

 A series of novel 7-azabenzofurans exhibiting pan-genotype inhibition of HCV NS5B polymerase via binding to the primer grip site is presented.
A series of novel 7-azabenzofurans exhibiting pan-genotype inhibition of HCV NS5B polymerase via binding to the primer grip site is presented.
Abstract
The development of a series of novel 7-azabenzofurans exhibiting pan-genotype inhibition of HCV NS5B polymerase via binding to the primer grip site is presented. Many challenges, including poor oral bioavailability, high clearance, bioactivation, high human serum shift, and metabolic stability were encountered and overcome through SAR studies. This work culminated in the selection of BMS-986139 (43) as a preclinical candidate.
Introduction
Hepatitis is a condition characterized by inflammation of the liver. Viral infection is the most predominant cause of hepatitis, and hepatitis C virus (HCV) is currently the leading cause of liver cirrhosis and liver cancer across the globe.1 HCV is a blood-borne virus that specifically targets the liver. Of those who become infected with HCV, nearly 20% will clear the disease without medical intervention while the remaining 80% face chronic infection, which persists for a lifetime without treatment with antiviral therapy. Progression of the disease may ultimately lead to cirrhosis, hepatocellular carcinoma or both.1 Furthermore, estimates have suggested that more than 15 000 HCV-related deaths occur annually in the US, and upwards of 1 000 000 deaths per year globally, with an inordinate percentage of deaths occurring in underdeveloped nations.
HCV has a wide genetic diversity with seven major genotypes (GT) designated 1–7, of which the most recently identified is GT-7,2 with each comprised of subtypes. The predominant GT varies from country to country with GT-1a predominating in the in the U.S. and Western Europe while GT-1b predominates in Japan and China. The HCV NS5B polymerase is error-prone due to the lack of an effective proof reading function. The high replication rate, estimated to be up to 1 trillion viral particles a day, 10- to 100-fold higher than HIV-1, with a median rate of mutation recently estimated at 2.5 × 10–5 or 1 nucleotide change for every 10 genomes replicated, anticipated the need for combination therapy.3,4 Combination therapy, in which two or three direct-acting antiviral agents (DAAs) with complementary mechanisms of action are co-administered in order to minimize the emergence of resistance, has become the therapeutic standard.1
Until 2014, the standard of care (SOC) for the treatment of HCV relied upon once-weekly injections of the immune modulator pegylated interferon-α (PEG-IFN) in combination with twice-daily dosing of the nucleoside analogue ribavirin (RBV). This regimen was only effective in curing GT-1 infections approximately 45% of the time after 48 weeks of therapy and was associated with a high incidence of side effects that included severe flu-like symptoms, depression, insomnia, arthritis and gastrointestinal complications. The addition of either of the NS3 protease inhibitors telaprevir or boceprevir (Fig. 1) to PEG-IFN/RBV therapy improved the sustained virological response (SVR) in GT-1 patients to nearly 70% under the idealized conditions of clinical trials, although additional treatment-specific side effects including anemia, neutropenia, and severe rash were often observed. The 2014 approval of the dual combination of the NS5A inhibitor daclatasvir5 and the NS3 protease inhibitor asunaprevir6 in Japan represented the first all oral, interferon-free regimen for the treatment of GT-1b patients. Harvoni®, the combination of the nucleoside analogue sofosbuvir and the NS5A inhibitor ledipasvir, was approved in the USA in 2014 but is not optimized for the treatment of GT-2 and GT-3-infected patients. More recently, the approval of Eplcusa®, a combination of sofosbuvir and velpatasvir, provides an interferon-free treatment regimen for all HCV GTs.
Fig. 1. Former and currently marketed HCV drugs.

Of the non-structural (NS) genes comprising the HCV genome, Bristol-Myers Squibb has explored inhibitors of NS5A,5 the NS3 protease,6 NS5B site I7 and NS5B primer grip, also known as site II.8 This communication details further efforts toward the design of inhibitors of site II of the RNA-dependent RNA polymerase NS5B. Essential for replication, the NS5B polymerase has multiple allosteric sites, with site II located approximately 10 Å from the active site and characterized as being both large and flexible in nature. The most prominent site II inhibitor to date is HCV-796 which embodies a benzofuran core and was progressed into phase II clinical trials before being terminated due to liver toxicity.9,10 Influenced by the efficacy of HCV-796 and the promise of the benzofuran chemotype, our own early studies merged this chemotype with learnings from our anthranilic acid inhibitor investigations11 to arrive at a family of benzofuran-based compounds represented generically by 1 that embodies variably substituted phenyl substituents at C5 and no substitution at C6.8,12–14 A single representative from this chemotype was advanced into clinical studies where it demonstrated excellent pharmacokinetic (PK) properties in normal healthy volunteers. However, a notable drawback of this early family of compounds was the lack of pan-genotype coverage, which focused further optimization of this chemotype on broadening the GT coverage.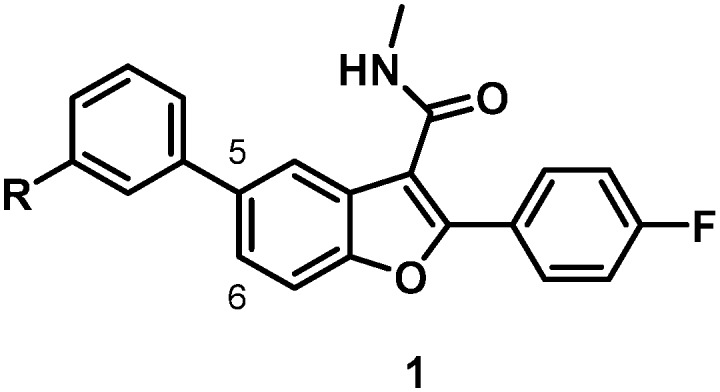
Results and discussion
The single antiviral deficiency associated with inhibitors based on 1 is the lack of inhibition of GT-2a virus. Addressing this gap in GT-2 coverage became the key objective for a backup program which was part of a broader effort to identify a pan-GT-inhibiting drug combination. The inhibitory profile targeted was one that encompassed the major GTs 1–6 with extension to the pre-existing GT-1b C316N mutant, with EC50 values of less than 20 nM in replicons. From the perspective of human PK properties, a low human dose projection to facilitate fixed-dose combinations with once daily administration was sought.
With an aim towards maintaining a C5 aryl-substituted fused 6/5 bicyclic core silhouette similar to 1, a pyrazolopyridine as well as the 4-, 6-, and 7-azabenzofuran cores were evaluated from which the most promising with respect to targeted inhibitory activity and PK properties was the 7-azabenzofuran (Fig. 2). Early explorations of a series of pyrazolopyridine analogues yielded compounds with EC50 values toward the C316N mutant replicon in excess of 500 nM and poor PK properties, while the 4- and 6-azabenzofuran analogues showed disappointing levels of inhibitory activity toward GT-1a, 1b, 2a and GT-1b C316N mutant replicons.
Fig. 2. Alternative cores evaluated.

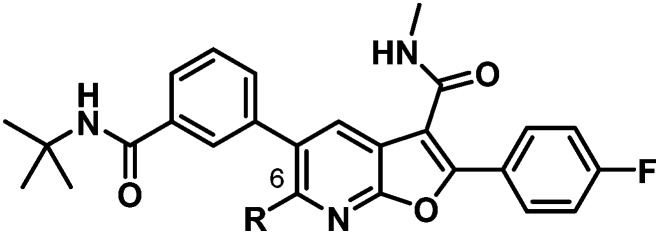
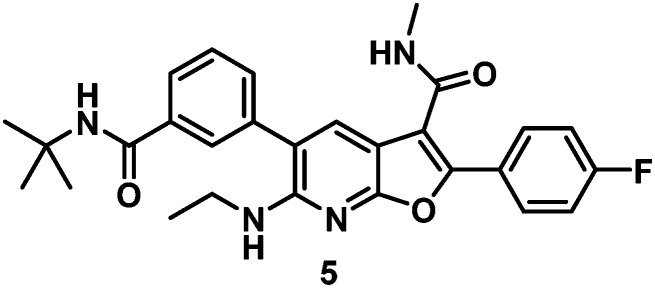
A critical discovery toward achieving GT-2a inhibitory activity in both the benzofuran and the 7-azabenzofuran series was the introduction of substitution at C6 of the core heterocycle. In the absence of C6 substitution GT-2a replicon inhibitory activity was well beyond the targeted upper end EC50 value of 20 nM (compound 2, Table 1). Introduction of a C6 ethyl ether (3) improved GT-2a inhibitory activity presumably through increased van der Waals contact with the protein via partially filling a narrow pocket abutting glutamine 414. Further improvements to GT-2a replicon inhibition were achieved with the incorporation of a propyl (4) or an N-ethyl (5) substituent, resulting in EC50 values of 23 nM and 18 nM, respectively. The near 3-fold improvement in GT-2a inhibition for both 4 and 5 may, in part, be rationalized by hydrogen-bonding to glutamine 414, a weak hydrogen-bond with one of the polarized benzylic protons of 4 and a stronger hydrogen-bond with the NH of 5 (Fig. 3).
Table 1. SARs associated with C6 azabenzofuran substitution.
| |||||
| Cmpd | R | GT-1a EC50 | GT-2a EC50 | GT-1b C316N IC50 a | CC50 b |
| 2 | H | 600 nM | 346 nM | 447 nM | 13 μM |
| 3 | CH3CH2O | 112 nM | 57 nM | 61 nM | >100 μM |
| 4 | CH3CH2CH2 | 45 nM | 23 nM | 5 nM | >100 μM |
| 5 | CH3CH2NH | 22 nM | 18 nM | 17 nM | >100 μM |
aEnzyme data (polyC:pGpG).
bGT-1b LUC CC50: see ESI for experimental details.
Fig. 3. Co-crystal structure of 5 bound to NS5B GT-2a L30S protein. Compound 5 is depicted in ball and stick representation with orange carbon atoms. Protein is shown with a green backbone cartoon and specific residues are displayed in stick representation with either green or cyan (Q414) carbon atoms. Hydrogen-bonds are denoted as black dotted lines. The omitted Fo–Fc electron density is contoured at 3 rmsd in magenta mesh. Image generated with the PyMOL molecular graphics system (v. 1.8, Schrödinger, LLC).15.

With satisfactory replicon EC50 values of less than 20 nM across GTs 1, 2 and the GT-1b C316N mutant and with a >5000-fold in vitro therapeutic window, 5 was examined in a rat PK study. The low peak (Cmax) to trough (C24) ratio, resulting in a nearly flat PK curve, and the long half-life associated with 5 were encouraging data toward the potential for achieving sustained efficacious drug concentrations with a low dose and an infrequent dosing schedule (Table 2).
Table 2. Rat PK data for 5.
| |||||||
| Cmpd a | C max (μM) IV/PO | IV clearance (mL min–1 kg–1) | t 1/2 (h) IV/PO | PO (μM) 24 h liver | PO (μM) 24 h plasma | PO AUC (μM h) | % oral F |
| 5 | 1.7/0.6 | 12 | 14/29 | 1.6 | 0.56 | 18 | 64 |
aIV dose = 2 mg kg–1; PO dose = 6 mg kg–1.
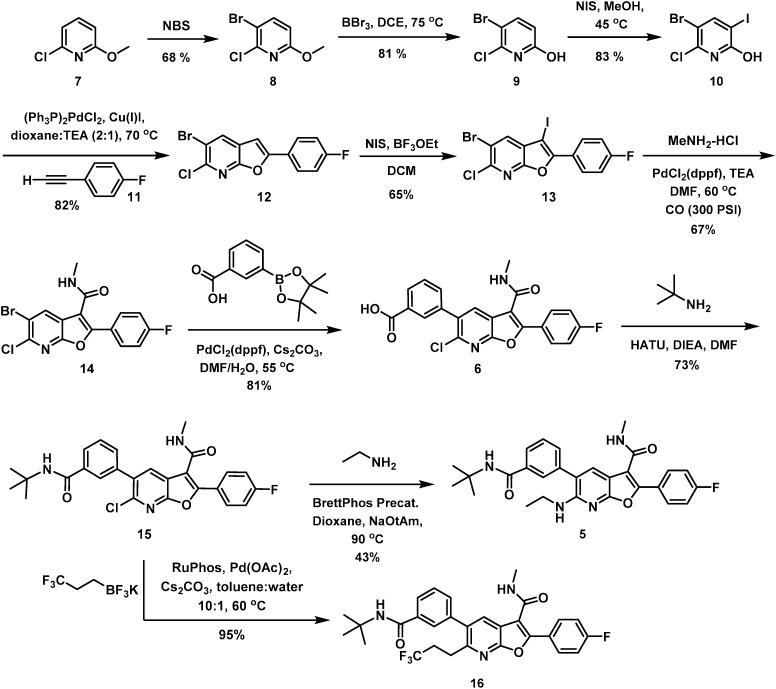
An in vitro metabolism study in an Aroclor-induced rat liver S9 (ref. 16) assay was the next step in the progression of 5. In addition to the observation of amide demethylation, amine demethylation, and oxidation of the tBu amide moiety, 12% of the material was converted to a glutathione (GSH) adduct. This observation heightened concern for the production of metabolism-induced reactive intermediates in vivo that precipitated a systematic evaluation of differential functionality on the western portion of the 7-azabenzofuran core. At this juncture of the program, the eastern portion of the chemotype had been subjected to extensive optimization with the C2 pF-phenyl and C3 methyl amide determined to be optimal. This led to a focus on derivatization of the acid 6, a versatile synthetic intermediate that was prepared by a sequence beginning with 2-chloro-6-methoxypyridine (7). Site-selective bromination of 7 afforded the bromopyridine 8 in 68% yield which was deprotected using BBr3 at 75 °C in DCE to effect clean conversion to the hydroxypyridine 9. N-Iodosuccinimide-mediated iodination of 9 afforded 10 in good yield and was followed by a Sonogashira coupling with the fluoro-substituted phenyl acetylene 11, which proceeded with concomitant cyclization to afford the 7-azabenzofuran 12. C3 iodination with NIS in the presence of BF3(OEt)2 furnished 13 in acceptable yield. Carbonylative coupling at C3 of 13 in the presence of MeNH2 resulted in the formation of the C3 methyl amide 14 in 67% yield. Finally, a selective Suzuki coupling at C5 of 14 produced 6. For the purposes of illustrating the preparation of 5, HATU-mediated coupling between 6 and tBu amine gave 15 which was subsequently substituted at C6 under standard Buchwald coupling conditions to give 5 in 43% yield. Similarly, Pd-mediated coupling between 15 and a suitable coupling partners such as potassium trifluoro(trifluoroethyl) borate furnished C6 carbon analogues such as 16 in excellent yield.
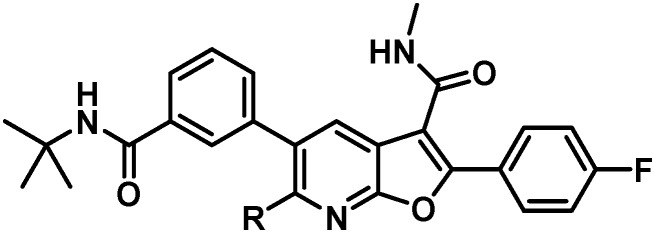
With the observation of in vitro oxidation of the tBu amide and GSH conjugation, the identification of a suitable combination of amide and C6 substitution that maintained all of the positive aspects of 5 while improving metabolic stability in vitro and reducing the extent of bioactivation became the objective. Attention was first directed toward optimizing C6, with a particular focus toward nitrogen- and carbon-linked substituents. With a reliable route to attain significant quantities of 6, SAR evaluation on the western portion of the 7-azabenzofuran core could be rapidly evaluated through amide coupling followed by Pd-mediated derivatization at C6. Table 3 summarizes the key SARs associated with a select set of C6 substitutions that were evaluated.
Table 3. Optimization of C6 azabenzofuran substitution.
| |||||
| Cmpd | R | GT-1a EC50 | GT-2a EC50 | GT-1b C316N IC50 a | CC50 b |
| 5 | CH3CH2NH | 18 nM | 13 nM | 16 nM | >100 μM |
| 16 | CF3CH2CH2 | 14 nM | 6 nM | 10 nM | >100 μM |
| 17 | CF3CH2NH | 13 nM | 5 nM | 10 nM | >100 μM |
| 18 | CH3CH2CH2NH | 29 nM | 15 nM | 35 nM | >100 μM |
| 19 | CF3CH2CH2NH | 13 nM | 13 nM | 20 nM | >100 μM |
| 20 | CH3CF2CH2NH | 55 nM | 39 nM | 96 nM | >100 μM |
| 21 | CH3CH2NCH3 | 77 nM | 31 nM | 31 nM | 92 μM |
| 22 | CH3 | 337 nM | 139 nM | 362 nM | 77 μM |
| 23 | i-Pr | 109 nM | 43 nM | 64 nM | >100 μM |
| 24 | n-Butyl | 142 nM | 54 nM | 66 nM | >100 μM |
| 25 | HO(CH2)3CH2 | 145 nM | 359 nM | 187 nM | >100 μM |
aEnzyme data (polyC:pGpG).
bGT-1b LUC CC50: see ESI for experimental details.
Compared to the ethyl amine, a small improvement in GT-2a inhibition was observed in association with the slight increase size and lipophilicity associated with terminal fluorination, as exemplified by 17, or with the installation of the trifluoropropyl substituent in 16. Attempts to shorten the C6 substituent resulted in a significant loss in replicon inhibitory activity (22). Extending the C6 N-substituents (18) resulted in slightly decreased inhibitory activity toward GT-1a and GT-1b C316N while the terminally fluorinated 19 maintained the activity of 5. Lengthening the chain in the carbon series (24) or adding terminal polarity (25) had detrimental effects, significantly decreasing antiviral activity in all 3 assays. Branching the C6 chain with fluoro (20) or alkyl (23) substituents or alkylating the nitrogen atom (21) all proved detrimental to activity. Taken together, these data are consistent with a narrow and shallow hydrophobic pocket (Fig. 4) that appears to be optimally filled with either C6 trifluoroethyl amine (17) or C6 trifluoropropyl (16) substitution.
Fig. 4. Co-crystal structure of 5 bound to NS5B GT-1b WT protein. Compound 5 is depicted in ball and stick representation with orange carbon atoms. Protein is shown with a green backbone cartoon and specific residues are displayed in stick representation with either green or cyan (M414) carbon atoms. Hydrogen-bonds are denoted as black dotted lines. The surface of the binding site in the vicinity of C6 of the 7-azabenzofuran is shown in light gray. The omitted Fo–Fc electron density is contoured at 2 rmsd in magenta mesh. Image generated with the PyMOL molecular graphics system (v. 1.8, Schrödinger, LLC).15.

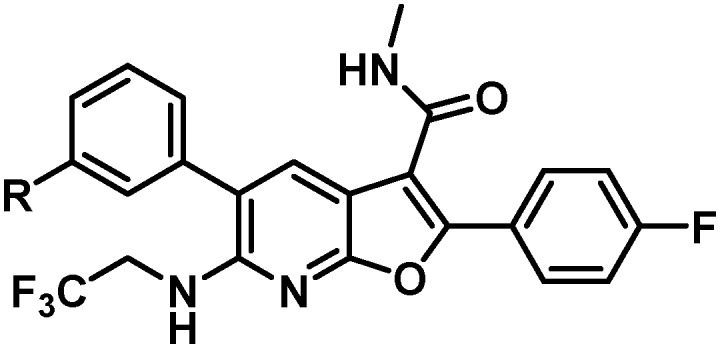
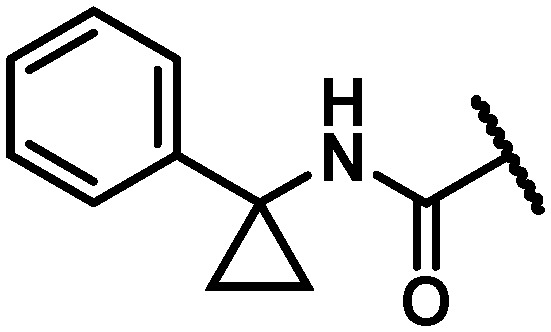
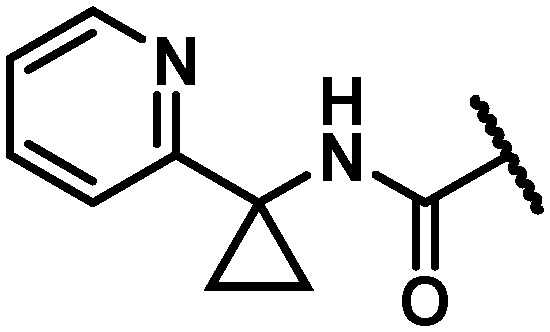
With maintenance or improvement in antiviral activity accompanying two structural permutations at C6 (16 and 17), focus was directed at the amide substitution. Successful deployment of a cyclopropylpyrimidine amide in the context of efforts toward 1, which offered favorable metabolic stability, warranted inquiry into similarly-substituted amides in the 7-azabenzofuran context. The data presented in Table 4 demonstrates that there are minimal differences between cyclopropyl-phenyl (26), -pyridine (27), and -pyrimidine (28) amides with respect to activity toward the GT-1a and GT-2a replicons and the GT-1b C316N mutant. While consistent for this SAR comparison, it should be noted that the cyclopropyl moiety does not universally impart an activity advantage across all of the western aryls evaluated, vide infra. Clearer differences between the amide substitution pattern emerge when evaluating liver microsomal (LM) half-life data, with 26 demonstrating unsatisfactory half-life values in rat, dog and cynomolgus monkey LMs and 27 similarly falling short of a desired half-life in dog. Most differentiated was the cyclopropylpyrimidine 28 with a t1/2 of >120 minutes across all four species of LMs evaluated. It should be mentioned at this point that a five membered heterocycle appended to the cyclopropyl amide that was evaluated early in the program, oxadiazole 29, offered borderline antiviral activity that taken together with relatively low LM half-life data contributed to this chemotype not being pursued at this point of the study. The oxadiazole moiety would, however, reemerge later in the program as possessing a unique balance of PK and antiviral activity (vide infra).
Table 4. Antiviral activity and liver microsomal half-life data for variably substituted C6 aryl amides 26–36.
| |||||||||
| Cmpd | R | GT-1a EC50 (nM) | GT-2a EC50 (nM) | GT-1b C316N IC50 a (nM) | CC50 b (μM) | Liver microsomal half-life
c
(min) |
|||
| Human | Rat | Dog | Cyno | ||||||
| 26 |
|
6 | 10 | 18 | >100 | 60 | 16 | 19 | 8 |
| 27 |
|
17 | 12 | 18 | >100 | 120 | 71 | 37 | 84 |
| 28 |
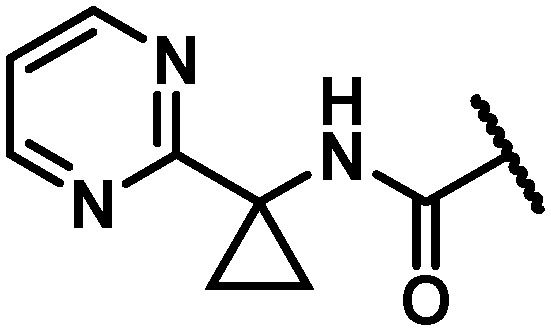
|
12 | 6 | 17 | 13 | 120 | 120 | 120 | 120 |
| 29 |
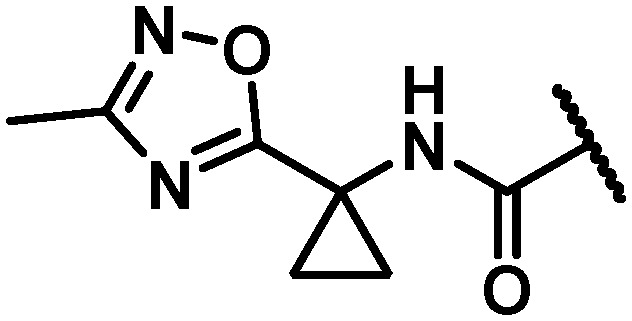
|
25 | 20 | 32 | >100 | 100 | 46 | 11 | 22 |
| 30 |
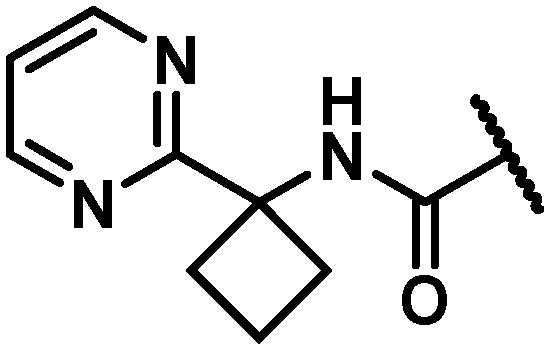
|
4 | 4 | 5 | 2.2 | 50 | 18 | 96 | 1.2 |
| 31 |
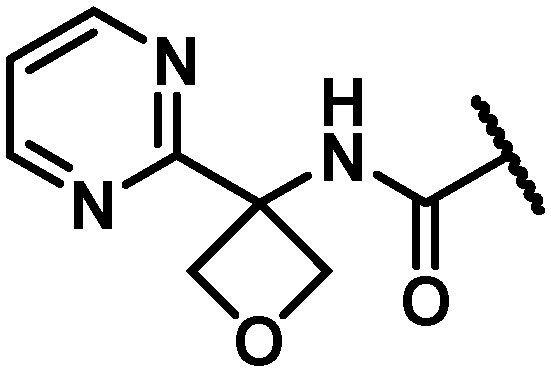
|
35 | 16 | 43 | 9.5 | 64 | 99 | 112 | 13 |
| 32 |
|
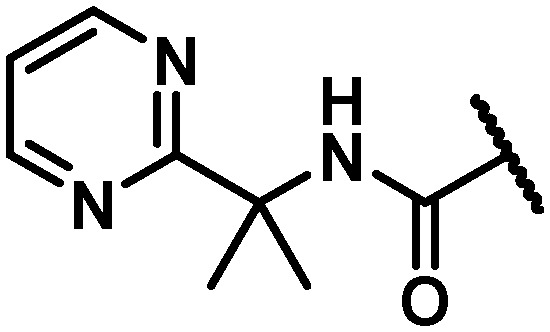
46 | 8 | 50 | 4.2 | >120 | 112 | 20 | 22 |
| 17 |
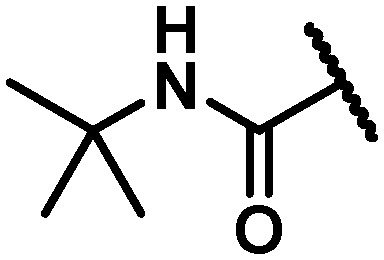
|
11 | 5 | 9 | >100 | 44 | 96 | >120 | 7.9 |
| 33 |
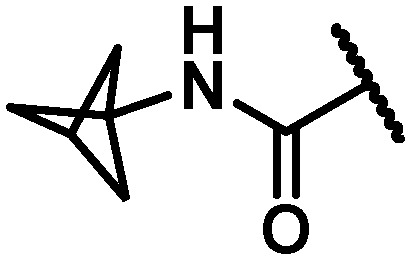
|
10 | 5 | 15 | >100 | >120 | 106 | >120 | 36 |
| 34 |
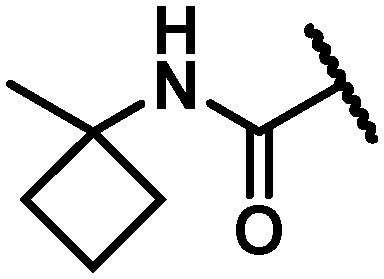
|
8 | 4 | 11 | >100 | 18 | 46 | 1.7 | 13 |
| 35 |
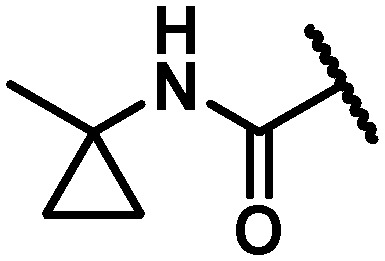
|
32 | 87 | 305 | 13.5 | >120 | >120 | >120 | 39 |
aEnzyme data (polyC:pGpG).
bGT-1b LUC CC50: see ESI for experimental details.
cHuman, rat, dog and cynomolgus monkey liver microsome half-life: see ESI for experimental details.
With the identification of a cyclopropylpyrimidine moiety imparting a good balance of antiviral activity and metabolic stability, efforts were undertaken to identify additional binding interactions through alternative functionalization of the benzylic cyclopropyl element. Table 4 shows that the cyclobutane 30 achieved a 3-fold improvement in GT-1a and GT-1b C316N mutant inhibitory activity compared to 28; however, a short half-life in rat and cynomolgus monkey LMs coupled with the reduced in vitro therapeutic index due to a CC50 value of 2.2 μM eliminated 30 from consideration. Neither the oxetane 31 nor the gem-dimethyl substituted homologue 32 offered an advantage in GT-1a replicon or GT-1b C316N mutant inhibition, thus establishing the cyclopropyl as the preferred substituent for pairing with the pyrimidine.
In an attempt to replicate the PK observed with tBu amide 5 and avoid the metabolic liability associated with this functionality, several tBu isosteres were evaluated. In the context of the C6 trifluoroethylamine substituent, acceptable antiviral activity was observed with the tBu amide 17, the bicyclo[1.1.1]pentane12,17,18 33 and the ipso-substituted methyl cyclobutane 34, while the ipso-substituted methyl cyclopropane 35 fell just outside of the targeted activity value in the GT-1a replicon (Table 4). The tBu amide 17 exhibited poor stability in human and cynomolgus monkey LMs while the t1/2 for the ipso-substituted methyl cyclobutane amide 34 was short in all species LMs. The bicyclo[1.1.1]pentane-based amide 33 exhibited very good stability in human, rat and dog LMs, and while the half-life in cynomolgus monkey LMs was 36 minutes, the overall profile was considered acceptable for further progression, thus positioning the bicyclo[1.1.1]pentane as a tBu isostere suitable for further evaluation.
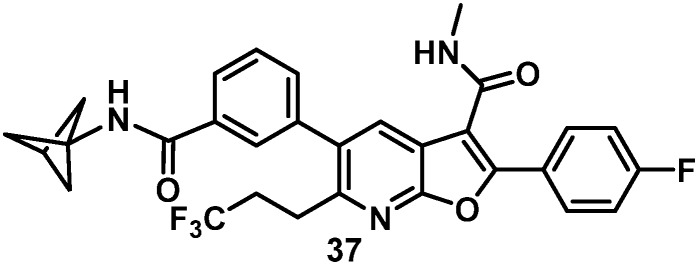
As a result of the SAR evaluation and optimization of C6 substitution and the western amide functionality, the cyclopropylpyrimidine amides 28 and 36 and the bicyclo[1.1.1]pentane amides 33 and 37 were identified as the leading candidates for further study. All four compounds expressed replicon EC50 values of less than 15 nM toward all GTs tested. Both of the C6 trifluoroethylamine analogues 28 and 33 demonstrated time-dependent inhibition (TDI) of cytochrome P450 (CYP) 3A4 when evaluated in human LMs. Detection of 5% GSH adducts upon evaluation of 36 in an in vitro Aroclor-induced rat liver S9 assay halted progression of this compound. Combining the bicyclo[1.1.1]pentane and the C6 trifluoropropane substituents in 37 resulted in a compound with undetectable bioactivation, no CYP liability and an excellent balance of pan genotype activity (Table 5) and good in vitro metabolic stability.
Table 5. Rat PK comparison of 37 and 5 and full GT activity for 37.
| ||||||||||||
| Cmpd a |
C
max (μM) IV/PO |
IV clearance (mL min–1 kg–1) |
t
1/2 (h) IV/PO |
PO (μM) 24 h liver |
PO (μM) 48 h liver | PO (μM) 24 h plasma | PO AUC (μM h) | % oral F | ||||
| 5 | 1.7/0.6 | 12 | 14/29 | 1.6 | na | 0.56 | 18 | 64 | ||||
| 37 | 3.2/0.9 | 7.3 | 14/20 | 0.9 | 0.82 | 0.16 | 10 | 44 | ||||
| cmpd | EC50 (nM) |
CC50 c | EC50 (nM) |
|||||||||
| GT: 1a | 1b | 2a | 2b | 3a | 4a | 5a | 6a | 1b C316N b | 1aFBS | 1aHS | ||
| 37 | 9.6 | 4 | 8 | 3 | 4 | 2 | 0.2 | 10 | 9.6 | >100 μM | 16 | 158 |
aIV dose = 2 mg kg–1; PO dose = 6 mg kg–1.
bEnzyme data (polyC:pGpG).
cGT-1b LUC CC50: see ESI for experimental details.
The rat PK profile of 37 (Table 5) was promising, demonstrating lower clearance and a PO half-life similar to 5 as well as a flat oral PK curve that resulted in very similar Cmax and 24 h liver levels that were sustained for an extended period, declining by an additional 10% at 48 h. When evaluated in the presence of 40% human serum (HS), the EC50 value in the GT-1a replicon increased ten-fold to 158 nM and target exposure in vivo is typically set as a multiple of this value. The HS-adjusted EC50 in the human dose prediction gave an estimated 150 mg QD for 37, twice that of our previous clinical candidate.8 Consequently, further modification of amide substitution was initiated in an effort to improve upon the HS shift and thus lower the human dose prediction, based on the anticipation of final deployment within a fixed dose combination.
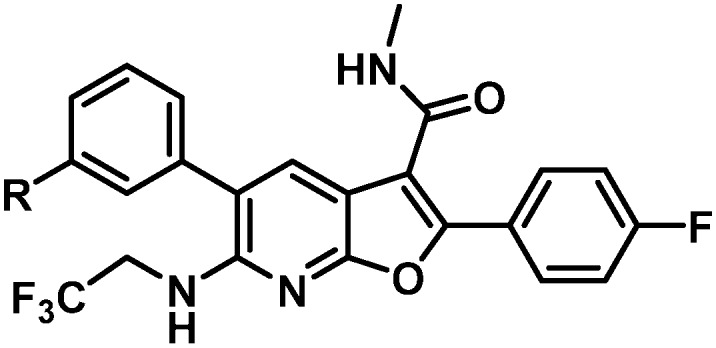
The direction of the next phase of the program emerged from an analysis of the shift in antiviral activity in the presence of HS for a series of analogues. These data indicated that aliphatic or phenyl (26, Table 6) substituents experienced a higher shift in the presence of HS while hetero aromatic substituents were more generally associated with lower HS shifts. While the antiviral potency of both the pyridine 27 and the pyrimidine 28 shifted by 3.6-fold in the presence of HS, the pyridine series was dropped from consideration due to its less than optimal metabolic stability in LMs from several species. Despite a good balance of antiviral activity and metabolic stability, challenges with bioactivation, human liver microsome (HLM) CYP TDI, as well as universally poor rat PK properties in the 7-azabenzofuran series rendered pyrimidines as suboptimal. 1,2,4-oxadiazole 29 showed a similarly low HS shift as the pyridine and pyrimidine and optimization of compounds incorporating this ring system were pursued in an effort to identify compounds with the targeted antiviral profile, a favorable HS shift, and maintenance of all other beneficial aspects of 37.
Table 6. FBS and HS activity data imparted by western aromatic substituents.
| ||||
| Cmpd | R | EC50 (nM) |
HS shift | |
| GT-1a (FBS) | GT-1a (HS) | |||
| 26 |
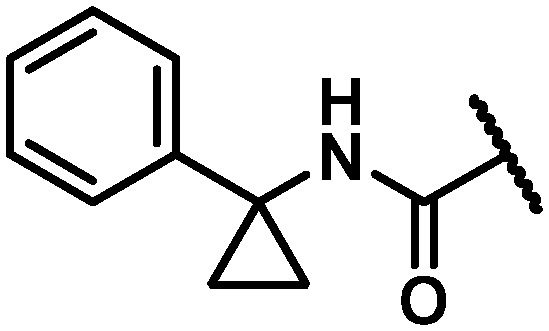
|
8 | 102 | 12.8 |
| 27 |
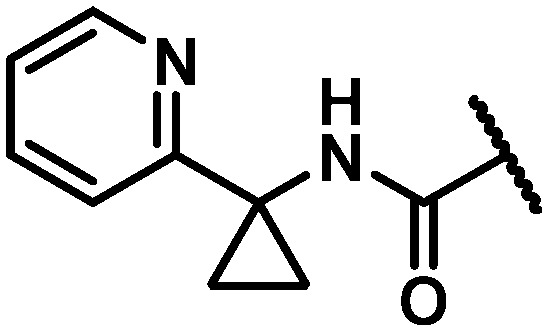
|
22 | 80 | 3.6 |
| 28 |
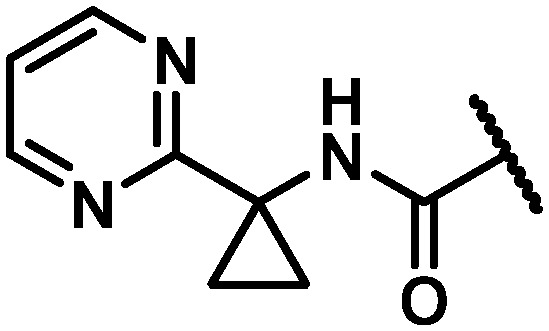
|
31 | 112 | 3.6 |
| 29 |
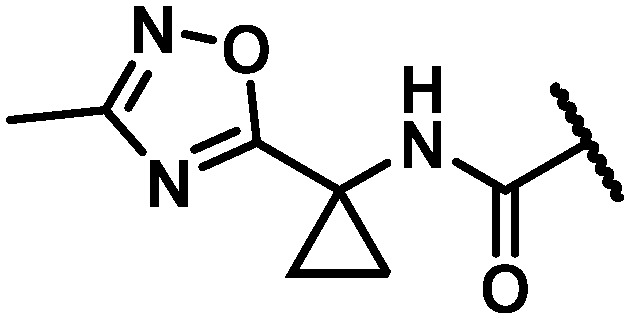
|
56 | 226 | 4 |
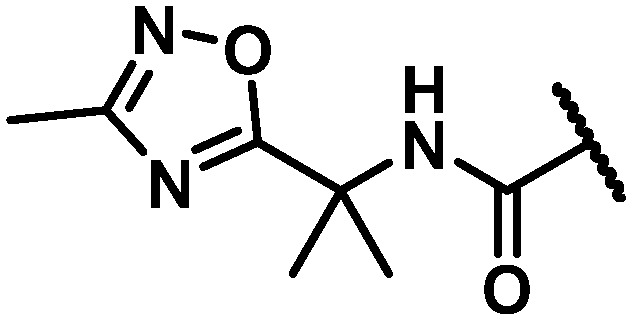
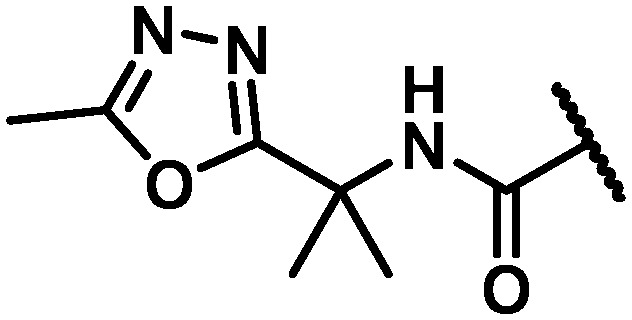
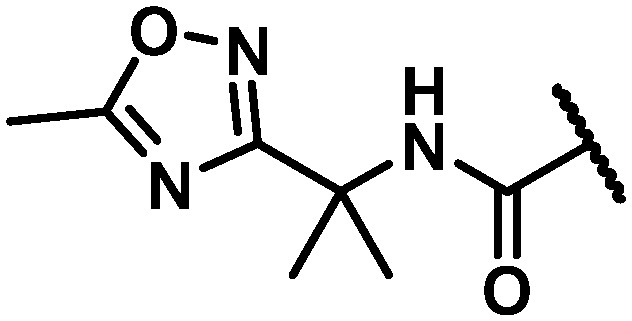
Data for the three oxadiazoles 38–40 evaluated in the context of the more active gem-dimethyl benzylic substitution are compiled in Table 7. Both the methylated 1,2,4-oxadiazole 38 and its topological isomer 40 emerged as possessing excellent antiviral activity. However, LM stability for both of these compounds was marginal while the 2-methyl-1,3,4-oxadiazole 39 failed to meet targeted antiviral activity. In an effort to block suspected oxidative metabolism on the C3 methyl of 40, the trifluoromethylated 1,2,4-oxadiazole 41 was prepared. Unfortunately, fluorination proved detrimental to antiviral activity. A key observation in this series was that removal of substitution from the 1,2,4-oxadiazole offered the best balance of properties, as exemplified by the C6 trifluoroethylamine analogue 42 and the C6 trifluoropropyl analogue 43, both of which exhibited EC50 values of less than 10 nM across all 3 assays, a HS shift of only 5-fold and targeted in vitro metabolic stability.
Table 7. Antiviral activity and liver microsomal half-life data for a series of substituted oxadiazole derivatives.
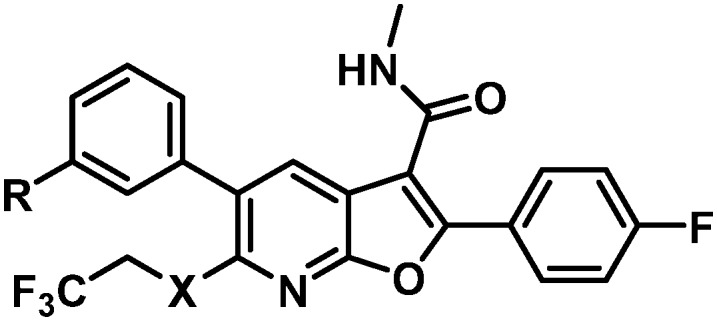
| ||||||||||
| Cmpd | R | x | GT-1a EC50 (nM) | GT-2a EC50 (nM) | GT-1b C316N IC50 a (nM) | CC50 b (μM) | Liver microsomal half-life
c
(min) |
|||
| Human | Rat | Dog | Cyno | |||||||
| 38 |
|
NH | 5 | 3 | 15 | 8.8 | 13 | 25 | 7 | 5 |
| 39 |
|
NH | 21 | 49 | 27 | 5.0 | na | na | na | na |
| 40 |
|
NH | 4 | 2 | 17 | 14.7 | 108 | 59 | 3 | 13 |
| 41 |
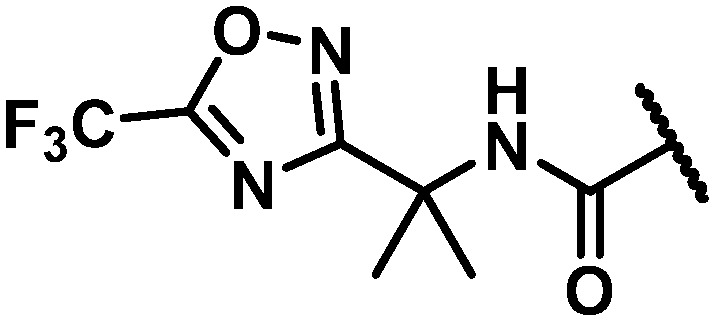
|
NH | 39 | 53 | 99 | >100 | na | na | na | na |
| 42 |
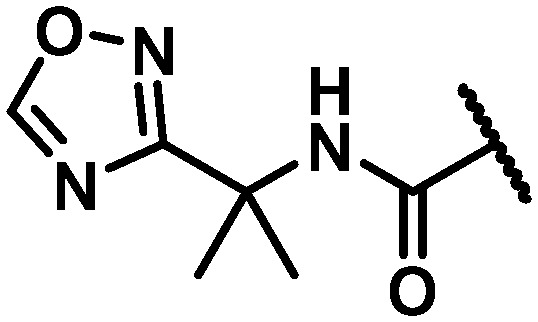
|
NH | 9 | 3 | 9 | 5.2 | >120 | >120 | >120 | >120 |
| 43 |
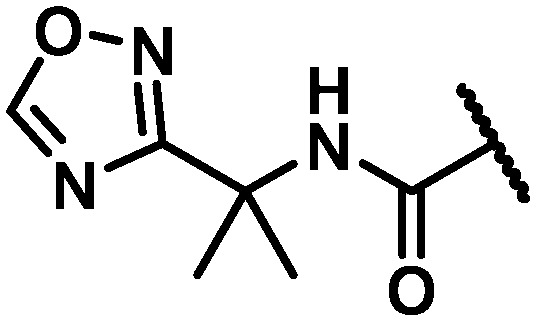
|
CH2 | 9 | 4 | 6 | >100 | >120 | >120 | >120 | 67 |
aEnzyme data (polyC:pGpG).
bGT-1b LUC CC50: see ESI for experimental details.
cHuman, rat, dog and cynomolgus monkey liver microsome half-life: see ESI for experimental details.
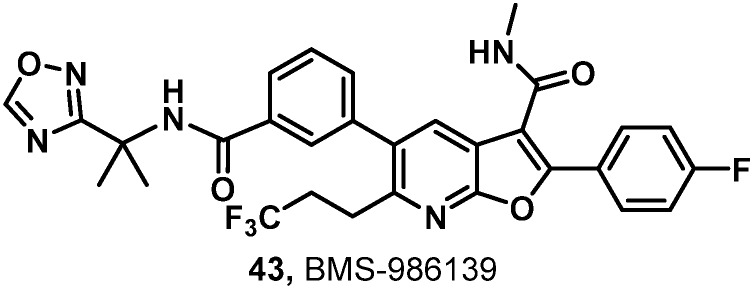
The rat PK results for both 42 and 43 are compared to 37 in Table 8. Trifluoroethylamine 42 demonstrated a slightly lower Cmax, higher IV clearance, and a shorter half-life than 37, while drug levels in plasma and liver 24 hours post-dose were low and oral bioavailability was modest at 31%. With a slightly lower IV and comparable oral Cmax, 43 marked an improvement in clearance and exhibited a similar IV half-life to 37. The oral half-life was comparably short; however, when viewed in context of the high liver concentration of 1.5 μM at 24 hours, nearly matching the benchmark oral liver concentration of 37, 43 was well positioned for further evaluation. When assayed in Aroclor-treated rat liver S9 cells, 43 suffered no observable bioactivation. Additionally, 43 exhibited a dose-proportional increase of exposure across the dosing range of 5, 15 and 30 mg kg–1 in rats while 37 did not.
Table 8. Rat PK comparison of 42 and 43 with 37.
| ||||||
| cmpd a | C max (μM) IV/PO | IV clearance (mL min–1 kg–1) | t 1/2 (h) IV/PO | PO (μM) 24 h plasma | PO AUC (μM h) | % oral F |
| 37 | 3.2/0.9 | 7.3 | 14/20 | 0.91 | 0.16 | 44 |
| 42 | 2.5/0.6 | 9.3 | 5.0/5.4 | 0.38 | 0.039 | 31 |
| 43 | 1.7/1.1 | 5.7 | 12/6.7 | 1.5 | 0.15 | 58 |
aIV dose = 2 mg kg–1; PO dose = 6 mg kg–1.
Table 9 summarizes the measured antiviral activity data as well as the rat, dog and cynomolgus monkey PK for 43. The predicted human dose calculated based on allometric scaling of rat, dog and monkey PK using the HS-adjusted EC50 value of 52 nM was 35 mg QD, less than half of that for the previous clinical compound.8 Clean in both Ames and genotoxicity assays, 43 was progressed into investigational new drug (IND) toxicology studies where its progress was halted do to highly unexpected microcrystallization in multiple tissues at elevated doses in both rats and dogs.
Table 9. Full GT activity data and three species PK for 43.
| ||||||||||||
| Cmpd | EC50 (nM) |
CC50 b | EC50 (nM) |
|||||||||
| GT: 1a | 1b | 2a | 2b | 3a | 4a | 5a | 6a | 1b C316N a | 1a (FBS) | 1a (HS) | ||
| 43 | 9 | 3 | 4 | 3 | 3 | 2 | 0.3 | 6 | 6 | >100 μM | 11 | 52 |
| Species |
IV clearance (mL min–1 kg–1) |
Vss (L kg–1) |
t
1/2 (h) IV/PO |
PO AUC (μM h) |
% oral F |
|||||||
| Rat c | 5.7 | 5.6 | 12/6.7 | 13 | 58 | |||||||
| Dog d | 1.2 | 3.3 | 34/35 | 25 | 85 | |||||||
| Cyno d | 5.8 | 2.8 | 6.1/6.4 | 8.3 | 61 | |||||||
aEnzyme data (polyC:pGpG).
bGT-1b LUC CC50: see ESI for experimental details.
cIV dose = 2 mg kg–1; PO dose 6 mg kg–1.
dIV dose = 1 mg kg–1; PO dose 3 mg kg–1.
Conclusion
With C6 unsubstituted benzofurans 1 as a starting point, a promising new azabenzofuran core was discovered that addressed the absence of HCV GT-2 antiviral activity exhibited by 1 through the introduction of C6 functionality. Systematic evaluation of combinations of substituents at the western amide and C6 resulted in many compounds meeting antiviral activity requirements. The 1,1,1-bicyclopentane amide derivative 37 emerged as a promising lead in all respects. However, stimulated by a desire to minimize the predicted human dose, further efforts identified a 1,2,4-oxadiazole heterocycle as a promising western amide appendage that balanced HS binding while maintaining benchmark levels of antiviral activity and PK properties. Ultimately, the effort was rewarded with the discovery of 43 which met all program objectives and was progressed into IND toxicology studies. While an unusual finding of microcrystallization in many tissues at elevated doses during the toxicological studies halted progression of 43, these data informed a parallel backup effort that focused on improved physicochemical properties. The nature of the structural changes implemented to successfully impart further improvements in PK and solubility will be disclosed in due course.
Ethical statement
All these studies were approved by local institutional review boards and conducted in accordance with Good Clinical Practice, as defined by the International Conference on Harmonization, in accordance with the ethical principles underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50, and in accordance with the ethical principles that have their origin in the Declaration of Helsinki. Informed written consent was obtained from all subjects.
Supplementary Material
Acknowledgments
Use of the IMCA-CAT beamline 17-ID and 17-BM at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00636a
References
- Viral Hepatitis - Hepatitis C Information, Centers for Diseas Control and Prevention, http://www.cdc.gov/hepatitis/hcv/, (October 1, 2016).
- Murphya D. G., Sablonb E., Chamberlanda J., Fourniera E., Dandavinoc R., Tremblaya C. L. J. Clin. Microbiol. 2015;53:967–972. doi: 10.1128/JCM.02831-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelson A. S., Herrmann E., Micol F., Zeuzem S. Hepatology. 2005;42:749–754. doi: 10.1002/hep.20882. [DOI] [PubMed] [Google Scholar]
- Ribeiro R. M., Li H., Wang S., Stoddard M. B., Learn G. H., Korber B. T., Bhattacharya T., Guedj J., Parrish E. H., Hahn B. H., Shaw G. M., Perelson A. S. PLoS Pathog. 2012;8:e1002881. doi: 10.1371/journal.ppat.1002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belema M., Nguyen V. N., Bachand C., Deon D. H., Goodrich J. T., James C. A., Lavoie R., Lopez O. D., Martel A., Romine J. L., Ruediger E. H., Snyder L. B., Laurent D. R. S., Yang F., Zhu J., Wong H. S., Langley D. R., Adams S. P., Cantor G. H., Chimalakonda A., Fura A., Johnson B. M., Knipe J. O., Parker D. D., Santone K. S., Fridell R. A., Lemm J. A., O'Boyle D. R., Colonno R. J., Gao M., Meanwell N. A., Hamann L. G. J. Med. Chem. 2014;57:2013–2032. doi: 10.1021/jm401836p. [DOI] [PubMed] [Google Scholar]
- Scola P. M., Sun L.-Q., Wang A. X., Chen J., Sin N., Venables B. L., Sit S.-Y., Chen Y., Cocuzza A., Bilder D. M., D'Andrea S. V., Zheng B., Hewawasam P., Tu Y., Friborg J., Falk P., Hernandez D., Levine S., Chen C., Yu F., Sheaffer A. K., Zhai G., Barry D., Knipe J. O., Han Y.-H., Schartman R., Donoso M., Mosure K., Sinz M. W., Zvyaga T., Good A. C., Rajamani R., Kish K., Tredup J., Klei H. E., Gao Q., Mueller L., Colonno R. J., Grasela D. M., Adams S. P., Loy J., Levesque P. C., Sun H., Shi H., Sun L., Warner W., Li D., Zhu J., Meanwell N. A., McPhee F. J. Med. Chem. 2014;57:1730–1752. doi: 10.1021/jm500297k. [DOI] [PubMed] [Google Scholar]
- Gentles R. G., Ding M., Bender J. A., Bergstrom C. P., Grant-Young K., Hewawasam P., Hudyma T., Martin S., Nickel A., Regueiro-Ren A., Tu Y., Yang Z., Yeung K.-S., Zheng X., Chao S., Sun J.-H., Beno B. R., Camac D. M., Chang C.-H., Gao M., Morin P. E., Sheriff S., Tredup J., Wan J., Witmer M. R., Xie D., Hanumegowda U., Knipe J., Mosure K., Santone K. S., Parker D. D., Zhuo X., Lemm J., Liu M., Pelosi L., Rigat K., Voss S., Wang Y., Wang Y.-K., Colonno R. J., Gao M., Roberts S. B., Gao Q., Ng A., Meanwell N. A., Kadow J. F. J. Med. Chem. 2014;57:1855–1879. doi: 10.1021/jm4016894. [DOI] [PubMed] [Google Scholar]
- Yeung K.-S., Parcella K. E., Beno B. R., Bender J. A., Grant-Young K., Rigat K., Wang Y.-K., Liu M., Lemm J., Mosure K., Hanumegowda U., Zhuo X., Parker D., Sinz M., Santone K., Smith D., Li J., Fraunhoffer K. J., Delmonte A., Colston E., Pasquinelli C., Gao M., Meanwell N. A., Roberts S., Knipe J. and Kadow J. F., presented in part at the 250th ACS National Meeting & Exposition, Boston, MA, United States, August 16–20, 2015, 2015.
- Kneteman N. M., Howe A. Y. M., Gao T., Lewis J., Pevear D., Lund G., Douglas D., Mercer D. F., Tyrrell D. L. J., Immermann F., Chaudhary I., Speth J., Villano S. A., O'Connell J., Collett M. Hepatology. 2009;49:745–752. doi: 10.1002/hep.22717. [DOI] [PubMed] [Google Scholar]
- Feldstein A., Kleiner D., Kravetz D., Buck M. J. Clin. Gastroenterol. 2009;43:374–381. doi: 10.1097/MCG.0b013e318178d91f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcella K., Nickel A., Beno B. R., Sheriff S., Wan C., Wang Y.-K., Roberts S. B., Meanwell N. A., Kadow J. F. Bioorg. Med. Chem. Lett. 2017;27:295–298. doi: 10.1016/j.bmcl.2016.11.054. [DOI] [PubMed] [Google Scholar]
- Yeung K.-S. and Grant-Young K. A., WO2015088958 A1, A Novel Compound for the Treatment of Hepatitis C, 2015.
- Yeung K.-S. and Kadow J. F., US9303020 B2, Novel Compounds for the Treatment of Hepatitis C, 2015.
- Yeung K.-S., Eastman K. J. and Parcella K. E., US 20160024103 A1, Fused Furans for the Treatment of Hepatitis C, 2016.
- The structures of NS5B 2a L30S with compound 5 and NS5B 1b WT with compound 5 and PDB ID ligand 23E and the crystallographic data were deposited with the PDB with IDs 5TWM and 5TWN, respectively
- Easterbrook J., Fackett D., Li A. P. Chem.-Biol. Interact. 2001;134:243–249. doi: 10.1016/s0009-2797(01)00159-4. [DOI] [PubMed] [Google Scholar]
- Bunker K. D., Sach N. W., Huang Q., Richardson P. F. Org. Lett. 2011;13:4746–4748. doi: 10.1021/ol201883z. [DOI] [PubMed] [Google Scholar]
- Zhuo X., Cantone J. L., Wang Y., Leet J. E., Drexler D. M., Yeung K.-S., Huang X. S., Eastman K. J., Parcella K. E., Mosure K. W., Soars M. G., Kadow J. F., Johnson B. M. Drug Metab. Dispos. 2016;44:1332–1340. doi: 10.1124/dmd.115.069062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.