

 (E)-3-[3-(Pyridin-3/4-yl)-1-(phenyl/sulfonylmethylphenyl)-1H-pyrazol-4-yl]acrylamides were synthesized and their COX-1 and COX-2 inhibitory, antiplatelet and cytotoxic activities were evaluated.
(E)-3-[3-(Pyridin-3/4-yl)-1-(phenyl/sulfonylmethylphenyl)-1H-pyrazol-4-yl]acrylamides were synthesized and their COX-1 and COX-2 inhibitory, antiplatelet and cytotoxic activities were evaluated.
Abstract
With the aim of achieving new compounds possessing both anti-inflammatory and antiplatelet activities, we synthesized (E)-3-[3-(pyridin-3/4-yl)-1-(phenyl/sulfonylmethylphenyl)-1H-pyrazol-4-yl]acrylamides, and evaluated their COX-1 and COX-2 inhibitory and antiplatelet activities. Since COX-2 inhibitory and antiplatelet compounds have anticancer potential, we also screened their antiproliferative effects against three human cancer cell lines. Compounds 5n, 5p, 5s, 10d, 10g and 10i were determined as dual COX-2 inhibitor/antiplatelet compounds. Compound 10h appeared to be a compound that exhibited antiplatelet activity without inhibiting the COX enzyme. Compounds 5h, 10a and 10i were the most effective derivatives which displayed antiproliferative activity against Huh7, MCF7 and HCT116 cells. Particularly, compound 10i, as the compound exhibiting the highest cytotoxic, antiplatelet and COX-2 inhibitory activity, was remarkable.
Introduction
Studies towards better tolerated and potent non-steroidal anti-inflammatory drugs (NSAIDs) with fewer side effects as compared to current NSAIDs have been of interest for many years. The non-selective inhibition of the two isoforms of COX is thought to be responsible for the gastric side effects associated with the chronic use of NSAIDs. It was thought that more selective COX-2 inhibitors would have reduced side effects.1,2 Recently, research has focused on the development of COX-2-selective inhibitors, which are demonstrated to possess significantly enhanced gastric safety compared to non-selective NSAIDs. Celecoxib and rofecoxib are two well-known selective COX-2 inhibitors belonging to the COXIB class. However, rofecoxib and other COX-2 inhibitors have been withdrawn from the market due to their adverse cardiovascular side effects.3–7 While a selective COX-2 inhibitor causes the reduction of the prostacyclin (PGI2) amount, thromboxane A2 (TXA2) production is still continued in platelets by the COX-1 isoform.8
TXA2 is a cellular lipidic mediator resulting from metabolization of arachidonic acid (AA) by cyclooxygenases and thromboxane synthase9 and produced by platelets, macrophages and lung parenchyma.10 TXA2 is a potent vasoconstrictor as well as a labile platelet aggregation inducer. Conversely, PGI2 is synthesized in the walls of the vasculature and inhibits thrombus formation. It is thought that the cardiovascular toxicity of coxibs was generated because of the imbalance between PGI2 and TXA2 levels.11 In a patient who requires effective anti-inflammatory treatment and has a high risk for both gastrointestinal bleeding and cardiovascular thrombosis, the use of selective COX-2 inhibitors with antiplatelet agents like thromboxane synthase inhibitors (TxSI) is being recommended.12,13 This aspect has encouraged the search for dual inhibitors of both COX-2 and thromboxane synthase (TxS) which should display an enhanced anti-inflammatory potency with fewer side effects. Compared to COX or TxS pathways as single inhibitors, dual inhibitors shall present at least two major advantages: first, dual inhibitors, by acting on the AA metabolic pathway, possess a wide range of anti-inflammatory activities; second, dual inhibitors appear to be almost exempt from gastric and cardiac toxicity, which are the most troublesome side effects of COX inhibitors.14
There are several reviews which are mainly focused on the molecular and functional bases of the inhibition of COX enzymes by non-selective and COX-2-selective inhibitors.15,16 The pharmacophore for most of the selective COX-2 inhibitors consists of a central 1,2-diaryl-substituted five-membered heterocyclic ring.2,17–19 Some 1,3-diarylpyrazole derivatives have also been reported as selective COX-2 inhibitors.20,21 The essential structural features of TXA2 synthase inhibitors are a basic nitrogen atom of a substituted pyridine or imidazole ring and a carboxylic acid group separated by an unsaturated trans-alkyl chain.11 It is found that there should be an appropriate length between the nitrogen atom of the pyridine residue, which is known to selectively inhibit TXA2 synthase via chelating with iron, and the carboxylic acid moiety for selective TxSI.22,23
It is widely recognized that inflammation may trigger cancer initiation and progression. COX-2 is overexpressed in most solid tumors.24–27 COX-2 has been recognized as an important molecule promoting tumor progression and angiogenesis. Due to the activities of COX pro-inflammatory metabolites, this enzyme is a relevant target for anticancer therapies.28 COX-2 inhibitors have anticancer effects which include inducing apoptosis, suppressing proliferation, reducing angiogenesis and weakening invasiveness.29,30
Platelets play major roles in cancer progression by providing surface and granular contents for several interactions as well as behaving like immune cells.31,32 Therefore, the anticancer potential of antiplatelet therapy has been intensively investigated for many years.33,34 Anti-platelet agents may prevent cancer and decrease tumor growth and metastatic potential, as well as improve the survival of cancer patients. Advanced knowledge about the molecular and functional aspects of platelet-mediated tumor dissemination motivated scientists to search for drugs with anticancer potential.28
Our research includes the design, synthesis, and analgesic, anti-inflammatory, antiplatelet and anticancer screening of bioactive compounds.35–40 In our previous work,41 a series of (E)-3-[3-(2,3-dihydro-3-methyl-2-oxo-3H-benzoxazol-6-yl)-1-phenyl-1H-pyrazol-4-yl]acrylamides 1 (Fig. 1) has been synthesized using both conventional and microwave-assisted methods and evaluated for their in vitro inhibitory activities on COX-1 and COX-2 isoforms using human whole blood assay as well as their antiplatelet profiles against human platelet aggregation using arachidonic acid, as agonists. (E)-3-[3-(Pyridin-4-yl)-1-phenyl-1H-pyrazol-4-yl]acrylamides 2 were also synthesized and evaluated as inhibitors of AA-induced and collagen-induced platelet aggregation with a focus on the role of the pyridine replacement of one of the aryl groups in the diarylpyrazole template.42
Fig. 1. Bioactive compounds belonging to our chemical library.

In our other works, a series of novel thiophene-containing 1,3-diarylpyrazole derivatives and ester and amide derivatives of pyridine-containing 1,3-diarylpyrazoles were synthesized and their anticancer activities against MCF7, MDA-MB-231, HeLa, Raji and HL60 human cancer cell growth were investigated by MTT assay. Derivative 3 bearing a benzylpiperidine group at the amide portion in the thiophene-containing 1,3-diarylpyrazole series showed significant inhibitory effects against the Raji and HL60 cell lines.43 In the series of ester and amide derivatives of 1,3-diarylpyrazoles, two derivatives were found as the most promising anticancer agents in Raji cells.44 In another study, we synthesized a series of pyrazolic chalcone derivatives 4 and evaluated their anti-proliferative activities in comparison with clinically used chemotherapeutics such as 5-FU and cladribine against human cancer cell lines. Among the tested compounds, four derivatives bearing a 3/4-pyridyl moiety at the C(3)-position of the central pyrazole ring were the most effective derivatives, which displayed anti-proliferative activity with IC50 values smaller than 5 μM against HCC cells. Moreover, these compounds were less active on transformed and normal cells (MCF12A and MRC-5). We showed that two derivatives in these series caused cell cycle arrest at the G2/M phase, induced apoptotic cell death and decreased the levels of phospho-cyclin B1 at the Ser147 residue.45
In continuation of our previous works, we describe herein the design, synthesis, and COX-1 and COX-2 inhibitory, antiplatelet and anticancer activities of new (E)-3-[3-(pyridin-3/4-yl)-1-(phenyl/sulfonylmethylphenyl)-1H-pyrazol-4-yl]acrylamides, bearing a central pyrazole ring substituted with phenyl, pyridine and 3-oxoprop-1-en-1-yl fragments at 1, 3 and 4 positions, respectively (Fig. 2).
Fig. 2. Newly synthesized 1,3-diarylpyrazole derivatives.

Results and discussion
Chemistry
The synthesis of (E)-3-[1-phenyl-3-(pyridin-3/4-yl)-1H-pyrazol-4-yl]acrylamides has been accomplished as outlined in Scheme 1. Firstly, the hydrazone derivatives 2 were generated by carrying out condensation in the presence of an appropriate acetylpyridine derivative, phenylhydrazine and acetic acid in refluxing ethanol. Using Vilsmeier–Haack reaction conditions, these hydrazone derivatives were then reacted with POCl3 and DMF resulting in 1,3-diarylpyrazoles (3a, b; 8) with an aldehyde group at the 4 position. The 1-substituted phenyl-3-(pyridin-3/4-yl)-1H-pyrazole-4-carbaldehydes (3a, b; 8) were then treated with malonic acid in pyridine to prepare the corresponding α,β-unsaturated carboxylic acid derivatives (2E)-3-[1-phenyl-3-(pyridin-3/4-yl)-1H-pyrazol-4-yl]prop-2-enoic acids (4a, b; 9), via the Knoevenagel condensation reaction. By treatment of 4a, b and 9 with appropriate amines in the presence of DMAP and EDCI, which was used as the carboxylate activator, the resulting eighteen 3-[3-(pyrid-3-yl)-1-phenyl-1H-pyrazol-4-yl]acrylamide (5a–s) and nine 3-[3-(pyrid-4-yl)-1-phenyl-1H-pyrazol-4-yl]acrylamide (10a–i) derivatives were prepared in good yield (24–88%). The compounds were purified by automated flash chromatography and checked for purity with UPLC before being tested in biological assays (purity was >97%). The structures of these compounds were confirmed by high-resolution mass spectrometry (HRMS), IR, 1H-NMR spectral data and elemental analysis. Detailed synthetic methods and analytical results for the obtained compounds are given in the Experimental section.
Scheme 1. Synthesis pathway to achieve the title compounds.

Structure–activity studies for the tricyclic class of selective COX-2 inhibitors have shown that a SO2Me or SO2NH2 substituent at the para position of one aryl ring usually provides optimal COX-2 inhibitory potency.46 The sulfonylmethyl COX-2 pharmacophore is a suitable scaffold to design COX-2 inhibitors and anti-cancer agents. Therefore, we aimed to introduce a sulfonylmethyl pharmacophore on the para position of the phenyl ring at position 1 of the pyrazole ring. In the 4-pyridyl derivatives of the synthesized compounds, the phenyl ring is located at position 1 of the pyrazole ring. During the synthesis studies, the pyrazole derivative bearing the sulfonylmethyl group at position 1 and the aldehyde functional group at position 4 cannot be obtained. Cyclization of some ketone hydrazones to 1-substituted 4-formylpyrazoles by using the Vilsmeier–Haack reagent (POCl3–DMF) involves double formylation and its mechanistic pathways are not certain. We obtained 4-(1-(4-(methylsulfonyl)phenyl)-1H-pyrazol-3-yl)pyridine 12 by the Vilsmeier–Haack reaction. Further formylation of compound 12 was not achieved, consistent with the results of Kira et al.47 Therefore, the synthesis of the amide derivatives intended to be obtained using this starting material has not been realized.
The C O bands of the ketone carbonyl group of the starting compounds at 1675 cm–1 were not observed in the IR spectrum of the synthesized hydrazone derivatives (2a, b; 7). In the IR spectrum of the aldehyde derivatives (3a, b; 8), the C O stretching band of the aldehyde structure was observed at 1673, 1690 and 1669 cm–1, respectively. In the 1H-NMR spectra of these compounds, the aldehyde proton was recorded at 10.06, 10.02 and 10.04 ppm, while the pyrazole ring proton was recorded at 8.58, 9.59 and 9.43 ppm, respectively. Compounds 2a, 3a, 4a, 7, 8 and 9 were previously reported.44
Biological evaluations
In vitro purified COX enzyme inhibition studies
The COX-1 and COX-2 inhibitory activities of the compounds were examined by the EIA-COX inhibitor screening method (Cayman Chemical).48 Preliminary screening of the inhibitory effects on the COX-1 and COX-2 isoforms of the compounds was carried out at a concentration of 10 μM. Indomethacin (INDO) was used as the reference compound. The results are given in Table 1.
Table 1. The in vitro inhibitory effects of the synthesized compounds on purified COX-1 and COX-2 enzymes and on platelet aggregation.
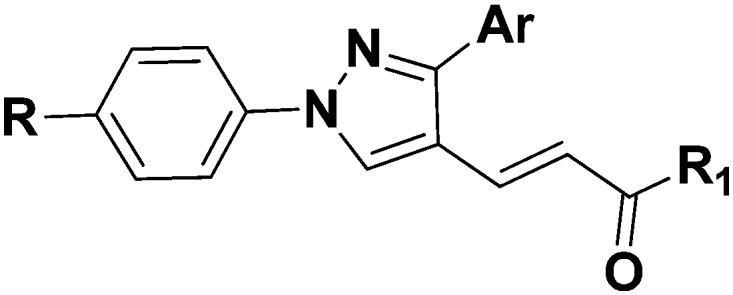
| |||||||
| Comp. | Ar | R | R1 | Inhibition % |
Inhibition of platelet aggregation % |
||
| COX-1 | COX-2 | AA | Collagen | ||||
| 5a | 3-Pyridyl | H |
|
0.12 | –30.52 | 16.14 | 86.52 |
| 5b | 3-Pyridyl | H |
|
0.32 | 11.17 | 1.24 | 60.26 |
| 5c | 3-Pyridyl | H |

|
2.46 | 2.55 | 0.0 | 15.52 |
| 5d | 3-Pyridyl | H |
|
11.32 | –3.88 | 0.0 | 56.52 |
| 5e | 3-Pyridyl | H |
|
5.56 | 3.79 | 1.26 | 63.3 |
| 5f | 3-Pyridyl | H |
|
11.60 | 15.36 | 4.54 | 36.62 |
| 5g | 3-Pyridyl | H |

|
3.21 | 10.22 | 3.68 | 21.89 |
| 5h | 3-Pyridyl | H |
|
0.45 | –5.65 | 0.0 | 76.59 |
| 5i | 3-Pyridyl | H |

|
10.15 | –3.54 | 11.29 | 75.89 |
| 5j | 3-Pyridyl | SO2CH3 |
|
40.55 | 65.16 | 82.40 | 52.65 |
| 5k | 3-Pyridyl | SO2CH3 |
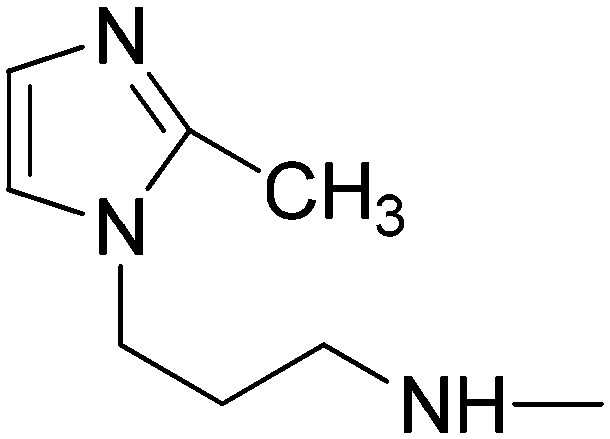
|
36.76 | 35.93 | 19.53 | 11.07 |
| 5l | 3-Pyridyl | SO2CH3 |

|
29.57 | 36.92 | 14.3 | 8.60 |
| 5m | 3-Pyridyl | SO2CH3 |
|
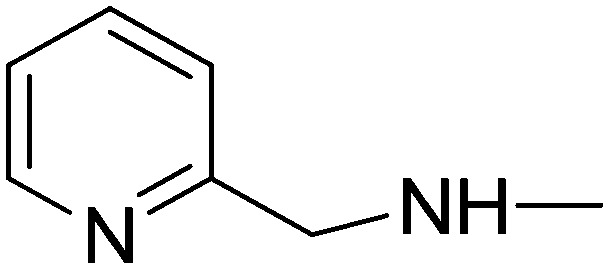
10.7 | 15.85 | 8.92 | 59.84 |
| 5n | 3-Pyridyl | SO2CH3 |

|
13.1 | 58.85 | 89.98 | 64.11 |
| 5o | 3-Pyridyl | SO2CH3 |

|
12.53 | –29.64 | 6.20 | 40.87 |
| 5p | 3-Pyridyl | SO2CH3 |
|
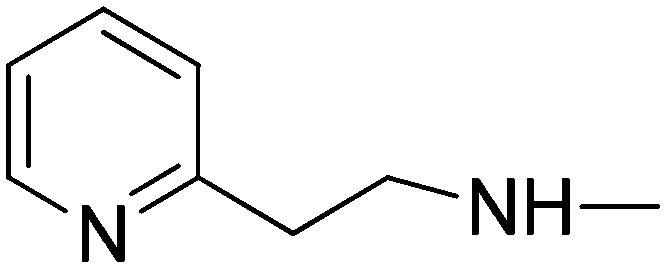
54.6 | 66.27 | 90.69 | 78.66 |
| 5r | 3-Pyridyl | SO2CH3 |

|
1.31 | 5.43 | 14.55 | 70.26 |
| 5s | 3-Pyridyl | SO2CH3 |

|
7.95 | 66.01 | 100 | 71.79 |
| 10a | 4-Pyridyl | H |
|
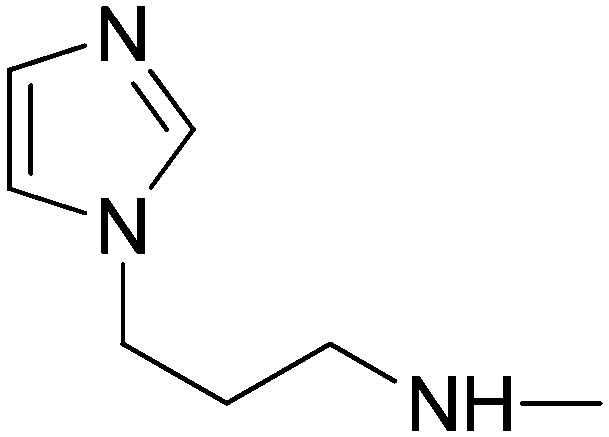
12.58 | 36.08 | 90.57 | 88.93 |
| 10b | 4-Pyridyl | H |
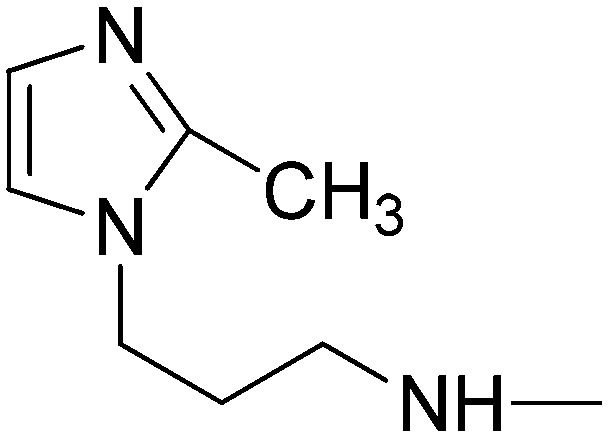
|
5.41 | 6.06 | 7.91 | 41.30 |
| 10c | 4-Pyridyl | H |
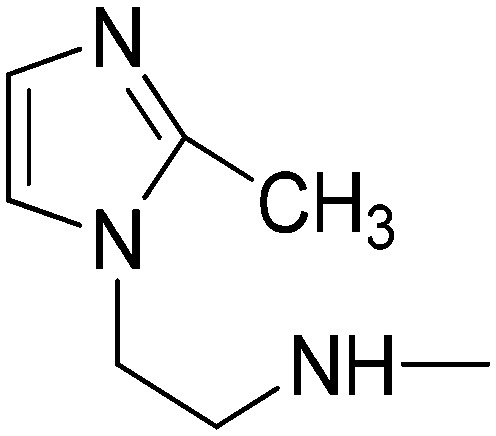
|
16.89 | 12.61 | 2.54 | 35.03 |
| 10d | 4-Pyridyl | H |
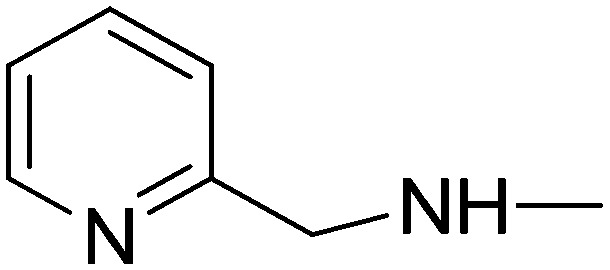
|
5.96 | 43.13 | 100 | 41.57 |
| 10e | 4-Pyridyl | H |
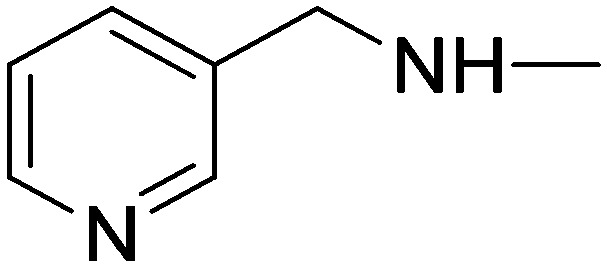
|
13.66 | 20.67 | 43.30 | 63.37 |
| 10f | 4-Pyridyl | H |
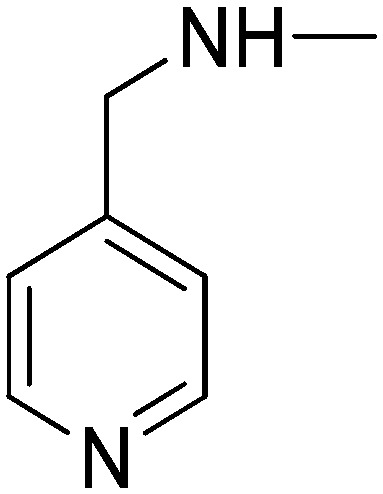
|
5.82 | –2.74 | 4.96 | 50.09 |
| 10g | 4-Pyridyl | H |
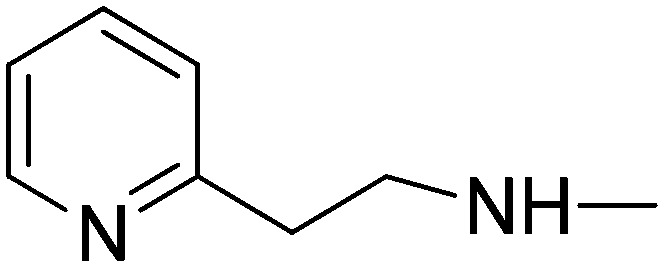
|
13.06 | 49.67 | 100 | 41.87 |
| 10h | 4-Pyridyl | H |

|
9.37 | 16.27 | 93.67 | 86.90 |
| 10i | 4-Pyridyl | H |
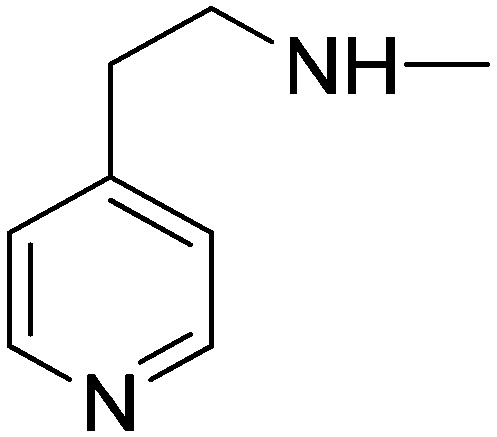
|
26.55 | 25.45 | 71.43 | 74.58 |
| INDO | 69.52 | 84.27 | — | — | |||
| ASA | — | — | 100 | 100 | |||
In the 3-pyridyl derivatives, the inhibitory activity against COX-1 and COX-2 isoenzymes was not observed for derivatives bearing a phenyl ring at position 1 of the pyrazole ring. Compounds 5f and 5g showed the highest inhibitory potency in this series of compounds. Compound 5f has 4-pyridylmethylamine at the amide part and exhibited 11% and 15% inhibitory activity against COX-1 and COX-2, respectively. Compound 5g bearing a 2-pyridylethylamine group in the amide portion similarly showed poor inhibitory activity against COX-1 and COX-2 isozymes (3% and 10%, respectively).
In the 3-pyridyl derivatives, derivatives 5j–5s with 4-sulfonylmethylphenyl substitution at position 1 on the pyrazole ring exhibited stronger inhibitory effects on the COX-1 and COX-2 isoenzymes than their nonsubstituted counterparts 5a–5i. Derivatives having an imidazole moiety at the amide part (5j, 5k and 5l) have markedly enhanced inhibitory activity against both isoenzymes. In particular, compound 5j presented a high activity against the COX-2 enzyme (65% inhibition). This compound also showed 40% inhibitory activity against the COX-1 enzyme. The highest inhibitory activity of compounds 5p and 5s was also observed among compounds (5m, 5n, 5o, 5p, 5r and 5s) formed by conversion of cinnamic acid residues into the amide form with pyridylalkyl amine derivatives. These derivatives have a pyridylethylamine group at the amide part. Compounds bearing pyridylmethyl amine residues in the amide group are subject to a reduction in activity. Of these derivatives, only compound 5n (having the pyrid-3-yl-methylamine group) had a potent inhibitory effect on COX-1 and COX-2 enzymes (58%).
In the 4-pyridyl derivatives, the phenyl ring is located at position 1 of the pyrazole ring. In these derivatives, only compound 10a (possessing an imidazol-1-yl-propylamine residue) was found to be active against COX-1 and COX-2 enzymes (36%). Compounds carrying the pyridyl amine group in the amide moiety exhibit higher inhibitory activity against COX-1 and COX-2 enzymes when compared to those carrying the imidazole group. In particular, compounds 10d (carrying the pyrid-2-yl-methylamine group) and 10g (carrying the pyrid-2-yl-ethylamine group) had 43% and 49% inhibitory potency against the COX-2 enzyme, respectively. Compounds 10d and 10g showed weak inhibitory effects on the COX-1 enzyme.
Inhibition of platelet aggregation
The antiplatelet effects of the compounds were tested at a final concentration of 100 μM. As a reference, acetyl salicylic acid (ASA) was again used at the final concentration of 100 μM. The activity results of AA and collagen-induced platelet aggregation are given in Tables 1 and 2.
Table 2. IC50 (μM) of the most active compounds on AA-induced platelet aggregation.
| Compound | 5n | 5p | 5s | 10a | 10d | 10g | 10h | ASA |
| IC50 (μM) | 49.77 | 10.14 | 21.43 | 33.57 | 78.33 | 32.80 | 29.07 | 7.70 |
As seen in Table 1, in the 3-pyridyl derivatives of the synthesized compounds, no inhibitory effect on the AA-induced platelet aggregation was found in the derivatives bearing a phenyl group (5a–5i) at position 1 of the pyrazole ring. However, these compounds were tested for collagen-induced platelet aggregation at the same concentration and the inhibitory effects on collagen-induced platelet aggregation were determined. In particular, compounds 5a, 5b, 5d, 5e, 5h and 5i inhibited collagen-induced platelet aggregation in high percentages (86–56%).
In the 3-pyridyl derivatives, the inhibitory effect on AA-induced platelet aggregation of the derivatives containing a sulfonylmethylphenyl ring at position 1 of the pyrazole ring markedly increased. Compounds 5j, 5n 5p and 5s inhibited platelet aggregation induced by AA (82%, 89%, 90% and 100% inhibition, respectively). All of these derivatives were tested for collagen-induced platelet aggregation at the same concentration and were found to significantly inhibit collagen-induced platelet aggregation. Compound 5p inhibited collagen-induced platelet aggregation by 78%.
In the 4-pyridyl derivatives, compounds 10a, 10d, 10g, 10h and 10i strongly inhibited platelet aggregation induced by AA. Among them, compounds 10d and 10g inhibited AA-induced platelet aggregation at 100% percentage at 100 μM concentration. These derivatives also showed an inhibitory effect on collagen-induced platelet aggregation. Compounds 10a, 10h and 10i inhibited collagen-induced platelet aggregation by 88%, 86% and 74%, respectively.
Anticancer activity
The inducible COX-2 isoform is overexpressed in cancer tissues and is associated with critical events of tumorigenesis. COX-2 expression correlates with expression of angiogenic factors and new blood vessel formation. Inhibition of COX-2 favors apoptosis and causes a dose-dependent decline of tumor growth and metastasis.49 Platelets play major roles in cancer progression.31,32 Therefore, the anticancer potential of antiplatelet therapy has been intensively investigated for many years.33,34 Advanced knowledge about the molecular and functional aspects of platelet-mediated tumor dissemination motivated scientists to search for drugs with anticancer potential.28
Towards this goal, we carried out a cytotoxic drug-screening method based on a sulforhodamine B assay (SRB) in triplicate to determine their IC50 values50 (Table 3). The cytotoxic activity of the synthesized compounds 5a–s and 10a–i was investigated on liver (Huh7), breast (MCF7) and colon (HCT116) cancer cell lines, by means of SRB assays in triplicate.
Table 3. Cytotoxicity of compounds 5a–5s, 10a–10i and nucleoside analogue 5-FU assessed in different human cancer cells.
| IC50
a
(μM) |
|||
| Comp. | Huh7 | MCF7 | HCT116 |
| 5a | 14.3 | 15.4 | 17.6 |
| 5b | 13.4 | 25.1 | 21.4 |
| 5c | 25.6 | 21.8 | 20.0 |
| 5d | 18.5 | 27.8 | 24.5 |
| 5e | 21.0 | 30.9 | 21.6 |
| 5f | 24.4 | NI b | NI |
| 5g | 18.9 | 24.9 | 14.2 |
| 5h | 8.1 | 30.8 | 16.9 |
| 5i | 14.5 | NI | 26.7 |
| 5j | NI | NI | NI |
| 5k | NI | NI | NI |
| 5l | NI | NI | NI |
| 5m | 24.7 | NI | 27.0 |
| 5n | NI | NI | NI |
| 5o | NI | NI | 31.7 |
| 5p | NI | 14.3 | NI |
| 5r | NI | NI | NI |
| 5s | 38.7 | NI | 19.8 |
| 10a | 6.8 | NI | 18.5 |
| 10b | 21.1 | NI | 10.8 |
| 10c | 21.0 | NI | 12.6 |
| 10d | 14.7 | NI | 20.4 |
| 10e | NI | NI | 14.6 |
| 10f | 32.6 | NI | 19.9 |
| 10g | 27.3 | 12.2 | NI |
| 10h | NI | 16.1 | 14.6 |
| 10i | 6.5 | 11.0 | 7.1 |
| 5-FU | 21.0 | 1.4 | 18.4 |
aExperiments were performed in triplicate and R2 values were between 1–0.8.
bNI: no inhibition.
In general, compounds having a 3-pyridyl moiety at position 3 on the pyrazole ring (5a–5i) displayed moderate antiproliferative potency on all cancer cell lines. Compounds having an imidazole moiety at the amide part of the molecules (5a and 5b) showed good cytotoxicity with IC50 values of 13.4 and 14.3 μM in the Huh7 cell line. These two derivatives had slight cytotoxicity (IC50 ∼ 15.4–25.1 μM) in the MCF7 and HCT116 cell lines. Compound 5c, bearing a short alkyl chain compared to other imidazole derivatives 5a and 5b, had weak activity for all cell lines used in this study (IC50 ∼ 20.0–25.6 μM). Compounds 5d–5h have 2-, 3- and 4-pyrimidine moieties at the amide part. Among these derivatives, 5h was the most active compound against the Huh7 cell line with an IC50 value of 8.1 μM. 5h had decreased cytotoxicity against the MCF7 and HCT116 cancer cell lines.
Replacing the 3-pyridyl moiety (5a–5i) with a 4-pyridyl group (10a–10i), in general, resulted in a slight decrease in the cytotoxic activity of the compounds. Interestingly, the most active compounds among all synthesized derivatives belong to the 4-pyridyl series. Compounds 10a and 10i exhibited good cytotoxic activity especially against the Huh7 cell line (IC50 = 6.8 and 6.5 μM, respectively). Compound 10i was the most active derivative among all tested compounds on the MCF7 and HCT116 cell lines with IC50 values of 11.0 and 7.1 μM, respectively.
Introducing the 4-sulfonylmethyl group to phenyl at position 1 on the pyrazole ring (5j–5s) resulted in the loss of the cytotoxic activity of the compounds with the exception of derivatives 5m and 5s bearing a 2-pyrimidinylmethyl group and a 4-pyrimidinylethyl group at the amide part, respectively. Compounds 5m and 5s exhibited weak cytotoxic activity especially against Huh7 (IC50 = 24.7 and 38.7 μM, respectively) and HCT116 (IC50 = 27.0 and 19.8 μM, respectively).
Lipinski's rule of five and drug-likeness profile
Physicochemical properties of the synthesized compounds
The synthesized compounds 5a–s and 10a–10i were submitted to an in silico evaluation using a molecular modeling approach. To predict the drug-like properties of the synthesized compounds, we analyzed these derivatives according to the rule-of-five developed by Lipinski et al.51 Lipinski's ‘rule-of-five’ and the later addition of other parameters such as PSA52 describe the molecular properties important for a drug's pharmacokinetics in the human body. PSA has been shown to be a very good descriptor characterizing drug absorption, including intestinal absorption, bioavailability, Caco-2 permeability and blood–brain barrier penetration. This approach has been widely used as a filter for substances that are likely to be further developed in drug design programs. Log P, the measure of a compound's solubility and permeability, is believed to be very important. Very high lipophilicity and the resulting large log P values cause poor absorption or permeation and should be avoided.
Predictions of ADME properties for these compounds are given in Table 4. The calculated physicochemical properties53 showed that all of the compounds fulfilled Lipinski's ‘rule-of-five’. Theoretically, these compounds should present good passive oral absorption and differences in their bioactivity cannot be attributed to this property. However, introducing the 4-sulfonylmethyl group to phenyl at position 1 on the pyrazole ring (5j–5s) resulted in very polar compounds (clog P values of 0.48–1.35). Compounds 5j, 5k, 5l, 5m, 5n and 5o had very low clog P values of 0.67, 0.75, 0.48, 0.94, 0.88 and 0.82, respectively, which might be disadvantageous with regard to the pharmacokinetic properties of these molecules in biological systems. These compounds were found inactive in cytotoxicity screening against all cell lines except 5m, 5o, 5p and 5s demonstrating weak activity. The rest of the compounds exhibited higher clog P values. Along with this, compounds 5h, 10a and 10i, which showed good antitumor screening results (Huh7 cells, IC50 = 8.1, 6.8 and 6.5 μM, respectively), have optimal clog P values, compared to other compounds in the series. The total polar surface area (TPSA) was calculated based on the methodology published by Ertl et al.52 as sums of O- and N-centered polar fragment contributions. The PSA is closely related to the hydrogen bonding potential of a compound. The TPSAs of the synthesized compounds were relatively small in comparison with the average value for acceptable drug molecules (<90 Å2) except for compounds 5j–5s (TPSA = 106.85–111.78 Å2), which were found to be inactive in cytotoxicity screening as mentioned above. We were not able to find a direct correlation between the TPSA values and anti-inflammatory and antiplatelet activities of the tested compounds. However, it has to be kept in mind that log P and PSA values are the two most important features, although not sufficient for predicting oral absorption of a drug.
Table 4. Calculated physicochemical properties and the drug-likeness of the synthesized compounds.
| Compd. | Predicted oral bioavailability |
Drug-likeness | |||||
| HBA a | HBD b | M. W. c | clog P d | clog S e | TPSA f | ||
| 5a | 7 | 1 | 398.47 | 1.80 | –2.39 | 77.64 | 5.04 |
| 5b | 7 | 1 | 412.50 | 1.88 | –2.02 | 77.64 | 4.36 |
| 5c | 7 | 1 | 398.47 | 1.61 | –1.75 | 77.64 | 4.73 |
| 5d | 6 | 1 | 381.44 | 2.07 | –3.26 | 72.71 | 2.74 |
| 5e | 6 | 1 | 381.44 | 2.01 | –3.23 | 72.71 | 2.74 |
| 5f | 6 | 1 | 381.44 | 1.95 | –3.23 | 72.71 | 2.74 |
| 5g | 6 | 1 | 395.47 | 2.48 | –3.37 | 72.71 | 2.83 |
| 5h | 6 | 1 | 395.47 | 2.41 | –3.34 | 72.71 | 2.83 |
| 5i | 6 | 1 | 395.47 | 2.36 | –3.34 | 72.71 | 2.83 |
| 5j | 9 | 1 | 476.56 | 0.67 | –2.81 | 111.78 | 6.10 |
| 5k | 9 | 1 | 490.59 | 0.75 | –2.43 | 111.78 | 5.52 |
| 5l | 9 | 1 | 476.56 | 0.48 | –2.16 | 111.78 | 5.82 |
| 5m | 8 | 1 | 459.53 | 0.94 | –3.67 | 106.85 | 4.13 |
| 5n | 8 | 1 | 459.53 | 0.88 | –3.65 | 106.85 | 3.97 |
| 5o | 8 | 1 | 459.53 | 0.82 | –3.23 | 106.85 | 2.74 |
| 5p | 8 | 1 | 473.56 | 1.35 | –3.79 | 106.85 | 4.26 |
| 5r | 8 | 1 | 473.56 | 1.28 | –3.76 | 106.85 | 4.09 |
| 5s | 8 | 1 | 473.56 | 1.23 | –3.76 | 106.85 | 2.57 |
| 10a | 7 | 1 | 398.47 | 1.58 | –2.39 | 77.64 | 5.04 |
| 10b | 7 | 1 | 412.50 | 1.67 | –2.02 | 77.64 | 4.36 |
| 10c | 7 | 1 | 398.47 | 1.40 | –1.75 | 77.64 | 4.73 |
| 10d | 6 | 1 | 381.44 | 1.85 | –3.26 | 72.71 | 2.74 |
| 10e | 6 | 1 | 381.44 | 1.79 | –3.23 | 72.71 | 2.74 |
| 10f | 6 | 1 | 381.44 | 1.74 | –3.23 | 72.71 | 2.74 |
| 10g | 6 | 1 | 395.47 | 2.26 | –3.37 | 72.71 | 2.83 |
| 10h | 6 | 1 | 395.47 | 2.19 | –3.34 | 72.71 | 2.83 |
| 10i | 6 | 1 | 395.47 | 2.14 | –3.34 | 72.71 | 2.83 |
aNumber of hydrogen bond acceptors.
bNumber of hydrogen bond donors.
cMolecular weight.
dCalculated lipophilicity.
eSolubility parameter.
fTopological polar surface area (Å2).
Drug-likeness and toxicity profile of compounds 5a–s and 10a–10i
Currently, there are many approaches to assess a compound's drug-likeness based on topological descriptors, fingerprints of molecular drug-likeness structure keys.54 In this work, we used the DataWarrior program55 for calculating the physicochemical properties (solubility and drug-likeness) and the toxicity risks (mutagenicity, tumorigenicity, irritation and reproduction) of compounds 5a–s and 10a–10i (Table 4). The aqueous solubility of a compound significantly affects its absorption and distribution characteristics. Typically, a low solubility goes along with bad absorption and therefore, in general, the aim is to avoid poorly soluble compounds. More than 80% of the drugs on the market have a (estimated) log S value of greater than –4. The log S values of most of our compounds 5a–s and 10a–10i are around –3. Drug-likeness may be defined as a complex balance of various molecular properties and structural features which determine whether a particular molecule is similar to known drugs. These properties influence the behavior of a molecule in a living organism, including bioavailability, transport properties, affinity to proteins, reactivity, toxicity, metabolic stability and many others. It is interesting that most of our compounds demonstrated good drug-likeness values (from 6.10 to 2.57). A positive value states that the molecule contains predominantly fragments which are frequently present in commercial drugs. None of the compounds exhibited a toxic profile.
Conclusion
In conclusion, we have designed and synthesized compounds that are potential inhibitors of cyclooxygenase and thromboxane synthase enzymes, which play an important role in the arachidonic acid pathway. The antiplatelet and anti-inflammatory activities of the synthesized compounds were investigated in vitro. The design of the compounds was made according to the work done in this area in the literature and the results of the preliminary tests obtained in our laboratory. The aim is to combine both COX and antiplatelet activities on the same compound. The synthesized compounds were designed as a tricyclic structure carrying a nonsubstituted/para-methanesulfonyl-substituted phenyl ring at the 1 position of the central pyrazole ring, while having a 3/4-pyridyl group at the 3 position of the central pyrazole ring. Synthesis of the designed compounds and investigation of COX and antiplatelet effects revealed that compounds 5n, 5p, 5s, 10d, 10g and 10i were dual COX-2 inhibitor/antiplatelet compounds. Compound 10h appeared to be a compound that exhibited antiplatelet activity without inhibiting the COX enzyme.
With the knowledge of the anticancer potential of COX-2 inhibitors and antiplatelet agents, we screened the anticancer activity of the synthesized compounds. Compounds 5h, 10a and 10i were the most effective derivatives which displayed antiproliferative activity with IC50 values smaller than 10 μM against hepatocellular (Huh7), breast (MCF7) and colon carcinoma (HCT116) cells. Maximal inhibitory effects were observed with these three compounds, in accord with their antiplatelet effects. Particularly, compound 10i, as the compound exhibiting the highest anticancer, antiplatelet and COX-2 inhibitory activity, was remarkable.
The physicochemical properties of the synthesized compounds were also evaluated in silico, and it was found that some of our compounds should present good passive oral absorption. All synthesized compounds demonstrated good drug-likeness values. Thus, the results of this study have reached the conclusions that will form the basis of further studies. In particular, further optimization studies can be planned as a result of the structure–activity relationships.
Experimental
Chemistry
Chemicals were purchased from commercial vendors and were used without purification. Thin-layer chromatography (TLC) was performed on Merck 60F254 plates. Reactions were monitored by TLC on silica gel, with detection by UV light (254 nm) or charring Dragendorff reagent.56 Melting points were determined with an SMP-II digital melting point apparatus and are uncorrected (Schorpp Geraetetechnik, Germany). IR spectra were obtained in-house using a Perkin Elmer Spectrum 400 FTIR/FTNIR spectrometer equipped with a universal ATR sampling accessory. 1H-NMR spectra were recorded in CDCl3 or DMSO-d6 on a Varian Mercury 400 MHz FT-NMR spectrometer using tetramethylsilane as the internal standard at the NMR facility of the Faculty of Pharmacy, Ankara University. All chemical shifts were recorded as δ (ppm). High-resolution mass spectral data (HRMS) were collected in-house using a Waters LCT Premier XE mass spectrometer (high-sensitivity orthogonal acceleration time-of-flight instrument) operating in ESI (+) mode also coupled to an ACQUITY ultra performance liquid chromatography system (Waters Corporation, Milford, MA, USA). Flash chromatography was performed with a Combiflash® Rf automated flash chromatography system with RediSep columns (Teledyne-Isco, Lincoln, NE, USA) using dichloromethane–methanol solvent gradients.
N-(4-Methanesulfonylphenyl)-N′-(1-pyridin-3-yl-ethylidene)hydrazine (2b)
A solution of acetylpyridine derivative (0.052 mol), phenyl hydrazine derivative (0.058 mol) and acetic acid (2 ml, 0.035 mol) in ethanol was stirred for 2 h under reflux, and then evaporated. The precipitate was filtered off and dried. Yield 78%, mp 176.8–178 °C. IR (FTIR/FTNIR-ATR): 3299 cm–1 (N–H), 2988 cm–1 (aliphatic C–H). 1H-NMR (DMSO-d6) δ: 10.04 (1H, s), 9.01 (1H, d, J = 2 Hz), 8.53 (1H, dd, Ja = 1.6 Hz, Jb = 4.8 Hz), 8.17 (1H, dt, Ja = 1.6 Hz, Jb = 7.6 Hz), 7.74 (2H, d, J = 9.2 Hz), 7.45–7.40 (3H, m), 3.12 (3H, s), 2.34 (3H, s). HRMS C14H16N3O2S [M + H]+ calc. 290.0963, found m/z 290.0969. Anal. calc. (%) for C14H15N3O2S calc. % C: 58.11, H: 5.23, N: 14.52, S: 11.08, found % C: 58.02, H: 5.33, N: 14.38, S: 10.99.
1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazole-4-carbaldehyde (3b)
In a dry flask, phosphoroxychloride (POCl3) (0.124 mol) was added dropwise to an ice-cold stirred solution of hydrazone derivative (0.041 mol) in 80 ml DMF. The reaction mixture was allowed to attain room temperature, and then heated at 50 °C for 4 h. The resulting mixture was poured onto crushed ice, neutralized with dilute NaOH and left overnight. The yellow precipitate obtained was purified by crystallization in toluene. Yield 85%, mp 216.4–217 °C. IR (FTIR/FTNIR-ATR): 1690 cm–1 (C O). 1H-NMR (DMSO-d6) δ: 10.02 (1H, s), 9.59 (1H, s), 9.12 (1H, d, J = 2 Hz), 8.70 (1H, dd, Ja = 1.6 Hz, Jb = 5.2 Hz), 8.35 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 8.30 (2H, d, J = 8.8 Hz), 8.14 (2H, d, J = 8.4 Hz), 7.58–7.55 (1H, m), 3.42 (3H, s). HRMS C16H14N3O3S [M + H]+ calc. 328.0756, found m/z 328.0748. Anal. calc. (%) for C16H13N3O3S calc. % C: 58.70, H: 4.00, N: 12.84, S: 9.80, found % C: 58.98, H: 3.90, N: 12.86, S: 9.80.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]acrylic acid (4b)
To a solution of 1-phenyl-3-(pyridin-3/4-yl)-1H-pyrazole-4-carbaldehyde (8.72 mmol) in pyridine (20 ml), malonic acid (0.035 mol) and piperidine (0.0131 mol) were added, and the reaction mixture was refluxed for 4 h. Upon cooling, the reaction mixture was poured onto a solution (100 ml) of crushed ice and concentrated HCl (50% by volume), then the pH was adjusted to 5. The resulting precipitate was filtered off, washed with acidified water and dried. Yield 97%, mp 316–318 °C. IR (FTIR/FTNIR-ATR): 1677 cm–1 (C O). 1H-NMR (DMSO-d6) δ: 12.44 (1H, s), 9.45 (1H, s), 8.86 (1H, m), 8.72 (1H, m), 8.21 (2H, m), 8.14–8.07 (3H, m), 7.63–7.60 (1H, m), 7.47 (1H, d, J = 16 Hz), 6.49 (1H, d, J = 16 Hz), 3.25 (3H, s). HRMS C18H16N3O4S [M + H]+ calc. 370.0862, found m/z 370.0852.
General procedure for the preparation of 3-(3-(pyrid-3/4-yl)-1-phenyl-1H-pyrazol-4-yl)acrylamide derivatives (5a–s and 10a–i)
To the solution of acid derivatives (0.692 mmol) in dichloromethane were added EDCI (0.761 mmol) and DMAP (0.138 mmol). After addition of the appropriate amine derivatives (0.761 mmol), the mixture was stirred overnight. DCM was added, and the organic phase was washed with a 1% NaHCO3 solution and brine, dried over Na2SO4, and evaporated under vacuum. The residue was purified by flash column chromatography (Combiflash® Rf) using DCM–MeOH as eluents.
(2E)-N-(3-Imidazol-1-ylpropyl)-3-[1-phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]acrylamide (5a)
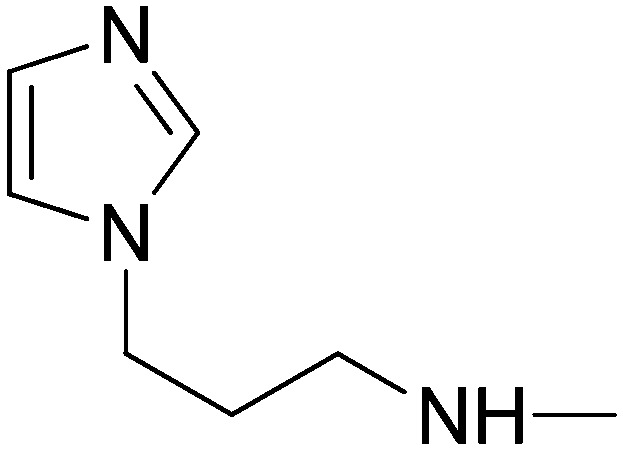
Elution with DCM–MeOH (0–10%) gave 5a as a white solid. Yield 48%, mp 116.5–118 °C. IR (FTIR/FTNIR-ATR): 1669 cm–1 (C O), 3249 cm–1 (N–H). 1H-NMR (CDCl3) δ: 8.95 (1H, d, J = 1.2 Hz), 8.65 (1H, m), 8.19 (1H, s), 7.99 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.75 (2H, d, J = 7.6 Hz), 7.62 (1H, d, J = 15.2 Hz), 7.52–7.48 (3H, m), 7.42–7.26 (2H, m), 7.06 (1H, s), 6.96 (1H, s), 6.18 (1H, d, J = 15.2 Hz), 5.87 (1H, t, J = 6 Hz), 4.03 (2H, t, J = 7 Hz), 3.38 (2H, q, J = 6.8 Hz), 2.06 (2H, m). HRMS C23H23N6O [M + H]+ calc. 399.1933, found m/z 399.1915. Anal. calc. (%) for C23H22N6O calc. % C: 69.33, H: 5.57, N: 21.09, found % C: 69.13, H: 5.92, N: 21.23.
(2E)-N-[3-(2-Methylimidazol-1-yl)propyl]-3-[1-phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]acrylamide (5b)
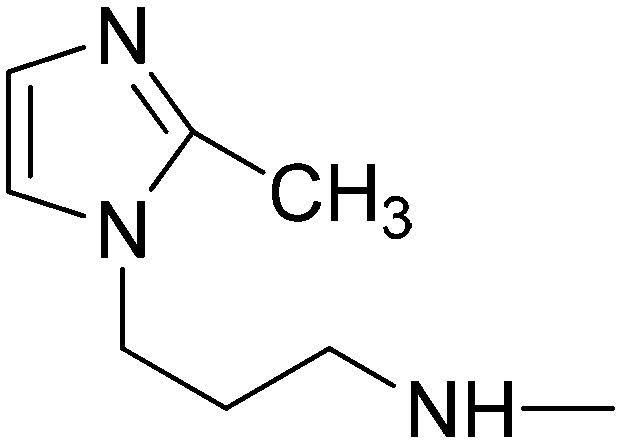
Elution with DCM–MeOH (0–20%) gave 5b as a white solid. Yield 30%, mp 153.2–154 °C. IR (FTIR/FTNIR-ATR): 1650 cm–1 (C O), 3281 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.05 (1H, s), 8.84 (1H, d, J = 2 Hz), 8.67 (1H, dd, Ja = 1.6 Hz, Jb = 4 Hz), 8.20 (1H, t, J = 5.6 Hz), 8.04 (1H, d, J = 2 Hz), 7.95 (2H, d, J = 8 Hz), 7.56 (3H, m), 7.39 (1H, t, J = 8 Hz), 7.37 (1H, d, J = 15.6 Hz), 7.07 (1H, s), 6.72 (1H, s), 6.49–6.45 (1H, d, J = 16 Hz), 3.88 (2H, t, J = 7 Hz), 3.16 (2H, q, J = 6.4 Hz), 2.26 (3H, s), 1.84 (2H, m). HRMS C24H25N6O [M + H]+ calc. 413.2090, found m/z 413.2074. Anal. calc. (%) for C24H24N6O calc. % C: 69.88, H: 5.86, N: 20.37, found % C: 69.84, H: 5.82, N: 20.26.
(2E)-N-[2-(2-Methylimidazol-1-yl)ethyl]-3-[1-phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]acrylamide (5c)
Elution with DCM–MeOH (0–20%) gave 5c as a white solid. Yield 24%, mp 123–124.8 °C. IR (FTIR/FTNIR-ATR): 1655 cm–1 (C O), 3139 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.06 (1H, s), 8.83 (1H, d, J = 1.6 Hz), 8.68 (1H, dd, Ja = 1.6 Hz, Jb = 4.4 Hz), 8.28 (1H, t, J = 2 Hz), 8.03 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.95 (2H, d, J = 8 Hz), 7.57 (3H, m), 7.40 (1H, m), 7.37 (1H, d, J = 15.6 Hz), 7.01 (1H, s), 6.72 (1H, s), 6.44 (1H, d, J = 16 Hz), 3.99 (2H, t, J = 6 Hz), 3.42 (2H, q, J = 6 Hz), 2.25 (3H, s). HRMS C23H23N6O [M + H]+ calc. 399.1933, found m/z 399.1918. Anal. calc. (%) for C23H22N6O·0.45H2O calc. % C: 67.95, H: 5.68, N: 20.67, found % C: 68.09, H: 5.65, N: 20.40.
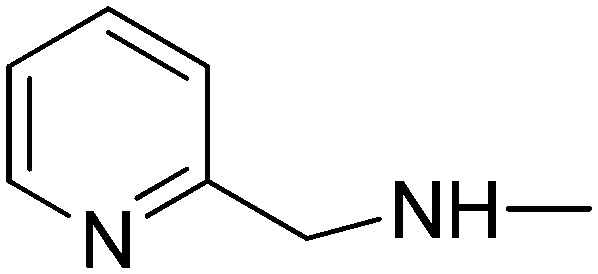
(2E)-3-[1-Phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(pyridin-2-ylmethyl)acrylamide (5d)
Elution with DCM–MeOH (0–10%) gave 5d as a white solid. Yield 54%, mp 197–198 °C. IR (FTIR/FTNIR-ATR): 1650 cm–1 (C O), 3278 cm–1 (N–H). 1H-NMR (CDCl3) δ: 8.96 (1H, d, J = 1.6 Hz), 8.65 (1H, dd, Ja = 1.6 Hz, Jb = 4.4 Hz), 8.54 (1H, d, J = 4.4 Hz), 8.21 (1H, s), 7.97 (1H, t, J = 2 Hz), 7.75 (2H, d, J = 7.6 Hz), 7.66 (1H, d, J = 15.2 Hz), 7.65 (1H, m), 7.50 (2H, t, J = 7.6 Hz), 7.40–7.30 (3H, m), 7.23–7.20 (1H, m), 7.03 (1H, t, J = 4.8 Hz), 6.38 (1H, d, J = 15.2 Hz), 4.68 (2H, d, J = 5.2 Hz). HRMS C23H20N5O [M + H]+ calc. 382.1668, found m/z 382.1656. Anal. calc. (%) for C23H19N5O calc. % C: 72.42, H: 5.02, N: 18.36, found % C: 72.58, H: 5.27, N: 18.31.
(2E)-3-[1-Phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(pyridin-3-ylmethyl)acrylamide (5e)
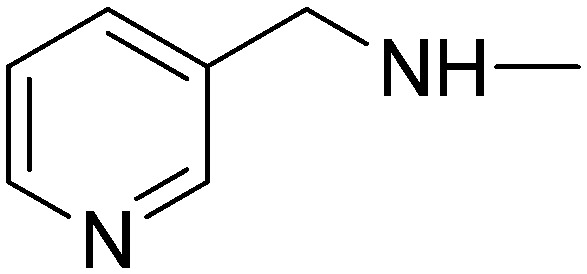
Elution with DCM–MeOH (0–10%) gave 5e as a white solid. Yield 55%, mp 197–198 °C. IR (FTIR/FTNIR-ATR): 1646 cm–1 (C O), 3275 cm–1 (N–H). 1H-NMR (CDCl3) δ: 8.93 (1H, d, J = 1.6 Hz), 8.62 (1H, dd, Ja = 1.6 Hz, Jb = 4.4 Hz), 8.53 (1H, d, J = 2 Hz), 8.50 (1H, dd, Ja = 1.6 Hz, Jb = 4.8 Hz), 8.19 (1H, s), 7.97 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.74 (2H, d, J = 7.6 Hz), 7.70–7.66 (2H, m), 7.49 (2H, t, J = 8 Hz), 7.40–7.33 (2H, m), 7.28–7.25 (1H, m), 6.29 (1H, t, J = 6 Hz), 6.25 (1H, d, J = 15.6 Hz), 4.56 (2H, d, J = 6 Hz). HRMS C23H20N5O [M + H]+ calc. 382.1668, found m/z 382.1678. Anal. calc. (%) for C23H19N5O calc. % C: 72.42, H: 5.02, N: 18.36, found % C: 72.10, H: 5.00, N: 18.10.
(2E)-3-[1-Phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(pyridin-4-ylmethyl)acrylamide (5f)
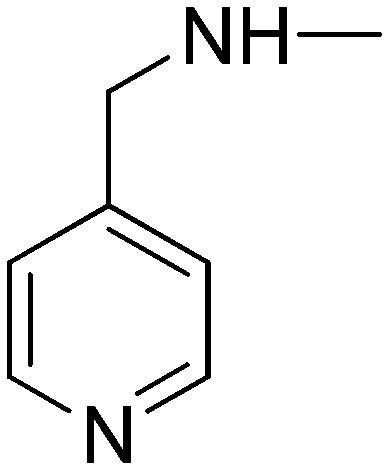
Elution with DCM–MeOH (0–10%) gave 5f as a white solid. Yield 50%, mp 217–218 °C. IR (FTIR/FTNIR-ATR): 1649 cm–1 (C O), 3272 cm–1 (N–H). 1H-NMR (CDCl3) δ: 8.94 (1H, d, J = 2 Hz), 8.64 (1H, dd, Ja = 1.6 Hz, Jb = 4.4 Hz), 8.53 (2H, d, J = 6.4 Hz), 8.21 (1H, s), 7.99 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.75 (2H, d, J = 7.6 Hz), 7.70 (1H, d, J = 15.6 Hz), 7.49 (2H, t, J = 7.8 Hz), 7.41–7.36 (2H, m), 7.21 (2H, d, J = 6 Hz), 6.28 (1H, d, J = 15.6 Hz), 6.22 (1H, m), 4.56 (2H, d, J = 6.4 Hz). HRMS C23H20N5O [M + H]+ calc. 382.1668, found m/z 382.1676. Anal. calc. (%) for C23H19N5O calc. % C: 72.42, H: 5.02, N: 18.36, found % C: 72.23, H: 5.32, N: 18.26.
(2E)-3-[1-Phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-2-ylethyl)acrylamide (5g)
Elution with DCM–MeOH (0–10%) gave 5g as a white solid. Yield 56%, mp 185–186.2 °C. IR (FTIR/FTNIR-ATR): 1669 cm–1 (C O), 3274 cm–1 (N–H). 1H-NMR (CDCl3) δ: 8.97 (1H, d, J = 2 Hz), 8.66 (1H, dd, Ja = 2 Hz, Jb = 4.4 Hz), 8.53 (1H, d, J = 4 Hz), 8.20 (1H, s), 7.98 (1H, dt, Ja = 1.6 Hz, Jb = 8 Hz), 7.75 (2H, d, J = 8 Hz), 7.64 (1H, m), 7.59 (1H, d, J = 15.2 Hz), 7.49 (2H, t, J = 7.6 Hz), 7.41–7.33 (2H, m), 7.20–7.15 (2H, m), 6.73 (1H, m), 6.23 (1H, d, J = 15.2 Hz), 3.78 (2H, q, J = 6 Hz), 3.04 (2H, t, J = 6.2 Hz). HRMS C24H22N5O [M + H]+ calc. 396.1824, found m/z 396.1806. Anal. calc. (%) for C24H21N5O calc. % C: 72.89, H: 5.35, N: 17.71, found % C: 72.56, H: 5.52, N: 17.52.
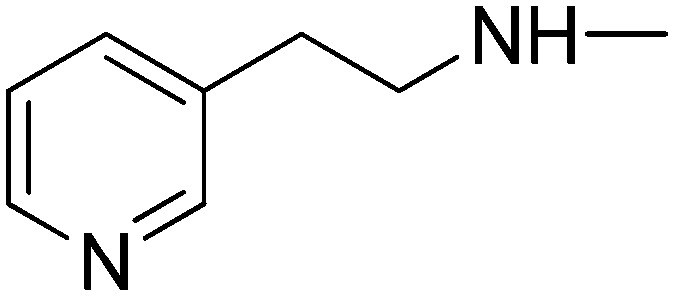
(2E)-3-[1-Phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-3-ylethyl)acrylamide (5h)
Elution with DCM–MeOH (0–10%) gave 5h as a white solid. Yield 63%, mp 211–213 °C. IR (FTIR/FTNIR-ATR): 1649 cm–1 (C O), 3279 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.04 (1H, s), 8.84 (1H, d, J = 1.6 Hz), 8.68 (1H, dd, Ja = 1.6 Hz, Jb = 4.4 Hz), 8.44 (1H, d, J = 1.6 Hz), 8.42 (1H, dd, Ja = 1.6 Hz, Jb = 6.4 Hz), 8.23 (1H, t, J = 5.6 Hz), 8.04 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.95 (2H, d, J = 8 Hz), 7.66–7.63 (1H, m), 7.60–7.54 (3H, m), 7.36 (1H, d, J = 15.6 Hz), 7.40–7.30 (2H, m), 6.45 (1H, d, J = 15.6 Hz), 3.43 (2H, q, J = 6.8 Hz), 2.80 (2H, t, J = 7 Hz). HRMS C24H22N5O [M + H]+ calc. 396.1824, found m/z 396.1811. Anal. calc. (%) for C24H21N5O calc. % C: 72.89, H: 5.35, N: 17.71, found % C: 73.24, H: 5.45, N: 17.59.
(2E)-3-[1-Phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-4-ylethyl)acrylamide (5i)
Elution with DCM–MeOH (0–10%) gave 5i as a white solid. Yield 58%, mp 182–184 °C. IR (FTIR/FTNIR-ATR): 1649 cm–1 (C O), 3282 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.04 (1H, s), 8.83 (1H, d, J = 2 Hz), 8.68 (1H, dd, Ja = 1.6 Hz, Jb = 4.4 Hz), 8.46 (2H, d, J = 5.6 Hz), 8.22 (1H, t, J = 5.6 Hz), 8.03 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.95 (2H, d, J = 8 Hz), 7.60–7.54 (3H, m), 7.39 (1H, t, J = 7.2 Hz), 7.36 (1H, d, J = 15.6 Hz), 7.25 (2H, d, J = 6 Hz), 6.44 (1H, d, J = 15.6 Hz), 3.44 (2H, q, J = 6 Hz), 2.80 (2H, m). HRMS C24H22N5O [M + H]+ calc. 396.1824, found m/z 396.1806. Anal. calc. (%) for C24H21N5O calc. % C: 72.89, H: 5.35, N: 17.71, found % C: 72.54, H: 5.46, N: 17.47.
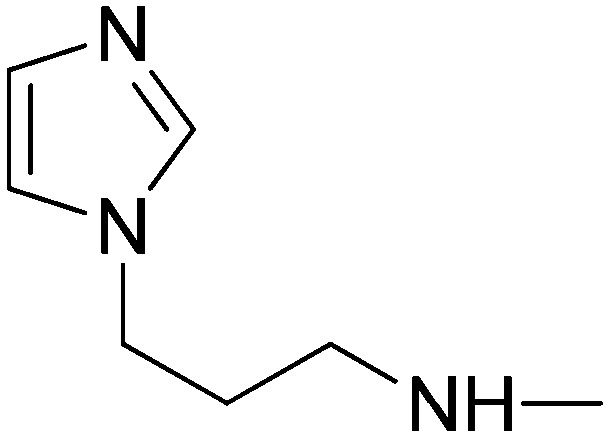
(2E)-N-(3-Imidazol-1-ylpropyl)-3-[1-(4-methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]acrylamide (5j)
Elution with DCM–MeOH (0–15%) gave 5j as a white solid. Yield 52%, 213–215 °C. IR (FTIR/FTNIR-ATR): 1649 cm–1 (C O), 3280 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.23 (1H, s), 8.86 (1H, d, J = 2.4 Hz), 8.70 (1H, dd, Ja = 1.6 Hz, Jb = 5 Hz), 8.26–8.22 (3H, m), 8.11 (2H, d, J = 7.2 Hz), 8.07 (1H, dt, Ja = 2 Hz, Jb = 8.4 Hz), 7.64 (1H, s), 7.59 (1H, q, J = 4.4 Hz), 7.37 (1H, d, J = 15.6 Hz), 7.19 (1H, s), 6.89 (1H, s), 6.49 (1H, d, J = 15.6 Hz), 3.98 (2H, t, J = 6.4 Hz), 3.29 (3H, s), 3.12 (2H, q, J = 5.6 Hz), 1.90–1.87 (2H, m). HRMS C24H25N6O3S [M + H]+ calc. 477.1709, found m/z 477.1704. Anal. calc. (%) for C24H24N6O3S calc. % C: 60.49, H: 5.08, N: 17.64, S: 6.73, found % C: 59.95, H: 5.24, N: 17.26, S: 6.63.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-[3-(2-methylimidazol-1-yl)propyl]acrylamide (5k)
Elution with DCM–MeOH (0–15%) gave 5k as a white solid. Yield 42%, mp 253–255 °C. IR (FTIR/FTNIR-ATR): 1649 cm–1 (C O), 3285 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.24 (1H, s), 8.86 (1H, d, J = 1.6 Hz), 8.71 (1H, dd, Ja = 1.6 Hz, Jb = 5 Hz), 8.27–8.22 (3H, m), 8.11 (2H, d, J = 8.8 Hz), 8.07 (1H, dt, Ja = 2 Hz, Jb = 8.4 Hz), 7.61–7.58 (1H, m), 7.37 (1H, d, J = 15.6 Hz), 7.08 (1H, d, J = 1.2 Hz), 6.74 (1H, d, J = 1.2 Hz), 6.49 (1H, d, J = 15.6 Hz), 3.89 (2H, t, J = 7 Hz), 3.29 (3H, s), 3.15 (2H, q, J = 6.4 Hz), 2.26 (3H, s), 1.85 (2H, m). HRMS C25H27N6O3S [M + H]+ calc. 491.1865, found m/z 491.1847. Anal. calc. (%) for C25H26N6O3S·0.9H2O calc. % C: 59.25, H: 5.53, N: 16.58, S: 6.33, found % C: 59.37, H: 5.4, N: 16.43, S: 6.17.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-[2-(2-methylimidazol-1-yl)ethyl]acrylamide (5l)
Elution with DCM–MeOH (0–15%) gave 5l as a white solid. Yield 74%, mp 165–168 °C. IR (FTIR/FTNIR-ATR): 1667 cm–1 (C O), 3306 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.26 (1H, s), 8.84 (1H, d, J = 1.6 Hz), 8.71 (1H, dd, Ja = 1.6 Hz, Jb = 4.8 Hz), 8.41 (1H, t, J = 5.8 Hz), 8.23 (2H, d, J = 8.8 Hz), 8.11 (2H, d, J = 8.8 Hz), 8.06 (1H, dt, Ja = 1.8 Hz, Jb = 8 Hz), 7.61 (1H, m), 7.36 (1H, d, J = 15.6 Hz), 7.30 (1H, d, J = 1.6 Hz), 7.12 (1H, d, J = 1.6 Hz), 6.46 (1H, d, J = 15.6 Hz), 4.10 (2H, t, J = 5.8), 3.52 (2H, t, J = 5.6 Hz), 3.29 (3H, s), 2.41 (3H, s). HRMS C24H25N6O3S [M + H]+ calc. 477.1709, found m/z 477.170. Anal. calc. (%) for C24H24N6O3S·2.5H2O calc. % C: 55.27, H: 5.6, N: 16.11, S: 6.15, found % C: 55.07, H: 5.29, N: 15.82, S: 6.06.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(pyridin-2-ylmethyl)acrylamide (5m)
Elution with DCM–MeOH (0–10%) gave 5m as a white solid. Yield 42%, mp 228–229 °C. IR (FTIR/FTNIR-ATR): 1655 cm–1 (C O), 3279 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.27 (1H, s), 8.87 (1H, d, J = 1.6 Hz), 8.78 (1H, t, J = 6 Hz), 8.70 (1H, dd, Ja = 1.6 Hz, Jb = 4.8 Hz), 8.50 (1H, d, J = 4.4 Hz), 8.24 (2H, d, J = 7.2 Hz), 8.11 (2H, d, J = 7.2 Hz), 8.08 (1H, dt, Ja = 2.4 Hz, Jb = 6 Hz), 7.77 (1H, td, Ja = 2 Hz, Jb = 7.6 Hz), 7.61–7.58 (1H, m), 7.42 (1H, d, J = 15.6 Hz), 7.31–7.26 (2H, m), 6.62 (1H, d, J = 16 Hz), 4.48 (2H, d, J = 6.4 Hz), 3.29 (3H, s). HRMS C24H22N5O3S [M + H]+ calc. 460.1443, found m/z 460.1447. Anal. calc. (%) for C24H21N5O3S calc. % C: 62.73, H: 4.61, N: 15.24, S: 6.98, found % C: 62.58, H: 4.70, N: 15.07, S: 6.93.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(pyridin-3-ylmethyl)acrylamide (5n)
Elution with DCM–MeOH (0–10%) gave 5n as a white solid. Yield 63%, mp 225–226 °C. IR (FTIR/FTNIR-ATR): 1648 cm–1 (C O), 3280 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.21 (1H, s), 8.83 (1H, d, J = 2 Hz), 8.73 (1H, t, J = 6 Hz), 8.66 (1H, dd, Ja = 1.6 Hz, Jb = 5 Hz), 8.47 (1H, d, J = 2 Hz), 8.42 (1H, dd, Ja = 2 Hz, Jb = 4.6 Hz), 8.19 (2H, d, J = 7.2 Hz), 8.07 (2H, d, J = 7.2 Hz), 8.04 (1H, dt, Ja = 2 Hz, Jb = 7.6 Hz), 7.65 (1H, dt, Ja = 1.6 Hz, Jb = 8 Hz), 7.57–7.54 (1H, m), 7.38 (1H, d, J = 16 Hz), 7.34–7.31 (1H, m), 6.51 (1H, d, J = 16 Hz), 4.37 (2H, d, J = 6 Hz), 3.25 (3H, s). HRMS C24H22N5O3S [M + H]+ calc. 460.1443, found m/z 460.1447. Anal. calc. (%) for C24H21N5O3S calc. % C: 62.73, H: 4.61, N: 15.24, S: 6.98, found % C: 62.72, H: 4.70, N: 15.12, S: 7.00.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(pyridin-4-ylmethyl)acrylamide (5o)
Elution with DCM–MeOH (0–10%) gave 5o as a white solid. Yield 50%, mp 265–266 °C. IR (FTIR/FTNIR-ATR): 1651 cm–1 (C O), 3279 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.28 (1H, s), 8.87 (1H, d, J = 1.6 Hz), 8.80 (1H, t, J = 6 Hz), 8.70 (1H, dd, Ja = 1.6 Hz, Jb = 5.2 Hz), 8.50 (2H, d, J = 6 Hz), 8.24 (2H, d, J = 7.2 Hz), 8.10 (2H, d, J = 7.2 Hz), 8.08 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.61–7.58 (1H, m), 7.43 (1H, d, J = 15.6 Hz), 7.26 (2H, d, J = 6 Hz), 6.59 (1H, d, J = 15.6 Hz), 4.41 (2H, d, J = 5.6 Hz), 3.29 (3H, s). HRMS C24H22N5O3S [M + H]+ calc. 460.1443, found m/z 460.1440. Anal. calc. (%) for C24H21N5O3S calc. % C: 62.73, H: 4.61, N: 15.24, S: 6.98, found % C: 63.08, H: 4.63, N: 15.23, S: 7.04.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-2-ylethyl)acrylamide (5p)
Elution with DCM–MeOH (0–10%) gave 5p as a white solid. Yield 61%, mp 237–238 °C. IR (FTIR/FTNIR-ATR): 1654 cm–1 (C O), 3280 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.22 (1H, s), 8.86 (1H, d, J = 1.2 Hz), 8.71 (1H, dd, Ja = 1.6 Hz, Jb = 4.8 Hz), 8.51 (1H, m), 8.24 (3H, m), 8.11–8.05 (3H, m), 7.73–7.68 (1H, m), 7.61–7.58 (1H, m), 7.35 (1H, d, J = 16 Hz), 7.27–7.21 (2H, m), 6.47 (1H, d, J = 15.6 Hz), 3.58 (2H, q, J = 6.8 Hz), 3.29 (3H, s), 2.93 (2H, t, J = 7.2 Hz). HRMS C25H24N5O3S [M + H]+ calc. 474.1600, found m/z 474.1592. Anal. calc. (%) for C25H23N5O3S calc. % C: 63.41, H: 4.90, N: 14.79, S: 6.77, found % C: 63.33, H: 4.79, N: 14.67, S: 6.78.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-3-ylethyl)acrylamide (5r)
Elution with DCM–MeOH (0–10%) gave 5r as a white solid. Yield 88%, mp 188–191 °C. IR (FTIR/FTNIR-ATR): 1652 cm–1 (C O), 3269 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.19 (1H, s), 8.82 (1H, d, J = 1.6 Hz), 8.67 (1H, dd, Ja = 1.6 Hz, Jb = 4.4 Hz), 8.41 (1H, d, J = 2 Hz), 8.39 (1H, dd, Ja = 1.6 Hz, Jb = 4.8 Hz), 8.24 (1H, t, J = 5.6 Hz), 8.19 (2H, d, J = 8.8 Hz), 8.06 (2H, d, J = 9.2 Hz), 8.03 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.63 (1H, dt, Ja = 2 Hz, Jb = 7.6 Hz), 7.58–7.54 (1H, m), 7.31 (1H, d, J = 15.6 Hz), 7.29 (1H, m), 6.43 (1H, d, J = 15.6 Hz), 3.38 (2H, q, J = 6.6 Hz), 3.25 (3H, s), 2.77 (2H, t, J = 6.8 Hz). HRMS C25H24N5O3S [M + H]+ calc. 474.1600, found m/z 474.1578. Anal. calc. (%) for C25H23N5O3S·2.05H2O calc. % C: 58.82, H: 5.35, N: 13.72, S: 6.28, found % C: 59.23, H: 5.18, N: 13.51, S: 5.84.
(2E)-3-[1-(4-Methanesulfonylphenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-4-ylethyl)acrylamide (5s)
Elution with DCM–MeOH (0–10%) gave 5s as a white solid. Yield 70%, mp 225–228 °C. IR (FTIR/FTNIR-ATR): 1653 cm–1 (C O), 3283 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.19 (1H, s), 8.82 (1H, d, J = 2 Hz), 8.67 (1H, dd, Ja = 1.6 Hz, Jb = 4.6 Hz), 8.43 (2H, d, J = 6.4 Hz), 8.23 (1H, m), 8.19 (2H, d, J = 8.8 Hz), 8.07 (2H, d, J = 9.2 Hz), 8.03 (1H, dt, Ja = 2 Hz, Jb = 8 Hz), 7.56 (1H, m), 7.31 (1H, d, J = 16 Hz), 7.22 (2H, d, J = 6 Hz), 6.42 (1H, d, J = 16 Hz), 3.41 (2H, q, J = 6.5 Hz), 3.25 (3H, s), 2.77 (2H, t, J = 7 Hz). HRMS C25H24N5O3S [M + H]+ calc. 474.1600, found m/z 474.1590. Anal. calc. (%) for C25H23N5O3S calc. % C: 63.41, H: 4.90, N: 14.79, S: 6.77, found % C: 62.99, H: 4.75, N: 14.49, S: 6.68.
(2E)-N-(3-Imidazol-1-ylpropyl)-3-[1-phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]acrylamide (10a)
Elution with DCM–MeOH (0–10%) gave 10a as a white solid. Yield 54%, mp 176.2–177 °C. IR (FTIR/FTNIR-ATR): 1666 cm–1 (C O), 3220 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.05 (1H, s), 8.73 (2H, d, J = 6 Hz), 8.23 (1H, t, J = 5.6 Hz), 7.95 (2H, d, J = 8 Hz), 7.66 (3H, d, J = 6 Hz), 7.57 (2H, t, J = 8 Hz), 7.43 (1H, d, J = 16 Hz), 7.39 (1H, t, J = 7.2 Hz), 7.20 (1H, s), 6.90 (1H, s), 6.49 (1H, d, J = 15.6 Hz), 3.99 (2H, m), 3.16–3.11 (2H, m), 1.92–1.87 (2H, m). HRMS C23H23N6O [M + H]+ calc. 399.1933, found m/z 399.1945. Anal. calc. (%) for C23H22N6O·1.1H2O calc. % C: 66.04, H: 5.83, N: 20.09, found % C: 66.27, H: 5.59, N: 19.75.
(2E)-N-[3-(2-Methylimidazol-1-yl)propyl]-3-[1-phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]acrylamide (10b)
Elution with DCM–MeOH (0–10%) gave 10b as a white solid. Yield 34%, mp 141–143 °C. IR (FTIR/FTNIR-ATR): 1665 cm–1 (C O), 3287 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.05 (1H, s), 8.73 (2H, d, J = 6 Hz), 8.24–8.21 (1H, m), 7.95 (2H, d, J = 7.6 Hz), 7.65 (2H, d, J = 6.4 Hz), 7.59–7.55 (2H, m), 7.43 (1H, d, J = 15.6 Hz), 7.39 (1H, t, J = 7.2 Hz), 7.07 (1H, d, J = 1.2 Hz), 6.72 (1H, d, J = 1.2 Hz), 6.49 (1H, d, J = 16 Hz), 3.90–3.87 (2H, m), 3.16 (2H, q, J = 6 Hz), 2.26 (3H, s), 1.86–1.83 (2H, m). HRMS C24H25N6O [M + H]+ calc. 413.2090, found m/z 413.2079. Anal. calc. (%) for C24H24N6O·1.75H2O calc. % C: 64.92, H: 6.24, N: 18.93, found % C: 65.15, H: 6.3, N: 18.66.
(2E)-N-[2-(2-Methylimidazol-1-yl)ethyl]-3-[1-phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]acrylamide (10c)
Elution with DCM–MeOH (0–10%) gave 10c as a white solid. Yield 33%, mp 203.3–205 °C. IR (FTIR/FTNIR-ATR): 1667 cm–1 (C O), 3192 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.02 (1H, s), 8.70 (2H, d, J = 6.4 Hz), 8.28 (1H, t, J = 6 Hz), 7.91 (2H, d, J = 8 Hz), 7.61 (2H, d, J = 6 Hz), 7.55–7.51 (2H, m), 7.41 (1H, d, J = 16 Hz), 7.36 (1H, t, J = 7.6 Hz), 6.99 (1H, d, J = 1.6 Hz), 6.69 (1H, d, J = 1.2 Hz), 6.42 (1H, d, J = 15.6 Hz), 3.98–3.94 (2H, m), 3.41 (2H, q, J = 5.2 Hz), 2.23 (3H, s). HRMS C23H23N6O [M + H]+ calc. 399.1933, found m/z 399.1944. Anal. calc. (%) for C23H22N6O·0.45H2O calc. % C: 67.95, H: 5.68, N: 20.67, found % C: 68.13, H: 5.53, N: 20.39.
(2E)-3-[1-Phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]-N-(pyridin-2-ylmethyl)acrylamide (10d)
Elution with DCM–MeOH (0–10%) gave 10d as a white solid. Yield 32%, mp 196–197 °C. IR (FTIR/FTNIR-ATR): 1650 cm–1 (C O), 3277 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.08 (1H, s), 8.77–8.72 (3H, m), 8.51 (1H, d, J = 1.6 Hz), 7.97 (2H, d, J = 5.6 Hz), 7.77 (1H, td, Ja = 2 Hz, Jb = 6 Hz), 7.66 (2H, d, J = 6.4 Hz), 7.57 (2H, t, J = 8 Hz), 7.50 (1H, d, J = 15.6 Hz), 7.42–7.39 (1H, m), 7.31–7.26 (2H, m), 6.62 (1H, d, J = 16 Hz), 4.48 (2H, d, J = 6 Hz). HRMS C23H20N5O [M + H]+ calc. 382.1668, found m/z 382.1650. Anal. calc. (%) for C23H19N5O calc. % C: 72.42, H: 5.02, N: 18.36, found % C: 72.35, H: 5.08, N: 18.07.
(2E)-3-[1-Phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]-N-(pyridin-3-ylmethyl)acrylamide (10e)
Elution with DCM–MeOH (0–10%) gave 10e as a white solid. Yield 51%, mp 186.3–187 °C. IR (FTIR/FTNIR-ATR): 1649 cm–1 (C O), 3267 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.07 (1H, s), 8.72 (3H, m), 8.52 (1H, d, J = 2 Hz), 8.46 (1H, dd, Ja = 1.6 Hz, Jb = 4.8 Hz), 7.95 (2H, d, J = 8 Hz), 7.70–7.67 (1H, m), 7.66 (2H, d, J = 6.4 Hz), 7.59–7.55 (2H, m), 7.49 (1H, d, J = 15.6 Hz), 7.42–7.35 (2H, m), 6.55 (1H, d, J = 16 Hz), 4.41 (2H, d, J = 5.6 Hz). HRMS C23H20N5O [M + H]+ calc. 382.1668, found m/z 382.1654. Anal. calc. (%) for C23H19N5O·0.6H2O calc. % C: 70.43, H: 5.19, N: 17.85, found % C: 70.61, H: 5.29, N: 17.71.
(2E)-3-[1-Phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]-N-(pyridin-4-ylmethyl)acrylamide (10f)
Elution with DCM–MeOH (0–10%) gave 10f as a white solid. Yield 41%, mp 186 °C. IR (FTIR/FTNIR-ATR): 1653 cm–1 (C O), 3278 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.10 (1H, s), 8.79–8.76 (1H, m), 8.73 (2H, d, J = 6 Hz), 8.50 (2H, d, J = 5.6 Hz), 7.95 (2H, d, J = 8 Hz), 7.66 (2H, d, J = 6 Hz), 7.59–7.55 (2H, m), 7.50 (1H, d, J = 16 Hz), 7.43–7.39 (1H, m), 7.27 (2H, d, J = 5.6 Hz), 6.57 (1H, d, J = 16 Hz), 4.41 (2H, d, J = 5.6 Hz). HRMS C23H20N5O [M + H]+ calc. 382.1668, found m/z 382.1663. Anal. calc. (%) for C23H19N5O calc. % C: 72.42, H: 5.02, N: 18.36, found % C: 72.32, H: 5.16, N: 18.20.
(2E)-3-[1-Phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-2-ylethyl)acrylamide (10g)
Elution with DCM–MeOH (0–10%) gave 10g as a white solid. Yield 48%, mp 200.4–202 °C. IR (FTIR/FTNIR-ATR): 1656 cm–1 (C O), 3289 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.04 (1H, s), 8.73 (2H, d, J = 5.6 Hz), 8.50 (1H, d, J = 4.8 Hz), 8.25–8.22 (1H, m), 7.95 (2H, d, J = 8 Hz), 7.71 (1H, td, Ja = 2 Hz, Jb = 6 Hz), 7.65 (2H, d, J = 5.6 Hz), 7.58–7.54 (2H, m), 7.42 (1H, d, J = 15.6 Hz), 7.40 (1H, t, J = 8.4 Hz), 7.28–7.21 (2H, m), 6.47 (1H, d, J = 15.6 Hz), 3.53 (2H, q, J = 7.2 Hz), 2.93 (2H, t, J = 7.2 Hz). HRMS C24H22N5O [M + H]+ calc. 396.1824, found m/z 396.1817. Anal. calc. (%) for C24H21N5O calc. % C: 72.89, H: 5.35, N: 17.71, found % C: 72.56, H: 5.34, N: 17.51.
(2E)-3-[1-Phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-3-ylethyl)acrylamide (10h)
Elution with DCM–MeOH (0–10%) gave 10h as a white solid. Yield 58%, mp 171.5–173 °C. IR (FTIR/FTNIR-ATR): 1651 cm–1 (C O), 3297 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.04 (1H, s), 8.73 (2H, d, J = 6 Hz), 8.45–8.42 (2H, m), 8.25 (1H, m), 7.95 (2H, d, J = 7.6 Hz), 7.65 (2H, m), 7.56 (3H, t, J = 8 Hz), 7.42 (1H, d, J = 16 Hz), 7.42–7.38 (1H, m), 7.38–7.31 (1H, m), 6.46 (1H, d, J = 16 Hz), 3.45–3.40 (2H, m), 2.82–2.79 (2H, m). HRMS C24H22N5O [M + H]+ calc. 396.1824, found m/z 396.1821. Anal. calc. (%) for C24H21N5O·0.25H2O calc. % C: 72.07, H: 5.42, N: 17.51, found % C: 72.29, H: 5.28, N: 17.28.
(2E)-3-[1-Phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]-N-(2-pyridin-4-ylethyl)acrylamide (10i)
Elution with DCM–MeOH (0–10%) gave 10i as a white solid. Yield 56%, mp 199.7–201 °C. IR (FTIR/FTNIR-ATR): 1652 cm–1 (C O), 3271 cm–1 (N–H). 1H-NMR (DMSO-d6) δ: 9.04 (1H, s), 8.73 (2H, d, J = 6 Hz), 8.47 (2H, d, J = 5.6 Hz), 8.25 (1H, t, J = 5.6 Hz), 7.95 (2H, d, J = 7.6 Hz), 7.64 (2H, d, J = 6 Hz), 7.56 (2H, t, J = 8 Hz), 7.42 (1H, d, J = 16 Hz), 7.40 (1H, t, J = 7.6 Hz), 7.26 (2H, d, J = 6 Hz), 6.46 (1H, d, J = 15.6 Hz), 3.45 (2H, q, J = 5.6 Hz), 2.83–2.79 (2H, m). HRMS C24H22N5O [M + H]+ calc. 396.1824, found m/z 396.1837. Anal. calc. (%) for C24H21N5O·0.55H2O calc. % C: 71.11, H: 5.50, N: 17.28, found % C: 71.11, H: 5.40, N: 17.13.
Biological studies
Cyclooxygenase (COX-1 and COX-2) enzyme inhibition assay
The inhibitory activities of the synthesized compounds on the COX-1 and COX-2 enzymes were determined by an enzyme immunoassay method (EIA) using EIA-COX activity screening kits (Cayman Chemical, No: 560101). In this method, recombinant sheep COX-1 and COX-2 enzymes were used, and the possible inhibitory activities of the compounds on both enzymes were determined by testing the inhibitory effects of these enzymes on the formation of PGE2 from arachidonic acid. PGE2 is the major source of arachidonic acid metabolism in many cells.
This experiment is based on competition of PGE2 and the PGE2–acetylcholinesterase conjugate (PGE2 tracer) to bind to PGE2 monoclonal antibody, which is limited in the assay medium. While the concentration of the PGE2–acetylcholinesterase conjugate, designated as PGE2 tracer in the assay, is constant, the amount of free PGE2 is variable and that of the PGE2 monoclonal antibody-bound PGE2 is inversely proportional to the amount of free PGE2 in the experimental environment. If the resulting antibody–PGE2 complex is bound to the “goat polyclonal anti-mouse IgG antibody” previously immobilized on the walls of the wells in microplates, then Ellman's reagent containing the substrate for acetylcholinesterase is added to the wells after microplate washing to remove unbound reagents. After Ellman's reagent is added, the resulting product (5-thio-2-nitrobenzoic acid) of the enzymatic reaction catalyzed by acetylcholine esterase is strongly yellow in color and is directly proportional to the PGE2 viewer attached to the color intensity wells. The intensity of the yellow color formed in the wells is determined spectrophotometrically from the absorbance at 412 nm and the amount of PGE2 present in the test medium during the incubation period is calculated.
Each compound was tested at a concentration of 10 μM and the % inhibition of enzyme activity for each compound was calculated from the inhibitory effects on PGE2 that would result from COX-1 and COX-2 enzymes. The amount of PGE2 in the wells was calculated from the standard curve generated by 8 different known quantities of PGE2.
Effect of compounds on platelet aggregation
For platelet isolation, human blood was collected into citrate Vacutainer (TM) tubes and used within 3 hours. Platelet aggregation was determined by a turbidimetric method on the found aggregometer (APAC 4004) of the Biochemistry Department of the Faculty of Pharmacy of Gazi University.
All compounds were tested at a final concentration of 100 μM. 1% DMSO was used as the carrier. As a reference compound, aspirin was used at a concentration of 100 μM. All procedures on human subjects were performed in accordance with the Helsinki Declaration with the approval of Gazi University Clinical Research Ethics Committee (Decision no. 304/2011). All subjects included in the study signed informed consent upon explanation of the study.
Preparation of platelet-rich plasma (PRP)
Freshly drawn venous human citrated blood (sodium citrate 3.2%, 1 : 9 v/v) from healthy subjects, who had not taken drugs with antiplatelet activity for 10 days, was centrifuged at 800 rpm for 10 min to obtain PRP. After separation, samples were centrifuged at 1500 rpm for 10 min to obtain PPP. Platelet count in PRP was adjusted to 3.8 × 105 platelets per ml using PPP.
Determination of platelet aggregation
Platelet aggregation was measured by using an aggregometer (APACT 4004, LABiTec, Ahrensburg, Germany) according to the turbidimetric method described by Born et al.57 The test compound (or the standard inhibitor, aspirin) was dissolved in DMSO. PRP was incubated at 37 °C with constant stirring at 1100 rpm and stimulated with arachidonic acid (AA, final concentration = 700 μM) or collagen (5 μg ml–1). A 199 μl sample of PRP was placed in the cuvette of the aggregometer and incubated for 5 min with 0.5 μl of test compound (or standard inhibitor, or DMSO) before addition of 10 μl AA. The changes in optical density were monitored for 3 min. All experiments were performed in triplicate. Inhibition of platelet aggregation was expressed as percentage of inhibition using the following equation:
Cytotoxic bioactivity
Cell culture
Hepatocellular (Huh7), breast cancer (MCF7) and colon cancer (HCT116) cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen GIBCO), 1% non-essential amino acids (GIBCO, Invitrogen) and 100 units per ml penicillin and streptomycin and cells were maintained at 37 °C in a humidified incubator under 5% CO2.
SRB assay
Cells were plated in 96-well plates (2000–3000 cells per well) and grown for 24 hours at 37 °C. Then, they were treated with the compounds (dissolved in DMSO) in a concentration range of 40, 20, 10, 5 and 2.5 μM. After 72 h, the cells were fixed with 10% (v/v) trichloroacetic acid (MERCK) and stained with SRB solution (50 μl of a 0.4% (m/v) SRB in 1% acetic acid solution). For the removal of unbound SRB, the cells were washed with 1% acetic acid and left for air-drying. Protein-bound SRB was solubilized in 10 mM Tris-base and absorbance measurements (515 nm) were performed using a 96-well plate reader. Cells treated with DMSO alone were used as controls for the IC50 calculations.50 All experiments were conducted in triplicate and R2 values were between 1–0.8.
Lipinski's rule of five and drug-likeness profile
In order to explore the bioavailability of the synthesized derivatives, theoretical calculations were carried out to predict some physicochemical properties of the synthesized compounds. The bioavailability of the compounds was assessed using ADME (absorption, distribution, metabolism and elimination) prediction methods. In particular, we calculated the compliance of compounds to Lipinski's rule of five.51 Lipinski's ‘rule-of-five’ and the later addition of other parameters such as polar surface area (PSA)52 describe the molecular properties important for a drug's pharmacokinetics in the human body. This approach has been widely used as a filter for substances that would likely be further developed in drug design programs. Poor absorption and permeation are more likely to occur when there are more than 5 hydrogen-bond donors (HBD) or more than 10 hydrogen-bond acceptors (HDA), the molecular mass (MM) is greater than 500, or the log P value (clog P) is greater than 5. The topological polar surface area (TPSA) should be smaller than 90 Å2.58 Molecules violating more than one of these rules may have problems with bioavailability.
Conflicts of interest
The authors declare no competing interest.
Supplementary Material
Acknowledgments
We greatly acknowledge The Scientific and Technological Research Council of Turkey (TÜBİTAK) for financial support (Grant No. 110S284).
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00022k
References
- Coruzzi G., Venturi N., Spaggiari S. Acta Biomed. 2007;78:96–110. [PubMed] [Google Scholar]
- van Ryn J., Trummlitz G., Pairet M. Curr. Med. Chem. 2000;7:1145–1161. doi: 10.2174/0929867003374255. [DOI] [PubMed] [Google Scholar]
- Mitchell J. A., Warner T. D. Nat. Rev. Drug Discovery. 2006;5:75–86. doi: 10.1038/nrd1929. [DOI] [PubMed] [Google Scholar]
- Fitzgerald G. A. N. Engl. J. Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- Funk C. D., Fitzgerald G. A. J. Cardiovasc. Pharmacol. 2007;50:470–479. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- Bresalier R. S., Sandler R. S., Quan H., Bolognese J. A., Oxenius B., Horgan K., Lines C., Riddell R., Morton D., Lanas A., Konstam M. A., Baron J. A. N. Engl. J. Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- Solomon S. D., McMurray J. J. V., Pfeffer M. A., Wittes J., Fowler R., Finn P., Anderson W. F., Zauber A., Hawk E., Bertagnolli M. N. Engl. J. Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- McGettigan P., Henry D. JAMA, J. Am. Med. Assoc. 2006;296:1633–1644. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- Hamberg M., Svensson J., Samuelsson B. Proc. Natl. Acad. Sci. U. S. A. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S., Vane J. R. Pharmacol. Rev. 1978;30:293. [PubMed] [Google Scholar]
- De Leval X., Hanson J., David J. L., Masereel B., Pirotte B., Dogne J. M. Curr. Med. Chem. 2004;11:1243–1252. doi: 10.2174/0929867043365279. [DOI] [PubMed] [Google Scholar]
- Krotz F., Schiele T. M., Klauss V., Sohn H.-Y. J. Vasc. Res. 2005;42:312–324. doi: 10.1159/000086459. [DOI] [PubMed] [Google Scholar]
- Michelson A. D. Nat. Rev. Drug Discovery. 2010;9:154–169. doi: 10.1038/nrd2957. [DOI] [PubMed] [Google Scholar]
- Leval Xd., Julemont F., Delarge J., Pirotte B., Dogne J. M. Curr. Med. Chem. 2002;9:941–962. doi: 10.2174/0929867024606713. [DOI] [PubMed] [Google Scholar]
- Michaux C., Charlier C. Mini-Rev. Med. Chem. 2004;4:603–615. doi: 10.2174/1389557043403756. [DOI] [PubMed] [Google Scholar]
- Blobaum A. L., Marnett L. J. J. Med. Chem. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- Dannhardt G., Laufer S. Curr. Med. Chem. 2000;7:1101–1112. doi: 10.2174/0929867003374237. [DOI] [PubMed] [Google Scholar]
- Penning T. D., Talley J. J., Bertenshaw S. R., Carter J. S., Collins P. W., Docter S., Graneto M. J., Lee L. F., Malecha J. W., Miyashiro J. M., Rogers R. S., Rogier D. J., Yu S. S., Anderson G. D., Burton E. G., Cogburn J. N., Gregory S. A., Koboldt C. M., Perkins W. E., Seibert K., Veenhuizen A. W., Zhang Y. Y., Isakson P. C. J. Med. Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- Prasit P., Wang Z., Brideau C., Chan C. C., Charleson S., Cromlish W., Ethier D., Evans J. F., Ford-Hutchinson A. W., Gauthier J. Y., Gordon R., Guay J., Gresser M., Kargman S., Kennedy B., Leblanc Y., Leger S., Mancini J., O'Neill G. P., Ouellet M., Percival M. D., Perrier H., Riendeau D., Rodger I., Zamboni R. Bioorg. Med. Chem. Lett. 1999;9:1773–1778. doi: 10.1016/s0960-894x(99)00288-7. [DOI] [PubMed] [Google Scholar]
- Sui Z., Guan J., Ferro M. P., McCoy K., Wachter M. P., Murray W. V., Singer M., Steber M., Ritchie D. M., Argentieri D. C. Bioorg. Med. Chem. Lett. 2000;10:601–604. doi: 10.1016/s0960-894x(00)00041-x. [DOI] [PubMed] [Google Scholar]
- Kim H. H., Park J. G., Moon T. C., Chang H. W., Jahng Y. Arch. Pharmacal Res. 1999;22:372–379. doi: 10.1007/BF02979060. [DOI] [PubMed] [Google Scholar]
- Lizuka K., Akahane K., Momose D., Nakazawa M. J. Med. Chem. 1981;24:1139–1148. doi: 10.1021/jm00142a005. [DOI] [PubMed] [Google Scholar]
- Tanouchi T., Kawamura M., Ohyama I., Kajiwara I., Iguchi Y., Okada T., Miyamoto T., Taniguchi K., Hayashi M., Lizuka K., Nakazawa M. J. Med. Chem. 1981;24:1149–1155. doi: 10.1021/jm00142a006. [DOI] [PubMed] [Google Scholar]
- Chan G., Boyle J. O., Yang E. K., Zhang F., Sacks P. G., Shah J. P., Edelstein D., Soslow R. A., Koki A. T., Woerner B. M., Masferrer J. L., Dannenberg A. J. Cancer Res. 1999;59:991–994. [PubMed] [Google Scholar]
- Hwang D., Byrne J., Schollard D., Levine E. J. Natl. Cancer Inst. 1998;90:455–460. doi: 10.1093/jnci/90.6.455. [DOI] [PubMed] [Google Scholar]
- Achiwa H., Yatabe Y., Hida T., Kuroishi T., Kozaki K., Nakamura S., Ogawa M., Sugiura T., Mitsudomi T., Takahashi T. Clin. Cancer Res. 1999;5:1001–1005. [PubMed] [Google Scholar]
- Wollf H., Saukkonen K., Anttila S., Karjalainen A., Vainio H., Ristimaki A. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- Wojtukiewicz M. Z., Hempel D., Sierko E., Tucker S. C., Honn K. V. Cancer Metastasis Rev. 2017;36:305–329. doi: 10.1007/s10555-017-9683-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S. L., Wu Y. L., Zhang Y. P., Qiao M. M., Chen Y. World J. Gastroenterol. 2004;10:1971–1974. doi: 10.3748/wjg.v10.i13.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Qu L., Yan S. Cancer Cell Int. 2015;15:106. doi: 10.1186/s12935-015-0260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menter D. G., Tucker S. C., Kopetz S., Sood A. K., Crissman J. D., Honn K. V. Cancer Metastasis Rev. 2014;33:231–269. doi: 10.1007/s10555-014-9498-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtukiewicz M. Z., Hempel D., Sierko E., Tucker S. C., Honn K. V. Cancer Metastasis Rev. 2016;35:213–233. doi: 10.1007/s10555-016-9626-0. [DOI] [PubMed] [Google Scholar]
- Sakai H., Suzuki T., Takahashi Y., Ukai M., Tauchi K., Fujii T., Horikawa N., Minamimura T., Tabuchi Y., Morii M., Tsukada K., Takeg N. FEBS Lett. 2006;580:3368–3374. doi: 10.1016/j.febslet.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Nie D., Che M., Zacharek A., Qiao Y., Li L., Li X., Lamberti M., Tang K., Cai Y., Guo Y., Grignon D., Honn K. V. Am. J. Pathol. 2004;6:429–439. doi: 10.1016/S0002-9440(10)63133-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banoglu E., Sukuroglu M., Ergun B. C., Baytas S. N., Aypar E., Ark M. Turk. J. Chem. 2007;31:677–687. [Google Scholar]
- Unlu S., Baytas S. N., Kupeli E., Yesilada E. Archiv. Pharm. Pharm. Med. Chem. 2003;336:310–321. doi: 10.1002/ardp.200300748. [DOI] [PubMed] [Google Scholar]
- Nacak S., Dogruer D. S., Sahin M. F. Farmaco. 1999;54:768–772. doi: 10.1016/s0014-827x(99)00100-7. [DOI] [PubMed] [Google Scholar]
- Baytas S. N., Inceler N., Yılmaz A., Olgac A., Menevse S., Banoglu E., Hamel E., Bortolozzi R., Viola G. Bioorg. Med. Chem. 2014;22:3096–3104. doi: 10.1016/j.bmc.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersan S., Nacak S., Noyanalpan N., Yeşilada E. Arzneim. Forsch. 1997;47:834–836. [PubMed] [Google Scholar]
- Baytas S., İnceler N., Mavaneh K. F., Uludağ M. O., Abacıoğlu N., Gökçe M. Turk. J. Chem. 2012;36:734–748. [Google Scholar]
- Baytas S., Dural N. N. T., Özkan Y., Simsek H. B., Gürsel T., Ünlü S. Turk. J. Chem. 2012;36:367–382. [Google Scholar]
- Baytas S., Inceler N., Ozkan Y., Unlu S., Sahin M. F. Med. Chem. Res. 2013;22:5922–5933. [Google Scholar]
- Inceler N., Yılmaz A., Baytas S. N. Med. Chem. Res. 2013;22:3109–3118. [Google Scholar]
- Baytas S. N., Inceler N., Yılmaz A. Med. Chem. Res. 2013;22:4893–4908. [Google Scholar]
- Hawash M. M. A., Kahraman D. C., Eren F., Cetin-Atalay R., Baytas S. N. Eur. J. Med. Chem. 2017;129:12–26. doi: 10.1016/j.ejmech.2017.02.002. [DOI] [PubMed] [Google Scholar]
- Khanna I. K., Weier R. M., Yu Y., Xu X. D., Koszyk F. J., Collins P. W., Koboldt C. M., Veenhuizen A. W., Perkins W. E., Casler J. J., Masferrer J. L., Zhang Y. Y., Gregory S. A., Seibert K., Isakson P. C. J. Med. Chem. 1997;40:1634–1647. doi: 10.1021/jm9700225. [DOI] [PubMed] [Google Scholar]
- Kira M. A., Abdel-Rahman M. O., Gadalla K. Z. Tetrahedron Lett. 1969;10:109–110. [Google Scholar]
- Pradelles P., Grassi J., Maclouf J. Anal. Chem. 1985;57:1170–1177. doi: 10.1021/ac00284a003. [DOI] [PubMed] [Google Scholar]
- Stoehlmacher J., Lenz H. J. Semin. Oncol. 2003;30:10–16. doi: 10.1016/s0093-7754(03)00120-9. [DOI] [PubMed] [Google Scholar]
- Monks A., Scudiero D., Skehan P., Shoemaker R., Paull K., Vistica D., Hose C., Langley J., Cronise P., Vaigro-Wolff A., Gray-Goodrich M., Campbell H., Mayo J., Boyd M. J. Natl. Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. Adv. Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Ertl P., Rohde B., Selzer P. J. Med. Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- http://www.molinspiration.com .
- Tetko I. V. Drug Discovery Today. 2005;10:1497–1500. doi: 10.1016/S1359-6446(05)03584-1. [DOI] [PubMed] [Google Scholar]
- http://www.openmolecules.org .
- Stahl E., Thin-layer Chromatography, Springer, New York, 1969. [Google Scholar]
- Born G. V., Cross M. J. J. Physiol. 1963;168:178–195. doi: 10.1113/jphysiol.1963.sp007185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemeier R. W., Hackett J. C., Diaz-Cruz E. S. Endocr. Rev. 2005;26:331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.