

 In this review, inhibitors of the cell wall biosynthetic pathway are presented with a focus on the recent advances in glycopeptide antibiotics.
In this review, inhibitors of the cell wall biosynthetic pathway are presented with a focus on the recent advances in glycopeptide antibiotics.
Abstract
Cell wall biosynthesis inhibitors (CBIs) have historically been one of the most effective classes of antibiotics. They are the most extensively used class of antibiotics and their importance is exemplified by the β-lactams and glycopeptide antibiotics. However, this class of antibiotics has not received impunity from resistance development. In the wake of this predicament, this review presents the progress of CBIs, especially glycopeptide derivatives as antibiotics to confront antibacterial resistance. The various strategies used for the development of CBIs, their clinical status and possible directions in which this field can evolve have also been discussed.
Introduction
In the antibiotic armamentarium, cell wall biosynthesis inhibitors represent the most successful class of compounds. Glycopeptide antibiotics, a subset of CBIs, have been traditionally used to treat drug-resistant pathogens. In fact, ever since its introduction, vancomycin, the first glycopeptide antibiotic approved, was used as a “drug of last resort” for a long time. However, resistance against glycopeptides and other CBIs has compromised the armamentarium greatly. This is of grave concern because it is estimated that around 700 000 patients die due to antimicrobial resistance (AMR) every year globally.1 This has been predicted to rise to 10 million by 2050 if not tackled immediately.1 In this regard, several health agencies like the World Health Organization, the Infectious Disease Society of America, and the European Centre for Disease Prevention and Control have iterated on the development of antibiotics with a novel mechanism of action. Despite the efforts of these organizations, the scientific community is yet to add a new class of drug into the arsenal after linezolid, daptomycin, mupirocin and retapamulin.2
Development of semi-synthetic antibiotics is another way to tackle the problem of AMR. Understanding the molecular mechanisms of natural product antibiotics helps in designing newer and more effective derivatives. This is reflected in the fact that most of the new drugs undergoing clinical trials are synthetic modifications of natural products. In this review, we have introduced the readers to the process of bacterial cell wall biosynthesis and the different classes of antibiotics that target this extremely important process. The different strategies that have been used in the development of new glycopeptide antibiotics have then been described. The different mechanisms by which bacteria develop resistance against this class of compounds have also been covered. After briefly touching on the recent approaches taken towards the development of new glycopeptide antibiotics, we provided our perspective on the direction the field should foray into.
Bacterial cell wall biogenesis
The bacterial cell envelope is composed of the cell membrane and the cell wall. The cell membrane and the cytoplasm are fortified by the bacterial cell wall, which has the primary function, as in other organisms, of providing rigidity to the cell.3 This section will deal, primarily, with the composition and biogenesis of the cell wall of bacteria. Cell wall biosynthesis along with bacterial cell division is an important physiological process for bacterial survival. Since mammalian cells are devoid of a cell wall, inhibition of cell wall biosynthesis is an important approach for antibiotic discovery.4 For the development of drugs, it is imperative to understand the process in depth. Towards understanding the process, it is essential to know the chemistry of the bacterial cell wall. The rigidity of the bacterial cell wall is provided by the peptidoglycan backbone. It consists of repeating units of N-acetylglucosamine (NAG) linked with N-acetylmuramic acid (NAM, β 1 → 4 glycosidic linkages) which are cross-linked by pentapeptides (l-Ala–d-Glu–l-Lys–d-Ala–d-Ala) that hang from the layers. Generally, the pentapeptide of the cell wall in Gram-positive bacteria (GPB) is l-Ala–d-Glu–l-Lys–d-Ala–d-Ala and in Gram-negative bacteria (GNB), the third amino acid (l-lysine) is generally replaced with diaminopimelic acid (DAP). However, the sequence of the stem peptide, the type of peptide crosslinks and the presence of secondary modifications in both the glycan chains and peptides differ among species.5 The classical way to classify bacteria, Gram-staining, is based on the difference between the components of the cell wall. Gram-negative bacteria possess an additional outer membrane rich in lipopolysaccharides, in addition to a thin peptidoglycan layer.6 It is this outer membrane that confers these bacteria with resistance to some of the common antibiotics used against GPB.6 As mentioned earlier, the cell wall distinguishes a bacterium from the host cells, thus a molecule, which can target a protein involved in cell wall biosynthesis, will not have a mammalian counterpart. An idea of the different proteins involved in the process of synthesizing the cell wall is pertinent for designing new drugs.
Biosynthesis of the cell wall is a complex process that occurs in three stages involving multiple proteins (Fig. 1). The three stages have been categorised as the cytoplasmic stage, the membrane-associated stage and the exocytoplasmic stage.7
Fig. 1. Bacterial cell wall biosynthetic pathway.

Stage I: the cytoplasmic stage
The first stage occurs in the cytoplasm wherein the monomer units uridine diphosphate-N-acetylmuramyl pentapeptide (UDP-MurNAc-pp) and UDP-N-acetyl glucosamine (UDP-GlcNAc) are formed.4 Initially, UDP-GlcNAc is synthesized from fructose-6-phosphate in a four-step process catalyzed sequentially by GlmS, GlmM and GlmU in the last two steps. UDPGlcNAc is an important precursor for various macromolecules present in the cell wall such as teichoic acid (in Gram-positive bacteria), UDP-MurNAc (pentapeptide) and lipopolysaccharides (in Gram-negative bacteria). UDP-MurNAc is then formed from UDP-GlcNAc in a two-step process catalyzed by MurA and MurB successively. This is followed by the assembly of the pentapeptide on the d-lactoyl group on UDP-MurNAc. The components l-alanine, d-glutamic acid, a diamino acid (like DAP or l-lysine) and a dipeptide (usually d-Ala–d-Ala) are incorporated in successive steps with each step being catalysed by enzymes MurC, MurD, MurE and MurF, respectively (Fig. 1).8 The d-Ala–d-Ala that is incorporated in the last step is synthesized by the action of two enzymes: first, alanine racemase and second, d-alanyl–d-alanine ligase. The enzyme alanine racemase catalyzes the conversion of l-Ala to d-Ala, while the d-Ala–d-Ala ligase (Ddl) catalyzes the formation of d-Ala–d-Ala.4
Stage II: the membrane-associated stage
The second stage involves the formation of the precursor lipid intermediates (Fig. 1). These lipid intermediates transport the monomer units from the cytoplasm to the outer side of the cytoplasm, where the final stage of the process occurs. The pyrophosphoryl–MurNAc–pentapeptide moiety of UDP-MurNAc-pentapeptide is transferred to a membrane lipid C55-undecaprenyl phosphate through catalysis by the MraY enzyme. The oxygen of the phosphate group of the C55-lipid (bactoprenol) attacks the pyrophosphate linkage of the UDP moiety to form MurNAc-pentapeptide pyrophosphoryl undecaprenol with the release of UMP. This MurNAc-pentapeptide pyrophosphoryl undecaprenol is also known as lipid I. This step is followed by the addition of N-acetylglucosamine to lipid I to yield GlcNAc-MurNAc(pentapeptide)phosphoryl undecaprenol (also known as lipid II) catalyzed by the MurG enzyme. The lipid II is then transferred to the outer side of the cytoplasmic membrane by the enzyme MurJ.9 The lipophilic molecule bactoprenol helps in the transport of the hydrophilic precursor molecules from the aqueous environment (in the cytoplasm) to the exterior of the cell membrane for incorporation into the growing peptidoglycan chain.
Stage III: the extracytoplasmic stage
In the third stage, glycosyltransferases catalyze the formation of linear peptidoglycan chains which are then cross-linked by transpeptidases to produce a mesh-like structure (Fig. 1). These transpeptidases consist of enzymes such as the penicillin-binding proteins (PBPs) and RodA. First, the 4′-OH group of the terminal GlcNAc moiety of the elongating glycan chain breaks the muramyl-C1–O–PO3 bond, to release the C55 lipid pyrophosphate. In order to re-enter the cytoplasm and continue the cycle, the pyrophosphate must be hydrolysed from the starting lipid phosphate by a membrane-bound phosphatase enzyme. The formation of the linear peptidoglycan chains is catalyzed by transglycosylases. These linear peptidoglycan chains are then cross-linked by transpeptidases. First, the serine in the active site of the transpeptidase attacks the amide bond between the terminal d-Ala groups of the pentapeptide moiety, forming an acyl-O-Ser enzyme with the release of a free d-Ala amino acid. This enzyme–substrate complex then undergoes a nucleophilic attack by the ε-amine group of either diaminopimelic acid (DAP) or lysine at the third position of the pentapeptide of an adjacent chain.
The components of the cell wall biosynthesis pathway that are commonly targeted are lipid I and lipid II, the Mur enzymes, the transglycosylases and the transpeptidases. In the next section, we summarize the different antibiotics which target the various stages involved in cell wall biosynthesis.
Antibiotics that target bacterial cell wall biosynthesis
There are numerous classes of antibiotics that target the cell wall biosynthesis in bacteria (Table 1). This review focuses primarily on the clinically relevant antibiotics; other antibiotics which are used as growth promoters in poultry animals have not been covered in detail in this review. We direct the readers to some relevant papers covering that aspect.10 In this section we cover the various antibiotics targeting different stages of bacterial cell wall biosynthesis.
Table 1. Cell wall biosynthesis inhibitors and their targets.
| Cell wall biosynthetic stages | Antibiotics | Target |
| Stage I: the cytoplasmic stage | d-Cycloserine | d-Ala–d-Ala ligase, alanine racemase |
| Fosfomycin | MurA | |
| Stage II: the membrane-associated stage | Uridyl peptides (tunicamycin) | MraY |
| Ramoplanin | MurG, lipid II | |
| Stage III: the extracytoplasmic stage | β-Lactams | PBPs |
| Glycopeptides | Lipid II (d-Ala–d-Ala terminal) | |
| Moenomycin | Transglycosylase | |
| Mannopeptimycins | Lipid II | |
| Lantibiotics (nisin) | Lipid II | |
| Defensin (plectasin) | Lipid II | |
| Bacitracin | Undecaisoprenyl pyrophosphate |
Molecules targeting stage I (cytoplasmic stage)
d-Ala–d-Ala ligase and d-Ala racemase inhibitors
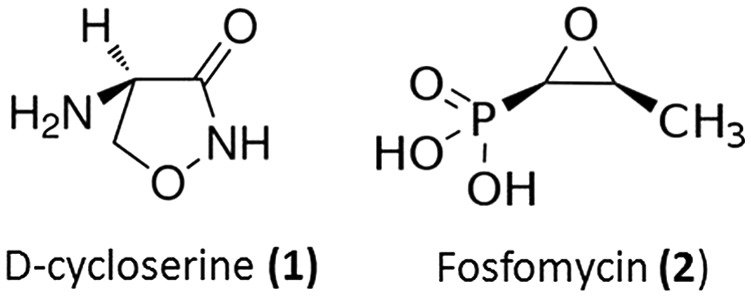
An example of such inhibitors is d-cycloserine (seromycin, 1), a cyclic small molecule that mimics d-alanine and inhibits the enzymes d-Ala–d-Ala ligase and alanine racemase (Fig. 2).11 This molecule is produced by Streptomyces. The inhibition of these enzymes starves the cells of d-Ala thereby inhibiting cell wall biosynthesis. It is a second-line antibiotic for the treatment of tuberculosis.12
Fig. 2. Inhibitors of the cytoplasmic stage of cell wall biosynthesis (stage I).
Mur enzyme inhibitors
As has been discussed earlier, the Mur enzymes are crucial components of the bacterial cell wall biosynthetic pathway. Inhibiting these enzymes is expected to cripple this process leading to weakening of the cell wall and subsequent bactericidal activity. These Mur proteins are widely conserved in most bacterial species and their inhibition is expected to have broad spectrum activity. They are attractive targets for the development of new antibiotics. Currently, there is only one clinically used antibiotic, fosfomycin (2), which inhibits the MurA enzyme (Fig. 2). Although numerous inhibitors of these enzymes have been identified through various screening processes, these have not shown potential as clinical candidates. Literature reports of inhibitors of this class of enzymes have been reviewed elsewhere.4,13,14
Fosfomycin (2) is a broad spectrum bactericidal antibiotic which is highly effective against Gram-positive pathogens such as Staphylococcus aureus and Enterococcus sp., and against Gram-negative pathogens such as Escherichia coli, Pseudomonas aeruginosa and Klebsiella pneumoniae. It mimics the substrate phosphoenolpyruvate and binds to the enzyme UDP-N-acetylglucosamine-3-enolpyruvyl transferase (MurA) thereby inhibiting the first step of cell wall biosynthesis.15 The antibiotic binds to the active site of MurA via a thioether bond to the key residue Cys115 and inhibits this enzyme. Due to its unique mechanism of action, synergism with other classes of antibiotics including β-lactams, aminoglycosides and fluoroquinolones is possible. Oral fosfomycin is used mainly in the treatment of urinary tract infections, like those caused by Escherichia coli and Enterococcus faecalis.16 Resistance to fosfomycin is conferred by reduced uptake, target modification, overexpression of MurA and antibiotic modification.
Molecules targeting stage II (membrane-associated stage)
MraY inhibitors
There are no clinically approved drugs that target this enzyme. Various uridyl peptides such as tunicamycin (3, Fig. 3), liposidomycins, and mureidomycin have been reported as competitive inhibitors of MraY.17 Tunicamycin is not selective, in that it interferes with mammalian glycoprotein biosynthesis as well. However, the next generation uridyl peptides (liposidomycins, mureidomycins) were able to overcome this problem.17
Fig. 3. Inhibitors of the membrane-associated stage of cell wall biosynthesis (stage II).

Agents targeting lipid II
Lipid II is another important component of the cell wall biosynthetic pathway consisting of the complete peptidoglycan precursor subunit. In fact, it is the target of four classes of antibiotics which are mannopeptimycins, lantibiotics (such as ramoplanin, nisin and mersacidin), defensins (such as plectasin) and the clinically important glycopeptide antibiotics. The structures of ramoplanin (4) and nisin (5) are illustrated in Fig. 3.
Ramoplanin (4) was first isolated in 1984 from a strain of Actinoplanes.18 It is a complex consisting of three components A1, A2, and A3 each having a common cyclic peptide part composed of 17 amino acids and a dimannosyl unit but differs in the nature of the di-unsaturated fatty acid chain. Ramoplanin is a bactericidal glycolipodepsipeptide antibiotic which is under clinical trials for aerobic and anaerobic Gram-positive bacteria including Clostridium difficile and vancomycin-resistant Enterococci (VRE). It acts by inhibiting the late stage membrane-associated reactions catalyzed by the transglycosylase and MurG enzymes. Ramoplanin was found to inhibit the MurG enzyme, albeit this is not the only factor responsible for its antibacterial activity.19 It has been found to bind to lipid II and the transglycosylases found on the outer side of the bacterial membrane.20 The site of binding of ramoplanin to lipid II is different from that of the glycopeptide antibiotics.21 Nisin (5) is known to act through pore-formation post binding to lipid II. It was approved by the FDA for use as a food preservative. Lipid II inhibitors have been extensively reviewed elsewhere.22,23 Teixobactin is a new potent antibiotic which was discovered through screening of previously uncultured bacteria.24 It was found to act by binding to both lipid II and the wall teichoic acid (WTA) precursor undecaprenyl-PP-GlcNAc (lipid III).
Molecules targeting stage III
Agents targeting the penicillin-binding proteins: β-lactam antibiotics
β-Lactam antibiotics are the most widely used bactericidal agents which inhibit cell wall biosynthesis by binding to PBPs.25 These PBPs are enzymes distributed on the outer side of the cytoplasm, which exhibit peptidoglycan transpeptidase, endopeptidase, d,d-carboxypeptidase and even transglycosylase activity and are present in both Gram-positive and Gram-negative bacteria.26
This antibiotic class consists of a large number of mostly semi-synthetic compounds that can be classified into bicyclic penicillins, cephalosporins, carbapenems, and monocyclic monobactams.25 The common feature of this class of antibiotics is a four-membered lactam ring fused through the nitrogen and the adjacent tetrahedral carbon to a secondary ring structure except for the monobactams. They are further sub-classified based on the structure of the secondary ring and the hetero-atom present in it. The susceptibility of bacteria towards this class of antibiotics varies widely with various factors, such as the binding affinity to the target site (PBPs), stability to β-lactamases and outer membrane permeability (in the case of Gram-negative bacteria). Over the years, various semi-synthetic derivatives of penicillin and cephalosporin have been developed to overcome resistance against older β-lactams. These antibiotics displayed improved antibacterial activity, pharmacokinetic properties and a broader spectrum of activity. This structural class has been extensively reviewed elsewhere.25,27,28
The β-lactam ring mimics the d-Ala–d-Ala moiety of the pentapeptide terminal and the PBPs mistake them as their substrate. The serine residue of the active site of the PBP attacks the carbonyl of the β-lactam ring, cleaving the ring and forming an inactive acyl-enzyme which has a long lifetime. Since there is no leaving group, the β-lactam completely blocks the active site from a further nucleophilic attack. This inhibits these enzymes from catalyzing the crosslinking of peptidoglycan layers. Although the biosynthesis of the cell wall is halted, autolysis of the murein sacculus continues thereby compromising this component and ultimately lysing the cell.7
Bacterial resistance to this class of antibiotics is conferred through i) production of β-lactamase enzymes (the most common mechanism of resistance);29 ii) modification of target PBPs (changes in the active site result in proteins with lower affinity to PBPs);30 iii) impaired penetration of drug to target PBPs and iv) efflux pumps (responsible for the carbapenem resistance of A. baumannii).31
Scientists have designed effective strategies to combat such resistance mechanisms. Production of β-lactamases is one of the major causes of resistance to β-lactam antibiotics. β-Lactamases are broadly classified into two categories – the serine β-lactamases (SBL) and the metallo-β-lactamases (MBL) – differing from each other in terms of their hydrolytic mechanisms and the rate of hydrolysis.29 They have been further divided into classes A, B, C and D. Classes A, C and D are serine β-lactamases while class B belongs to metallo-β-lactamases. The serine β-lactamases hydrolyse the β-lactam ring through an attack by the serine residue present in the active site while the MBLs utilize the zinc ions. The NDM enzyme was first reported in 2009 in a Klebsiella pneumoniae isolate and was referred to as NDM-1.32 MBLs can hydrolyse all β-lactam families including penicillins, cephalosporins, carbapenems and SBL inhibitors, with the exception of the monobactams (aztreonam). Various strategies have been developed to combat lactamase-mediated resistance.7
Semi-synthetic β-lactams that are substrates with a slow reaction rate with β-lactamases have been designed.7 These can bind to the enzyme with high affinity and forms an acyl-enzyme which leads to unfavourable steric interactions thereby preventing hydrolysis. In these semi-synthetic compounds, the β-lactam is modified such that β-lactamase binding or catalysis is inhibited but does not interfere with PBP-binding and acylation. Aztreonam, imipenem and meropenem are successful examples of this strategy. The other strategy involves the development of mechanism-based irreversible suicide inhibitors.29 Molecules (β-lactamase inhibitors) that occupy the active site of the lactamase enzymes longer than the β-lactam antibiotic have been designed (Fig. 4). This property is by virtue of their high acylation rates, slow deacylation rates and stability towards consequent hydrolysis. At present, there are seven formulations of β-lactams and β-lactamase inhibitors available in the market (Table 2). Clavulanic acid (6), sulbactam (7) and tazobactam (8) are some of the older β-lactam based β-lactamase inhibitors. Recently avibactam (9), a non-β-lactam inhibitor, was approved for use in combination with ceftazidime.33 Other β-lactamase inhibitors such as relebactam (10), zidebactam (11), vaborbactam (12) and OP0595 (13) are currently in various stages of clinical trials (Table 2). However, there are no clinically approved MBL inhibitors. Moreover, since most bacteria have acquired both SBLs and MBLs, there is a need for broad spectrum inhibitors for both classes of inhibitors. Recently, novel cyclic boronates as dual action inhibitors of SBLs and MBLs which also potently inhibit some PBPs were reported.34 These molecules act by forming complexes with the enzymes that mimic the high-energy tetrahedral intermediate common to both classes of lactamases and the PBPs.34
Fig. 4. β-Lactamase inhibitors.

Table 2. Combination therapy of β-lactams with β-lactamase inhibitors.
| Inhibitor | In combination with | Generic name | Treatment for | Status |
| Clavulanic acid | i) Amoxicillin | Augmentin | Sinusitis, pneumonia, ear infection, bronchitis, urinary tract infections and infection of the skin | Approved |
| ii) Ticarcillin | Timentin | Septicemia, lower respiratory tract infections, bone and joint infections, urinary tract infections, infections of the skin, gynecologic infections, intra-abdominal infections caused by Gram +ve and Gram –ve bacteria | Approved | |
| Tazobactam | i) Ceftolozane | Zerbaxa | Complicated intra-abdominal infections (cIAI) and complicated urinary tract infections (cUTI) | Approved |
| ii) Piperacillin | Zosyn | Urinary tract infections, bone and joint infections, severe vaginal infections, stomach infections, skin infections and pneumonia | Approved | |
| Sulbactam | Ampicillin | Unasyn | Skin and skin structure infections, intra-abdominal infections, gynecological infections | Approved |
| Avibactam | i) Ceftazidime | Avycaz | cUTI including acute pyelonephritis, caused by E. coli (including cases with concurrent bacteremia), K. pneumoniae, Citrobacter koseri, Enterobacter aerogenes, E. cloacae, Citrobacter freundii, Proteus spp. (including P. mirabilis and indole-positive Proteus) and P. aeruginosa | Approved |
| ii) Ceftazidime and Metronidazole | cIAI, caused by Escherichia coli (including cases with concurrent bacteremia), Klebsiella pneumoniae, Proteus mabilis, Providencia stuartii, Enterobacter cloacae, K. oxytoca, Pseudomonas aeruginosa and P. stutzeri; and polymicrobial infections caused by aerobic and anaerobic organisms including Bacteroides spp. | Approved | ||
| iii) Ceftaroline | Bacterial infections | Phase 2 | ||
| iv) Aztreonam | cIAI | Phase 2 | ||
| Vaborbactam | Biapenem | cUTI, acute pyelonephritis | Phase 3 | |
| Hospital acquired bacterial pneumonia | ||||
| Ventilator-associated bacterial pneumonia | ||||
| Bacteremia | ||||
| Meropenem | Carbavance | cUTI and serious bacterial infections due to confirmed or suspected carbapenem-resistant Enterobacteriaceae | Phase 3 | |
| Relebactam | Imipenem | Imipenem-resistant bacterial infections | Phase 3 | |
| Hospital-acquired bacterial pneumonia (HABP), ventilator-associated bacterial pneumonia (VABP), cIAI and cUTI | ||||
| Zidebactam | Cefepime | cUTI, hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia | Phase 1 | |
| OP0595 | — | G +ve/G –ve bacterial infections | Phase 1 | |
| AAI0595 | Cefepime | G –ve bacterial infections | Phase 1 |
In order to overcome the resistance due to reduced permeability through the outer membrane, siderophore–β-lactam conjugates have been developed (Fig. 5). One of these derivatives is GSK-2696266 (S-649266) (14) which contains a catechol moiety attached to the 3-position side chain and is currently undergoing clinical trials.35,36 This molecule which binds mainly to PBP-3 similar to other cephalosporins was found to exhibit significant activity against a wide range of clinical isolates, including NDM expressing Gram-negative bacteria. This molecule is currently in phase 2 of clinical trials. BAL30072 (15) is another novel β-lactam wherein tigemonam is substituted with dihydroxypyridone, a siderophoric moiety.37,38 This molecule was found to bind to PBP1a and PBP1b as well as PBP 3.37 This is currently undergoing clinical trials (phase 1) for the treatment of Gram-negative pathogens in combination with meropenem.
Fig. 5. Strategies to overcome β-lactam resistance due to reduced permeability through the outer membrane.

Agents that target the d-Ala–d-Ala terminal of pentapeptides of cell wall precursors: glycopeptides
Glycopeptide antibiotics are an important class of antibiotics that prevent this step. These antibiotics bind to the C-terminal d-Ala–d-Ala of the murein precursor, lipid II and immature peptidoglycan, through five H-bonds and thereby inhibit transglycosylation and/or transpeptidation during cell wall biosynthesis.39 This leads to weakening of the peptidoglycan, leaving the bacteria susceptible to lysis due to changes in the osmotic pressure. The first generation of glycopeptides, vancomycin and teicoplanin, were the first of this class to be approved for clinical use. Vancomycin was widely used as “the drug of last resort” against complicated multidrug-resistant Gram-positive infections for about thirty years before resistance was observed. After resistance development, three new glycopeptides, telavancin, dalbavancin and oritavancin, were approved for clinical use by the FDA. This class of antibiotics has been covered in detail in the next sections of the review.
Agents targeting the transglycosylase enzyme
Moenomycin (16), which is used as a growth promoter in animal feed, is known to inhibit the transglycosylase enzyme (Fig. 6). Its poor absorption properties made it unsuitable for clinical use in human beings.40
Fig. 6. Stage III inhibitors.

Inhibitors of dephosphorylation of C55-isoprenyl pyrophosphate
Bacitracin (17) is a metal-dependent antibiotic produced and obtained from Bacillus licheniformis and Bacillus subtilis (Fig. 6). This antibiotic is used topically in combination with other antibiotics for the treatment of skin and eye infections. Due to its systemic toxicity, its uses are limited. In order to bind to its target, bacitracin requires a divalent metal ion such as Zn2+. The metallo-bacitracin forms a 1 : 1 complex with undecaisoprenyl pyrophosphate, thus preventing it from being converted to its corresponding phosphate.41 This phosphate is the carrier for building blocks of the peptidoglycan from the cytoplasm to the cell wall region. In the absence of this phosphate, the cell wall precursors are no longer available.
Glycopeptide antibiotics
This class of antibiotics are glycosylated tricyclic or tetracyclic heptapeptides. They have been divided into five distinct structural subclasses (I–V) based on the substitution and the residues at positions 1 and 3 of the heptapeptide; this has been described in detail in other reviews.42 Ristocetin, avoparcin, complestatin, vancomycin, and teicoplanin are representative molecules of these classes. The first member of this class of antibiotics, vancomycin (Fig. 6), was discovered by Elli Lilly in 1956 and is still vital for the treatment of Gram-positive bacterial infections caused by methicillin-resistant S. aureus (MRSA) and Clostridium difficile. The other natural glycopeptide that was approved for use in Europe (in 1980) for treating similar Gram-positive infections is teicoplanin (Fig. 6). This molecule was discovered in the Lepitit Research Center (Milan, Italy). Avoparcin is structurally similar to vancomycin and was used as a growth promoter in livestock feed. However, its use was banned due to its association with the transfer of vancomycin resistance to Enterococcus faecium. Ristocetin was discontinued from clinical use because it caused platelet aggregation in patients missing a platelet factor in platelet-type von Willebrand disease. The next generation semi-synthetic glycopeptides were majorly lipoglycopeptides such as telavancin (Vibativ®), dalbavancin (Dalvance®) and oritavancin (Orbactiv®). These demonstrated high activity against multidrug-resistant (MDR) Gram-positive pathogens. A description of the biosynthesis is beyond the scope of the review and the readers are directed to other reviews for further reading.7,43,44 In this review, we have described the evolution of the glycopeptides.
First generation glycopeptides
Vancomycin and teicoplanin
Vancomycin (22) and teicoplanin (23) (Fig. 6) are the two naturally occurring, first generation glycopeptides. The structures of teicoplanin and vancomycin are closely related. Teicoplanin is a lipoglycopeptide with a fatty acid chain linked to the glucosamine sugar. These two inhibit cell wall biosynthesis by binding to the d-Ala–d-Ala moiety of the cell wall. Vancomycin is known to dimerize in solution and this results in an improved binding affinity to the substrate. However, unlike other glycopeptide antibiotics, teicoplanin does not form dimers. The lipophilic moiety of teicoplanin imparts membrane-anchoring properties to it. Overall, these two glycopeptides bear similar toxicity and activity profiles.45
After studying the various antibacterial agents which target bacterial cell wall biosynthesis, we feel that glycopeptide antibiotics have the following advantages over the other CBIs due to several reasons. The enzyme-targeting approach is prone to high frequency of resistance development. Glycopeptides, on the other hand, targets the substrate, which makes it difficult for bacteria to develop rapid resistance to. Furthermore, the structure allows modifications at various places without compromising the main mode of action. The low absorption of glycopeptides juxtaposed with their stability in the gastrointestinal tracts makes them suitable for treating infections caused by Clostridium difficile.
Resistance to glycopeptides
It had been noticed earlier that bacteria found it difficult to develop resistance to vancomycin. In a comparative study of penicillin and vancomycin, Ziegler et al. found that the minimum inhibitory concentration (MIC) of penicillin against S. aureus increased by more than 100 000-fold after 25 serial passages while that of vancomycin showed only an 8-fold increase.46 Hence, it was hypothesized that bacteria could not alter the target of glycopeptides (the d-Ala–d-Ala terminus of lipid II and/or the immature peptidoglycan) easily as the process would involve simultaneous modifications to multiple enzymes in the pathway to peptidoglycan synthesis. The mode of action of glycopeptide antibiotics made the development of high-level resistance almost impossible.47 Vancomycin-resistant Enterococci (VRE) appeared in hospitals 30 years after the approval for use of vancomycin.48 The incidence of VRE in hospitalized patients with enterococcal infections in the US had increased to 30%. The extensive use of vancomycin for the treatment of MRSA infections resulted in reduced vancomycin susceptibility and led to the emergence of hetero-resistant vancomycin-intermediate S. aureus (VISA) and the first vancomycin-resistant S. aureus (VRSA) was reported in 2001.49,50 Resistance to vancomycin in Enterococci (VRE) was not a result of spontaneous mutations in clinically relevant microorganisms but due to the transfer of resistance genes from other glycopeptide-resistant bacteria (such as those resulting from the overuse of avoparcin as an animal growth promoter).51 The mechanism of vancomycin resistance of Enterococci was elucidated by Courvalin and Walsh groups in the 1990s. Subsequent work on glycopeptide resistance of producer organisms has revealed that they consist of the same resistance genes as the resistant enterococcal strains.52 The mechanism of vancomycin resistance of Enterococci and Staphylococci is described below. Nine gene clusters that confer resistance to vancomycin in Enterococci have been identified and these are vanA, vanB, vanC, vanD, vanE, vanF, vanG, vanL, vanM and vanN.53–57 The vanC, vanE, vanG, vanL and vanN gene clusters confer resistance by replacement of the d-Ala–d-Ala terminal of the cell wall precursor pentapeptide with d-Ala–d-Ser while vanA, vanB, vanD and vanM encode for the replacement of the same with d-Ala–d-Lac. The VanA and VanB phenotypes are more prominent and initially differentiated by their susceptibility to vancomycin vs. teicoplanin. The VanA phenotype shows reduced sensitivity to both teicoplanin and vancomycin whereas the VanB phenotype is sensitive to teicoplanin.57 Resistance of these phenotypes involves the replacement of the d-Ala–d-Ala terminal of the cell wall precursor pentapeptide with d-Ala–d-Lac leading to a 1000-fold loss of the binding affinity.58 A cluster of genes, vanRSHAX, encode for an alternate biosynthetic pathway that produces the mutated cell wall precursors.59 In these resistant phenotypes, the VanR protein (the response regulator) and the VanS protein (a histidine kinase sensor) form a two protein-component regulatory system (TCS).60 When the cell wall precursors are perturbed due to vancomycin or teicoplanin, the VanS protein is autophosphorylated, which in turn phosphorylates VanR.60 The phosphorylated VanR protein then binds to the promoter region of vanHAX and regulates its expression. The VanH protein catalyzes the conversion of pyruvate to d-lactate and the VanA ligase catalyses the formation of d-Ala-d-Lac.61 Two other enzymes, VanX, a d,d-dipeptidase and VanY, a d,d-carboxypeptidase, deplete the pool of d-Ala-d-Ala available, which is crucial for preventing the interaction of vancomycin with its target.62 VanX hydrolyses the d-Ala–d-Ala dipeptide, while VanY hydrolyses the terminal d-Ala residue from the PG precursor.63 The function of the VanZ protein, which confers resistance to teicoplanin in the VanA phenotype, is not yet understood.64 Another accessory protein VanXY is capable of performing the functions of both VanX and VanY.65 These modified precursors are polymerized into the functional cell wall, similar to the native cell wall.
In the case of VISA, the thickening of the bacterial cell wall imparts resistance. This thickening leads to an increase in the number of binding sites for vancomycin binding and hence a decrease in susceptibility.50 Genotypic analysis of VRSA indicated that the resistance genes were acquired from VRE.66 In some strains of VRSA, resistance was found to be due to both thickening of cell wall as well as modified pentapeptide termini.50
Second generation glycopeptides
The second-generation glycopeptides are semi-synthetic lipoglycopeptide antibiotics (Fig. 7) which exhibit enhanced antibacterial activity over vancomycin against both vancomycin-sensitive and vancomycin-resistant strains.44,67 They also demonstrate better pharmacological properties.68 There are three antibiotics which belong to this generation – telavancin, oritavancin and dalbavancin. The common feature of this class of glycopeptides is the presence of a hydrophobic side chain which serves as a membrane anchor.68 This leads to a superior binding affinity to the pentapeptide termini (d-Ala–d-Ala or d-Ala–d-Lac). All the three antibiotics are briefly discussed below.
Fig. 7. Clinically approved second generation glycopeptide antibiotics.

Telavancin
Telavancin (20) is a semi-synthetic vancomycin derivative. It consists of a lipophilic decylaminoethyl moiety conjugated to the vancosamine sugar of vancomycin and a (phosphomethyl)aminomethyl moiety substituted at the para position of the aromatic ring of the C-terminal dihydroxyphenylglycine residue.69 The lipophilic moiety provides membrane interaction properties leading to permeabilization and depolarization of the bacterial cell membrane.70 Like all glycopeptides, it inhibits cell wall biosynthesis and hence has a dual mechanism of action. The bactericidal properties of this compound may be attributed to its membrane activity. The hydrophilic moiety enhances tissue distribution and clearance, thereby reducing nephrotoxicity.71 This leads to improved activity in comparison to the first-generation of glycopeptides against MRSA and MSSA. Further, it is active against vancomycin-resistant strains such as VISA, VRSA, and VRE. In 2009, it was approved by the Food and Drug Administration (FDA) Agency of the United States of America (USA) for the treatment of complicated skin and skin-structure infections (cSSSi). Later, in 2013, it was approved for treatment of hospital-acquired and ventilator-associated pneumonia caused by S. aureus.
Dalbavancin
Dalbavancin (21) is a semi-synthetic derivative of a glycopeptide A40926 which belongs to the teicoplanin family. The parent drug is modified through the amidation of the C-terminal carboxyl group with a N,N-dimethylpropylamine group.72,73 This improved the antibacterial activity against Staphylococci. Like teicoplanin, it possesses a terminally branched dodecyl fatty acid chain connected through an amide linkage with the glucosamine moiety. This helps in membrane anchoring. It binds to d-Ala–d-Ala with higher affinity than its parent compound, by virtue of its abilities to form dimers and anchor into the membrane.44,74,75 This lipoglycopeptide exhibits better potency than vancomycin and teicoplanin against MRSA and susceptible Enterococci. It shows an MIC of ∼0.1 μg mL–1 against VRE (VanB phenotype), although it shows poor activity against VRE (VanA phenotype).76 It was approved by the FDA in 2014 for the treatment of acute skin and skin-structure infections caused by methicillin-susceptible and resistant S. aureus (MSSA and MRSA), Streptococci and vancomycin sensitive E. faecalis. Its half-life, ranging from 149 to 250 h in human beings, permits once-weekly dosing.68
Oritavancin
Oritavancin (22) is an N-acyl derivative of the naturally occurring product chloroeremomycin.77 It consists of a lipophilic 4-chloro-biphenyl group attached to the parent drug through the amino group of the epivancosamine moiety. This lipophilic biphenyl moiety is capable of interacting with the bacterial cell membrane leading to its permeabilization.78 It promotes dimer formation prior to binding to the target peptides resulting in an enhanced binding affinity to both d-Ala–d-Ala and d-Ala–d-Lac. Like all other glycopeptides, oritavancin inhibits transglycosylation, but it also has a significant inhibitory effect on transpeptidation.79 Its ability to bind to the pentaglycyl bridging segment of the peptidoglycan of Enterococci contributes to its activity against vancomycin-resistant strains.80 This multimodal mechanism of action results in MICs of <1 μg mL–1 against MRSA, VRSA, VRE (VanA phenotype) and VRE (VanB).81 Under conditions where vancomycin has a static effect, this antibiotic exhibits strong bactericidal properties. It was approved by the FDA in 2014 for the treatment of acute skin and tissue infections caused by MRSA, MSSA, Streptococci and vancomycin-susceptible E. faecalis.68 Its terminal half-life is about 393 h which enables treatment with just a single dose.82
In general, the lipoglycopeptides anchor to the bacterial membrane and enhance the binding affinity to the membrane-associated lipid II. They strongly inhibit both transglycosylation and transpeptidation. The lipophilic nature of the second-generation glycopeptides permits strong interactions with proteins within the body (93–98% and 86–90% of dalbavancin and oritavancin, respectively, are protein bound).68 This results in the prolonged terminal half-life and high retention times in mammalian cells and tissues.
Recent developments in strategies to tackle acquired resistance to glycopeptides
Although the second generation of glycopeptides has shown activity against vancomycin-resistant strains, these glycopeptides are yet to be approved for treating vancomycin-resistant infections. In order to overcome the resistance to glycopeptide antibiotics, numerous strategies have been developed over the years. These large complex molecules have several functional groups including amino, carboxyl and hydroxyl groups that facilitate further modification. Even the chloro-substituents of the triarylbiether backbone have been considered as sites for modification. The various modifications of glycopeptides have been extensively reviewed elsewhere.83,84 In this section we will focus on the strategies that have been successful in overcoming bacterial resistance to glycopeptides. The strategies to overcome glycopeptide resistance involve:
a) Enhanced binding affinity to both the sensitive and mutated terminii of the pentapeptide; and
b) Introduction of additional membrane interaction properties;
In this section, we will cover the recent developments towards overcoming glycopeptide resistance.
a) Enhanced binding affinity to both the sensitive and mutated terminii of the target pentapeptide
To achieve enhanced binding affinity to the target pentapeptide, two approaches have been developed so far. The approaches involve the modification of the heptapeptide backbone, attachment of moieties (that can form H-bonds with the target) and through the development of homomeric multivalent analogues.
Modifications of the peptide backbone
To improve the lost binding affinity to the target peptide, numerous modifications were made to the peptide backbone of vancomycin. These included modifications of the carboxamide of residue 3 (l-asparagine), substitutions at the residue 1 (l-leucine), and modification of the carboxamide of residue 4 (d-hydroxyphenylglycine).84 This section will focus primarily on those strategies that could successfully overcome the acquired resistance. Other modifications have been described in other reviews.83,84 The Boger group found that the repulsive lone pair interactions between vancomycin and the mutated target peptides (100-fold) is responsible for the largest share of the lost binding affinity (1000-fold), and not the H-bond loss (10-fold).58 This indicated that the designs that focus on eliminating the destabilizing lone pair interaction rather than the reintroduction of the lost H-bond are more effective. It was hypothesized that modifications to the binding pocket of vancomycin could improve the lost binding affinity to the target peptides.85 Therefore, a vancomycin analogue that lacks residue 4 amide carbonyl and instead contains a methylene group ([Ψ[CH2NH]Tpg4]-vancomycin aglycon) was developed by Boger et al.86 The association constant (Ka) of vancomycin to the d-Ala–d-Ala terminal was 4.4 × 105 M–1 and 4.3 × 102 M–1 to the mutated terminal, d-Ala–d-Lac. The ([Ψ[CH2NH]Tpg4]-vancomycin aglycon) analogue demonstrated a 40-fold increase in affinity for d-Ala–d-Lac and a 35-fold reduction in affinity for d-Ala–d-Ala. It exhibited an MIC of 31 μg mL–1 against VRE, confirming its improved binding affinity to the mutated peptide. Later, an amidine group was introduced in place of the carbonyl at residue 4 of vancomycin.87,88 This [Ψ[C( NH)NH]Tpg4]vancomycin aglycon (23) not only binds to the unaltered peptidoglycan d-Ala–d-Ala but also binds to the altered ligand d-Ala–d-Lac by virtue of its ability to serve as a hydrogen-bond donor or acceptor.87 This amidine derivative of the vancomycin aglycon displayed an MIC of <0.5 μg mL–1 against sensitive and resistant bacteria. Later, in order to incorporate both enhanced binding affinity and favourable hydrophobicity in the same molecule, Boger et al. developed the (4-chlorobiphenyl)methyl derivative of [Ψ[C( NH)NH]Tpg4]vancomycin (24).89 The two independent and synergistic mechanisms of action resulted in high activity against vancomycin-resistant bacteria with MICs in the range of 0.005–0.06 μg mL–1. However, since this strategy involved the total synthesis of the compounds, it involves significant synthetic challenges.
Attachment of H-bond forming moieties to the periphery of vancomycin
In the crystal structure of vancomycin–ligand complex, Nitanai et al. observed that a water molecule bridged the carboxylic group of vancomycin and the ligand.90 It was thus hypothesized that modification of the C-terminus with moieties that can form hydrogen bonds with the target peptide could stabilize the vancomycin–ligand complex and enhance the binding affinity. In a strategy developed in our lab, various conjugates of vancomycin and cyclic and acyclic sugars such as maltose, lactobionic acid, gluconic acid and cellobiose were synthesized (Fig. 8).91 The sugars were conjugated to vancomycin to increase the binding affinity to the mutated target peptide (d-Ala–d-Lac). All the derivatives showed binding affinities similar to that of vancomycin. The derivatives with lactobionic acid and gluconic acid conjugated to the carboxylic acid group of vancomycin (25) exhibited improved affinity to the mutated pentapeptide terminal. The lactobionic acid–vancomycin conjugate showed a ∼150-fold (Ka = 8.8 × 104) higher affinity for N,N-diacetyl-Lys–d-Ala–d-Lac than vancomycin. This improved binding affinity resulted in its improved antibacterial activity, wherein the MIC value was reduced from 750 to 36 μM against VRE (VanA phenotype). In order to improve the activity against vancomycin-resistant strains, alkyl chains (octyl to dodecyl) were appended to the amino group of vancosamine of compound 25 to yield the lipophilic vancomycin–sugar conjugates. This was expected to impart additional membrane-anchoring properties.91 Based on the antibacterial activity and toxicity, the derivative with a decyl chain (26) was selected as the lead compound. It (26) exhibited an improved activity of more than 1000-fold (MIC ∼0.7 μM) and 250-fold (MIC ∼1 μM) against the VanA and VanB strains of VRE, respectively, compared to vancomycin. The incorporation of lipophilicity into this glycopeptide scaffold could provide favourable additional properties resulting in improved activity against vancomycin-resistant Enterococci (VanA and VanB phenotypes of VRE). This compound showed a significant reduction in bacterial titre against VISA in a murine thigh infection model.92 The compound exhibited improved single-dose pharmacodynamic and pharmacokinetic properties compared to vancomycin.92
Fig. 8. Strategies to enhance the binding affinity to the mutated target pentapeptide terminal d-Ala–d-Lac.

Development of homomeric multivalent analogues
Glycopeptide antibiotics are known to form dimers through hydrogen bonding and hydrophobic interactions.93 These non-covalent dimers are co-operative and enhance the antibacterial efficacy.94 Intrigued by this observation, scientists devised various strategies to increase the binding affinity through multivalency (Fig. 9). In 1996, Griffin's group developed bis(vancomycin)carboxamides with variable linkers (27a–27d, Fig. 9).95 Derivatives 27a–27c exhibited improved activity and binding affinity towards VRE compared to monomeric vancomycin. The MIC of the best derivative (27b) showed a ∼60-fold decrease (MIC ∼11 μM) against VRE compared to vancomycin. Later in 1998, trivalent vancomycin derivatives (28) were developed by the Whitesides group.96 These trimers showed an enhanced binding affinity compared to monomeric vancomycin by virtue of polyvalency. This report illustrated the practicality of the concept of polyvalency. Arimoto et al. synthesized a multivalent polymer of vancomycin via ring opening metathesis polymerization (29).97 This exhibited a ∼60 fold increase in antibacterial activity against VRE (MIC ∼31 μg mL–1 against the VRE VanA phenotype and 2 μg mL–1 against the VRE VanB phenotype) compared to vancomycin. In 2000, Nicolaou's group selected vancomycin dimers through target-accelerated combinatorial synthesis (31). These dimers exhibited improved activity against drug-resistant bacteria.98 The dimers with disulphide linkers exhibited the highest activity with a MIC of ∼1–2 μg mL–1 against VRE.98 It had been found that the avidity of multivalent binding in dimers with flexible organic linkers was reduced due to a loss in conformational entropy. Bing Xu et al. hypothesized that a metal complex with a specific geometry and structural rigidity could increase the binding affinity by reducing this loss in conformational entropy. They developed dimers of vancomycin (30), in which two vancomycin molecules were complexed with [Pt(en)2(H2O)2].99 This derivative showed a ∼720-fold increase (MIC ∼0.8 μg mL–1) in activity compared to vancomycin. Our group developed and synthesized vancomycin aglycon dimers with linkers varying in lipophilicity and charge. The vancomycin dimer containing two positively charged centres in the octylene linker (32) exhibited a 15-fold (MIC ∼48 μM) and 130-fold (MIC ∼0.1 μM) higher activity than vancomycin against VRE and VISA, respectively. These dimers increased cell wall biosynthesis inhibition and possess higher binding affinity to the target peptides.100 Further, a vancomycin aglycon dimer with a pendant octyl lipophilic moiety in the linker (33) was developed to impart membrane anchoring properties to the design. This system showed a 300-fold higher activity than vancomycin against VRE with an MIC ∼2.5 μM.
Fig. 9. Multivalency approach: homomeric multivalent analogues of vancomycin.

b) Introduction of additional membrane interaction properties
Epitomized by teicoplanin and later the second generation of glycopeptides, vancomycin and other glycopeptides were appended with hydrophobic chains. The attachment of lipophilic moieties to vancomycin was expected to enhance membrane interaction properties. Reports on the introduction of lipophilic moieties to glycopeptides are well documented in the literature and will be briefly discussed in this section.83,84,101 Herein, we will cover some of the recent derivatives which exhibited activity against vancomycin-resistant strains although their membrane interaction properties have not been reported. Thorson et al. used a chemoenzymatic approach to incorporate lipophilic 6-azido glucose onto the vancomycin aglycon for subsequent glycorandomization (34, Fig. 10).102 This compound was found to be more effective than vancomycin against the VanB phenotype of VRE (MIC ∼1 μg mL–1). Arimoto et al. (35) reported the application of a Suzuki–Miyaura coupling to replace the chloro groups of vancomycin with carbon substituents. Upon replacement of the chloro group of amino acid residue 2 with a lipophilic group, the antibacterial activity against the VanB phenotype of VRE was found to be enhanced (MIC 0.5 μg mL–1) but it remained inactive against the VanA phenotype of vancomycin-resistant Enterococci (VRE).103 However, the additional introduction of such a group at the amino acid residue 6 led to a reduction in activity even against vancomycin-susceptible strains. In 2015, Miller et al. developed three regio-selective peptide catalysts that specifically substitute the aliphatic hydroxyls on vancomycin, generating three lipidated vancomycin analogues (40a–40c).104 The compounds exhibited good activity against both VanA and VanB phenotypic VRE with MIC ∼0.25 μg mL–1.
Fig. 10. Strategies to overcome resistance through introduction of membrane interaction properties to vancomycin.

Those derivatives whose membrane interaction properties have been investigated and reported will be discussed in detail in this section.
Conjugation of cationic lipophilic moieties to impart membrane-disruption properties
It was hypothesized that the conjugation of a permanent positively charged lipophilic moiety to vancomycin could enhance antibacterial activity through enhanced interactions with the negatively charged bacterial membrane. With this idea, our group developed a series of lipophilic cationic vancomycin derivatives (37).105 Cationic lipophilic quaternary ammonium groups with an alkyl chain length varying from hexyl to tetradecyl were attached to the carboxylic acid group of vancomycin. Compared to vancomycin, compound 37a with an octyl chain appended to vancomycin demonstrated a ∼32-fold (MIC ∼0.4 μM), 320-fold (MIC 0.3 μM) and 60-fold (MIC ∼12.5 μM) greater activity against VISA (vancomycin intermediate S. aureus), VRSA (vancomycin-resistant S. aureus) and vancomycin-resistant Enterococci (VRE) VanA phenotypes.105,106 As the length of the hydrophobic chain increased, activity against vancomycin-resistant bacteria increased. Compound 37b, with the tetradecyl chain, was found to be the most potent, with an MIC of 0.7 μM (>1000-fold more active than the parent drug). The attachment of a lipophilic cationic moiety imparted membrane-disruption properties to vancomycin. These derivatives were found to permeabilize and depolarise the bacterial membrane. Based on the activity and toxicity profiles, 37a was found to be the optimum compound. Further, this compound (37a) exhibited a significant reduction in murine models of infection against drug-resistant Staphylococci and also displayed significant intracellular antibacterial activity.106 Further, to impart strong membrane disruption properties at lower concentrations in addition to an enhanced binding affinity, a cationic lipophilic moiety was conjugated to the vancomycin–sugar conjugate (25) resulting in a lipophilic cationic vancomycin–sugar conjugate (38).107 This promising synergistic approach resulted in an 8000-fold (MIC ∼0.09 μM) increase in in vitro activity compared to vancomycin and promising in vivo activity against VRE. Both 37a and 38 exhibited increased cell wall biosynthesis inhibition compared to vancomycin, in addition to membrane-disruption properties. These compounds showed no propensity to induce resistance in bacteria (MRSA) even after multiple serial passages although vancomycin showed an increase in MIC after the seventh passage.
Conjugation of groups to enhance interactions with the other components of the bacterial membrane
To restore the activity of vancomycin in resistant strains, a Zn2+ binding ligand, dipicolyl-1,6-hexadiamine, was conjugated to the C-terminal of vancomycin (39, Fig. 10). The dipicolyl amine moiety captures the divalent zinc ion (Zn2+) with high selectivity. Thus, this dipicolyl–vancomycin conjugate (39) forms a complex with the pyrophosphate groups of cell wall lipids in addition to binding to the cell wall precursor peptides. This molecule exhibited a 375-fold (MIC ∼2 μM) higher in vitro activity than vancomycin against vancomycin-resistant Enterococci (VRE) and enhanced cell wall biosynthesis inhibition. Further, this compound reduced the bacterial burden in mice in VRE kidney infection.108 It did not show any increase in MIC when bacteria were subjected to serial passages of the compound indicating no propensity of resistance development.
c) Strategies to delay resistance development: glycopeptide hybrids with other antibiotics for multiple target binding
A multipronged approach is an attractive strategy in medicinal chemistry as it results in enhanced target affinity. In order to acquire resistance to such antibacterial agents, mutations leading to modifications at two biological targets would be less feasible. In one report, vancomycin–nisin conjugates were developed by linking the carboxylic acid group of vancomycin to the C-terminus of nisin.109 It was hypothesized that the loss of binding affinity of vancomycin to its target could be compensated by the additional binding of nisin to the pyrophosphate of lipid II. One of the developed conjugates showed a ∼40-fold (MIC ∼2.3 μM) increase in activity against VRE.
Both β-lactams and glycopeptides bind to targets in close proximity. It was hypothesized that by conjugating a glycopeptide with a β-lactam into a single molecule would result in enhanced bactericidal activity due to multiple target binding. With this concept, Theravance Biopharma, Inc. developed vancomycin–cephalosporin hybrids (Fig. 11). These molecules, cefilavancin (40, phase 2 completed) and TD-1607 (41, phase I), are currently in various phases of clinical trials.110,111 Although these derivatives were not effective against vancomycin-resistant strains, their initial studies exhibited enhanced bactericidal activity.
Fig. 11. Development of heterodimeric vancomycin analogues.

Glycopeptide antibiotics – the unmet needs
Glycopeptides, specifically vancomycin, have been used extensively over the decades. However, there are some aspects that need to be addressed with respect to the use of these antibiotics. In this respect, the abilities of the glycopeptides and their derivatives to i) overcome the intrinsic resistance in Gram-negative bacteria; ii) treat biofilm-associated infections and iii) treat intracellular infections have been discussed in the subsequent sections.
i) Ability to overcome intrinsic resistance in Gram-negative bacteria
Gram-negative bacteria are intrinsically resistant to glycopeptides due to the presence of an outer membrane. This membrane serves as a permeability barrier for these larger molecules. Various strategies have been developed to overcome the intrinsic resistance to vancomycin. Silver has been known to exhibit antibacterial activity through various mechanisms of action, one of them being permeabilization of the bacterial membrane in Gram-negative bacteria. In 2013, Collins' group found that on using silver, the Gram-negative bacteria were sensitized to various antibiotics including vancomycin in vitro.112 They also demonstrated that Ag+ potentiated vancomycin against E. coli in a mouse peritonitis model. In another instance, vancomycin was conjugated to gold nanoparticles.113 These multivalent Au–vancomycin conjugates were found to exhibit some activity against E. coli with an MIC of 8 μg mL–1. It was also found that upon loading vancomycin into fusogenic liposomes, the antibacterial activity against clinical isolates of E. coli and A. baumannii was obtained at concentrations at ∼6–10 μg mL–1.113 Our group developed a lipophilic cationic vancomycin derivative bearing a tetradecyl chain (37b) which permeabilises the outer and inner membranes of bacteria, depolarises the inner-membrane and inhibits cell wall biosynthesis in Gram-negative bacteria (A. baumannii).114 It was also found that serial exposure of A. baumannii to the compound did not induce resistance although the currently used antibiotic colistin showed an increase in MIC from the fifth passage itself. Compound 37b shows potent activity against clinically relevant Gram-negative pathogens and has in vivo efficacy against A. baumannii in a murine infection model. The glycopeptides vancomycin, teicoplanin and telavancin have been reported to show synergistic activity with colistin against A. baumannii in vitro.115,116 This aspect for the treatment of Gram-negative bacteria must be explored further.
ii) Ability to treat biofilm-associated infections
Most of the antibiotics belonging to this class kill planktonic bacteria but fail to kill non-growing cells. However, unlike vancomycin, the second generation glycopeptides such as oritavancin and telavancin have shown bactericidal activity against stationary phase cells.117,118 This bactericidal activity has been attributed to their ability to interact with the bacterial membrane. Oritavancin and telavancin have also been reported to disrupt biofilms of S. aureus.117,119 The cationic lipophilic vancomycin–sugar conjugate (38) developed by our group was found to disrupt bacterial biofilms of MRSA.107
iii) Ability to treat intracellular infections
The abilities of some bacteria to evade the host immune system and survive within the cells have been a growing concern. Diseases such as osteomyelitis and endocarditis are examples of such infections. These intracellular infections are difficult to treat as antibiotics are often inactive against such infections.120 Some glycopeptides have been shown to have intracellular activity. However, more research needs to be dedicated towards this aspect. Glycopeptide antibiotics exhibit intracellular antibacterial activity against S. aureus. Oritavancin and telavancin are more potent than vancomycin in treating intracellular infections.121,122 This enhanced intracellular activity was attributed to the lipophilic nature of the glycopeptides. Recently, a lipophilic cationic derivative of vancomycin was found to significantly reduce the burden of intracellular MRSA.123
Future directions
Cell wall biosynthesis inhibitors (CBIs) are an important class of antibacterial agents. The development of resistance to these CBIs calls for the addition of newer agents to this class.124
Given that there are few options for the treatment of Gram-negative superbugs such as those expressing the NDM enzymes, there is a dire need to develop inhibitors. Strategies to extend the applicability of this class of inhibitors (glycopeptides) to Gram-negative bacteria (GNB) need to be developed. Some of the new derivatives of glycopeptides have shown promising activity against some GNB, although a lot more is yet to be achieved towards obtaining broad-spectrum activity. We believe that, given the success of vancomycin as a drug, further derivatization could lead to drugs active against GNB. Further, the in vivo efficacy of the newly developed derivatives against various infection models of GNB must be tested in order to achieve clinical translation. Apart from monotherapy with these derivatives, we feel that combination therapy might indeed lead to an effective solution towards the treatment of acute and chronic Gram-negative infections. The β-lactam antibiotics are the most extensively used class of antibiotics but are prone to resistance development. In order to prevent this class from becoming obsolete, new dual inhibitors of serine-β-lactamase and metallo-β-lactamases need to be urgently developed and brought into the clinics.
Achievement of selectivity of antibiotics over commensal bacteria remains a universal challenge. Although vancomycin has been traditionally used to treat gut infections caused by C. difficile, the drug does not have selectivity over the beneficial gut bacteria. Another point that needs to be covered in this context is the inability of new glycopeptide derivatives to clear C. difficile spores at a low concentration. The literature does not have too many reports, which target this growing medical problem.
The non-inherited resistance to antibiotics in bacterial infections associated with biofilms and slow-growing or dormant bacteria is a growing health concern.125 Given the mechanism of their action, most cell wall biosynthesis inhibitors are ineffective against slowly dividing cells and dormant bacteria. Since these slowly-dividing or dormant bacteria downregulate their metabolic processes, most of these antibiotics are rendered ineffective. This is not only a problem for CBIs but also other antibiotics that target metabolic processes within bacteria. These dormant cells are often associated with biofilms of bacteria. 65–80% of all infections are biofilm mediated. With no dedicated treatment towards bacterial biofilms, the field needs to address this problem.126 Membrane-active compounds have been proved to be an effective solution to this problem.107,127–129
An interesting aspect that has not been often looked at is the ability or inability of antibiotics to act on the immune system. Some of the compounds have been shown to promote the stimulation of pro-inflammatory cytokines which ultimately leads to sepsis.130 Sepsis has claimed several lives in hospital settings and again, there is no dedicated treatment for this problem. Cell wall biosynthesis inhibitors such as glycopeptides and β-lactams are known to aggravate sepsis.130 It is imperative to study the effect of CBIs on the immune system.
Natural products have been playing a crucial role in antibiotic discovery. The emerging synthetic biology techniques offer unique opportunities to create natural product analogues by altering the biosynthetic enzymes and to expand nature's antibiotic chemical diversity. Further, this approach could solve the difficulties that are associated with synthetic chemistry in developing antibiotic analogues.
Lastly, we find that most of these compounds have been confined to treating bacterial infections. Glycopeptides and some of their derivatives have been reported to show antiviral activity against influenza virus and hepatitis virus,131 and have also been shown to inhibit Ebola pseudovirus infection in a cell culture.132 Therefore, we believe that the applicability of the glycopeptide antibiotics and their derivatives can be extended to other infectious diseases as well.
Conclusion
The use of bacterial cell wall biosynthesis inhibitors continues to be an effective way to combat bacterial infections. The approval of three new glycopeptide antibiotics in the past seven years bears testimony to this fact. Although some of the recent examples on derivatives of glycopeptides are yet to go beyond the laboratory, the results bear much promise. Some of the recent reports have also treaded on unconventional paths by using these derivatives against Gram-negative bacteria, biofilms and slowly dividing bacteria. The incorporation of moieties that confer newer/additional mechanisms of action to vancomycin might open up newer directions that this field can foray into. Indeed, glycopeptide antibiotics might be the future of cell wall biosynthesis inhibitors.
Acknowledgments
The authors thank Prof. C. N. R. Rao for his constant support and encouragement.
Biographies

Paramita Sarkar
Paramita Sarkar received her bachelor's degree in Chemistry from St. Xavier's College, Kolkata. She subsequently joined the New Chemistry Unit at the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR) as an integrated Ph.D. student and is currently pursuing her Ph.D. under the supervision of Prof. Jayanta Haldar. Her current research focuses on the development of semi-synthetic antibiotics to overcome acquired resistance in bacteria.

Venkateswarlu Yarlagadda
Venkateswarlu Yarlagadda obtained his Ph.D. degree from the New Chemistry Unit, JNCASR under the supervision of Prof. Jayanta Haldar. His research interests include antimicrobial resistance, discovery & development of novel antimicrobials using rationalized chemical and synthetic biology strategies, and understanding the role of bacterial infections in the disease progression of autoimmune diseases.

Chandradhish Ghosh
Chandradhish Ghosh received his bachelor's degree in Chemistry from St. Xavier's College (University of Calcutta) and subsequently completed his masters' degree in organic chemistry from the University of Delhi, India. His research at the Haldar Lab (JNCASR) deals with the design and development of small-molecule membrane-active agents with diverse biological applications.

Jayanta Haldar
Jayanta Haldar received his doctoral degree from the Indian Institute of Science in Bangalore. After completing postdoctoral research at the Massachusetts Institute of Technology, he joined the New Chemistry Unit, JNCASR as an assistant professor. He is currently an associate professor at the institute. His group's research focuses on the development of innovative chemical- and nanotechnology-based strategies towards the prevention and treatment of infectious diseases.
Footnotes
†The authors declare no competing interests.
References
- Neill J. O., The Review on Antimicrobial Resistance, 2014. [Google Scholar]
- Butler M. S., Blaskovich M. A., Cooper M. A. J. Antibiot. 2017;70:3–24. doi: 10.1038/ja.2016.72. [DOI] [PubMed] [Google Scholar]
- Heijenoort J. V. Glycobiology. 2001;11:25R–36R. doi: 10.1093/glycob/11.3.25r. [DOI] [PubMed] [Google Scholar]
- Barreteau H., Kovac A., Boniface A., Sova M., Gobec S., Blanot D. FEMS Microbiol. Rev. 2008;32:168–207. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- Vollmer W., Blanot D., De Pedro M. A. FEMS Microbiol. Rev. 2008;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- Zgurskaya H. I., Lopez C. A., Gnanakaran S. ACS Infect. Dis. 2015;1:512–522. doi: 10.1021/acsinfecdis.5b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C., Antibiotics: Actions, origins, resistance, ASM Press, Washington DC, USA, 2003. [Google Scholar]
- Smith C. A. J. Mol. Biol. 2006;362:640–655. doi: 10.1016/j.jmb.2006.07.066. [DOI] [PubMed] [Google Scholar]
- Sham L. T., Butler E. K., Lebar M. D., Kahne D., Bernhardt T. G., Ruiz N. Science. 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibner J. J., Richards J. D. Poult. Sci. 2005;84:634–643. doi: 10.1093/ps/84.4.634. [DOI] [PubMed] [Google Scholar]
- Lambert M. P., Neuhaus F. C. J. Bacteriol. 1972;110:978–987. doi: 10.1128/jb.110.3.978-987.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser G. A., de Carvalho L. P. FEBS J. 2013;280:1150–1166. doi: 10.1111/febs.12108. [DOI] [PubMed] [Google Scholar]
- El Zoeiby A., Sanschagrin F., Levesque R. C. Mol. Microbiol. 2003;47:1–12. doi: 10.1046/j.1365-2958.2003.03289.x. [DOI] [PubMed] [Google Scholar]
- Hrast M., Sosic I., Sink R., Gobec S. Bioorg. Chem. 2014;55:2–15. doi: 10.1016/j.bioorg.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Kahan F. M., Kahan J. S., Cassidy P. J., Kropp H. Ann. N. Y. Acad. Sci. 1974;235:364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- Castaneda-Garcia A., Blazquez J., Rodriguez-Rojas A. Antibiotics. 2013;2:217–236. doi: 10.3390/antibiotics2020217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandish P. E., Kimura K. I., Inukai M., Southgate R., Lonsdale J. T., Bugg T. D. Antimicrob. Agents Chemother. 1996;40:1640–1644. doi: 10.1128/aac.40.7.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S., Chen L., Hu Y., Rew Y., Shin D., Boger D. L. Chem. Rev. 2005;105:449–476. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- Helm J. S., Chen L., Walker S. J. Am. Chem. Soc. 2002;124:13970–13971. doi: 10.1021/ja021097n. [DOI] [PubMed] [Google Scholar]
- Hu Y., Helm J. S., Chen L., Ye X. Y., Walker S. J. Am. Chem. Soc. 2003;125:8736–8737. doi: 10.1021/ja035217i. [DOI] [PubMed] [Google Scholar]
- Cudic P., Kranz J. K., Behenna D. C., Kruger R. G., Tadesse H., Wand A. J., Veklich Y. I., Weisel J. W., McCafferty D. G. Proc. Natl. Acad. Sci. U. S. A. 2002;99:7384–7389. doi: 10.1073/pnas.102192099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breukink E., de Kruijff B. Nat. Rev. Drug Discovery. 2006;5:321–332. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- Ng V., Chan W. C. Chemistry. 2016;22:12606–12616. doi: 10.1002/chem.201601315. [DOI] [PubMed] [Google Scholar]
- Ling L. L., Schneider T., Peoples A. J., Spoering A. L., Engels I., Conlon B. P., Mueller A., Schaberle T. F., Hughes D. E., Epstein S., Jones M., Lazarides L., Steadman V. A., Cohen D. R., Felix C. R., Fetterman K. A., Millett W. P., Nitti A. G., Zullo A. M., Chen C., Lewis K. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K., Bradford P. A. Cold Spring Harbor Perspect. Med. 2016;6:a025247. doi: 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopapadakou N. H. Antimicrob. Agents Chemother. 1993;37:2045–2053. doi: 10.1128/aac.37.10.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. Future Med. Chem. 2016;8:921–924. doi: 10.4155/fmc-2016-0076. [DOI] [PubMed] [Google Scholar]
- Kong K. F., Schneper L., Mathee K. APMIS. 2010;118:1–36. doi: 10.1111/j.1600-0463.2009.02563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawz S. M., Bonomo R. A. Clin. Microbiol. Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers H. F. J. Infect. Dis. 1999;179 Suppl 2:S353–S359. doi: 10.1086/513854. [DOI] [PubMed] [Google Scholar]
- Rumbo C., Gato E., Lopez M., Ruiz de Alegria C., Fernandez-Cuenca F., Martinez-Martinez L., Vila J., Pachon J., Cisneros J. M., Rodriguez-Bano J., Pascual A., Bou G., Tomas M. Antimicrob. Agents Chemother. 2013;57:5247–5257. doi: 10.1128/AAC.00730-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomo R. A. Clin. Infect. Dis. 2011;52:485–487. doi: 10.1093/cid/ciq179. [DOI] [PubMed] [Google Scholar]
- Zasowski E. J., Rybak J. M., Rybak M. J. Pharmacotherapy. 2015;35:755–770. doi: 10.1002/phar.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem J., Cain R., Cahill S., McDonough M. A., Clifton I. J., Jimenez-Castellanos J. C., Avison M. B., Spencer J., Fishwick C. W., Schofield C. J. Nat. Commun. 2016;7:12406. doi: 10.1038/ncomms12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji M. E. A., S-649266, a novel siderophore cephalosporin: mechanisms of enhanced activity and beta-lactamase stability, Poster 40, 2015, (https://idsa.confex.com/idsa/2014/webprogram/Handout/id2651/POSTER40).
- Kohira N., West J., Ito A., Ito-Horiyama T., Nakamura R., Sato T., Rittenhouse S., Tsuji M., Yamano Y. Antimicrob. Agents Chemother. 2016;60:729–734. doi: 10.1128/AAC.01695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page M. G., Dantier C., Desarbre E. Antimicrob. Agents Chemother. 2010;54:2291–2302. doi: 10.1128/AAC.01525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushtaq S., Warner M., Livermore D. J. Antimicrob. Chemother. 2010;65:266–270. doi: 10.1093/jac/dkp425. [DOI] [PubMed] [Google Scholar]
- Reynolds P. E. Eur. J. Clin. Microbiol. Infect. Dis. 1989;8:943–950. doi: 10.1007/BF01967563. [DOI] [PubMed] [Google Scholar]
- Lovering A. L., de Castro L. H., Lim D., Strynadka N. C. Science. 2007;315:1402–1405. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]
- Stone K. J., Strominger J. L. Proc. Natl. Acad. Sci. U. S. A. 1971;68:3223–3227. doi: 10.1073/pnas.68.12.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C., Boddy C. N., Brase S., Winssinger N. Angew. Chem., Int. Ed. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Hubbard B. K., Walsh C. T. Angew. Chem., Int. Ed. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- Kahne D., Leimkuhler C., Lu W., Walsh C. Chem. Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- Svetitsky S., Leibovici L., Paul M. Antimicrob. Agents Chemother. 2009;53:4069–4079. doi: 10.1128/AAC.00341-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire J. M., Wolfe R. N., Ziegler D. W. Antibiot. Annu. 1955;3:612–618. [PubMed] [Google Scholar]
- Given D. B. C. G. L., Vancomycin, A Comprehensive Review of 30 Years of Clinical Experience, Park Row Publishers, 1986. [Google Scholar]
- Murray B. E. N. Engl. J. Med. 2000;342:710–721. doi: 10.1056/NEJM200003093421007. [DOI] [PubMed] [Google Scholar]
- Hiramatsu K., Hanaki H., Ino T., Yabuta K., Oguri T., Tenover F. C. J. Antimicrob. Chemother. 1997;40:135–136. doi: 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]
- Hiramatsu K. Lancet Infect. Dis. 2001;1:147–155. doi: 10.1016/S1473-3099(01)00091-3. [DOI] [PubMed] [Google Scholar]
- Bager F., Madsen M., Christensen J., Aarestrup F. M. Prev. Vet. Med. 1997;31:95–112. doi: 10.1016/s0167-5877(96)01119-1. [DOI] [PubMed] [Google Scholar]
- Courvalin P. Clin. Infect. Dis. 2006;42:S25–S34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- Boyd D. A., Willey B. M., Fawcett D., Gillani N., Mulvey M. R. Antimicrob. Agents Chemother. 2008;52:2667–2672. doi: 10.1128/AAC.01516-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Lin D., Yan G., Ye X., Wu S., Guo Y., Zhu D., Hu F., Zhang Y., Wang F. Antimicrob. Agents Chemother. 2010;54:4643–4647. doi: 10.1128/AAC.01710-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKessar S. J., Berry A. M., Bell J. M., Turnidge J. D., Paton J. C. Antimicrob. Agents Chemother. 2000;44:3224–3228. doi: 10.1128/aac.44.11.3224-3228.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton F., Depardieu F., Bourdon N., Fines-Guyon M., Berger P., Camiade S., Leclercq R., Courvalin P., Cattoir V. Antimicrob. Agents Chemother. 2011;55:4606–4612. doi: 10.1128/AAC.00714-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda E., Marinelli F., Marcone G. L. Antibiotics. 2014;3:572–594. doi: 10.3390/antibiotics3040572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComas C. C., Crowley B. M., Boger D. L. J. Am. Chem. Soc. 2003;125:9314–9315. doi: 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- Walsh C. T., Fisher S. L., Park I. S., Prahalad M., Wu Z. Chem. Biol. 1996;3:21–28. doi: 10.1016/s1074-5521(96)90079-4. [DOI] [PubMed] [Google Scholar]
- Wright G. D., Holman T. R., Walsh C. T. Biochemistry. 1993;32:5057–5063. doi: 10.1021/bi00070a013. [DOI] [PubMed] [Google Scholar]
- Arthur M., Reynolds P., Courvalin P. Trends Microbiol. 1996;4:401–407. doi: 10.1016/0966-842X(96)10063-9. [DOI] [PubMed] [Google Scholar]
- Reynolds P. E., Depardieu F., Dutka-Malen S., Arthur M., Courvalin P. Mol. Microbiol. 1994;13:1065–1070. doi: 10.1111/j.1365-2958.1994.tb00497.x. [DOI] [PubMed] [Google Scholar]
- Arthur M., Depardieu F., Cabanie L., Reynolds P., Courvalin P. Mol. Microbiol. 1998;30:819–830. doi: 10.1046/j.1365-2958.1998.01114.x. [DOI] [PubMed] [Google Scholar]
- Arthur M., Depardieu F., Molinas C., Reynolds P., Courvalin P. Gene. 1995;154:87–92. doi: 10.1016/0378-1119(94)00851-i. [DOI] [PubMed] [Google Scholar]
- Reynolds P. E., Arias C. A., Courvalin P. Mol. Microbiol. 1999;34:341–349. doi: 10.1046/j.1365-2958.1999.01604.x. [DOI] [PubMed] [Google Scholar]
- Weigel L. M., Clewell D. B., Gill S. R., Clark N. C., McDougal L. K., Flannagan S. E., Kolonay J. F., Shetty J., Killgore G. E., Tenover F. C. Science. 2003;302:1569–1571. doi: 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- Butler M. S., Hansford K. A., Blaskovich M. A., Halai R., Cooper M. A. J. Antibiot. 2014;67:631–644. doi: 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- Zhanel G. G., Calic D., Schweizer F., Zelenitsky S., Adam H., Lagace-Wiens P. R., Rubinstein E., Gin A. S., Hoban D. J., Karlowsky J. A. Drugs. 2010;70:859–886. doi: 10.2165/11534440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Saravolatz L. D., Stein G. E., Johnson L. B. Clin. Infect. Dis. 2009;49:1908–1914. doi: 10.1086/648438. [DOI] [PubMed] [Google Scholar]
- Higgins D. L., Chang R., Debabov D. V., Leung J., Wu T., Krause K. M., Sandvik E., Hubbard J. M., Kaniga K., Schmidt D. E. Antimicrob. Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter M. R., Adams S. M., Bazzini B., Fatheree P. R., Karr D. E., Krause K. M., Lam B. M., Linsell M. S., Nodwell M. B., Pace J. L., Quast K., Shaw J. P., Soriano E., Trapp S. G., Villena J. D., Wu T. X., Christensen B. G., Judice J. K. J. Antibiot. 2004;57:326–336. doi: 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- Candiani G., Abbondi M., Borgonovi M., Romano G., Parenti F. J. Antimicrob. Chemother. 1999;44:179–192. doi: 10.1093/jac/44.2.179. [DOI] [PubMed] [Google Scholar]
- Anderson V. R., Keating G. M., Drugs, 2008, 68 , 639 –648 , ; discussion 649–651 . [DOI] [PubMed] [Google Scholar]
- Malabarba A., Goldstein B. P. J. Antimicrob. Chemother. 2005;55 Suppl 2:ii15–ii20. doi: 10.1093/jac/dki005. [DOI] [PubMed] [Google Scholar]
- Malabarba A., Ciabatti R., Scotti R., Goldstein B. P., Ferrari P., Kurz M., Andreini B. P., Denaro M. J. Antibiot. 1995;48:869–883. doi: 10.7164/antibiotics.48.869. [DOI] [PubMed] [Google Scholar]
- Billeter M., Zervos M. J., Chen A. Y., Dalovisio J. R., Kurukularatne C. Clin. Infect. Dis. 2008;46:577–583. doi: 10.1086/526772. [DOI] [PubMed] [Google Scholar]
- Bouza E., Burillo A. Int. J. Antimicrob. Agents. 2010;36:401–407. doi: 10.1016/j.ijantimicag.2010.06.048. [DOI] [PubMed] [Google Scholar]
- Allen N. E., Nicas T. I. FEMS Microbiol. Rev. 2003;26:511–532. doi: 10.1111/j.1574-6976.2003.tb00628.x. [DOI] [PubMed] [Google Scholar]
- Patti G. J., Kim S. J., Yu T. Y., Dietrich E., Tanaka K. S., Parr, Jr. T. R., Far A. R., Schaefer J. J. Mol. Biol. 2009;392:1178–1191. doi: 10.1016/j.jmb.2009.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G., Schweizer F., Karlowsky J. A. Clin. Infect. Dis. 2012;54(Suppl 3):S214–S219. doi: 10.1093/cid/cir920. [DOI] [PubMed] [Google Scholar]
- Arhin F. F., Draghi D. C., Pillar C. M., Parr, Jr. T. R., Moeck G., Sahm D. F. Antimicrob. Agents Chemother. 2009;53:4762–4771. doi: 10.1128/AAC.00952-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravolatz L. D., Stein G. E. Clin. Infect. Dis. 2015;61:627–632. doi: 10.1093/cid/civ311. [DOI] [PubMed] [Google Scholar]
- Ashford P. A., Bew S. P. Chem. Soc. Rev. 2012;41:957–978. doi: 10.1039/c1cs15125h. [DOI] [PubMed] [Google Scholar]
- Malabarba A., Nicas T. I., Thompson R. C. Med. Res. Rev. 1997;17:69–137. doi: 10.1002/(sici)1098-1128(199701)17:1<69::aid-med3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- James R. C., Pierce J. G., Okano A., Xie J., Boger D. L. ACS Chem. Biol. 2012;7:797–804. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley B. M., Boger D. L. J. Am. Chem. Soc. 2006;128:2885–2892. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Pierce J. G., James R. C., Okano A., Boger D. L. J. Am. Chem. Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Okano A., Pierce J. G., James R. C., Stamm S., Crane C. M., Boger D. L. J. Am. Chem. Soc. 2012;134:1284–1297. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A., Nakayama A., Schammel A. W., Boger D. L. J. Am. Chem. Soc. 2014;136:13522–13525. doi: 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitanai Y., Kikuchi T., Kakoi K., Hanamaki S., Fujisawa I., Aoki K. J. Mol. Biol. 2009;385:1422–1432. doi: 10.1016/j.jmb.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Konai M. M., Manjunath G. B., Ghosh C., Haldar J. J. Antibiot. 2015;68:302–312. doi: 10.1038/ja.2014.144. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Konai M. M., Paramanandham K., Nimita V. C., Shome B. R., Haldar J. Int. J. Antimicrob. Agents. 2015;46:446–450. doi: 10.1016/j.ijantimicag.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Mackay J. P., Gerhard U., Beauregard D. A., Williams D. H., Westwell M. S., Searle M. S. J. Am. Chem. Soc. 1994;116:4581–4590. [Google Scholar]
- Williams D. H., Bardsley B. Angew. Chem., Int. Ed. 1999;38:1172–1193. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Sundram U. N., Griffin J. H., Nicas T. I. J. Am. Chem. Soc. 1996;118:13107–13108. [Google Scholar]
- Rao J., Lahiri J., Isaacs L., Weis R. M., Whitesides G. M. Science. 1998;280:708–711. doi: 10.1126/science.280.5364.708. [DOI] [PubMed] [Google Scholar]
- Arimoto H., Nishimura K., Hayakawa I., Kinumi T., Uemura D. Chem. Commun. 1999:1361–1362. [Google Scholar]
- Nicolaou K., Hughes R., Cho S. Y., Winssinger N., Smethurst C., Labischinski H., Endermann R. Angew. Chem., Int. Ed. 2000;39:3823–3828. doi: 10.1002/1521-3773(20001103)39:21<3823::AID-ANIE3823>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Xing B., Yu C. W., Ho P. L., Chow K. H., Cheung T., Gu H., Cai Z., Xu B. J. Med. Chem. 2003;46:4904–4909. doi: 10.1021/jm030417q. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Sarkar P., Manjunath G. B., Haldar J. Bioorg. Med. Chem. Lett. 2015;25:5477–5480. doi: 10.1016/j.bmcl.2015.10.083. [DOI] [PubMed] [Google Scholar]
- Nagarajan R. J. Antibiot. 1993;46:1181–1195. doi: 10.7164/antibiotics.46.1181. [DOI] [PubMed] [Google Scholar]
- Fu X., Albermann C., Zhang C., Thorson J. S. Org. Lett. 2005;7:1513–1515. doi: 10.1021/ol0501626. [DOI] [PubMed] [Google Scholar]
- Nakama Y., Yoshida O., Yoda M., Araki K., Sawada Y., Nakamura J., Xu S., Miura K., Maki H., Arimoto H. J. Med. Chem. 2010;53:2528–2533. doi: 10.1021/jm9017543. [DOI] [PubMed] [Google Scholar]
- Yoganathan S., Miller S. J. J. Med. Chem. 2015;58:2367–2377. doi: 10.1021/jm501872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarlagadda V., Akkapeddi P., Manjunath G. B., Haldar J. J. Med. Chem. 2014;57:4558–4568. doi: 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Konai M. M., Manjunath G. B., Prakash R. G., Mani B., Paramanandham K., Ranjan S. B., Ravikumar R., Chakraborty S. P., Roy S., Haldar J. Int. J. Antimicrob. Agents. 2015;45:627–634. doi: 10.1016/j.ijantimicag.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Samaddar S., Paramanandham K., Shome B. R., Haldar J. Angew. Chem., Int. Ed. 2015;54:13644–13649. doi: 10.1002/anie.201507567. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Sarkar P., Samaddar S., Haldar J. Angew.Angew. Chem., Int. Ed.Chem., Int. Ed. 2016;27:7836–7840. doi: 10.1002/anie.201601621. [DOI] [PubMed] [Google Scholar]
- Arnusch C. J., Bonvin A. M., Verel A. M., Jansen W. T., Liskamp R. M., de Kruijff B., Pieters R. J., Breukink E. Biochemistry. 2008;47:12661–12663. doi: 10.1021/bi801597b. [DOI] [PubMed] [Google Scholar]
- Long D. D., Aggen J. B., Chinn J., Choi S. K., Christensen B. G., Fatheree P. R., Green D., Hegde S. S., Judice J. K., Kaniga K., Krause K. M., Leadbetter M., Linsell M. S., Marquess D. G., Moran E. J., Nodwell M. B., Pace J. L., Trapp S. G., Turner S. D. J. Antibiot. 2008;61:603–614. doi: 10.1038/ja.2008.80. [DOI] [PubMed] [Google Scholar]
- Sader H. S., Rhomberg P. R., Farrell D. J., Flamm R. K. and Jones R. N., 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), Poster F-970, Washington, DC, USA, 2014.
- Morones-Ramirez J. R., Winkler J. A., Spina C. S., Collins J. J. Sci. Transl. Med. 2013;5:190ra181. doi: 10.1126/scitranslmed.3006276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolosi D., Scalia M., Nicolosi V. M., Pignatello R. Int. J. Antimicrob. Agents. 2010;35:553–558. doi: 10.1016/j.ijantimicag.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Manjunath G. B., Sarkar P., Akkapeddi P., Paramanandham K., Shome B. R., Ravikumar R., Haldar J. ACS Infect. Dis. 2016;2:132–139. doi: 10.1021/acsinfecdis.5b00114. [DOI] [PubMed] [Google Scholar]
- Hornsey M., Longshaw C., Phee L., Wareham D. W. Antimicrob. Agents Chemother. 2012;56:3080–3085. doi: 10.1128/AAC.05870-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareham D. W., Gordon N. C., Hornsey M. J. Antimicrob. Chemother. 2011;66:1047–1051. doi: 10.1093/jac/dkr069. [DOI] [PubMed] [Google Scholar]
- Belley A., Neesham-Grenon E., McKay G., Arhin F. F., Harris R., Beveridge T., Parr, Jr. T. R., Moeck G. Antimicrob. Agents Chemother. 2009;53:918–925. doi: 10.1128/AAC.00766-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde S. S., Reyes N., Skinner R., Difuntorum S. J Antimicrob. Chemother. 2008;61:169–172. doi: 10.1093/jac/dkm417. [DOI] [PubMed] [Google Scholar]
- LaPlante K. L., Mermel L. A. Antimicrob. Agents Chemother. 2009;53:3166–3169. doi: 10.1128/AAC.01642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B., Quie P. G., Windhorst D. B., Pollara B., Good R. A. Nature. 1966;210:1131–1132. doi: 10.1038/2101131a0. [DOI] [PubMed] [Google Scholar]
- Barcia-Macay M., Seral C., Mingeot-Leclercq M. P., Tulkens P. M., Van Bambeke F. J. Antimicrob. Chemother. 2006;50:841–851. doi: 10.1128/AAC.50.3.841-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia-Macay M., Lemaire S., Mingeot-Leclercq M. P., Tulkens P. M., Van Bambeke F. J. Antimicrob. Chemother. 2006;58:1177–1184. doi: 10.1093/jac/dkl424. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Samaddar S., Haldar J. J. Glob. Antimicrob. Resist. 2016;5:71–74. doi: 10.1016/j.jgar.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Boucher H. W., Talbot G. H., Bradley J. S., Edwards J. E., Gilbert D., Rice L. B., Scheld M., Spellberg B., Bartlett J. Clin. Infect. Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Levin B. R., Rozen D. E. Nat. Rev. Microbiol. 2006;4:556–562. doi: 10.1038/nrmicro1445. [DOI] [PubMed] [Google Scholar]
- Davies D. Nat. Rev. Drug Discovery. 2003;2:114–122. doi: 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- Ghosh C., Manjunath G. B., Konai M. M., Uppu D. S., Hoque J., Paramanandham K., Shome B. R., Haldar J. PLoS One. 2015;10:e0144094. doi: 10.1371/journal.pone.0144094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konai M. M., Haldar J. ACS Infect. Dis. 2015;1:469–478. doi: 10.1021/acsinfecdis.5b00056. [DOI] [PubMed] [Google Scholar]
- Hoque J., Konai M. M., Gonuguntla S., Manjunath G. B., Samaddar S., Yarlagadda V., Haldar J. J. Med. Chem. 2015;58:5486–5500. doi: 10.1021/acs.jmedchem.5b00443. [DOI] [PubMed] [Google Scholar]
- Uppu D. S., Ghosh C., Haldar J. Microb. Pathog. 2015;80:7–13. doi: 10.1016/j.micpath.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Maieron A., Kerschner H. J. Antimicrob. Chemother. 2012;67:2537–2538. doi: 10.1093/jac/dks217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Cui R., Li G., Gao Q., Yuan S., Altmeyer R., Zou G. Antiviral Res. 2016;125:1–7. doi: 10.1016/j.antiviral.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]