region to reduce H-bonding character and PSA, and alterations to the head group structure to improve permeability.
Head group SAR
Consistent with the arginine-rich putative binding site for this series, it was highly likely that we would require an acidic head group for activity at Nav1.3. The aryl sulfonyl amino-thiazole head group in 22 had an acidic pKa of 6.4. We explored a number of other head groups (Table 3) that covered a range of disparate pKa's from more to less acidic than the thiazole in 22. There did not appear to be a strong correlation between the acidic pKa of the head group and Nav1.3 potency, with the substantially more acidic thiadiazole group 24 being equipotent with the thiazole congener, while the similarly acidic pyrimidines 25 and 26 were substantially weaker. Head groups of intermediate acidity such as the isomeric pyrimidine 27 and the pyridazine 28 were very weak inhibitors of Nav1.3.
Table 3. Head group variations on the aryl amide core of 22.
| ||||
| Compound | R | clog P (log D) | pKa (cpKa) | Nav1.3 IC50 (nM) |
| 22 |
|
4.5 (3.3) | 6.4 (6.6) | 13 |
| 24 |
|
3.7 (2.1) | 3.3 (4.3) | 13 |
| 25 |
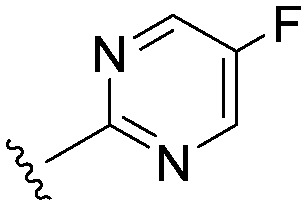
|
3.9 (2.1) | (3.8) | 993 |
| 26 |
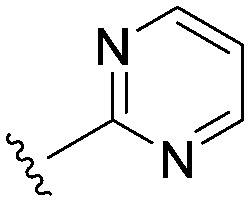
|
3.8 (1.8) | (3.9) | 374 |
| 27 |
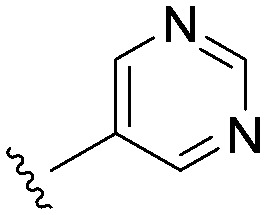
|
3.8 (2.0) | 4.6 (4.7) | >3000 |
| 28 |
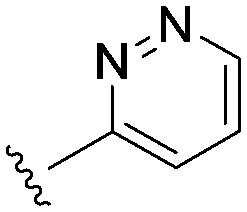
|
3.6 (2.4) | (4.1) | 1793 |
It seemed more likely that the binding region of the head group required a combination of an acidic grouping of sufficient compactness to be tolerated, with the activity observations for those head groups investigated being quite subtle. These SAR had identified the thiadiazole as an equivalent head group to the thiazole in terms of potency at Nav1.3.
Sulfonamide linkers
In parallel to the above head group alterations, we also returned to explore a number of sulfonamide linkers which in the early analogues were of similar potency to the amides. These were made as both secondary and tertiary sulfonamides; a selection of the tertiary sulphonamides made lacking an H-bond donor are shown in Table 4 with different linked saturated heterocycles (29, 30, 31 and 32) and vectors of attachment (33 and 34) but all were significantly weaker than 22. These data were replicated in the case of secondary sulphonamides (data not shown) and all subsequent analogues did not feature this linker.
Table 4. Sulfonamide linker analogues of 22.
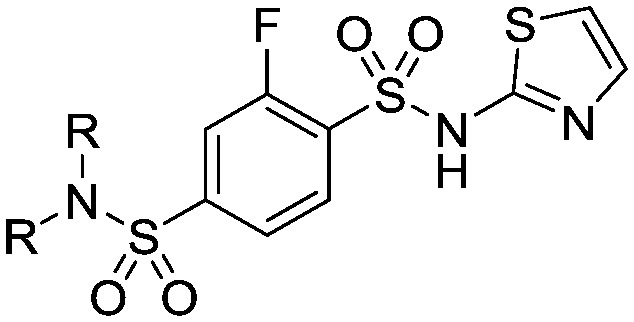
| ||||
| Compound | R–N–R | clog P (log D) | MWt. | Nav1.3 IC50 (nM) |
| 29 |
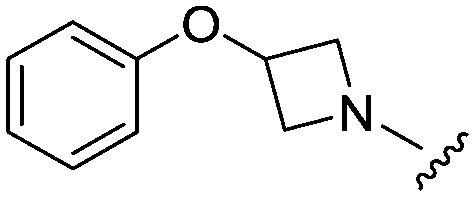
|
3.0 (1.4) | 451.0 | 959 |
| 30 |
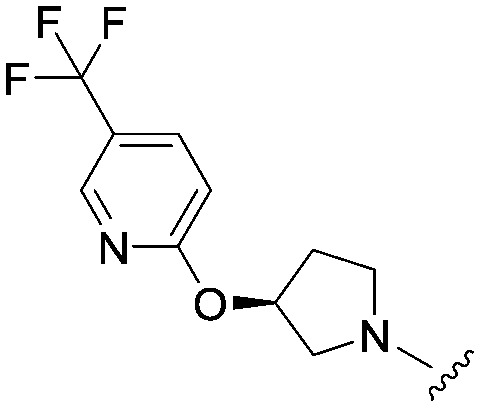
|
3.3 (ND) | 534.0 | 851 |
| 31 |
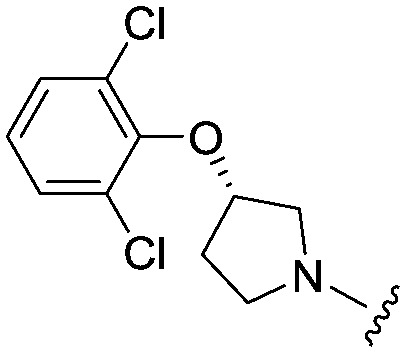
|
4.1 (2.6) | 534.4 | 945 |
| 32 |
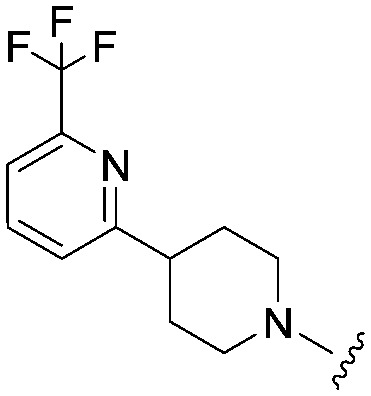
|
3.1 (ND) | 532.6 | 1640 |
| 33 |
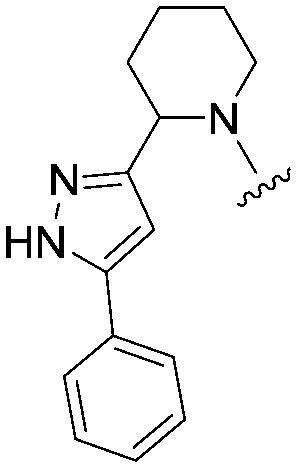
|
4.1 (ND) | 529.6 | 1830 |
| 34 |
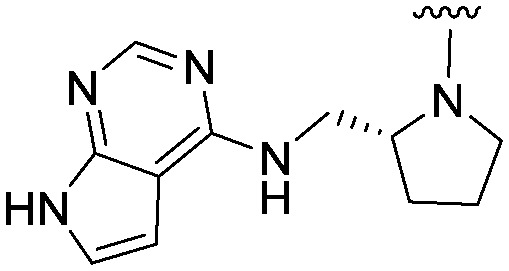
|
2.8 (ND) | 519.6 | >3000 |
Other linker variations
We then explored a number of alternative linker variations in the thiadiazole series, designed to reduce the H-bonding character of the linking group and improve membrane permeability (Table 5). For example the amide linker of 24 was capped off with a methyl group (35) or replaced with a phenyl ether linker (36), changes which resulted in more than 20 fold losses in potency. A number of tertiary amides were made (37 and 38 and 40), which continued the trend of large potency losses seen for the tertiary sulfonamides. However, we found that benzyl ether linkers were equipotent with the amide linker, while being of similar lipophilicity. The ether 39 had an IC50 at Nav1.3 of 22 nM. Other expressions of an ether linker were less successful, for example 41 in which we had incorporated the ether into an azetidine-linked amide which was some 10 fold weaker than the benzyl ether.
Table 5. Linker variations of the thiadiazole head group derivative 13.
| ||||
| Compound | Structure | clog P (log D) | MWt. | Nav1.3 IC50 (nM) |
| 24 |
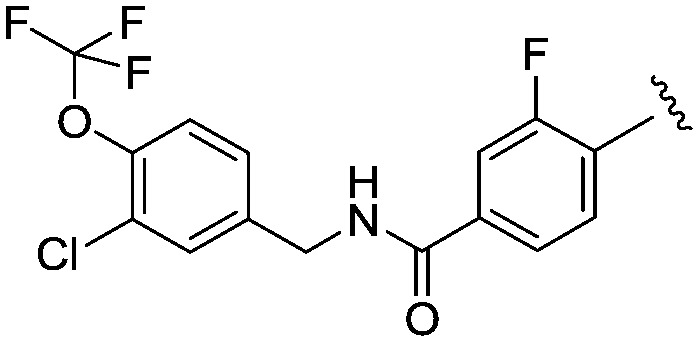
|
3.7 (2.1) | 510.9 | 13 |
| 35 |
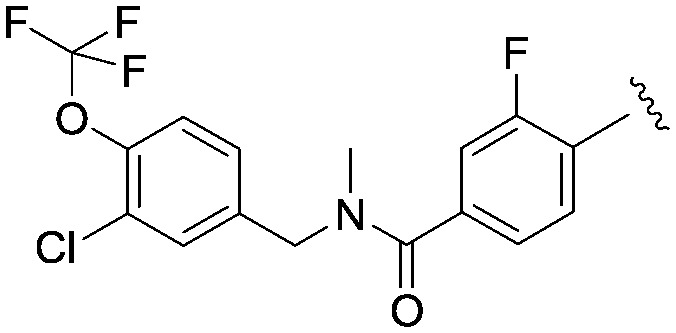
|
3.6 (1.3) | 524.9 | 522 |
| 36 |
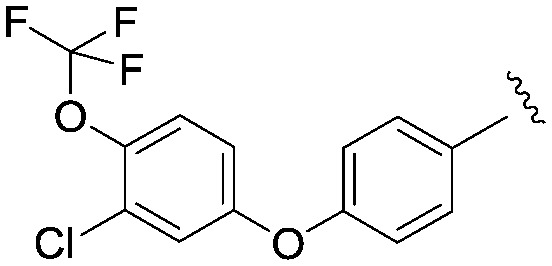
|
4.65 (2.5) | 451.8 | 296 |
| 37 |
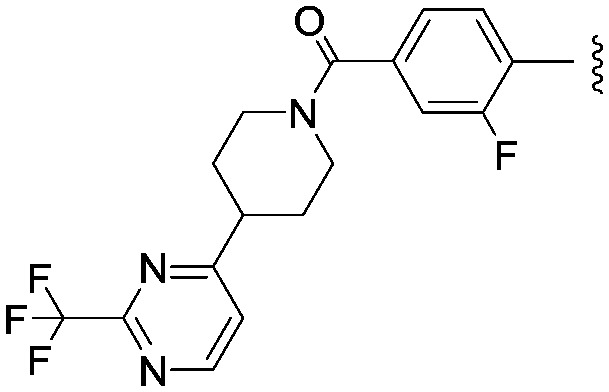
|
0.9 | 516.5 | 6675 |
| 38 |
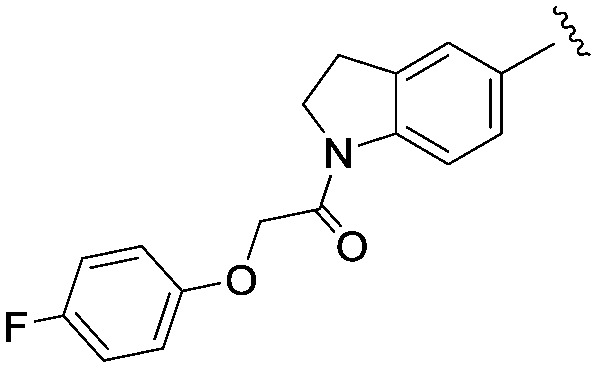
|
2.20 (0.4) | 434.5 | 1273 |
| 39 |
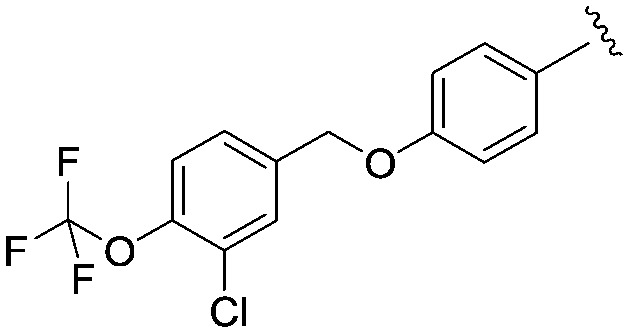
|
4.40 (2.6) | 465.9 | 22 |
| 40 |
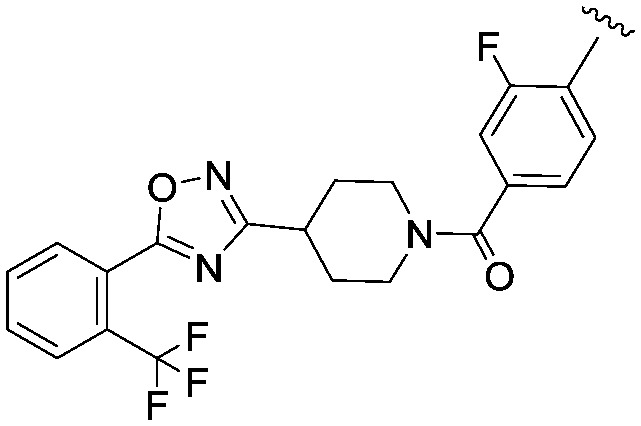
|
2.68 | 582.6 | 1955 |
| 41 |
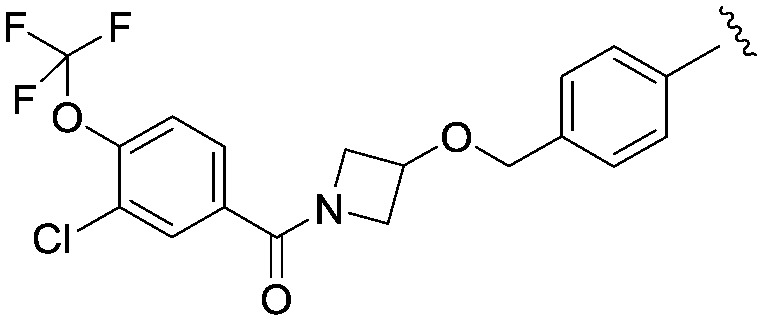
|
3.71 (1.30) | 548.9 | 237 |
Further work on 39 investigated its physicochemical and pharmacokinetic properties (Fig. 7). The compound showed good metabolic stability and much improved aqueous solubility compared to 22.
Fig. 7. Profile of the optimised non-amino thiazole tool compound 39.

Its broader Nav selectivity profile was also improved with the window over Nav1.1 now increased to 20 fold with good windows retained over the other Nav channels Nav1.2, Nav1.7 and Nav1.8. 39 showed no appreciable activity at the hERG channel.
Compound 39 had a measured pKa of 4.1, similar to that of previous thiadiazoles investigated in this series. The compound was of low clearance in the rat (2 mL min–1 kg–1, Vd 1 L kg–1), of high bioavailability (F 73%) and had an elimination half-life of some 7 h, a pharmacokinetic profile that was not typical of other thiadiazoles in the series. It is interesting to compare 39 to the previous thiadiazole 24. The latter compound is larger and of higher TPSA and has very low oral bioavailability compared to 39. It is likely that controlling molecular weight, H-bond count and TPSA in this series of acidic compounds helps to modulate transporter affinity, particularly within the more acidic thiadiazole series. Therefore, whilst the thiadiazole head group typically brings poor oral bioavailability, by careful control of physical and structural properties, a well absorbed compound can be identified.
Being a moderately lipophilic acid, the free fraction in rat of 39 was 0.00024. It was very selective in a broad panel of enzymes, channels and receptors (<35% inhibition across the panel at 10 μM), although it retained inhibition of CYP2C9 (85% inhibition @ 3 μM) and CYP3A4 (75% inhibition @ 3 μM) in line with both 22 and 24.
Conclusions
Through a focussed SAR exercise starting from a previous Nav1.3 active compound 11, we were rapidly able to identify a potent and selective and more aqueous soluble compound 39 devoid of the toxicity-suspect amino-thiazole headgroup, which was well-absorbed upon oral dosing. These properties gave a more attractive overall profile than our initial tool compound 22 in a non-thiazole series with similar levels of Nav1.3 potency. Further studies with compound 39 will be disclosed in due course.
Experimental
An example of the synthetic routes followed to prepare the compounds contained herein is shown below for compounds 22 and 39.
Synthesis of compound 22

4-(Benzylthio)-3-fluorobenzoic acid
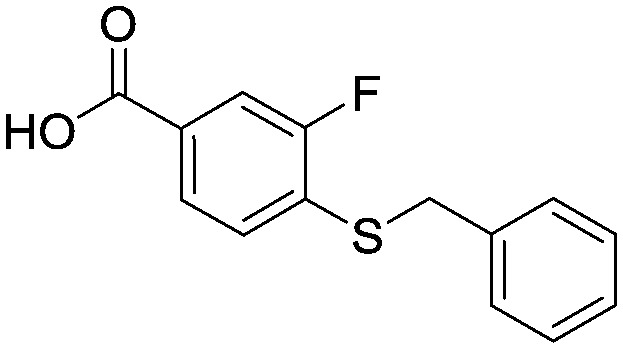
3,4-Difluorobenzoic acid (50 g, 320 mmol), caesium carbonate (206 g, 632 mmol) and benzyl mercaptan (37.4 mL, 320 mmol) were combined in DMSO (500 mL) and the reaction mixture heated at 70 °C for 3 hours. The reaction mixture was cooled to room temperature and then poured into water (1.5 L) before extraction with EtOAc (2 × 750 mL). The aqueous was acidified to pH = 1 with 4N HCI (aq) and the precipitate filtered and dried in vacuo to provide the title compound as a pale pink coloured solid (84.7 g, 320 mmol). 1HNMR (CDCI3): 4.2 (s, 2H), 7.3 (m, 6H), 7.75 (m, 2H). LCMS (0.05% formic acid in MeCN) Rt = 1.55 min. MS m/z 261 [M – H]–.
Methyl 4-(benzylthio)-3-fluorobenzoate
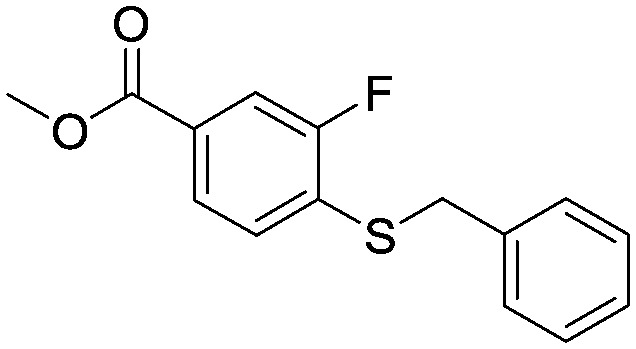
A suspension of 4-(benzylthio)-3-fluorobenzoic acid (81.4 g, 0.310 mol) in MeOH (800 mL) and concentrated H2SO4 (2 mL) was heated at reflux for 36 hours. The mixture was cooled to room temperature and the resulting precipitate filtered and washed with hexane. The filtrate was concentrated in vacuo and the resulting solid filtered and washed with hexane. The combined solids were dried in vacuo to provide the title compound as a white solid (60.9 g, 0.217 mol). 1HNMR (CDCI3): 3.90 (s, 3H), 4.18 (s, 2H), 7.22–7.35 (m, 6H), 7.65–7.72 (m, 2H).
Methyl 4-(chlorosulfonyl)-3-fluorobenzoate
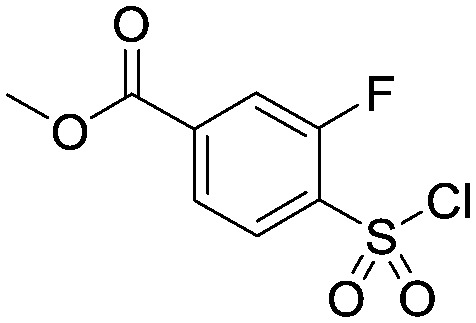
To a vigorously stirred solution of methyl 4-(benzylthio)-3-fluorobenzoate (30.4 g, 0. 110 mol) in DCM (860 mL) and 4N HCI (aq) (860 mL) at 5 °C was added sodium hypochlorite (445 mL) dropwise, keeping the temperature below 10 °C throughout. Upon complete addition the reaction mixture was allowed to warm to room temperature over 1 hour. Two reactions were carried out on identical scale in parallel (2 × 0.11 mol scale) and combined at this point for work-up and purification. The layers were separated and the aqueous extracted with DCM (500 mL). The combined organics were washed with a 10% aqueous solution of sodium metabisulfite (2 × 200 mL), brine (200 mL), dried (MgSO4) and concentrated in vacuo. The residue was purified via column chromatography on silica gel eluting with hexane/EtOAc (95 : 5 to 4 : 1) to provide the title compound as a white solid (53.1 g, 0.210 mol). 1HNMR (CDCI3): 3.99 (s, 3H), 7.95–8.08 (m, 3H).
Methyl 3-fluoro-4-[(1,3-thiazol-2-ylamino)sulfonyl]benzoate
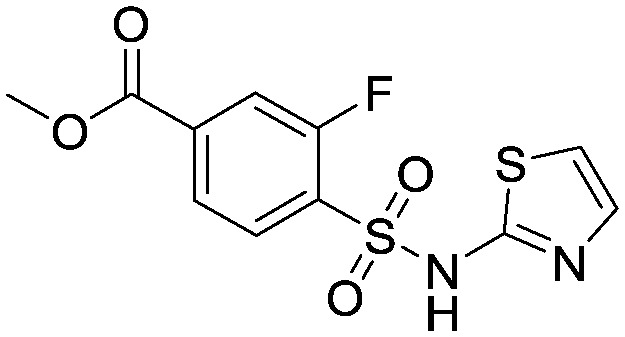
To a solution of 2-aminothiazole (105.2 g, 1.05 mol) in pyridine (250 mL) was added methyl 4-(chlorosulfonyl)-3-fluorobenzoate (53.1 g, 0.21 mol) and the mixture stirred at 50 °C for 18 hours. The reaction was then allowed to cool to room temperature and added portionwise into 2N HCI (aq) (300 mL) and the mixture acidified to pH 1 with 4N HCI (aq). The resulting precipitate was filtered, washed with water and dried in vacuo to provide the title compound as a tan coloured solid (31.97 g, 0.10 mol). This material was used crude for subsequent reactions. 1HNMR (d6-DMSO): 3.88 (s, 3H), 6.88 (d, 1H), 7.29 (d, 1H), 7.77–7.99 (m, 3H).
3-Fluoro-4-[(1,3-thiazol-2-ylamino)sulfonyl]benzoic acid
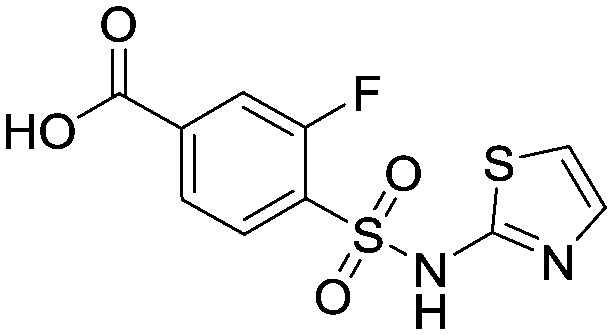
To a suspension of methyl 3-fluoro-4-[(1,3-thiazol-2-ylamino)sulfonyl] benzoate (32.0 g, 0.101 mol) in dioxane : water (1 : 1, 150 mL) at 5 °C was added lithium hydroxide (12.1 g, 0.505 mol). The mixture was allowed to warm to room temperature and stirred for 4.5 hours. The dioxane was then removed in vacuo and the aqueous washed with EtOAc (2 × 100 mL). The aqueous was acidified by addition to 2N HCI (aq) (1000 mL) and the resulting precipitate filtered and washed with water. Purification was accomplished via recrystallisation from DCM : MeOH (10 : 1) to provide the title compound as a tan coloured solid (12.9 g, 0.043 mol). 1HNMR (d6-DMSO): 6.88 (d, 1H), 7.29 (d, 1H), 7.72–7.98 (m, 3H). LCMS (0.05% formic acid in MeCN) Rt = 1.44 min. MS m/z 303 [M + H]+.
1-[3-Chloro-4-(trifluoromethoxy)phenyl]methanamine
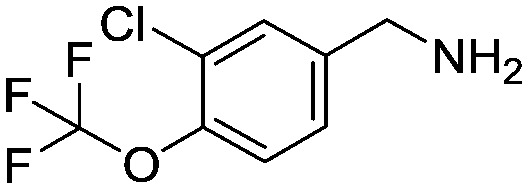
To a solution of 3-chloro-4-(trifluoromethoxy)benzyl bromide (10.0 g, 34.5 mmol) in DMF (100 mL) was added phthalimide (5.6 g, 38.0 mmol) and potassium carbonate (7.15 g, 51.8 mmol) and the mixture stirred at 80 °C for 5 hours. The mixture was cooled and treated with water (100 mL) and the resulting white precipitate filtered, washed with water and dried in vacuo. This crude solid (16 g) was suspended in methylamine (100 mL of a 40% aqueous solution) and water (100 mL) and heated in a steel bomb at 100 °C for 3 hours. Additional methylamine (100 mL of a 40% aqueous solution) was added and heating at 100 °C continued for 8 hours. The reaction was then cooled to room temperature and diluted with brine (300 mL) and TBME (300 mL). The organic layer was separated, dried (Na2SO4) then concentrated in vacuo to provide the title compound as a yellow oil (9.0 g, crude quant.). This material was used crude for subsequent reactions. 1H NMR (d6-DMSO): 3.71 (s, 2H), 7.37–7.41 (m, 1H), 7.44–7.48 (m, 1H), 7.63 (d, 1H).
N-[3-Chloro-4-(trifluoromethoxy)benzyl]-3-fluoro-4-[(1,3-thiazol-2-ylamino)sulfonyl]benzamide, compound 22
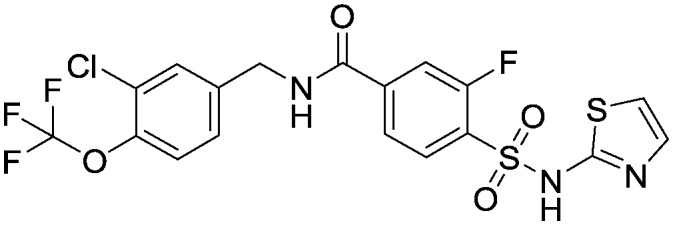
To a suspension of 3-fluoro-4-[(1,3-thiazol-2-ylamino)sulfonyl]benzoic acid (2.88 g, 9.54 mmol) and 1-[3-chloro-4-(trifluoromethoxy)phenyl]methanamine (3.00 g, 11.4 mmol) and Et3N (4.81 ml, 34.5 mmol) in DMSO (30 mL) was added HBTU (4.34 g, 34.5 mmol) and the mixture stirred at room temperature for 18 hours. The reaction was then diluted with EtOAc (100 ml) before washing with water (100 mL) and brine (3 × 100 mL). The EtOAc layer was dried (Na2SO4), filtered and concentrated in vacuo leaving a brown solid (6.38 g). The crude product was triturated with MTBE, dried in vacuo and stirred in EtOAc for 20 minutes and the resulting off-white solid filtered off and dried in vacuo overnight to provide the title compound (3.97 g, 7.8 mmol). 1H NMR (d6-DMSO): 4.50 (d, 2H), 6.90 (d, 1H), 7.30 (d, 1H), 7.40 (d, 1H), 7.50 (d, 1H), 7.60 (s, 1H), 7.75 (s, 1H), 7.80 (d, 1H), 7.90 (t, 1H), 9.25 (t, 1H). LCMS (0.05% formic acid in MeCN) Rt = 1.49 min. MS m/z 510 [M + H]+.
Synthesis of phosphate prodrug 23

Compound 12 (294 mg, 0.58 mmol) was dissolved in 6 mL DMF and cesium carbonate (595 mg, 1.7 mmol) and di-tert-butyl chloromethyl-phosphate (242 mg, 0.94 mmol) added to the mixture. The whole was stirred for 18 h at 60 °C, allowed to cool to room temperature, diluted with EtOAc and then washed with saturated NaHCO3. The mixture was extracted with DCM and the combined organic layers were dried over MgSO4, filtered and evaporated to give a yellow oil. Purification using automated flash column chromatography with 0–10% MeOH/DCM as eluant gave the product as a pale yellow oil. This was dissolved in 5 mL DCM and 1 mL TFA, and then stirred for 2 h. The mixture was then purified directly by preparative HPLC using MeCN and H2O as the mobile phase to give a white solid of the title compound (102 mg, 59%). 1HNMR (d4-MeOH): 4.61 (s, 2H), 5.78 (d, 2H), 6.87 (m, 1H), 7.42 (m, 3H), 7.80 (m, 1H), 7.78 (m, 2H), 8.09 (m, 1H). 19FNMR (d4-MeOH): 60.0. LCMS (0.05% formic acid in MeCN) Rt = 1.16 min. MS m/z 618 [M – H].
Synthesis of compound 39

4-((3-Chloro-4-(trifluoromethoxy)benzyl)oxy)benzenesulfonic acid
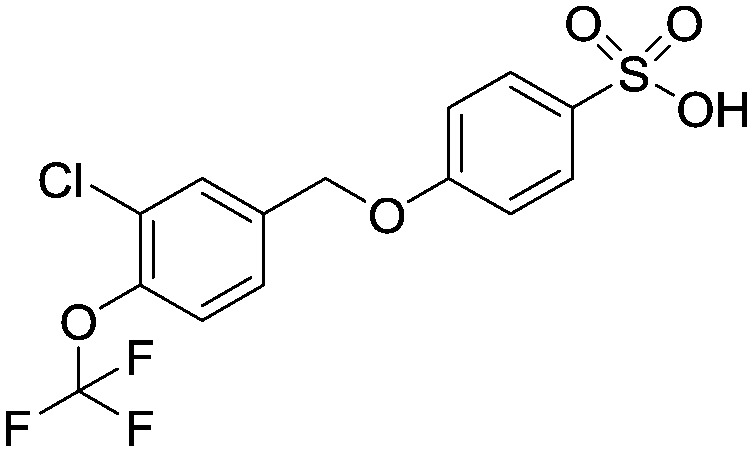
To a solution of sodium 4-hydroxybenzenesulfonate (836 mg, 3.60 mmol) in aqueous sodium hydroxide solution (1 M, 5.0 mL) at room temperature was added (dropwise) 4-(bromomethyl)-2-chloro-1-(trifluoromethoxy)benzene (1.0 g, 3.60 mmol) in ethanol (5 mL). The reaction mixture was heated up to 100 °C and allowed to stir for 16 hours. It was then cooled to room temperature and an aqueous solution of hydrogen chloride (2 M, 10 mL) was added (pH = 1). The organic phase was extracted into ethyl acetate (3 × 10 mL), these were combined and washed with brine (2 × 20 mL), dried over MgSO4, filtered and evaporated to afford the title compound as a white solid (1.80 g, 0.0047 mol). 1HNMR (CDCl3): 5.10 (s, 1H), 6.55 (d, 2H), 7.20 (br s, 1H), 7.45 (d, 2H), 7.50–7.60 (m, 2H), 7.85 (d, 1H). 19FNMR (CDCl3) –57 (s). LCMS (0.05% formic acid in MeCN) Rt = 2.63 min. MS m/z 381 [MH]+.
4-((3-Chloro-4-(trifluoromethoxy)benzyl)oxy)benzene-1-sulfonyl chloride
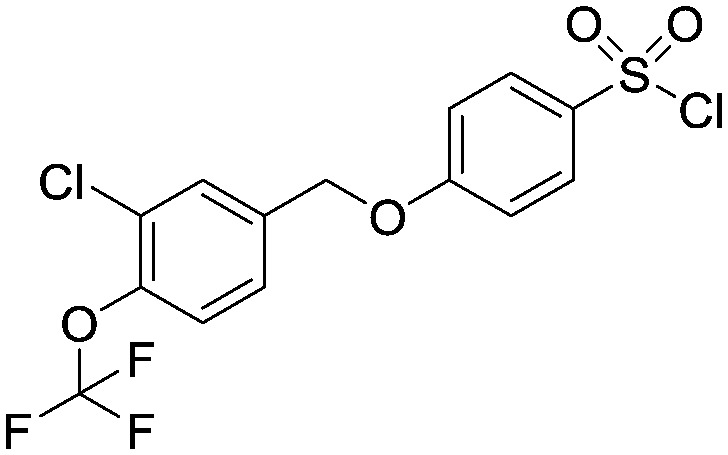
To a solution of 4-((3-chloro-4-(trifluoromethoxy)benzyl)oxy)benzenesulfonic acid (1.80 g, 4.70 mmol) in dichloromethane/dimethylformamide (50 mL/5 mL) was added thionyl chloride (2.88 mL, 47 mmol) dropwise at room temperature. The reaction mixture was stirred at room temperature for 16 hours then concentrated in vacuo and the solid remaining was dried with a high vacuum pump for 3 hours to afford the title compound as a green solid (4.1 g) which was taken on crude into the next step. LCMS (0.05% formic acid in MeCN) Rt = 4.03 min.
4-((3-Chloro-4-(trifluoromethoxy)benzyl)oxy)-N-(2,4-dimethoxybenzyl)-N-(1,2,4-thiadiazol-5-yl)benzenesulfonamide
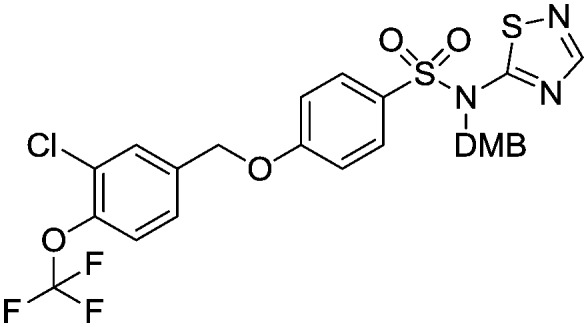
To a solution of N-(2,4-dimethoxybenzyl)-1,2,4-thiadiazol-5-amine (714 mg, 3.0 mmol) in anhydrous dry tetrahydrofuran (15 mL) at –60 °C was added a solution of sodium hexamethyldisilazane (1 M in tetrahydrofuran, 3.0 mL, 3.0 mmol) and the reaction mixture was allowed to warm up to 0 °C and stirred for 1 hour. The reaction mixture was then cooled to –60 °C and a solution of 4-((3-chloro-4-(trifluoromethoxy)benzyl)oxy)benzenesulfonic acid (1.0 g, 2.5 mmol) in tetrahydrofuran/dimethyformamide (5 mL/1 mL) was added dropwise and the reaction mixture was allowed to warm up and stir at room temperature for 16 hours. An aqueous solution of ammonium chloride (20 mL) was added to the reaction mixture and the organics were extracted into ethylacetate (3 × 20 mL). The combined organic phases were washed with brine (3 × 30 mL), dried over MgSO4 filtered and evaporated to give the title compound as a yellow oil (860 mg). This material was taken to the final deprotection step without any further purification.
4-((3-Chloro-4-(trifluoromethoxy)benzyl)oxy)-N-(1,2,4-thiadiazol-5-yl)benzenesulfonamide, compound 39
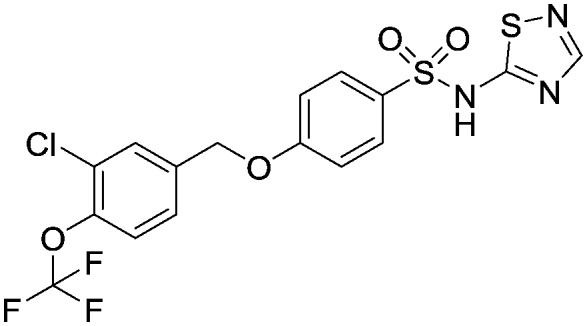
To a solution of 4-((3-chloro-4-(trifluoromethoxy)benzyl)oxy)-N-(2,4-dimethoxybenzyl)-N-(1,2,4-thiadiazol-5-yl)benzenesulfonamide (860 mg, 1.40 mmol) in dioxan (3 mL) at room temperature was added dropwise, hydrogen chloride in dioxan (4 M, 3 mL). The reaction mixture was stirred at room temperature for 2 hours. The solvent was then evaporated in vacuo and the crude oil was purified by reverse phase chromatography (eluting with acetonitrile/water + 0.1% formic acid, 5% to 95% gradient) followed by HTAP using basic conditions to afford the title compound as the diethylamine salt; white solid (18.9 mg, 0.04 mmol). 1HNMR (d4-MeOH): 1.21 (t, 2H), 3.0 (t, 1H), 5.15 (s, 2H), 7.05 (m, 2H), 7.40 (d, 1H), 7.45 (d, 1H), 7.65 (s, 1H), 7.82 (d, 2H), 7.95 (s, 1H). 19FNMR (d4-MeOH): –60.0. LCMS (0.05% formic acid in MeCN) Rt = 2.90 min. MS m/z 464 [MH]+.
Cell line construction and maintenance
Human embryonic kidney (HEK) cells were transfected with an hSCN3A construct using lipofectamine reagent (Invitrogen), using standard techniques. Cells stably expressing the hSCN3A constructs were identified by their resistance to G-418 (400 μg ml–1). Clones were screened for expression using the whole-cell voltage-clamp technique.
Cell culture
HEK cells stably transfected with hSCN3A were maintained in DMEM medium supplemented with 10% heat-inactivated fetal bovine serum and 400 μg ml–1 G418 sulfate in an incubator at 37 °C with a humidified atmosphere of 10% CO2. For HTS, cells were harvested from flasks by trypsinization and replated in an appropriate multi-well plate (typically 96 or 384 wells per plate) such that confluence would be achieved within 24 hours of plating. For electrophysiological studies, cells were removed from the culture flask by brief trypsinization and replated at low density onto glass cover slips. Cells were typically used for electrophysiological experiments within 24 to 72 h after plating.
Electrophysiological recording
Cover slips containing HEK cells expressing hSCN3A were placed in a bath on the stage of an inverted microscope and perfused (approximately 1 ml min–1) with extracellular solution of the following composition: 138 mM NaCI, 2 mM CaCI2, 5.4 mM KCI, 1 mM MgCI2, 10 mM glucose, and 10 mM HEPES, pH 7.4, with NaOH. Pipettes were filled with an intracellular solution of the following composition: 135 mM CsF, 5 mM CsCI2, 2 mM MgCI2, 10 mM EGTA, 10 mM HEPES, pH 7.3 to 7.4, and had a resistance of 1 to 2 mega ohms. The osmolarity of the extracellular and intracellular solutions was 300 mmol kg–1 and 295 mmol kg–1, respectively. All recordings were made at room temperature (22–24 °C) using AXOPATCH 200B amplifiers and PCLAMP software (Axon Instruments, Burlingame, CA) or PatchXpress 7000 hardware and associated software (Axon Instruments, Burlingame, CA). hSCN3A currents in HEK cells were measured using the whole-cell configuration of the patch-clamp technique. Uncompensated series resistance was typically 2 to 5 mega ohms and >85% series resistance compensation (50% for PatchXpress) was routinely achieved. As a result, voltage errors were negligible and no correction was applied. Current records were acquired at 20 to 50 kHz and filtered at 5 to 10 kHz. HEK cells stably transfected with hSCN3A were viewed under Hoffman contrast optics and placed in front of an array of flow pipes emitting either control or compound-containing extracellular solutions. All compounds were dissolved in dimethyl sulfoxide to make 10 mM stock solutions, which were then diluted into extracellular solution to attain the final concentrations desired. The final concentration of dimethyl sulfoxide (<0.3% dimethyl sulfoxide) was found to have no significant effect on hSCN3A sodium currents. The voltage-dependence of inactivation was determined by applying a series of depolarizing prepulses (8 s long in 10 mV increments) from a negative holding potential. The voltage was then immediately stepped to 0 mV to assess the magnitude of the sodium current. Currents elicited at 0 mV were plotted as a function of prepulse potential to allow estimation of the voltage midpoint of inactivation (V1/2). Cells were then voltage clamped at the empirically determined compounds were tested for their ability to inhibit hSCN3A sodium channels by activating the channel with a 20 ms voltage step to 0 mV following an 8 second conditioning prepulse to the empirically determined V1/2. Compound effect (% inhibition) was determined by difference in current amplitude before and after application of test compounds. For ease of comparison, “estimated IC50” (EIC50) values were calculated from single point electrophysiology data by the following equation, (tested concentration, μM) × (100% inhibition/% inhibition). Inhibition values <20% and >80% were excluded from the calculation. In some cases electrophysiological assays were conducted with PatchXpress 7000 hardware and associated software (Molecular Devices Corp). All assay buffers and solutions were identical to those used in conventional whole-cell voltage clamp experiments described above. hSCN3A cells were grown as above to 50–80% confluency and harvested by trypsinization. Trypsinized cells were washed and resuspended in extracellular buffer at a concentration of 1 × 106 cells per ml. The onboard liquid handling facility of the PatchXpress was used for dispensing cells and application of test compounds. Determination of the voltage midpoint of inactivation was as described for conventional whole-cell recordings. Cells were then voltage-clamped to the empirically determined V1/2 and current was activated by a 20 ms voltage step to 0 mV. Electrophysiological assays were also conducted using the lonworks Quattro automated electrophysiological platform (Molecular Devices Corp). Intracellular and extracellular solutions were as described above with the following changes, 100 mg ml–1 amphotericin was added to the intracellular solution to perforate the membrane and allow electrical access to the cells. hSCN3A cells were grown and harvested as for PatchXpress and cells were resuspended in extracellular solution at a concentration of 3–4 × 106 cells per ml. The onboard liquid handling facility of the lonworks Quattro was used for dispensing cells and application of test compounds. A voltage protocol was then applied that comprised of a voltage step to fully inactivate the sodium channels, followed by a brief hyperpolarized recovery period to allow partial recovery from inactivation for unblocked sodium channels, followed by a test depolarized voltage step to assess magnitude of inhibition by test compound. Compound effect was determined based on current amplitude difference between the pre-compound addition and post-compound addition scans.
In vivo electrophysiology
All experimental procedures were conducted in accordance with UK Animals (Scientific Procedures) Act 1986 and followed the guidelines of the International Association for the Study of Pain. All studies were reviewed and approved by the internal Pfizer Institutional Animal Care and Use committee prior to any in vivo experiments commencing. Animals received surgery for tibial nerve transection (TNT) 2 to 3 weeks prior to recordings. All surgical procedures were conducted under sterile conditions in a dedicated surgery suite. Animals were assigned randomly to each treatment group and all drugs were given in a blinded fashion. Fresh compound was prepared each week. For in vivo electrophysiology, animals were anesthetized with isoflurane (5% in O2) and placed in a stereotaxic frame. Body temperature was maintained at 37 °C using a homeothermic heating blanket and a laminectomy was performed to expose the L4–5 segments of the spinal cord. Extracellular spinal cord recordings were made from single dorsal horn neurones using parylene-coated tungsten electrodes (A-M Systems, Sequim, WA, USA). Neurones responsive to light touch, mechanical punctate stimuli, noxious pinch and heat (50 °C) were selected for this study. Neuronal activity evoked by different modalities was quantified over 10 seconds. Once stable baseline responses were obtained, compound was administered via an IV bolus and drug effects were assessed every 15 min for up to 30 minutes per dose. Data were captured and analyzed by a CED 1401 interface coupled to a computer with Spike 2 software (Cambridge Electronic Designs). Dry blood spot samples were obtained for measurement of compound exposure in plasma.
Gene expression data
Ipsi and contralateral L5 DRG were harvested from SNT rats 14 days post injury. RNA was isolated from the dissected tissue using the RNeasy micro plus kit (Qiagen). Given the small quantities of RNA isolated, amplified cDNA was generated from the purified RNA using the Quantitech whole transcriptome amplification kit (Qiagen). Quantitative PCR analysis was performed on amplified cDNA using TaqMan assays (Life technologies) for rat Nav1.3, ATF3 and beta actin genes. Assays were performed in Applied Biosystems 7900HT real time PCR instrument to determine cycle threshold (Ct) values for each gene in each cDNA sample.
Acknowledgments
The authors would like to thank Kiyoyuki Omoto and Andy Pike for helpful discussions during the course of these studies.
Footnotes
†All authors are, or were at the time of the work being undertaken, employees of Pfizer.
References
- (a) Agnew W. S., Levinson S. R., Brabson J. S., Raftery M. A. Proc. Natl. Acad. Sci. U. S. A. 1978;75:2606. doi: 10.1073/pnas.75.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bagal S. K., Chapman M. L., Marron B. E., Prime R., Storer R. I., Swain N. A. Bioorg. Med. Chem. Lett. 2014;24:3690. doi: 10.1016/j.bmcl.2014.06.038. [DOI] [PubMed] [Google Scholar]
- (a) Bagneris C., Naylor C. E., McCusker E. C., Wallace B. A. J. Gen. Physiol. 2014;145:5. doi: 10.1085/jgp.201411242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Catterall W. A. Exp. Physiol. 2014;99:35. doi: 10.1113/expphysiol.2013.071969. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Payandeh J., Minor D. L. J. Mol. Biol. 2015;427:3. doi: 10.1016/j.jmb.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagal S. K., Brown A. D., Cox P. J., Omoto K., Owen R. M., Pryde D. C., Sidders B., Skerratt S. E., Stevens E. B., Storer R. I., Swain N. A. J. Med. Chem. 2013;56:593. doi: 10.1021/jm3011433. [DOI] [PubMed] [Google Scholar]
- De Lera Ruiz M., Kraus R. L. J. Med. Chem. 2015;58(18):7093. doi: 10.1021/jm501981g. [DOI] [PubMed] [Google Scholar]
- Cestele S., Catterall W. A. Biochimie. 2000;82(9–10):883. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- (a) Payandeh J., Scheuer T., Zheng N., Catterall W. A. Nature. 2011;475:353. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Payandeh J., Gamal T. M., Scheuer T., Zheng N., Catterall W. A. Nature. 2012;486:135. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bagneris C., DeCaen P. G., Naylor C., Pryde D. C., Nobeli I., Clapham D. E., Wallace B. A. Proc. Natl. Acad. Sci. U. S. A. 2014;111:8428. doi: 10.1073/pnas.1406855111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ahuja S., Mukund S., Deng L., Khakh K., Chang E., Ho H., Shriver S., Lin S., Johnson J. P., Wu P., Li J., Coons M., Tam C., Brillantes B., Sampang H., Mortara K., Bowman K. K., Clark K. R., Estevez A., Xie Z., Verschoof H., Grimwood M., Dehnhardt C., Andrez J.-C., Focken T., Sutherlin D. P., Safina B. S., Starovasnik M. A., Ortwine D. F., Franke Y., Cohen C. J., Hackos D. H., Koth C. M., Payandeh J. Science. 2015;350:1491. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- (a) Ulmschneider M. B., Bagneris C., McCusker E. C., DeCaen P. G., Delling M., Clapham D. E., Ulmschneider J. P., Wallace B. A. Proc. Natl. Acad. Sci. U. S. A. 2013;110:6364. doi: 10.1073/pnas.1214667110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kaczmarski J. A., Corry B. Channels. 2014;8:264. doi: 10.4161/chan.28136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S., Mukund S., Deng L., Khakh K., Chang E., Ho H., Shriver S., Young C., Lin S., Johnson J. P., Wu P., Li J., Coons M., Tam C., Brillantes B., Sampang H., Mortara K., Bowman K. K., Clark K. R., Estevez A., Xie Z., Verschoof H., Grimwood M., Dehnhardt C., Andrez J.-C., Focken T., Sutherlin D. P., Safina B. S., Starovasnik M. A., Ortwine D. F., Franke Y., Cohen C. J., Hackos D. H., Koth C. M., Payandeh J. Science. 2015;350:aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- (a) Bagal S. K., Bungay P. J., Denton S. M., Gibson K. R., Glossop M. S., Hay T. L., Kemp M. I., Lane C. A. L., Lewis M. L., Maw G. N., Million W. A., Payne C. E., Poinsard C., Rawson D. J., Stammen B. L., Stevens E. B., Thompson L. R. ACS Med. Chem. Lett. 2015;6:650. doi: 10.1021/acsmedchemlett.5b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang L., Zellmer S. G., Printzenhoff D. M., Castle N. A. Br. J. Pharmacol. 2015;172:4905. doi: 10.1111/bph.13259. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sheets P. L., Heers C., Stoehr T., Cummins T. R. J. Pharmacol. Exp. Ther. 2008;326:89. doi: 10.1124/jpet.107.133413. [DOI] [PubMed] [Google Scholar]
- Zimmermann N., Grootenhuis P. D. J., Numa M. M. D., Stamos D., Anderson C. D. and Whitney T., WO2010002956, January 7, 2010.
- (a) Beaudoin S., Johnson M. S., Marron B. E. and Suto M. J., WO2009012241, January 22, 2009.; (b) Marron B. E., Fritch P. C., Markworth C. J., Maynard A. T. and Swain N. A., WO2008118758, October 2, 2008.; (c) Markworth C. J., Marron B. E. and Swain N. A., WO2010035166, April 1, 2010.
- McCormack K., Santos S., Chapman M. L., Krafte D. S., Marron B. E., West C. W., Krambis M. J., Antonio B. M., Zellmer S. G., Printzenhoff D., Padilla K. M., Lin Z., Wagoner P. K., Swain N. A., Stupple P. A., de Groot M., Butt R. P., Castle N. A. Proc. Natl. Acad. Sci. U. S. A. 2013:E2724. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribb A. E., Miller M., Leeder J. S., Hill J., Spielberg S. P. Drug Metab. Dispos. 1991;19(5):900–906. [PubMed] [Google Scholar]
- Focken T., Liu S., Chahal N., Dauphinais M., Grimwood M. E., Chowdhury S., Hemeon I., Bichler P., Bogucki D., Waldbrook M., Bankar G., Sojo L. E., Young C., Lin S., Shuart N., Kwan R., Pang J., Chang J. H., Safina B. S., Sutherlin D. P., Johnson J. P., Dehnhardt C. M., Mansour T. S., Oballa R. M., Cohen C. J., Robinette C. L. ACS Med. Chem. Lett. 2016;7(3):277–282. doi: 10.1021/acsmedchemlett.5b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) He G.-X., Krise J. P., Oliyai R. Biotechnol.: Pharm. Aspects. 2007;5:223. [Google Scholar]; (b) Landis M. S. Ther. Delivery. 2013;4(2):225. doi: 10.4155/tde.12.150. [DOI] [PubMed] [Google Scholar]
- Hains B. C., Waxman S. G. Prog. Brain Res. 2007;161:195. doi: 10.1016/S0079-6123(06)61013-3. [DOI] [PubMed] [Google Scholar]
- (a) Tan A. M., Samad O. A., Dib-Hajj S. D., Waxman S. G. Mol. Med. 2015;21:544. doi: 10.2119/molmed.2015.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Samad O. A., Tan A. M., Cheng X., Foster E., Dib-Hajj S. D., Waxman S. G. Mol. Ther. 2013;21:49. doi: 10.1038/mt.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Estacion M., Gasser A., Dib-Hajj S. D., Waxman S. G. Exp. Neurol. 2010;224:362. doi: 10.1016/j.expneurol.2010.04.012. [DOI] [PubMed] [Google Scholar]; (d) Hains B. C., Klein J. P., Saab C. Y., Craner M. J., Black J. A., Waxman S. G. J. Neurosci. 2003;23:8881. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R., Liu D., Chhoa M., Li C. M., Luo Y., Zang M., Lehto S. G., Immke D. C., Moyer B. D. Int. J. Neurosci. 2016;126:182. doi: 10.3109/00207454.2015.1004172. [DOI] [PubMed] [Google Scholar]
- Xu W., Zhang J., Wang Y., Wang L., Wang X. Neuroreport. 2016;27:929. doi: 10.1097/WNR.0000000000000632. [DOI] [PubMed] [Google Scholar]
- Nassar M. A., Baker M. D., Levato A., Ingram R., Mallucci G., McMahon S., Wood J. Mol. Pain. 2006;2:33. doi: 10.1186/1744-8069-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Kalgutkar A. S., Didiuk M. T. Chem. Biodiversity. 2009;6:2115. doi: 10.1002/cbdv.200900055. [DOI] [PubMed] [Google Scholar]; (b) Dalvie D. K., Kalgutkar A. S., Khojasteh-Bakht S. C., Obach R. S., O'Donnell J. P. Chem. Res. Toxicol. 2002;15:269. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]