

 This review article summarizes the recent knowledge about the enzyme NPP1 and its inhibitors.
This review article summarizes the recent knowledge about the enzyme NPP1 and its inhibitors.
Abstract
Ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1, EC 3.1.4.1) is a metalloenzyme that belongs to the NPP family, which comprises seven subtypes (NPP1-7). NPP1 hydrolyzes a wide range of phosphodiester bonds, e.g. in nucleoside triphosphates, (cyclic) dinucleotides, and nucleotide sugars yielding nucleoside 5′-monophosphates as products. Its main substrate is ATP which is cleaved to AMP and diphosphate. The enzyme is involved in various biological processes including bone mineralization, soft-tissue calcification, insulin receptor signalling, cancer cell proliferation and immune modulation. Therefore, NPP1 inhibitors have potential as novel drugs, e.g. for (immuno)oncology. In the last two decades several inhibitors of NPP1 derived from nucleotide- or non-nucleotide scaffolds have been developed. The most potent and selective NPP1-inhibitory substrate analog is adenosine 5′-α,β-methylene-γ-thiotriphosphate (Ki = 20 nM vs. p-Nph-5′-TMP, human membrane-bound NPP1). Non-nucleotide-derived NPP1 inhibitors comprise polysulfonates, polysaccharides, polyoxometalates and small heterocyclic compounds. The polyoxometalate [TiW11CoO40]8– (PSB-POM141) is the most potent and selective NPP1 inhibitor described to date (Ki = 1.46 nM vs. ATP, human soluble NPP1); it displays an allosteric mechanism of inhibition and represents a useful pharmacological tool for evaluating the potential of NPP1 as a novel drug target.
Introduction
Extracellular nucleotides are important signaling molecules which regulate a variety of biological effects via cell-surface receptors termed purinergic receptors.1 There are two main families of purinergic receptors, P1 receptors activated by the nucleoside adenosine and P2-receptors – subdivided into P2X- and P2Y receptors – activated by nucleotides (e.g. ADP, ATP, UDP, and UTP).1,2 Purinergic signaling pathways play crucial roles in many biological processes, e.g. neurotransmission, neuroprotection in hypoxia and ischemia, regulation of cardiovascular function, platelet aggregation, smooth muscle contraction, secretion of hormones, modulation of immune response, control of cell proliferation, differentiation, and apoptosis.3–5 Due to the relevance of nucleosides and nucleotides in cell signaling, the extracellular levels of nucleotides are tightly regulated by catalyzing their hydrolysis via cell surface-bound ecto-nucleotidases, i.e., ecto-nucleoside triphosphate diphosphohydrolases (NTPDases, EC 3.6.1.5), ecto-nucleotide pyrophosphatases/phosphodiesterases (NPPs, EC 3.1.4.1 and EC 3.6.1.9), alkaline phosphatases (APs, EC 3.1.3.1), and ecto-5′-nucleotidase (eN, CD73, EC 3.1.3.5), see Fig. 1.6–8 The NTPDases hydrolyze nucleoside 5′-triphosphates (NTPs) to nucleoside 5′-diphosphates (NDPs), as well as nucleoside 5′-diphosphates (NDPs) to nucleoside 5′-monophosphates (NMPs), releasing inorganic phosphate (Pi).9 The NPPs degrade NTPs in a single step to NMPs releasing diphosphate (previous nomenclature pyrophosphate; PPi).10 In a subsequent hydrolyzing step, the extracellular nucleoside 5′-monophosphates (NMPs) are hydrolyzed by eN (e.g. AMP to adenosine).11 Alkaline phosphatases are unique enzymes, which can hydrolyze a broad variety of phosphoric acid ester bonds, e.g. NTPs to NDPs, NDPs to NMPs, and NMPs to nucleosides.12
Fig. 1. Metabolism of nucleotides by ecto-nucleotidases (modified from Zimmermann6). NTPDases, ecto-nucleoside triphosphate diphosphohydrolases; NPPs, ecto-nucleotide pyrophosphatases/phosphodiesterases; APs, alkaline phosphatases; eN, ecto-5′-nucleotidase (CD73); NTP, nucleoside triphosphate; NDP, nucleoside diphosphate; NMP, nucleoside monophosphate; Nuc, nucleoside.

As shown in Fig. 1, ecto-nucleotidases have a potential to terminate purinergic signaling of certain P2X and P2Y receptors by hydrolyzing nucleoside tri-, di- or monophosphates, but on the other hand the newly formed nucleotides like UDP or ADP can also activate certain P2Y receptors (e.g. activation of P2Y1, P2Y12 or P2Y13 by ADP; activation of P2Y6 by UDP), and the formed adenosine can further stimulate P1 receptors (A1, A2A, A2B and A3 receptor subtypes).13,14
Nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1)
The NPP family includes seven structurally related isoenzymes (NPP1-7) that are numbered according to their order of discovery.10 Four members of this family are known to be capable of hydrolyzing nucleotides: NPP1 (PC-1), NPP2 (autotaxin), NPP3 (CD203c) and NPP4.15–18 They can hydrolyze a variety of the nucleotides including, besides nucleoside triphosphates, dinucleoside polyphosphates, cyclic (di-)nucleotides, and nucleotide sugars, releasing nucleoside monophosphates (e.g. AMP and GMP) as products.7,10,17,18 Moreover, it had been suggested that NPP1 can also hydrolyze ATP to ADP and monophosphate (Pi).7,10 In contrast to NPP1, 3 and 4, NPP2 has only a weak nucleotide-metabolizing activity,19 and like some other members of the NPP family, i.e., NPP6 and NPP7 (alkaline sphingomyelinase), NPP2 preferably hydrolyzes phosphodiester bonds of phospholipids; hence, those subtypes can be characterized as phospholipases rather than nucleotidases.20–24 A physiological substrate for NPP5 still remains to be identified. NPP1 is the most important member of the NPP family, which was originally discovered by Takahashi et al. as a plasma cell differentiation antigen 1 (PC-1) on the surface of mouse lymphocytes.25 This glycoenzyme is highly expressed in bone, cartilage and adipose tissue,26 and moderately in heart, liver, placenta, and testis.27–30
Structure and function of NPP1
NPP1 is a homodimeric type II transmembrane glycoprotein characterized by an N-terminal transmembrane domain, two somatomedin-B-like domains, a catalytic domain and a C-terminal nuclease-like domain (see Fig. 2).7,10,16,31,32 The transmembrane domain dictates the subcellular localization of the enzyme and is also essential for the dimerization between monomers via multiple disulfide bonds.31 NPP1 contains two somatomedin-B (SMB) like domains, SMB1 and SMB2 (see Fig. 2).16,31,33 Somatomedin-B is a serum peptide which is proteolytically derived from vitronectin, a serum and extracellular-matrix protein, that is involved in cell adhesion.34,35 The function of somatomedin-B like domains are largely unclear. It has been proposed that these domains contribute to the stabilization between the transmembrane and the catalytic domain.33,36 It is also notable that the SMB2 domain of NPP1 has been postulated to be the residue for the interaction with the insulin receptor.7,32 The catalytic domain of NPP1 consists of about 400 amino acid residues and sharing 24–60% identity between the different human NPP isoforms (NPP1-7).10,37–39 This catalytic domain is homologous to the family of alkaline phosphatases (APs).40 NPPs belong to the superfamily of phospho-/sulfo-coordinating metalloenzymes.41 As in the APs, two Zn2+ ions are tightly bound in the active site by a set of six conserved Asp/His residues.31,32 In addition, the catalytic domain is connected to the nuclease-like domain by a “lasso loop”.32 Mutation of this linker region in NPP1 abolishes catalytic activity and thus, the interaction between the catalytic and nuclease-like domains through the lasso-loop seems to be relevant for the catalytic activity.31,32 The nuclease-like domain reveals no catalytic activity itself, but it is required for the translocation of NPPs from the endoplasmic reticulum to the Golgi-apparatus since it is required for the correct folding of NPPs.7 Furthermore, this domain contains a putative ‘EF-hand (a hand-form helix-loop-helix structure with E- and F-helices)’ Ca2+-binding motif (DXD/NXDGXXD) and this is essential for the catalytic activity of NPP1.42,43
Fig. 2. Structure of the NPP1 dimer, modified from Stefan et al.16.

Substrate specificity of NPP1
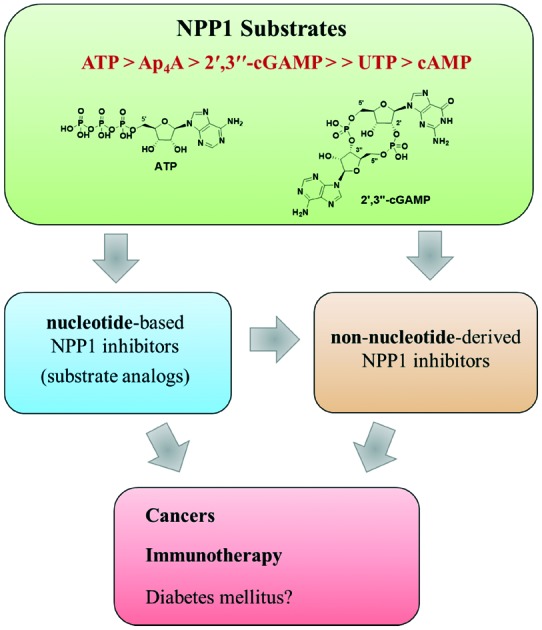
NPP1 accepts a remarkably broad range of substrates. The main substrate of NPP1 is ATP (Fig. 3).44 Besides, NPP1 metabolizes other nucleotides, e.g. nicotinamide adenine dinucleotide (NAD+), 3′-phosphoadenosine-5′-phosphosulfate (PAPS), diadenosine polyphosphates like diadenosine triphosphate (AP3A) or diadenosine tetraphosphate (AP4A), and UDP-glucose.7,33,45–47 Both, purine and pyrimidine nucleotides, serve as substrates.6 Adenosine 3′,5′-monophosphate (3′,5′-cAMP) has been found to be a poor substrate of NPP1.44 Recently, the cyclic dinucleotide, cyclic guanosine-(2′,5′)-monophosphate-adenosine-(3′′,5′′)-monophosphate (2′,3′′-cGAMP), has been reported as a substrate of NPP1.48 2′,3′′-cGAMP is known as an agonist of STING (stimulator of the interferon gene), which plays a pivotal role in innate immunity by inducing type I interferon production.44,49,50 In contrast to 2′,3′′-cGAMP, the 3′,3′′-bridged cyclic dinucleotides such as cyclic guanosine-(3′,5′)-monophosphate-adenosine-(3′′,5′′)-monophospate (3′,3′′-cGAMP), cyclic guanosine-(3′,5′)-monophosphate-guanosine-(3′′,5′′)-monophosphate (3′,3′′-cdiGMP) and cyclic inosine-(3′,5′)-monophosphate-inosine-(3′′,5′′)-monophosphate (3′,3′′-cdiIMP) were found to be no substrates of NPP1; thus, NPP1 displays specificity for 2′,3′′-cGAMP (see Fig. 4).44
Fig. 3. Natural substrates of NPP1. The site of hydrolysis is marked with a dashed red line.

Fig. 4. 3′,3′′-Bridged cyclic dinucleotides which are not hydrolyzed by NPP1.

Moreover, a number of artificial substrates have been described (Fig. 5). p-Nitrophenyl 5′-thymidine monophosphate (p-Nph-5′-TMP) is frequently used as a synthetic substrate for NPP1.51,52 p-Nitrophenyl phenyl phosphate (p-NPPP) and bis(p-nitrophenyl) phosphate (bis(p-NP)P) are other artificial substrates for NPP1, but their application is now obsolete due to its low binding affinity for the enzyme (Km > 800 μM).45 These artificial substrates allow colorimetric monitoring of the enzymatic reaction through the formation of intensively yellow-coloured p-nitrophenolate, which absorbs at 400 nm.51,52 The advantage of such artificial substrates is their suitability for high-throughput screening of compound libraries. Most recently, a new artificial substrate, p-nitrophenyl 5′-adenosine monophosphate (p-Nph-5′-AMP), has been proposed as an alternative substrate instead of p-Nph-5′-TMP, since it is structurally more similar to the natural substrate ATP than the latter.53 An unique artificial substrate, etheno-diadenosine diphosphate (εAP2A), was used for the biochemical analysis of NPP1 activity in rat brain membranes.54 Another unique artificial substrate, thiamine diphosphate (TPP) was applied for the cytochemical analysis of NPP1 activity in diverse rodent tissues, i.e., murine brain, pancreas and gastrointestinal tract tissues as well as rat liver.11,55
Fig. 5. Artificial substrates of NPP1. The site of hydrolysis is marked with a dashed red line.

Except for the new artificial substrate p-Nph-5′-AMP, the other natural and artificial substrates are all commercially available.
The kinetic parameters for a variety of substrates described in the literature are listed in the Table 1. The substrate specificity constants (kcat/Km) of natural and artificial substrates of the human NPP1 increase according to the following rank order: p-Nph-5′-AMP < UTP < p-Nph-5′-TMP < 2′,3′′-cGAMP < AP4A < ATP.44,53 This indicates that ATP is the best substrate for human NPP1 with the highest kcat/Km value. Interestingly, dinucleotides like AP4A and 2′,3′′-cGAMP are also well accepted as substrates with high kcat/Km values. Enzyme kinetic studies on mouse and human NPP1 showed that purine nucleotides (ATP and GTP) are better substrates than pyrimidine nucleotides (CTP and UTP).32,44 Km values for p-Nph-5′-TMP varied in different species. While at bovine and rat NPP1 lower Km values were determined than at human NPP1,45,56–58 mouse NPP1 displayed a similar Km for the artificial substrate as the human enzyme.59
Table 1. Kinetic parameters of nucleotide pyrophosphatase/phosphodiesterase 1 reactions with different substrates.
| K m (μM) | k cat (s–1) | k cat/Km (×103 M–1 s–1) | |
| Natural substrates | |||
| ATP | 6.2 (ref. 60), 8.17 (ref. 44), 46 (ref. 32) | 5.51 (ref. 44), 16 (ref. 32) | 674 (ref. 44), 348 (ref. 32) |
| UTP | 56.6 (ref. 44), 4300 (ref. 32) | 1.96 (ref. 44), 200 (ref. 32) | 34.6 (ref. 44), 46.5 (ref. 32) |
| GTP | 4200 (ref. 32) | 820 (ref. 32) | 95 (ref. 32) |
| CTP | 1200 (ref. 32) | 8.7 (ref. 32) | 7.25 (ref. 32) |
| AP3A | 5.1 (ref. 61) | n.d. | n.d. |
| AP4A | 20.5 (ref. 44) | 5.65 (ref. 44) | 276 (ref. 44) |
| NAD+ | 330 (ref. 47) | n.d. | n.d. |
| UDP-glucose | 270 (ref. 47) | n.d. | n.d. |
| PAPS | 120 (ref. 47) | n.d. | n.d. |
| 2′,3′′-cGAMP | 32.6 (ref. 44) | 5.36 (ref. 44) | 164 (ref. 44) |
| cAMP | 114 (ref. 44) | 2.16 (ref. 44) | 18.9 (ref. 44) |
| Artificial substrates | |||
| p-NPPP | 11 100 (ref. 45) | n.d. | n.d. |
| bis(p-NP)P | 850 (ref. 45) | n.d. | n.d. |
| p-Nph-5′-TMP | 43 (ref. 45), 61.8 (ref. 56), 91.4 (ref. 57), 101 (ref. 58), 210 (ref. 59), 222 (ref. 44), 280 (ref. 51), 281 (ref. 52) | 22.3 (ref. 44) | 100 (ref. 44) |
| p-Nph-5′-AMP | 188 (ref. 53) | 2.51 (ref. 53) | 13.4 (ref. 53) |
| εAP2A | 6.0 (ref. 54) | n.d. | n.d. |
| TPP | n.d. | n.d. | n.d. |
Nucleotide-based NPP1 inhibitors
Several NPP1 inhibitors have been reported in the literature. They have generally been investigated vs. different substrates (e.g. p-Nph-5′-AMP, p-Nph-5′-TMP, p-NPPP, bis(p-NP)P, ATP, [γ-32P]ATP (ATP with a radioactive phosphorus atom at the γ-position of the triphosphate for radiodetection) or εAP2A) and with different species and preparations of the enzyme (e.g. human soluble NPP1, human membrane-bound NPP1 or unspecified NPPs obtained from rat or human sources), so that a comparison of their inhibitory potencies is often difficult.44,53,54,60,62–69 Among the different substrates, p-Nph-5′-TMP has been the most frequently used one for the monitoring of NPP1 enzyme reactions since it allows simple and fast measurements with colorimetric detection of the product.51,58,70
The most intensively investigated inhibitors of NPP1 are substrate analogs, namely adenine nucleotide analogs and derivatives (see Fig. 6 and Table 2). They can be roughly divided into six groups: (i) α,β-methylene analogs, (ii) β,γ-methylene analogs, (iii) 2-methylthio-adenine derivatives, (iv) dialdehyde derivatives, (v) diadenosine derivatives and (vi) 2′(3′)-O-benzoylbenzoyl derivatives. These nucleotide analogs and derivatives generally exhibit – due to their structural similarity with natural substrates – a competitive mechanism of NPP1 inhibition.52,53,56,71–73 However, the nucleotide dialdehydes such as adenosine 5′-diphosphate-2′,3′-dialdehyde (dialADP) and adenosine 5′-triphosphate-2′,3′-dialdehyde (dialATP) are non-competitive (dialADP) or un-competitive (dialATP) inhibitors, respectively.52,53,56,71
Fig. 6. Inhibitors of NPP1 with nucleotide structure.

Table 2. Nucleotide-based inhibitors of NPP1.
| Inhibitor | Substrate | Enzyme | K i (μM) | Inhibition type | Selectivity | Ref. | |
| α,β-Methylene analogs | |||||||
| α,β-metADP | ATP | Human, soluble | 16.5, 24.3 | Competitive | More potent for eN (CD73) | 53, 71 | |
| p-Nph-5′-TMP | Human, membrane-bound | 16.5 | Competitive | 52 | |||
| Human, soluble | 1.28 | Competitive | 53 | ||||
| Unspecified rat NPPs | 9.6 | Mixed | 56 | ||||
| p-Nph-5′-AMP | Human, soluble | 25.8 | Competitive | 53 | |||
| α,β-metATP | ATP | Human, soluble | 13.0, 26.9 | Competitive | Not determined | 53, 71 | |
| Unspecified NPPs in human 1321N1 astrocytes | ca. a | Competitive | |||||
| 72 | |||||||
| p-Nph-5′-TMP | Human, soluble | 3.32 | Competitive | 53 | |||
| Unspecified rat NPPs | 2.2 | Competitive | 56 | ||||
| p-Nph-5′-AMP | Human, soluble | 8.19 | Competitive | 53 | |||
| γ-S-α,β-metATP derivative | p-Nph-5′-TMP | Human, membrane-bound | 0.02–4.5 | Competitive | Selective vs. NTPDase1–3 and –8, and vs. NPP3 | 75 | |
| β,γ-Methylene analogs | |||||||
| ARL 67156 | p-Nph-5′-TMP | Human, membrane-bound | 12.0 | Competitive | Non-selective vs. NTPDase1 and –3 | 77 | |
| α-borano-β,γ-metATP derivative | p-Nph-5′-TMP | Human, membrane-bound | 0.5–56 | Competitive | Selective vs. NTPDase1–3 and –8, and vs. NPP3 | 73 | |
| 2-Methylthio-adenine derivatives | |||||||
| 2-MeSADP | ATP | Human, soluble | 32.8, 23.7 | Competitive | Not determined | 53, 71 | |
| p-Nph-5′-TMP | Human, soluble | 2.18 | Competitive | 53 | |||
| p-Nph-5′-AMP | Human, soluble | 35.4 | Competitive | 53 | |||
| 2-MeSATP | ATP | Human, soluble | 25.3, 21.3 | Competitive | Not determined | 53, 71 | |
| p-Nph-5′-TMP | Human, soluble | 4.47 | Competitive | 53 | |||
| Unspecified rat NPPs | 20.5 | Competitive | 56 | ||||
| p-Nph-5′-AMP | Human, soluble | 39.9 | Competitive | 53 | |||
| Dialdehyde derivatives | |||||||
| dialADP | ATP | Human, soluble | 5.62, 5.14 | Noncompetitive | Not determined | 53, 71 | |
| p-Nph-5′-TMP | Human, membrane-bound | 3.42 | Noncompetitive | 52 | |||
| Human, soluble | 5.03 | Noncompetitive | 53 | ||||
| p-Nph-5′-AMP | Human, soluble | 5.09 | Noncompetitive | 53 | |||
| dialATP | ATP | Human, soluble | 6.82, 6.96 | Uncompetitive | Not determined | 53, 71 | |
| p-Nph-5′-TMP | Human, soluble | 4.09 | Uncompetitive | 53 | |||
| Rat NPPs, unspecified subtypes | 9.8 | Competitive | 56 | ||||
| p-Nph-5′-AMP | Human, soluble | 5.08 | Uncompetitive | 53 | |||
| Diadenosine derivatives | |||||||
| Diadenosine boranophosphate derivative | p-Nph-5′-TMP | Human, membrane-bound | 9–51 | Competitive | Selective vs. NTPDase1–3 and -8, vs. NPP3 and vs. eN | 78 | |
| 2′(3′)-O-Benzoylbenzoyl derivatives | |||||||
| bzATP | ATP | Human, soluble | 14.6 | Competitive | Not determined | 71 | |
| p-Nph-5′-TMP | Human, membrane-bound | 2.96 | Competitive | 52 | |||
| Unspecified rat NPPs | 20.5 | Competitive | 56 | ||||
aIC50 values.
Adenosine 5′-(α,β-methylene)diphosphate (α,β-metADP), also known as AOPCP, an ADP analogue was reported to be a weak inhibitor of human soluble and membrane-bound NPP1 as well as unspecified NPPs in rat serum, determined with p-Nph-5′-AMP, p-Nph-5′-TMP or ATP as substrates.52,53,56,71 AOPCP was shown to be not selective for NPP1, since it is known to be more potent as an inhibitor of eN (CD73).74 Adenosine 5′-(α,β-methylene)triphosphate (α,β-metATP) was also described as a weak inhibitor of human soluble NPP1 and unspecified NPPs isolated from rat serum as well as from human 1321N1 astrocytoma cells, as determined vs. p-Nph-5′-AMP, p-Nph-5′-TMP or ATP as substrates.53,56,71,72 Interestingly, replacement of one oxygen atom in the terminal phosphate group by a sulfur atom (in γ-S-α,β-metATP derivative) significantly increased the inhibitory potency at human membrane-bound NPP1 reaching the low nanomolar range, when tested vs. p-Nph-5′-TMP as a substrate.75 Introduction of a sulfur atom into the γ-phosphate group may increase ionic interactions between the triphosphate group and amino acids of the active site by increasing the ionization grade of the triphosphate.75 This scaffold displayed high selectivity against various human ecto-nucleotidases including NTPDase1–3 and -8 and NPP3. 6-N,N-Diethyl-d-β,γ-dibromomethylene ATP (ARL 67156), originally named FPL 67156, structurally belongs to the group of β,γ-methylene ATP analogs. ARL 67156 is known as a non-selective inhibitor of NTPDase1 and -3, and it inhibited weakly the activity of membrane-associated NPP1 vs. p-Nph-5′-TMP as a substrate.76,77 Another β,γ-methylene analog, α-borano-β,γ-metATP, in which a borano group was introduced into the α-phosphate, was found to inhibit human membrane-associated NPP1 more potently than ARL 67156 when tested vs. p-Nph-5′-TMP as a substrate.73 One reason for its better potency may be the removal of the halogen atoms present at the β,γ-methylene position in ARL 67156. One derivative of α-borano-β,γ-metATP with two chloro atoms at the β,γ-methylene showed reduced inhibitory potency, similar to the potency of ARL 67156.73 The α-borano-β,γ-metATP derivative showed high selectivity vs. other ecto-nucleotidases (e.g. NTPDase1–3 and -8 and NPP3). Both 2-methylthioADP (2-MeSADP) and 2-methylthioATP (2-MeSATP) are 2-methylthio-adenine derivatives, which displayed weak inhibition of human soluble NPP1 as well as unspecified rat NPPs, when tested vs. p-Nph-5′-AMP, p-Nph-5′-TMP or ATP as substrates.56,71 The oxidized derivatives, dialADP and dialATP, showed low micromolar potency for human soluble/memebrane-bound NPP1 as well as unspecified rat NPPs, when tested with p-Nph-5′-AMP, p-Nph-5′-TMP or ATP as substrates.52,53,56,71 Diadenosine boranophosphate derivatives have a unique scaffold among the nucleotidic inhibitors which is not based on ATP or ADP, but rather on another natural substrate diadenosine pentaphosphate (AP5A). These dinucleotide analogs were poor inhibitors for human membrane-associated NPP1 when tested vs. p-Nph-5′-TMP as a substrate, but they were highly selective for NPP1 vs. NTPDase1–3 and -8, NPP3 and eN.78 2′(3′)-O-(4-Benzoylbenzoyl)ATP (bzATP) belongs to the 2′(3′)-O-benzoylbenzoyl derivatives and it was found to be a weak inhibitor of human soluble/memebrane-bound NPP1 as well as unspecified rat NPPs, when tested vs. p-Nph-5′-TMP or ATP as substrates.52,56,71 It was observed that some nucleotide-derived inhibitors (e.g.α,β-metADP, α,β-metATP, 2-MeSATP, dialATP and bzATP) showed 2- to 8-fold higher Ki values at rat NPP1 as compared to human NPP1 tested versus p-Nph-5′-TMP as a substrate.52,53,56
Because the nucleotide-based inhibitors have similar structures as the natural substrates their development is straightforward. Many of the nucleotidic inhibitors are commercially available. However, they are only moderately potent and their selectivity for NPP1 vs. other ectonucleotidases has not been fully investigated. Only recently, nucleotide-based inhibitors with high inhibitory potency and selectivity were described among the γ-S-α,β-metATP and the α-borano-β,γ-metATP derivatives.73,75 The most potent inhibitor was adenosine 5′-α,β-methylene-γ-thiotriphosphate with a Ki value of 20 nM tested vs. p-Nph-5′-TMP as a substrate, represented in the Fig. 6.75 Nucleotide-derived inhibitors may have limited applicability due to their high acidity precluding peroral bioavailability, and because of their often moderate chemical and metabolic stability. Due to their structural similarity with the natural substrates, they may also have additional biological effects, e.g. activating P2 purinergic receptors. In summary, due to multiple drawbacks of the nucleotide-based inhibitors they may not represent the ideal lead structures for the development of NPP1 inhibitors as drugs.
Non-nucleotide-based NPP1 inhibitors
Several non-nucleotidic NPP1 inhibitors have been reported. Their scaffolds are structurally largely different. They can be widely divided into four categories: (i) polysulfonates, (ii) polysaccharides, (iii) polyoxometaletes and (iv) diverse small heterocyclic compounds (see Fig. 7 and Table 3). Polysulfonates such as reactive blue 2 and suramin are known as P2 receptor antagonists.79,80 They also showed strong inhibition for human soluble/membrane-bound NPP1 with Ki values in a nanomolar to micromolar range, when used ATP, p-Nph-5′-AMP or p-Nph-5′-TMP as substrates.53,60 Contrary to this, both polysulfonates showed rather weak inhibition with membrane preparations of unspecified NPPs in rat C6 glioma cells (IC50 values in the range of 10 to 80 μM) vs. [γ-32P]ATP as a substrate.62Reactive blue 2 was found to display a non-competitive mechanism of NPP1 inhibition, while suramin was characterized as an un-competitive inhibitor.53,60 The selectivity of reactive blue 2 and suramin for NPP1 was poor, since both compounds also inhibit NPP3 strongly as well as various P2 receptors (reactive blue 2 additionally inhibits eN (CD73)).60,79–81 The well-known anti-coagulant heparin was reported to moderately inhibit human soluble NPP1 with an IC50 value of ca. 100 μM vs. ATP as a substrate or ca. 1 μM vs. p-Nph-5′-TMP as a substrate,63 and unspecified rat NPPs with a Ki value of 25 μM vs. εAP2A as a substrate.54Heparin is non-selective for NPP1 considering its potency vs. its main target thrombin (Ki value 2.65 μM).82 This polysaccharide was reported to be a competitive inhibitor of NPP1.54,63
Fig. 7. A series of non-nucleotidic inhibitors of NPP1.

Table 3. Non-nucleotidic inhibitors of NPP1.
| Inhibitor | Substrate | Enzyme | K i (μM) | Inhibition type | Selectivity | Ref. |
| Polysulfonates | ||||||
| Reactive blue 2 | ATP | Human, membrane-bound | 0.52 | Not defined | Selective vs. NTPDase1–3, but non-selective vs. P2 receptors, vs. NPP3 and vs. eN | 60 |
| Human, soluble | 0.141 | Noncompetitive | 53 | |||
| p-Nph-5′-TMP | Human, soluble | 0.198 | Noncompetitive | 53 | ||
| p-Nph-5′-AMP | Human, soluble | 0.176 | Noncompetitive | 53 | ||
| [γ-32P]ATP a | Unspecified NPPs in rat C6 glioma cells | 12 b | Not defined | 62 | ||
| Suramin | ATP | Human, membrane-bound | 0.26 | Not defined | Selective vs. NTPDase1–3, but non-selective vs. P2 receptors and NPP3 | 60 |
| Human, soluble | 0.780 | Uncompetitive | 53 | |||
| p-Nph-5′-TMP | Human, membrane-bound | 8.67 b | Not defined | 92 | ||
| Human, soluble | 1.07 | Uncompetitive | 53 | |||
| p-Nph-5′-AMP | Human, soluble | 1.03 | Uncompetitive | 53 | ||
| [γ-32P]ATP a | Unspecified NPPs in rat C6 glioma cells | 72 b | Not defined | 62 | ||
| Polysaccharides | ||||||
| Heparin | ATP | Human, soluble | ca. 100 b | Competitive | Non-selective vs. thrombin | 63 |
| p-Nph-5′-TMP | Human, soluble | ca. 1.0 b | Competitive | 63 | ||
| εAP2A c | Unspecified rat NPPs | 25 | Competitive | 54 | ||
| Polyoxometalates | ||||||
| [TiW11CoO40]8– | ATP | Human, soluble | 0.00146 | Noncompetitive | Selective vs. NTPDase1–3, vs. NPP2–3, vs. eN and vs. TNAP | 64 |
| Diverse heterocyclic compounds | ||||||
| PPADS | [γ-32P]ATP a | Unspecified NPPs in rat C6 glioma cells | 12 b | Not defined | Non-selective vs. P2 receptors and vs. NTPDase1–3 | 62 |
| Oxadiazole derivative I | p-NPPP | Human, soluble | 150–850 | Noncompetitive | Not determined | 65 |
| bis(p-NP)P d | Snake venom | 100–1400 | Noncompetitive | Not determined | 65 | |
| Oxadiazole derivative II | p-Nph-5′-TMP | Human, membrane-bound | 1.9–5.5 b | Noncompetitive | Not selective vs. NPP3 | 85 |
| Biscoumarin derivative | p-NPPP e | Human, soluble | 50–1000 | Noncompetitive | Not determined | 66 |
| bis(p-NP)P d | Snake venom | 8–1150 | Noncompetitive | 66 | ||
| Triazole derivative | bis(p-NP)P d | Snake venom | 132–1164 b | Not defined | Not determined | 86 |
| Quinazoline derivative | ATP | Human, soluble | 0.215 e | Competitive | Selective vs. NTPDase1–3, vs. NPP3, vs. eN and vs. TNAP, but non-selective vs. hERG potassium channels | 53 |
| Not described | 0.0362–5.98 b | Not defined | 87 | |||
| p-Nph-5′-TMP | Human, membrane-bound | 0.0593–0.110 | Noncompetitive Competitive | 67 | ||
| Human, soluble | 0.0642 f | 53 | ||||
| p-Nph-5′-AMP | Human, soluble | 0.420 f | Competitive | 53 | ||
| Thioacetamide derivative | ATP | Human, soluble | 5.34–89.7 | Competitive | Selective vs. NPP2–3 | 68 |
| p-Nph-5′-TMP | Human, soluble | 0.00500–11.0 | Competitive | 68 | ||
| p-Nph-5′-AMP | Human, soluble | 14.9 g | Competitive | 53 | ||
| Thiazolobenzimidazolone derivative | ATP | Human, soluble | 0.467–0.981 | Uncompetitive | Selective vs. NTPDase1–3, vs. NPP2–3, vs. eN and vs. TNAP | 69 |
| Isoquinoline derivative | p-Nph-5′-TMP | Human, membrane-bound | 0.36–2.81 b | Competitive | Not determined | 91 |
| Thiadiazolopyrimidone derivative | p-Nph-5′-TMP | Human, membrane-bound | 0.31–2.26 b | Competitive | Selective vs. NTPDase1–3 and –8, and vs. NPP3 | 92 |
a[γ-32P]ATP: ATP with a radioactive phosphorus atom at γ-position of the triphosphate.
bIC50 values.
cεAP2A: etheno-diadenosine diphosphate.
dbis(p-NP)P: bis(p-nitrophenyl) phosphate.
e p-NPPP: p-nitrophenyl phenyl phospahte.
Polyoxometalates are negatively charged inorganic cluster compounds which contain heavy metal ions like tungsten (W), molybdenum (Mo) or vanadium (V) as a central atom, surrounded by oxygen atoms.83,84 A polyoxotungstate [TiW11CoO40]8– (PSB-POM141) has been described as the most potent inhibitor of human soluble NPP1 described so far with a Ki value of 1.46 nM vs. ATP as a substrate.64 PSB-POM141 revealed a non-competitive mechanism of inhibition. Furthermore, this inorganic cluster complex was shown to be highly selective vs. other human ecto-nucleotidases, including NTPDase1–3, NPP2–3, eN and TNAP (tissue non-specific alkaline phosphatase).64 However, an oral application of such polyanionic cluster compounds is limited because of their negative charge and their high molecular weight.
Diverse heterocyclic compounds have been also reported to act as NPP1 inhibitors. Pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) is known as a P2 receptor antagonist like reactive blue 2 or suramin, and it inhibited moderately unspecified NPPs in rat C6 glioma cells (IC50 value 12 μM) vs. [γ-32P]ATP as a substrate.62Oxadiazole (I and II) and biscoumarin derivatives were weak non-competitive inhibitors of human membrane-associated NPP1 vs. p-NPPP or p-Nph-5′-TMP as substrates, and of snake venom NPP1 vs. bis(p-NP)P as a substrate.65,66,85 Similarly to oxadiazole derivatives, triazole derivatives were reported to be weak inhibitors of snake venom NPP1 vs. bis(p-NP)P as a substrate.86 Its mechanism of inhibition has not been investigated. A series of quinazoline derivatives were shown to be potent inhibitors of human soluble/membrane-bound NPP1 vs. p-Nph-5′-AMP, p-Nph-5′-TMP or ATP as substrates, the best inhibitor (represented in the Fig. 7) displaying an IC50 value of 36.2 nM vs. ATP as a substrate and a Ki value of 59.3 nM vs. p-Nph-5′-TMP as a substrate.53,67,87 The quinazoline scaffold displayed high selectivity vs. other ecto-nucleotidases like NTPDase1–3, NPP3, eN and TNAP,67 but it showed high affinity binding to hERG (human ether-a-go-go related gene) potassium channels, which precluded their development as drugs due to expected cardiovascular side effects.87 The inhibition mechanism of quinazoline is still controversial. While Shayhidin et al. described a non-competitive mechanism of enzyme inhibition for quinazolines vs. p-Nph-5′-TMP as a substrate,67 our kinetic studies showed a competitive mechanism of inhibition vs. p-Nph-5′-AMP, p-Nph-5′-TMP and ATP as substrates.53Thioacetamide derivatives have been reported to be highly potent inhibitors of human soluble NPP1 vs. p-Nph-5′-TMP as a substrate with a competitive mechanism of inhibition.68 However, this class of compounds displayed significantly lower inhibitory potency when determined vs. ATP as a substrate of NPP1.53,68 The structure–activity relationship (SAR) results obtained with p-Nph-5′-TMP as a substrate can be summarized as follows: i) two methoxy substituents in the terminal aryl group are important for the inhibitory activity; ii) further modification on the central linker (i.e., elongation by a methylene unit on either side of the linker or branching of the methylene group) led to a loss in activity; iii) removal of the pyridine ring or the nitrogen atom at position 4 as well as methylation of the nitrogen atom at position 3 of the imidazopyridine ring led to a loss in activity; iv) introduction of electron-donating groups like a methoxy group at position 5 dramatically increased the inhibitory potency; v) introduction of electron-withdrawing groups like halogen atoms at position 6 or electron-donating groups like a methyl group at position 7 led to a slight increase in potency; vi) replacement of the imidazopyridine ring by adenine, hypoxanthine or xanthine structures led to a large increase in potency. However, the most potent inhibitor in this series vs. ATP as a substrate was derived from the imidazopyridine scaffold with a methoxy group at position 5 (showing a Ki value of 5.34 μM), represented in the Fig. 7. This compound showed high selectivity vs. human NPP2 and NPP3.68 Gao et al. have recently developed carbon-11-labeled thioacetamide derivatives as new potential positron emission tomography (PET) tracers for the imaging of NPP1.88
Recently, thiazolobenzimidazolone derivatives have been identified as potent un-competitive NPP1 inhibitors with Ki values ranging from 0.467 to 0.981 μM vs. ATP as a substrate.69 SAR studies showed that the cis-configurated isomers (Z-form) at the ethene linker – which is connected to the thiazolobenzimidazolone scaffold at position 2 – were generally more potent than the trans-configurated isomers (E-form), see Fig. 7. Moreover, the oxygen atom of the furanyl substituent at the ethene linker seems to serve as an important hydrogen bond acceptor. The introduction of electron-donating groups like methyl or N,N-dimethylamino at the 5′-position of the furan ring decreased the inhibitory potency, while the introduction of large electron-withdrawing bromine or iodine atoms at the same position significantly increased the inhibitory potency. The compound with a 5′-iodofuranyl substituent showed the highest inhibitory potency with a Ki value of 0.467 μM vs. ATP as a substrate (represented in the Fig. 7). This class of compounds exhibited high selectivity vs. diverse human ecto-nucleotidases, including NTPDase1–3, NPP2–3, eN and TNAP.69 Interestingly, thiazolo-benzimidazolones featured all properties required for peroral drugs according to Lipinski's rule of five (molecular weight < 500 g mol–1, number of hydrogen bond donors < 5, number of hydrogen bond acceptors < 10 and log P value (logarithm of the partition coefficient between octanol and water) < 5).69,89,90 However, there is still the requirement of improving the hydrolytic stability of this scaffold.69 Very recently, a series of isoquinoline derivative has been shown to act as competitive inhibitors of human membrane-bound NPP1 vs. p-Nph-5′-TMP as a substrate, the best inhibitor displaying an IC50 value of 360 nM (represented in the Fig. 7).91 However, selectivity data vs. other ecto-nucleotidases have not been provided. The SAR studies showed that two methoxy groups at the 2- and 3-positions of the isoquinoline scaffold are important for the inhibitory activity. The methylation at the 12-position of the isoquinoline scaffold and at the 3′-position of the indol substituent seems to increase the inhibitory potency. Molecular docking of the isoquinoline derivatives into a homology model of human NPP1 – based on the crystal structure of mouse NPP132 – showed an important π–π interaction of the indolo[2,1-a]isoquinoline structure with Tyr340.91 Furthermore, a series of thiadiazolopyrimidone derivatives have been reported to be competitive inhibitors of human membrane-bound NPP1 vs. p-Nph-5′-TMP as a substrate, the best inhibitor displaying an IC50 value of 310 nM (represented in the Fig. 7).92 SAR studies indicated that a fluorine atom at position 6 of the thiadiazolopyrimidone core as well as 3′,5′-dimethyl substitution of the aryl residue were important for the inhibitory activity. Molecular docking studies using a homology model of human NPP1 – the same model as for the isoquinoline derivatives – revealed that the carboxyl group at position 7 and the fluorine at position 6 formed hydrogen bonds with amino acid residues Asn277 and Leu290, respectively. The thiadiazolopyrimidone scaffold additionally formed two π–cation interactions with Lys278.92 The best inhibitor in this series (represented in Fig. 7) showed high selectivity vs. human NTPDases1–3 and 8 and moderate selectivity vs. human NPP3.92
Some of the non-nucleotidic inhibitors (e.g.reactive blue 2, suramin, heparin, PPADS and the thiazolobenzimidazolone derivatives) are commercially available, while the others are not. Comparison of inhibitory potencies of non-nucleotidic inhibitors at different species is currently not possible, because data for different species versus the same substrate are not available. The most potent NPP1 inhibitor described to date, [TiW11CoO40]8– (PSB-POM141, Ki = 1.46 nM vs. ATP), which is highly selective for NPP1 vs. other ecto-nucleotidases represents a useful pharmacological tool for in vitro and in vivo studies. The quinazoline derivatives are the most potent competitive inhibitors of NPP1 (the most potent one is (N-[2-[1-(6,7-dimethoxyquinazolin-4-yl)piperidin-4-yl]ethyl] sulfuric diamide, SAR 03004) showing a Ki value of 215 nM) when tested vs. the natural substrate ATP.53 Since the quinazolines display high selectivity vs. other ecto-nucleotidases but were found to bind to hERG channels, their use is limited to in vitro studies. The thiazolobenzimidazolones derivatives possess high potential to be perorally applicable, however, their hydrolytic stability will have to be optimized to allow broader application.
In conclusion, non-nucleotide based inhibitors of NPP1 are structurally highly diverse and their inhibitory potencies vary from moderate to high. In addition, they display different mechanisms of inhibition and different selectivity profiles vs. other target structures. Due to their drug-likeness they may serve as lead structures for the further development and optimization of novel NPP1 inhibitors as drugs.
Compounds lacking NPP1-inhibitory potency
According to Grobben et al. a series of P1 receptor antagonists (e.g. 9-chloro-2-(2-furanyl)-[1,2,4]triazolo[1,5-c]quinazolin-5-amine (CGS15943) and 8-cyclopentyl theophylline (CTP)), theophylline and a non-specific inhibitor of cAMP and cGMP phosphodiesterases like 3-isobutyl-1-methylxanthine (IBMX) were shown to be no inhibitors for the unspecified NPPs in rat C6 glioma cells, when tested vs. [γ-32P]ATP as a substrate.62 Moreover, levamisole, a potent and selective inhibitor of TNAP (tissue non-specific alkaline phosphatase), was reported to be no inhibitor for the unspecified NPPs obtained from rat heart by using p-Nph-5′-TMP as a substrate.57 Additionally, two inorganic compounds such as sodium azide (NaN3) and gadolinium chloride (GdCl3), which was reported to potently inhibit NTPDases,93 were shown to be not potent as inhibitors of unspecified rat NPPs vs. p-Nph-5′-TMP as a substrate.57
Substrate dependence of inhibitory potency of competitive NPP1 inhibitors
Several competitive inhibitors of NPP1 were found to show a moderate to strong substrate dependence of their inhibitory potency. Heparin had shown significantly higher potency vs. the artificial substrate p-Nph-5′-TMP as compared to the natural substrate ATP (ca. 100-fold more potent with the artificial substrate), see Fig. 7 and Table 3.63 Also, several nucleotidic inhibitors of NPP1 (e.g.α,β-metADP, α,β-metATP, 2-MeSADP and 2-MeSATP) had been found to be significantly less potent when investigated vs. the natural substrate ATP as compared to the artificial substrate p-Nph-5′-TMP (see Fig. 6 and Table 2).71 Moreover, the non-nucleotide-derived thioacetamides were discovered to be much more potent vs. p-Nph-5′-TMP as a substrate than vs. ATP (>100-fold difference, see Fig. 7 and Table 3).68 Further systematic evaluation of several nucleotidic inhibitors (including α,β-metADP, α,β-metATP, 2-MeSADP, 2-MeSATP, dialADP and dialATP) as well as non-nucleotidic NPP1 inhibitors (e.g.SAR 03004 (the most potent quinazoline derivative), reactive blue 2, suramin, PZB08513136A (the most potent thioacetamide derivative) and [TiW11CoO40]8– (PSB-POM141)) revealed significant differences in the determined Ki values for competitive, but not for non- and un-competitive inhibitors, depending on the employed substrate, the artificial substrate p-Nph-5′-TMP or the natural substrate ATP.53 Surprisingly, when the new artificial substrate p-nitrophenyl 5′-adenosine monophosphate (p-Nph-5′-AMP) was used instead of p-Nph-5′-TMP, the differences in the inhibition constants determined between the artificial and natural substrates were no longer observed (R2 = 0.9742 (p-Nph-5′-AMP) vs. 0.5722 (p-Nph-5′-TMP)).53
Such substrate-dependent inhibitory potencies of competitive enzyme inhibitors had been also noticed in several previous studies with various enzymes.68,71,94–96 For instance, captopril, a competitive inhibitor of the angiotensin-I converting enzyme (ACE), was shown to be significantly more potent when the synthetic substrates N-[3-(2-furyl)acryloyl]-Phe-Gly-Gly (FAPGG) or N-hippuryl-His-Leu hydrate (HHL) were used instead of the natural substrate angiotensin-I.94,95 Such a difference in inhibitory potencies depending on the substrate was also observed for NPP2 (autotaxin),96 which is an isoenzyme of NPP1, but preferentially hydrolyzes phospholipids rather than nucleotides.21
A possible explanation for the observed discrepancy between assays obtained with p-Nph-5′-TMP as compared to the natural substrate ATP might be that p-Nph-5′-TMP may act as an allosteric modulator in addition to being a substrate (see Fig. 8). A previous study with another nucleotide-metabolizing enzyme, bacterial UDP-N-acetylglucosamine 2-epimerase, described such an allosteric modulation by its substrate UDP-N-acetylglucosamine.97 The binding of p-Nph-5′-TMP to its allosteric binding site which may be near or even distant from the active site, may modulate the conformation of the active site, so that competitive inhibitors would then interact better with the active site resulting in increased affinity and accordingly in increased inhibitory potency (Fig. 8). This hypothesis remains to be examined in future studies.
Fig. 8. Hypothetical model of allosteric binding of p-Nph-5′-TMP. The artificial substrate p-Nph-5′-TMP acts as an allosteric modulator in addition to being a substrate.

Since p-Nph-5′-TMP is still frequently used as an artificial substrate for high-throughput screening, it is necessary that the identified hit compounds are subsequently retested with the natural substrate ATP. Alternatively, the new artificial substrate p-Nph-5′-AMP – which is, however, currently not commercially available yet – may be applied instead of p-Nph-5′-TMP for high-throughput screening.
Potential therapeutic applications of NPP1 inhibitors
Bone mineralization and soft-tissue calcification
The main physiological function of NPP1 appears to be bone mineralization. In this context, vascular smooth muscle cell calcification has also been observed.26,98–102 As shown in Fig. 9, NPP1 produces diphosphate (PPi) by hydrolysis of ATP, and PPi can be further converted to monophosphate (Pi) by tissue non-specific alkaline phosphatase (TNAP). Subsequently, monophosphate binds to extracellular calcium to form a tissue mineralization matrix, hydroxyapatite (HA, Ca10(PO4)6(OH)2), which facilitates both, bone mineralization and soft-tissue calcification.26,103 Further feedback response exists to prevent ectopic mineralization; PPi acts as an inhibitor of HA formation, by inducing the expression of osteopontin (OPN), a suppressor of tissue mineralization, and simultaneously it reduces the expression of osteocalcin (OCN) and TNAP, enhancers of tissue mineralization, which together lead to the suppression of HA crystal precipitation and growth.104,105 Several phenotypes of NPP1 knock-out mice with respect to bone mineralization and soft-tissue calcification have been described so far. They were hybrids of C57BL/6 × 129/J mouse strains.106,107 NPP1–/– mice displayed spontaneous calcification of soft-tissue, particularly vascular cells and articular cartilage.7,26,101,105 Furthermore, the skull-bones of NPP1–/– mice have been reported to be hyper-calcified, and calvarial osteoblasts derived from NPP1–/– mice reveal a decreased extracellular PPi concentrations that is associated with hyper-calcification.106 This phenotype could be rescued by transfection with NPP1 but not with NPP3, which demonstrated the isoenzyme specificity of this effect.108 Additionally, the ectopic calcification of soft-tissue could be also normalized by double knockouts for NPP1 and TNAP.107 NPP1-null mice also showed significant defects in long-bone mineralization, characterized by reduced trabecular bone mass and cortical thickness of both the femur and tibia.101 Recently, Nam et al. reported that NPP1 expression is also critical for osteoblast and chondrocyte differentiation, independently of its catalytic activity.105 That may be the reason why NPP1–/– mice display moderate to severe hypo-mineralization in the long bones.
Fig. 9. Physiological role of NPP1 in bone mineralization and soft-tissue calcification, modified from Stefan et al.10 Red minus: negative feedback of diphosphate (PPi).

Calcium pyrophosphate dihydrate deposition (CPPD) disease
Increased expression of NPP1 in the chondrocytes, which is age-related, leads to the over-production of diphosphate (PPi), see Fig. 10. Since the enzymatic activity of TNAP is limited, the excessive amounts of the formed diphosphate can react with the extracellular calcium, which leads to a formation of nearly in-soluble calcium diphosphate. Gradually, this calcium salt can accumulate in joints and cartilages, resulting in the so-called calcium pyrophosphate dihydrate deposition (CPPD) disease, which can result in inflammatory arthritis and joint pain.10,102,109 Therefore, NPP1 inhibitors have been suggested as novel drugs for the treatment of CPPD diseases.61,69,88 However, as described in the section above, NPP1-null mice display spontaneous calcification of soft-tissue, and moderate to severe hypo-mineralization in the long bones. Thus, NPP1 inhibitors may be effective for the treatment of CPPD disease, but chronic application may be limited by their moderate to severe undesired side effects in bones and soft-tissues.
Fig. 10. The calcium pyrophosphate dihydrate deposition (CPPD) disease by increased expression of NPP1 in the condrocytes, modified from Stefan et al.10.

Diabetes mellitus type 2
A role of NPP1 in insulin receptor signalling has been reported.30,110–114 Elevated NPP1 expression was found in dermal fibroblast cultures from patients with non-insulin-dependent diabetes mellitus type 2.110 Moreover, an overexpression of NPP1 has been reported to result in defective insulin-stimulated insulin receptor (IR) autophosphorylation in type 2 diabetes patients.30 Transgenic mice that overexpressed NPP1 in different tissues were insulin resistant and diabetic.115 Abate et al. reported that NPP1 binds directly to the α-subunit of the insulin receptor (Fig. 11).112 This interaction inhibits further signalling by decreasing its β-subunit autophosphorylation which finally leads to the desensitization of the insulin receptor and the occurrence of insulin resistence.30,112 It is well accepted that NPP1 inhibition of the insulin receptor is independent of the phosphodiesterase activity of NPP1, and its regulation of the insulin receptor results from simple protein–protein interaction.30,112,116 Moreover, the polymorphism K121Q of NPP1 was found to be associated with insulin resistance in some human populations.117–121 However, these findings were not consistently observed in some other populations.122–125 The phenotype Q121 of NPP1 was found to have a stronger physical interaction in vitro with the insulin receptor than the more common phenotype K121 of NPP1.126 This K121 residue has been reported to be located in the SMB2 domain of NPP1, therefore, this domain probably represents the binding site for the interaction with the insulin receptor.7,32 Recently, an animal study, in which NPP1 was knocked down with siRNA, showed that this greatly increased insulin-stimulated phosphorylation of tyrosine kinase in HuH7 human hepatoma cells.127 This study indicated that NPP1 malfunction may be helpful to treat type 2 diabetes mellitus. However it is still unknown, whether NPP1 inhibitors are useful for diabetes treatment, since the phosphodiesterase activity of NPP1 is not required for stimulating the insulin receptor. This remains to be clarified in future studies by investigating structurally diverse competitive and allosteric inhibitors.
Fig. 11. Binding of NPP1 to the insulin receptor, modified from Abate et al.112.

Brain cancer and cancer immunotherapy
NPP1 expression has been reported to be increased in neural brain tumors, e.g. in membranes of rat C6 glioma cells,128 human astrocytic brain tumors,129 and human glioblastoma stem-like cells,130 and its expression level was closely related to the aggressiveness of astrocytic brain tumors.128–131 NPP1 has also been reported to be expressed in N2a mouse neuroblastoma cells, its expression level substantially changing with cells differentiating into a neuronal-like phenotype.131 Lentiviral shRNA-knockdown of NPP1 expression in a human glioblastoma cell line (NCH421k cells) reduced cell proliferation, increased accumulation of cells in the G1/G0 cell cycle phase and induced cell death.130 The mechanism for this effect may be based on the increased activation of cell apoptosis inducing P2X7 receptor activation owing to the accumulation of un-degraded ATP (the main substrate of NPP1) by the knockdown of NPP1 function (see Fig. 12).132 Furthermore, the reduced production of extracellular adenosine by a blockade of NPP1 function may also reduce the adenosine induced angiogenesis as well as cell proliferation and differentiation by stimulating P1 receptors.2,133 Thus, NPP1 inhibitors may have antineoplastic effects on neuronal tumor cells.
Fig. 12. NPP1 inhibitors for the potential treatment of (brain) cancer. CD73, ecto-5′-nucleotidase; Ado, adenosine; NKT cells, natural killer cells; DCs, dendritic cells; Treg cells, regulatory T cells (CD4+); NLRP3 inflammasome, N[combining low line]ACHT, L[combining low line]R[combining low line]R and P[combining low line]YD domains-containing protein 3[combining low line].

Many previous studies have demonstrated that the presence of inflammatory infiltrate is involved in tumor progression.134,135 Particularly in gliomas, the presence of inflammatory infiltrate is closely correlated with the tumor malignancy grade.136 One of the most important immunosuppressive regulatory pathways is the phosphohydrolysis of extracellular ATP to adenosine by the high levels of ecto-nucleotidase expressed by tumour cells (e.g. CD39, NPP1 and CD73), which are therefore possible novel targets for cancer immunotherapy.137 Since the expression of CD39 has been reported to be negligibly low in gliomas,138 and meanwhile the expression of both NPP1 and CD73 has been shown to be increased in gliomas,128–130,139 the NPP1/CD73 pathway, not the CD39/CD73 pathway, may be the immunosuppressive regulatory pathway in many glioma cells. ATP is massively released in the tumor microenvironment by stressed, damaged or dying cells.132 Together with ecto-5′-nucleotidase (CD73) NPP1 can hydrolyze ATP via AMP to produce immunosuppressive adenosine (see Fig. 12),137 which is a critical regulator of both innate and adaptive immune responses by stimulating G-protein coupled P1 receptors (A2A- and A2B receptors).140,141 Adenosine inhibits phagocytosis of macrophages and neutrophils, T- or B cell receptor (TCR or BCR) triggered NF-κB activation, and production of a series of cytokines like interleukin-2 (IL-2), interleukin-4 (IL-4) or interferon-gamma (INF-γ) in diverse immune cells (e.g. natural killer T (NKT) cells, dendritic cells (DCs) and T lymphocytes).133,140–144 In addition, extracellular adenosine promotes regulatory T cells (Treg cells, CD4+), which leads to a drastically impaired antitumor immune response.140,142 Contrary to adenosine, extracellular ATP is a proinflammatory signal molecule.5 ATP serves as a ‘find-me’ signal for the recruitment of DCs (dendritic cells), monocytes and macrophages (“chemotaxis”), which present tumor antigens and thereby further participate in the immune response against the cancerous lesion.140 ATP also activates NLRP3 (N[combining low line]ACHT, L[combining low line]R[combining low line]R and P[combining low line]YD domains-containing protein 3[combining low line]) inflammasome, a multiprotein complex that triggers caspase-1 activation and secretion of the cytokines (e.g. interleukin-1b (IL-1b) and IL-18), important activators of innate and adaptive immune responses.145,146 In the tumor microenvironment, tumor-derived DNAs (likely released by dead cells) can access to the cytosol of intratumoral dendritic cells (DCs) and activate cGAS (2′,3′′-cGAMP synthase) in DCs to generate 2′,3′′-cGAMP, which leads to the activation of STING.147 Subsequently, STING promotes the production of type I interferons (e.g. interferon α, interferon β), which facilitates the innate immunity by activating cytotoxic T cells (CD8+).147,148 NPP1 can also hydrolyze 2′,3′′-cGAMP (see Fig. 12).44,48–50 The activation of STING by 2′,3′′-cGAMP occurs in the intracellular space, but its hydrolysis has been proposed to take place in the extracellular space by the extracellular membrane-bound NPP1.48 It appears likely that there is a specific transporter to import and export for 2′,3′′-cGAMP through the cell membrane, as observed for other nucleotides.48
The blockade of NPP1 can increase the concentration of ATP and 2′,3′′-cGAMP, but at the same time it can decrease the concentration of adenosine by reducing the concentration of its precursor AMP (Fig. 12). This all can lead to an enhancement of the immune response in the body. Therefore, NPP1 could be a promising target for cancer immunotherapy, especially in gliomas.
Conclusions
NPP1 is of increasing interest as a potential therapeutic target for diabetes mellitus and cancer, including brain cancers. NPP1 inhibitors are also expected to activate the immune response. In the last two decades significant advances have been made in the development of NPP1 inhibitors. The data collected in the present review article demonstrate the current state of the development of NPP1 inhibitors and pave the way to gain novel and drug-like inhibitors for human NPP1, which have potential as future drugs. Furthermore, this review article highlights the discrepancy of the inhibitory potencies determined in different assays; very different potencies may be measured with the frequently used artificial substrate p-Nph-5′-TMP in comparison to the natural substrate ATP. Therefore, we recommend particularly for investigating competitive inhibitors the re-evaluation of the initial screening results obtained with p-Nph-5′-TMP as a substrate by using ATP as a substrate, and the use of the new artificial substrate p-Nph-5′-AMP instead of p-Nph-5′-TMP for high-throughput screening.
Biographies

Sang-Yong Lee
Sang-Yong Lee, born in Seoul, received a Ph.D. degree in Pharmaceutical Chemistry at the University of Bonn in 2015. His current research interests are focused on the medicinal chemistry and pharmacology of ecto-nucleotidases, nucleotide hydrolyzing ecto-enzymes, including analytical biochemistry, and molecular biology.

Christa E. Müller
Christa E. Müller, born in Rottweil (South-Germany), received a Ph.D. degree in Pharmaceutical Chemistry at the University of Tübingen in 1988. She worked as a post-doctoral fellow at the NIH, Bethesda, MD, USA, in the group of J. W. Daly (1989–1990 and 1992), became an Assistant Professor at the University of Würzburg, Germany in 1994, and a Full Professor at the University of Bonn in 1998, where she is currently heading the Department of Pharmaceutical Chemistry I. Her research interests include medicinal chemistry and molecular pharmacology of purinergic targets (adenosine (P1), nucleotide (P2Y, P2X) and adenine (P0) receptors and ectonucleotidases, as well as orphan G protein-coupled receptors).
Footnotes
†The authors declare no competing interests.
References
- Ralevic V., Burnstock G. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- Müller C. E., Jacobson K. A. Biochim. Biophys. Acta. 2011;1808:1290–1308. doi: 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbracchio M. P., Burnstock G., Boeynaems J., Barnard E. A., Boyer L., Kennedy C., Knight G. E., Fumagalli M., Gachet C., Jacobson K. A., Weisman G. A. Pharmacol. Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh B. S., Burnstock G., Kennedy C., King B. F., North R. A., Séguéla P., Voigt M., Humphrey P. P. Pharmacol. Rev. 2001;53:107–118. [PubMed] [Google Scholar]
- Burnstock G. Trends Pharmacol. Sci. 2006;27:166–176. doi: 10.1016/j.tips.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Drug Dev. Res. 2001;52:44–56. [Google Scholar]
- Zimmermann H., Zebisch M., Sträter N. Purinergic Signalling. 2012;5:437–502. doi: 10.1007/s11302-012-9309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegutkin G. G. Biochim. Biophys. Acta. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Kukulski F., Lévesque S. A., Sévigny J. Adv. Pharmacol. 2011;61:263–299. doi: 10.1016/B978-0-12-385526-8.00009-6. [DOI] [PubMed] [Google Scholar]
- Stefan C., Jansen S., Bollen M. Trends Biochem. Sci. 2005;30:542–550. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Yegutkin G. G. Crit. Rev. Biochem. Mol. Biol. 2014;49:473–497. doi: 10.3109/10409238.2014.953627. [DOI] [PubMed] [Google Scholar]
- Al-Rashida M., Iqbal J. Mini-Rev. Med. Chem. 2015;15:41–51. doi: 10.2174/1389557515666150219113205. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Pharmacol. Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- Fredholm B. B., IJzerman A. P., Jacobson K. A., Klotz K. N., Linden J. Pharmacol. Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Goding J. W., Grobben B., Slegers H. Biochim. Biophys. Acta. 2003;1638:1–19. doi: 10.1016/s0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- Stefan C., Jansen S., Bollen M. Purinergic Signalling. 2006;2:361–370. doi: 10.1007/s11302-005-5303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright R. A., Ornstein D. L., Cao W., Chang W. C., Robert D., Tehan M., Hoyer D., Liu L., Stabach P., Yang G., De La Cruz E. M., Braddock D. T. J. Biol. Chem. 2014;289:3294–3306. doi: 10.1074/jbc.M113.505867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright R. A., Chang W. C., Robert D., Ornstein D. L., Cao W., Liu L., Redick M. E., Young J. I., De La Cruz E. M., Braddock D. T. Blood. 2012;120:4432–4440. doi: 10.1182/blood-2012-04-425215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen S., Andries M., Derua R., Waelkens E., Bollen M. J. Biol. Chem. 2009;284:14296–14302. doi: 10.1074/jbc.M900790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki J. Semin. Cell Dev. Biol. 2004;15:477–489. doi: 10.1016/j.semcdb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Aoki J., Inoue A., Okudaira S. Biochim. Biophys. Acta. 2008;1781:513–518. doi: 10.1016/j.bbalip.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Kawaguchi M., Okabe T., Okudaira S., Hanaoka K., Fujikawa Y., Terai T., Komatsu T., Kojima H., Aoki J., Nagano T. J. Am. Chem. Soc. 2011;133:12021–12030. doi: 10.1021/ja201028t. [DOI] [PubMed] [Google Scholar]
- Sakagami H., Aoki J., Natori Y., Nishikawa K., Kakehi Y., Natori Y., Arai H. J. Biol. Chem. 2005;280:23084–23093. doi: 10.1074/jbc.M413438200. [DOI] [PubMed] [Google Scholar]
- Duan R.-D. Biochim. Biophys. Acta. 2006;1761:281–291. doi: 10.1016/j.bbalip.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Old L. J., Boyse E. A. J. Exp. Med. 1970;131:1325–1341. doi: 10.1084/jem.131.6.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie N. C. W., Huesa C., Rutsch F., MacRae V. E. Bone. 2012;51:961–968. doi: 10.1016/j.bone.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Harahap A. R., Goding J. W. J. Immunol. 1988;141:2317–2320. [PubMed] [Google Scholar]
- Morley D. J., Hawley D. M., Ulbright T. M., Butler L. G., Culp J. S., Hodes M. E. J. Histochem. Cytochem. 1987;35:75–82. doi: 10.1177/35.1.3025290. [DOI] [PubMed] [Google Scholar]
- Razzell W. E. J. Biol. Chem. 1961;236:3028–3030. [PubMed] [Google Scholar]
- Goldfine I. D., Maddux B. A., Youngren J. F., Reaven G., Accili D., Trischitta V., Vigneri R., Frittitta L. Endocr. Rev. 2008;29:62–75. doi: 10.1210/er.2007-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen S., Perrakis A., Ulens C., Winkler C., Andries M., Joosten R. P., Van Acker M., Luyten F. P., Moolenaar W. H., Bollen M. Structure. 2012;20:1948–1959. doi: 10.1016/j.str.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Kato K., Nishimasu H., Okudaira S., Mihara E., Ishitani R., Takagi J., Aoki J., Nureki O. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16876–16881. doi: 10.1073/pnas.1208017109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollen M., Gijsbers R., Ceulemans H., Stalmans W., Stefan C. Crit. Rev. Biochem. Mol. Biol. 2000;35:393–432. doi: 10.1080/10409230091169249. [DOI] [PubMed] [Google Scholar]
- Heldin C. H., Wasteson A., Fryklund L., Westermark B. Science. 1981;213:1122–1123. doi: 10.1126/science.6973821. [DOI] [PubMed] [Google Scholar]
- Schvartz I., Seger D., Shaltiel S. Int. J. Biochem. Cell Biol. 1999;31:539–544. doi: 10.1016/s1357-2725(99)00005-9. [DOI] [PubMed] [Google Scholar]
- Bellacchio E. J. Cell. Physiol. 2012;227:3366–3374. doi: 10.1002/jcp.24058. [DOI] [PubMed] [Google Scholar]
- Cimpean A., Stefan C., Gijsbers R., Stalmans W., Bollen M. Biochem. J. 2004;381:71–77. doi: 10.1042/BJ20040465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers R., Ceulemans H., Stalmans W., Bollen M. J. Biol. Chem. 2001;276:1361–1368. doi: 10.1074/jbc.M007552200. [DOI] [PubMed] [Google Scholar]
- Massé K., Bhamra S., Allsop G., Dale N., Jones E. A. Int. J. Dev. Biol. 2010;54:181–193. doi: 10.1387/ijdb.092879km. [DOI] [PubMed] [Google Scholar]
- Zalatan J. G., Fenn T. D., Brunger A. T., Herschlag D. Biochemistry. 2006;45:9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- Galperin M. Y., Bairoch A., Koonin E. V. Protein Sci. 1998;7:1829–1835. doi: 10.1002/pro.5560070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K., Moffa A., Chen Y., Pritzker K., Goding J., Terkeltaub R. J. Bone Miner. Res. 1999;14:883–892. doi: 10.1359/jbmr.1999.14.6.883. [DOI] [PubMed] [Google Scholar]
- Petersen C., Nygård A., Viuff B., Fredholm M., Aasted B., Salomonsen J. Dev. Comp. Immunol. 2007;31:618–631. doi: 10.1016/j.dci.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Namasivayam V., Lee S.-Y., Müller C. E. Biochim. Biophys. Acta. 2017;1861:603–614. doi: 10.1016/j.bbagen.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Kelly S. J., Dardinger D. E., Butler L. G. Biochemistry. 1975;14:4983–4988. doi: 10.1021/bi00693a030. [DOI] [PubMed] [Google Scholar]
- Stefan C., Gijsbers R., Stalmans W., Bollen M. Biochim. Biophys. Acta. 1999;1450:45–52. doi: 10.1016/s0167-4889(99)00031-2. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Fukui S., Funakoshi I., Yamashina I. J. Biochem. 1983;93:1641–1648. doi: 10.1093/oxfordjournals.jbchem.a134303. [DOI] [PubMed] [Google Scholar]
- Li L., Yin Q., Kuss P., Maliga Z., Millán J. L., Wu H., Mitchison T. J. Nat. Chem. Biol. 2014;10:1043–1048. doi: 10.1038/nchembio.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber G. N. Trends Immunol. 2014;35:88–93. doi: 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Woo S. R., Corrales L., Gajewski T. F. Trends Immunol. 2015;36:250–256. doi: 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henz S. L., Fürstenau C. R., Chiarelli R. A., Sarkis J. J. F. Arch. Oral Biol. 2007;52:916–923. doi: 10.1016/j.archoralbio.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y., Lévesque S. A., Sévigny J., Müller C. E. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012;911:162–169. doi: 10.1016/j.jchromb.2012.10.044. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y., Sarkar S., Bhattarai S., Namasivayam V., De Jonghe S., Stephan H., Herdewijn P., El-Tayeb A., Müller C. E. Front. Pharmacol. 2017;8 doi: 10.3389/fphar.2017.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio A. C., Rodríguez-Ferrer C. R., Castañeyra-Perdomo A., Oaknin S., Rotllán P. Neurochem. Int. 2007;50:581–590. doi: 10.1016/j.neuint.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Langer D., Hammer K., Koszalka P., Schrader J., Robson S., Zimmermann H. Cell Tissue Res. 2008;334:199–217. doi: 10.1007/s00441-008-0681-x. [DOI] [PubMed] [Google Scholar]
- Laketa D., Bjelobaba I., Savic J., Lavrnja I., Stojiljkovic M., Rakic L., Nedeljkovic N. Mol. Cell. Biochem. 2010;339:99–106. doi: 10.1007/s11010-009-0373-1. [DOI] [PubMed] [Google Scholar]
- Rücker B., Almeida M. E., Libermann T. A., Zerbini L. F., Wink M. R., Sarkis J. J. F. Mol. Cell. Biochem. 2007;306:247–254. doi: 10.1007/s11010-007-9576-5. [DOI] [PubMed] [Google Scholar]
- Buffon A., Casali E. A., Cardoso V. V., Zerbini L. F., Robson S. C., Sarkis J. J. F., Wink M. R. Life Sci. 2010;86:435–440. doi: 10.1016/j.lfs.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Evans W. H., Hood D. O., Gurd J. W. Biochem. J. 1973;135:819–826. doi: 10.1042/bj1350819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J., Lévesque S. A., Sévigny J., Müller C. E. Electrophoresis. 2008;29:3685–3693. doi: 10.1002/elps.200800013. [DOI] [PubMed] [Google Scholar]
- Vollmayer P., Clair T., Goding J. W., Sano K., Servos J., Zimmermann H. Eur. J. Biochem. 2003;270:2971–2978. doi: 10.1046/j.1432-1033.2003.03674.x. [DOI] [PubMed] [Google Scholar]
- Grobben B., Claes P., Roymans D., Esmans E. L., Van Onckelen H., Slegers H. Br. J. Pharmacol. 2000;130:139–145. doi: 10.1038/sj.bjp.0703289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda N., Hoshino S. I., Kanda Y., Katada T. Eur. J. Biochem. 1999;265:763–770. doi: 10.1046/j.1432-1327.1999.00779.x. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y., Fiene A., Li W., Hanck T., Brylev K. A., Fedorov V. E., Lecka J., Haider A., Pietzsch H.-J., Zimmermann H., Sévigny J., Kortz U., Stephan H., Müller C. E. Biochem. Pharmacol. 2015;93:171–181. doi: 10.1016/j.bcp.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Khan K. M., Fatima N., Rasheed M., Jalil S., Ambreen N., Perveen S., Choudhary M. I. Bioorg. Med. Chem. 2009;17:7816–7822. doi: 10.1016/j.bmc.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Choudhary M. I., Fatima N., Khan K. M., Jalil S., Iqbal S., Rahman A.-U. Bioorg. Med. Chem. 2006;14:8066–8072. doi: 10.1016/j.bmc.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Shayhidin E. E., Forcellini E., Boulanger M.-C., Mahmut A., Dautrey S., Barbeau X., Lagüe P., Sévigny J., Paquin J.-F., Mathieu P. Br. J. Pharmacol. 2015;172:4189–4199. doi: 10.1111/bph.13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L., Lee S. Y., Leonczak P., Rozenski J., De Jonghe S. C., Hanck T., Müller C. E., Herdewijn P. J. Med. Chem. 2014;57:10080–10100. doi: 10.1021/jm501434y. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y., Perotti A., De Jonghe S., Herdewijn P., Hanck T., Müller C. E. Bioorg. Med. Chem. 2016;24:3157–3165. doi: 10.1016/j.bmc.2016.05.046. [DOI] [PubMed] [Google Scholar]
- Fürstenau C. R., Trentin D. S., Barreto-Chaves M. L. M., Sarkis J. J. F. Thromb. Res. 2007;120:877–884. doi: 10.1016/j.thromres.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y., Müller C. E. Electrophoresis. 2014;35:855–863. doi: 10.1002/elps.201300453. [DOI] [PubMed] [Google Scholar]
- Joseph S. M., Pifer M. A., Przybylski R. J., Dubyak G. R. Br. J. Pharmacol. 2004;142:1002–1014. doi: 10.1038/sj.bjp.0705865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecka J., Ben-David G., Simhaev L., Eliahu S., Oscar J., Luyindula P., Pelletier J., Fischer B., Senderowitz H., Sévigny J. J. Med. Chem. 2013;56:8308–8320. doi: 10.1021/jm400918s. [DOI] [PubMed] [Google Scholar]
- Bhattarai S., Freundlieb M., Pippel J., Meyer A., Abdelrahman A., Fiene A., Lee S.-Y., Zimmermann H., Yegutkin G. G., Sträter N., El-Tayeb A., Müller C. E. J. Med. Chem. 2015;58:6248–6263. doi: 10.1021/acs.jmedchem.5b00802. [DOI] [PubMed] [Google Scholar]
- Nadel Y., Lecka J., Gilad Y., Ben-David G., Förster D., Reiser G., Kenigsberg S., Camden J., Weisman G. A., Senderowitz H., Sévigny J., Fischer B. J. Med. Chem. 2014;57:4677–4691. doi: 10.1021/jm500196c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J., Vollmayer P., Braun N., Zimmermann H., Müller C. E. Purinergic Signalling. 2005;1:349–358. doi: 10.1007/s11302-005-8076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque S. A., Lavoie E. G., Lecka J., Bigonnesse F., Sévigny J. Br. J. Pharmacol. 2007;152:141–150. doi: 10.1038/sj.bjp.0707361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliahu S., Lecka J., Reiser G., Haas M., Bigonnesse F., Lévesque S. A., Pelletier J., Sévigny J., Fischer B. J. Med. Chem. 2010;53:8485–8497. doi: 10.1021/jm100597c. [DOI] [PubMed] [Google Scholar]
- Müller C. E., Iqbal J., Baqi Y., Zimmermann H., Röllich A., Stephan H. Bioorg. Med. Chem. Lett. 2006;16:5943–5947. doi: 10.1016/j.bmcl.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Iqbal J., Knowles A. F., Müller C. E. J. Chromatogr. A. 2010;1217:600–604. doi: 10.1016/j.chroma.2009.11.100. [DOI] [PubMed] [Google Scholar]
- Baqi Y., Lee S.-Y., Iqbal J., Ripphausen P., Lehr A., Scheiff A. B., Zimmermann H., Bajorath J., Müller C. E. J. Med. Chem. 2010;53:2076–2086. doi: 10.1021/jm901851t. [DOI] [PubMed] [Google Scholar]
- Zavyalova E., Kopylov A. Thromb. Res. 2015;135:212–216. doi: 10.1016/j.thromres.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Hasenknopf B. Front. Biosci. 2005;10:275–287. doi: 10.2741/1527. [DOI] [PubMed] [Google Scholar]
- Pope M. T. and Kortz U., Polyoxometalates. Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley, Chichester, 2012. [Google Scholar]
- Raza R., Akhtar T., Hameed S., Lecka J., Iqbal J., Sévigny J. Open Enzyme Inhib. J. 2011;4:17–22. [Google Scholar]
- Khan K. M., Siddiqui S., Saleem M., Taha M., Saad S. M., Perveen S., Choudhary M. I. Bioorg. Med. Chem. 2014;22:6509–6514. doi: 10.1016/j.bmc.2014.08.032. [DOI] [PubMed] [Google Scholar]
- Patel S. D., Habeski W. M., Cheng A. C., de la Cruz E., Loh C., Kablaoui N. M. Bioorg. Med. Chem. Lett. 2009;19:3339–3343. doi: 10.1016/j.bmcl.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Gao M., Wang M., Zheng Q. H. Bioorg. Med. Chem. Lett. 2016;26:1371–1375. doi: 10.1016/j.bmcl.2016.01.081. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. Adv. Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Leeson P. D., Springthorpe B. Nat. Rev. Drug Discovery. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Ausekle E., Ejaz S., Khan S., Ehlers P., Villinger A., Lecka J., Sevigny J., Iqbal J., Langer P. Org. Biomol. Chem. 2016;14:11402–11414. doi: 10.1039/c6ob02236g. [DOI] [PubMed] [Google Scholar]
- Jafari B., Yelibayeva N., Ospanov M., Ejaz S. A., Afzal S., Khan S. U., Abilov Z. A., Turmukhanova M. Z., Kalugin S. N., Safarov S., Lecka J., Sévigny J., Rahman Q., Ehlers P., Iqbal J., Langer P. RSC Adv. 2016;6:107556–107571. [Google Scholar]
- Fürstenau C. R., Trentin D. D. S., Barreto-Chaves M. L. M., Sarkis J. J. F. Platelets. 2006;17:84–91. doi: 10.1080/09537100500246641. [DOI] [PubMed] [Google Scholar]
- Michaud A., Williams T. A., Chauvet M., Corvol P. Mol. Pharmacol. 1997;1076:1070–1076. doi: 10.1124/mol.51.6.1070. [DOI] [PubMed] [Google Scholar]
- Ben Henda Y., Labidi A., Arnaudin I., Bridiau N., Delatouche R., Maugard T., Piot J. M., Sannier F., Thiery V., Bordenave-Juchereau S., Agric J. Food Chem. 2013;61:10685–10690. doi: 10.1021/jf403004e. [DOI] [PubMed] [Google Scholar]
- Schiemann K., Wienke D., Staehle W., Schultz M., Ross T., Busch M., Kober I., Hecht S., Zimmermann A., Musil D. and Bladt F., EFMC/ISMC Conference 2012 (Berlin), 2012, p. 57.
- Velloso L. M., Bhaskaran S. S., Schuch R., Fischetti V. A., Stebbins C. E. EMBO Rep. 2008;9:199–205. doi: 10.1038/sj.embor.7401154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K., Vaingankar S., Chen Y., Moffa A., Goldring M. B., Sano K., Jin-Hua P., Sali A., Goding J., Terkeltaub R. Arthritis Rheum. 1999;42:1986–1997. doi: 10.1002/1529-0131(199909)42:9<1986::AID-ANR26>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Johnson K. A., Hessle L., Vaingankar S., Wennberg C., Mauro S., Narisawa S., Goding W., Sano K., Millan J. L., Terkeltaub R. Am. J. Physiol. 2000;279:R1365–R1377. doi: 10.1152/ajpregu.2000.279.4.R1365. [DOI] [PubMed] [Google Scholar]
- Mackenzie N. C. W., Zhu D., Milne E. M., van 't Hof R., Martin A., Darryl Quarles L., Quarles D. L., Millán J. L., Farquharson C., MacRae V. E. PLoS One. 2012;7:e32177. doi: 10.1371/journal.pone.0032177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson H. C., Harmey D., Camacho N. P., Garimella R., Sipe J. B., Tague S., Bi X., Johnson K., Terkeltaub R., Millán J. L. Am. J. Pathol. 2005;166:1711–1720. doi: 10.1016/S0002-9440(10)62481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terkeltaub R. Purinergic Signalling. 2006;2:371–377. doi: 10.1007/s11302-005-5304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke Y., Hartmann S., Torsello G., Horstmann R., Seifarth H., Weissen-Plenz G., Rutsch F. J. Cell. Mol. Med. 2011;15:220–231. doi: 10.1111/j.1582-4934.2009.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke Y., Weissen-Plenz G., Terkeltaub R., Rutsch F. J. Cell. Mol. Med. 2012;15:2273–2283. doi: 10.1111/j.1582-4934.2011.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam H. K., Liu J., Li Y., Kragor A., Hatch N. E. J. Biol. Chem. 2011;286:39059–39071. doi: 10.1074/jbc.M111.221689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K., Goding J., Van Etten D., Sali A., Hu S., Milla L., Farley D., Krug H., Hessle L., Millán J. L., Terkeltaub R. Am. Soc. Bone Miner. Res. 2003;18:994–1004. doi: 10.1359/jbmr.2003.18.6.994. [DOI] [PubMed] [Google Scholar]
- Hessle L., Johnson K. A., Anderson H. C., Narisawa S., Sali A., Goding J. W., Terkeltaub R., Millan J. L. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmey D., Hessle L., Narisawa S., Johnson K. A., Terkeltaub R., Millán J. L. Am. J. Pathol. 2004;164:1199–1209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F., Vaingankar S., Johnson K., Goldfine I., Maddux B., Schauerte P., Kalhoff H., Sano K., Boisvert W. A., Superti-Furga A., Terkeltaub R. Am. J. Pathol. 2001;158:543–554. doi: 10.1016/S0002-9440(10)63996-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine I. D., Maddux B. A., Youngren J. F., Trischitta V., Frittitta L. Ann. N. Y. Acad. Sci. 1999;892:204–222. doi: 10.1111/j.1749-6632.1999.tb07797.x. [DOI] [PubMed] [Google Scholar]
- Youngren J. F. Cell. Mol. Life Sci. 2007;64:873–891. doi: 10.1007/s00018-007-6359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abate N., Chandalia M., Di Paola R., Foster D. W., Grundy S. M., Trischitta V. Nat. Clin. Pract. Endocrinol. Metab. 2006;2:694–701. doi: 10.1038/ncpendmet0367. [DOI] [PubMed] [Google Scholar]
- Frittitta L., Camastra S., Baratta R., Costanzo B. V., D'Adamo M., Graci S., Spampinato D., Maddux B. A., Vigneri R., Ferrannini E., Trischitta V. J. Clin. Endocrinol. Metab. 1999;84:3620–3625. doi: 10.1210/jcem.84.10.6050. [DOI] [PubMed] [Google Scholar]
- Dong H., Maddux B. A., Altomonte J., Meseck M., Accili D., Terkeltaub R., Johnson K., Youngren J. F., Goldfine I. D. Diabetes. 2005;54:367–372. doi: 10.2337/diabetes.54.2.367. [DOI] [PubMed] [Google Scholar]
- Maddux B. A., Chang Y., Accili D., Mcguinness O. P., Youngren J. F., Goldfine I. D., Betty A., Mcguinness P., Over I. D. G. Am. J. Physiol. Endocrinol. Metab. 2006;290:E746–E749. doi: 10.1152/ajpendo.00298.2005. [DOI] [PubMed] [Google Scholar]
- Grupe A., Alleman J., Goldfine I. D., Sadick M., Stewart T. A. J. Biol. Chem. 1995;270:22085–22088. doi: 10.1074/jbc.270.38.22085. [DOI] [PubMed] [Google Scholar]
- Mcateer J. B., Prudente S., Bacci S., Lyon H. N., Hirschhorn J. N., Trischitta V., Florez J. C. Diabetes. 2008;57:1125–1130. doi: 10.2337/db07-1336. [DOI] [PubMed] [Google Scholar]
- Abate N., Carulli L., Cabo-Chan A., Chandalia M., Snell P. G., Grundy S. M. J. Clin. Endocrinol. Metab. 2003;88:5927–5934. doi: 10.1210/jc.2003-030453. [DOI] [PubMed] [Google Scholar]
- González-Sánchez J. L., Zabena C., Martínez-Larrad M. T., Martínez-Calatrava M. J., Pérez-Barba M., Serrano-Ríos M. Clin. Endocrinol. 2008;68:724–729. doi: 10.1111/j.1365-2265.2007.03103.x. [DOI] [PubMed] [Google Scholar]
- Kubaszek A., Pihlajamäki J., Karhapää P., Vauhkonen I., Laakso M. Diabetes Care. 2003;26:464–467. doi: 10.2337/diacare.26.2.464. [DOI] [PubMed] [Google Scholar]
- Matsuoka N., Patki A., Tiwari H. K., Allison D. B., Johnson S. B., Gregersen P. K., Leibel R. L., Chung W. K. Int. J. Obes. 2006;30:233–237. doi: 10.1038/sj.ijo.0803132. [DOI] [PubMed] [Google Scholar]
- Willer C. J., Bonnycastle L. L., Conneely K. N., Duren W. L., Jackson A. U., Scott L. J., Narisu N., Chines P. S., Skol A., Stringham H. M., Petrie J., Erdos M. R., Swift A. J., Enloe S. T., Sprau A. G., Smith E., Tong M., Doheny K. F., Pugh E. W., Watanabe R. M., Buchanan T. A., Valle T. T., Bergman R. N., Tuomilehto J., Mohlke K. L., Collins F. S., Boehnke M. Diabetes. 2007;56:256–264. doi: 10.2337/db06-0461. [DOI] [PubMed] [Google Scholar]
- González-Sánchez J. L., Martínez-Larrad M. T., Fernández-Pérez C., Kubaszek A., Laakso M., Serrano-Ríos M. Obes. Res. 2003;11:603–605. doi: 10.1038/oby.2003.86. [DOI] [PubMed] [Google Scholar]
- Keshavarz P., Inoue H., Sakamoto Y., Kunika K., Tanahashi T., Nakamura N., Yoshikawa T., Yasui N., Shiota H., Itakura M. J. Hum. Genet. 2006;51:559–566. doi: 10.1007/s10038-006-0399-0. [DOI] [PubMed] [Google Scholar]
- Rasmussen S. K., Urhammer S. A., Pizzuti A., Echwald S. M., Ekstrøm C. T., Hansen L., Hansen T., Borch-johnsen K., Frittitta L., Trischitta V., Pedersen O. Diabetes. 2000;49:1608–1611. doi: 10.2337/diabetes.49.9.1608. [DOI] [PubMed] [Google Scholar]
- Di Paola R., Caporarello N., Marucci A., Dimatteo C., Iadicicco C., Del Guerra S., Prudente S., Sudano D., Miele C., Parrino C., Piro S., Beguinot F., Marchetti P., Trischitta V., Frittitta L. PLoS One. 2011;6:e19462. doi: 10.1371/journal.pone.0019462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. H., Chin C.-N., Wu M., Ni W., Quan S., Liu F., Dallas-Yang Q., Ellsworth K., Ho T., Zhang A., Natasha T., Li J., Chapman K., Strohl W., Li C., Wang I.-M., Berger J., An Z., Zhang B. B., Jiang G. Eur. J. Pharmacol. 2009;616:346–352. doi: 10.1016/j.ejphar.2009.06.057. [DOI] [PubMed] [Google Scholar]
- Grobben B., De Deyn P. P., Slegers H. Cell Tissue Res. 2002;310:257–270. doi: 10.1007/s00441-002-0651-7. [DOI] [PubMed] [Google Scholar]
- Aerts I., Martin J.-J., De Deyn P. P., Van Ginniken C., Van Ostade X., Kockx M., Dua G., Slegers H. Clin. Neurol. Neurosur. 2011;113:224–229. doi: 10.1016/j.clineuro.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Bageritz F., Puccio R. M., Hovestadt V., Phillips E., Pankert T., Lohr J., Herold-Mende C., Lichter P., Goidts V. Cell Death. Differ. 2014;21:929–940. doi: 10.1038/cdd.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Villafuertes R., Pintor J., Miras-Portugal M. T., Gualix J. J. Neurochem. 2014;131:290–302. doi: 10.1111/jnc.12794. [DOI] [PubMed] [Google Scholar]
- Burnstock G., Di Virgilio F. Purinergic Signaling. 2013;9:491–540. doi: 10.1007/s11302-013-9372-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan S. P., Eltzschig H. K., Eckle T., Thompson L. F. Purinergic Signalling. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komohara Y., Ohnishi K., Kuratsu J., Takeya M. J. Pathol. 2008;216:15–24. doi: 10.1002/path.2370. [DOI] [PubMed] [Google Scholar]
- Mora R., Abschuetz A., Kees T., Dokic I., Joschko N., Kleber S., Geibig R., Mosconi E., Zentgraf H., Martin-Villalba A., Régnier-Vigouroux A. Glia. 2009;57:561–581. doi: 10.1002/glia.20785. [DOI] [PubMed] [Google Scholar]
- Watters J. J., Schartner J. M., Badie B. J. Neurosci. Res. 2005;81:447–455. doi: 10.1002/jnr.20485. [DOI] [PubMed] [Google Scholar]
- Horenstein A. L., Chillemi A., Zaccarello G., Bruzzone S., Quarona V., Zito A., Serra S., Malavasi F. Oncoimmunology. 2013;2:e26246. doi: 10.4161/onci.26246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Shao Q., Sun J., Yang N., Xie Q., Wang D., Huang Q., Huang B., Wang X., Li X., Qu X. Neuro Oncol. 2013;15:1160–1172. doi: 10.1093/neuonc/not067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavaresco L., Bernardi A., Braganhol E., Cappellari A., Rockenbach L., Farias P., Wink M., Delgado-Cañedo A., Battastini A. Mol. Cell. Biochem. 2008;319:61–68. doi: 10.1007/s11010-008-9877-3. [DOI] [PubMed] [Google Scholar]
- Bastid J., Cottalorda-Regairaz A., Alberici G., Bonnefoy N., Eliaou J.-F., Bensussan A. Oncogene. 2013;32:1743–1751. doi: 10.1038/onc.2012.269. [DOI] [PubMed] [Google Scholar]
- Gessi S., Varani K., Merighi S., Fogli E., Sacchetto V., Benini A., Leung E., Mac-Lennan S., Borea P. A. Purinergic Signalling. 2007;3:109–116. doi: 10.1007/s11302-006-9042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J., Smyth M. J. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- Bergamin L. S., Braganhol E., Zanin R. F., Edelweiss M. I. A., Battastini A. M. O. J. Biomed. Biotechnol. 2012;2012:959848. doi: 10.1155/2012/959848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F., Bruchard M., Chalmin F., Rébé C. J. Biomed. Biotechnol. 2012;2012:473712. doi: 10.1155/2012/473712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo E.-K., Kim J. K., Shin D.-M., Sasakawa C. Cell. Mol. Immunol. 2015;13:1–12. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao B. Z., Xu Z. Q., Han B. Z., Su D. F., Liu C. Front. Pharmacol. 2015;6:1–9. doi: 10.3389/fphar.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L., Gajewski T. F. Clin. Cancer Res. 2015;21:4774–4779. doi: 10.1158/1078-0432.CCR-15-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria O., De Gassart A., Coso S., Gestermann N., Di Domizio J., Flatz L., Gaide O., Michielin O., Hwu P., Petrova T. V., Martinon F., Modlin R. L., Speiser D. E., Gilliet M. Proc. Natl. Acad. Sci. U. S. A. 2015;112:15408–15413. doi: 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]