

 A meta-analysis of Skp2 inhibitors based on bioactivity, structure, and medicinal chemistry was used to identify validated Skp2 pharmaceutical hits.
A meta-analysis of Skp2 inhibitors based on bioactivity, structure, and medicinal chemistry was used to identify validated Skp2 pharmaceutical hits.
Abstract
Skp2 is a member of the F-box family of proteins that serve as substrate-specific adaptors in Skp1–CUL1–ROC1–F-box (SCF) E3 ubiquitin ligases. Skp2 (Fbxl1) directly binds to the tumor suppressor p27 in the context of the SCFSkp2 E3 ubiquitin ligase to ubiquitylate and target-phosphorylated p27 for proteasomal degradation. As p27 is a powerful suppressor of growth in a variety of cells, and as Skp2 is also overexpressed in many human cancers, Skp2 is considered an oncogene and an intriguing drug target. However, despite 20 years of investigation, a valid chemical inhibitor of Skp2-mediated degradation of p27 has not been identified. Recently, an increasing number of compounds designed to have this bioactivity have been reported. Here, we conduct a meta-analysis of the evidence regarding bioactivity, structure, and medicinal chemistry in order to evaluate and compare these Skp2 inhibitor compounds. Despite chemically diverse compounds with a wide array of Skp2-mediated p27 ubiquitylation inhibition properties reported by several independent groups, no current chemical probe formally qualifies as a validated pharmaceutical hit compound. This finding suggests that our knowledge of the structural biochemistry of the Skp2–p27 complex remains incomplete and highlights the need for novel modes of inquiry.
Introduction
Posttranslational modification of proteins by polyubiquitylation is a highly-regulated process with a myriad of functional consequences for a protein's behavior or fate, but is most commonly associated with targeting of proteins for degradation by the proteasome. In this manner, cells are able to both fine-tune levels of specific proteins and irreversibly inhibit their function by catalyzing their destruction. Dysregulation of this process is associated with numerous and varied disease states. Ubiquitylation is a post-translational modification that is important for maintaining cellular homeostasis during cell cycle progression, proliferation, and apoptosis. Ubiquitylation marks proteins for degradation by the ubiquitin proteasome system (UPS). Dysregulation of the UPS has been implicated in the development of cancer, immunological disorders, and neurological conditions.1 These critical cellular roles explain why the UPS is highly regulated. Target proteins are ubiquitylated in a stepwise manner. First, the ubiquitin-activating enzyme (E1) forms a reactive thioester linkage to a single ubiquitin protein. This ubiquitin is exchanged (trans-esterified) to a ubiquitin-conjugating enzyme (E2). The ubiquitin-bearing E2 enzyme is then recruited to a third enzyme, the ubiquitin E3 ligase, which brings the substrate and the ubiquitin moiety in close proximity, enabling the ubiquitin transfer to the substrate protein.1 Polyubiquitination of a substrate by at least four ubiquitin moieties triggers protein degradation by the 26S proteasome, a multicatalytic enzyme that degrades proteins.
The development and approval of bortezomib (Velcade; Millennium Pharmaceuticals) revealed the potential of targeting the UPS therapeutically. Bortezomib is a 26S proteasome inhibitor used in the treatment of multiple myeloma and mantle cell lymphoma.2,3 However, new treatments are needed to alleviate bortezomib resistance and its adverse effects, which include peripheral neuropathy.4,5 An attractive group of targets for inhibitor development are the E3 ubiquitin ligases, which act upstream of the proteasome activity.6–11 Due to the existence of numerous E3 ubiquitin ligases, a specific ligase that recognizes specific substrates may be targeted, which is theorized to confer therapeutic benefit by minimizing off-target effects.12
The SKP1–CUL1–ROC1–F-box (SCF) complex is a multi-protein RING-finger E3 ubiquitin ligase. S-phase kinase-associated protein 1 (SKP1) bridges the scaffold cullin 1 (CUL1) to an interchangeable F-box protein. The F-box protein determines the specificity of the SCF complex. S-phase kinase-associated protein 2 (Skp2) is an F-box protein that targets the p27, p57, p130, Tob1, and c-myc substrates.13 While Skp2 together with its accessory protein cyclin-dependent kinases regulatory subunit 1 (Cks1), regulate the eukaryotic cell cycle by controlling ubiquitination-mediated degradation of p21 and p27. Overall, a functioning SCFSkp2 E3 ubiquitin ligase is generated upon the assembly of three components around Skp2: its substrate (p27), its accessory unit (Cks1), and its SCF adaptor (Skp1).1
Skp2 is a known oncogene and is overexpressed in various cancers.14–17 Likewise, patients with overexpression of Skp2 have a worse prognosis.17 It has been shown that excessive degradation of p27 is also seen in human cancers.1,18 Moreover, Skp2 deletions were found to delay tumor progression through the accumulation of p27.19 Hence, Skp2 is a potential therapeutic target for cancer treatment.
Inhibitors of the UPS cascade target upstream cyclin-dependent kinases, cullins (via neddylation), E1 or E2 enzymes and/or deubiquitinating enzymes (DUBs) and are not specific to Skp2 substrates. Similarly, Skp1 inhibitors and downstream proteasomal components are non-specific. Skp1/Skp2 interaction inhibitors would specifically diminish p21 and p27 degradation as well as the Cks1–Skp2 and Cks1 substrate interaction.1 However, this review focuses on small molecules that directly bind to Skp2. We conducted a meta-analysis of published Skp2 inhibitors with a focus on bioactivity, structure, and medicinal chemistry.
Inhibitors of Skp2-mediated p27 ubiquitylation targeting distinct interfaces of the SCFSkp2 complex
Efforts to identify inhibitors of the SCFSkp2 complex have often been successful in demonstrating inhibition of p27 ubiquitylation. High throughput screening (HTS) and in silico virtual library screening (VLS) have been used to identify the current generation of inhibitors of the SCFSkp2 complex. Different studies have identified chemical compounds purported to target each of the protein–protein interfaces on the SCFSkp2 complex (Fig. 1 and Table 1). We previously performed two in silico screens to identify the only reported inhibitors of the Skp2 and phospho-p27 interface. The best-characterized inhibitors from these screens were termed C1 and C2.6 It should be noted that subsequent studies referred to C1 as C5,20 but to prevent confusion, we will only refer to the representative inhibitor of this group as C1. Two groups have reported inhibitors of the Skp2–Skp1 interface: Chan et al. used an in silico screen to identified compound #25 while Chen et al. through HTS identified their lead compound, compound A.7,8 In addition, derivatives of compound A were further synthesized and interrogated by another group that also reported inhibition of p27 degradation.24 Several groups have also employed HTS to identify inhibitors that disrupt the Skp2–Cks1 interface. The most potent and well-characterized of the compounds identified by Singh and colleagues was assigned the identifier 22d; Ooi et al. identified two natural product compounds, linichlorin A and gentian violet; and Ungermannova et al. identified both NSC689857 and NSC681152 as their best compounds.9–11 Both compounds share similar chemical structures so we will focus our attention on NSC689857 due to its favorable results. Other inhibitors are known to indirectly attenuate p27 ubiquitylation. An additional inhibitor of Skp2, SMIP004, has been reported to downregulate the expression of Skp2, but it is not included in this review because there is no evidence of direct Skp2 binding.25
Fig. 1. SCFSkp2 complex with proposed inhibitor binding sites. ICM-Skin representation of PDB: ; 2AST. Compounds C1 and C2 target the Skp2 (cyan) and phospho-p27 peptide (green) interface. 22d targets the Skp2 and Cks1 (purple) interface preventing assembly of these subunits. Compound #25 and compound A both target the interface of Skp2 and Skp1 (orange).

Table 1. SCFSKP2 inhibitors.
| Compound | Proposed target site | Biophysical confirmation | Biochemical confirmation | Cell signaling confirmation (G1 arrest) | In vivo | PAINS |
| C1 (ref. 6 and 20–23) | Skp2–P–p27 | No | Yes | Yes | Yes | Yes |
| C2 (ref. 6 and 20) | Skp2–P–p27 | No | Yes | Yes | Yes | Yes |
| Compound #25 (ref. 7) | Skp1–Skp2 | No | Yes | No | Yes | Maybe |
| Compound A (ref. 8) | Skp1–Skp2 | No | Yes | Yes | No | No |
| 22d (ref. 9) | Skp2–Cks1 | No | Yes | No | No | No |
| Linichlorin A (ref. 11) | Skp2–Cks1 | No | Yes | Yes | No | Yes |
| Gentian violet (ref. 11) | Skp2–Cks1 | No | Yes | Yes | No | No |
| NSC689857 (ref. 10) | Skp2–Cks1 | No | Yes | No | No | Yes |
Validated hit requirements
Drug development follows sequential milestones; each of which has significant and multiplicative biomedical value. First, hit validation is followed by in vivo proof of mechanism where pharmacologic inhibition of Skp2 would demonstrate impairment of cancer cell growth in mice. Next, lead optimization is followed in the preclinical setting. Bona fide leads are usually licensed and proof of concept in phase III clinical trials is sought out. The early milestone of a validated hit is sufficiently challenging that commercial partners sometimes directly license the intellectual property of such hits. For a hit to be considered a validated hit it must (1) elicit a reproducible response in two orthogonal assays and also elicit a dose-response over a hundred-fold concentration range, (2) be analytically validated in terms of integrity and purity, (3) demonstrate potencies of IC50 ∼ 1 μM or less, and (4) be an attractable starting point of chemical optimization with no obvious major chemical liabilities.
An analysis of the current Skp2 inhibitors was done to determine if any of the compounds meet the validated hit criteria (Table 2). Most inhibitors lack the second orthogonal assay except for C1 and compound #25. C1 used both immunoprecipitation (IP) and differential scanning fluorimetry (DSF), although DSF does not directly identify protein binding partners, while compound #25 used IP and liquid chromatography-mass spectrometry (LC-MS/MS) to identify their protein-binding partners. Our lab has analytically validated and resynthesized both compounds C1 and C2. The data published for compound #25, compound A and 22d suggests that the compounds have been analytically verified as well. Both linichlorin A and gentian violet were attained from the RIKEN NPDepo (natural product depository), but these are well-known compounds, which suggests that they have been verified. The same assumption is made for NSC689857 since it was provided from the National Cancer Institute (NCI) library that holds fully characterized synthetic and natural products. All compounds except for NSC689857 have been tested on cells. NSC689857 only reported an in vitro IC50 of 36 μM that is notably above the minimum 1 μM requirement. The IC50 provided by the rest of the compounds were generated from cell proliferation or viability assays. Compounds C1, compound #25, 22d, linichlorin A, and gentian violet had at least one sample within the 1 μM range. Lastly, the only compounds that did not have chemical liabilities concomitant with pan assay interference compounds (PAINS) were compound A, 22d, and gentian violet (Table 3).26 In summary, none of the compounds meet the full criteria for a validated hit.
Table 2. Hit validation criteria.
| Compound | Assay 1 | Assay 2 | Analytically validated | Potency
a
|
Chemical liabilities | |
| Cell line | IC50 (μM) | |||||
| C1 (ref. 6 and 20–23) | IP | DSF | Yes | MCF-7 | 8.0 | Yes |
| T47D | 5.0 | |||||
| 501 Mel | 30.0 | |||||
| KSM-11 | 1.84 | |||||
| ARP-1 | 3.84 | |||||
| JeKo-1 | 4.8 b | |||||
| C2 (ref. 6 and 20) | IP | No | Yes | JeKo-1 | 9.3 | Unclear |
| ECC-1 | 14.3 b | |||||
| Compound #25 (ref. 7) | IP | LC-MS/MS | Yes | 8 cancer lines | 1.22–10.5 | Yes |
| Compound A (ref. 8) | IP | No | Yes | RPMI 8226 | 8.0 | No |
| U266 | 4.2 | |||||
| MM1.S | 5.4 | |||||
| ANBL6 | 10.0 | |||||
| RPMI 8226/LR5 | 6.3 | |||||
| RPMI 8226/Dox40 | 8.5 | |||||
| U266/LR6 | 4.3 | |||||
| MM1.RL | 5.4 | |||||
| ANBL-6/B7R | 8.0 | |||||
| 22d (ref. 9) | ELISA | No | Yes | H1299 | 1.05 | No |
| A549 | 1.49 | |||||
| Linichlorin A (ref. 11) | ELISA | No | Yes | HeLa | 3.2 | Yes |
| tsFT210 | 1.6 | |||||
| NIH3T3 | 12.7 | |||||
| Gentian violet (ref. 11) | ELISA | No | Yes | HeLa | 0.4 | No |
| tsFT210 | 0.6 | |||||
| NIH3T3 | 5.3 | |||||
| NSC689857 (ref. 10) | AlphaScreen | No | Yes | In vitro Skp2–Cks1 | 36.0 | Yes |
aPotency has been identified in cell proliferation or viability assays except where noted.
bEC50 value.
Table 3. Inhibitors of SCFSkp2.
| Entry | Structure | Biological characterization | Literature identifier | Ref. |
| 1 |
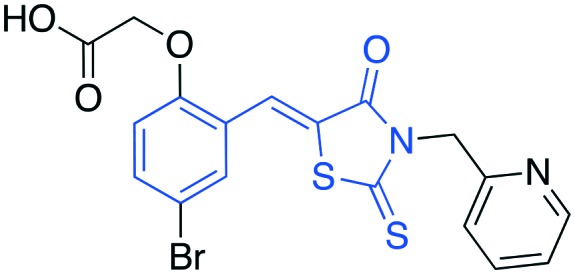
|
• In vitro inhibition of p27 ubiquitylation | C1 | 6, 20–23 |
| • Cell-based stabilization of p27 | ||||
| • Antiproliferation | ||||
| • G1 arrest | ||||
| 2 |
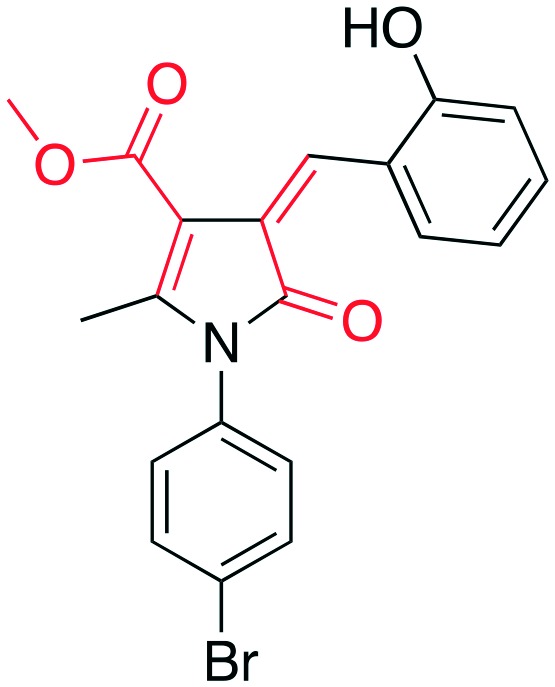
|
• In vitro inhibition of p27 ubiquitylation | C2 | 6, 20 |
| • Cell-based stabilization of p27 | ||||
| • Antiproliferation | ||||
| • G1 arrest | ||||
| 3 |
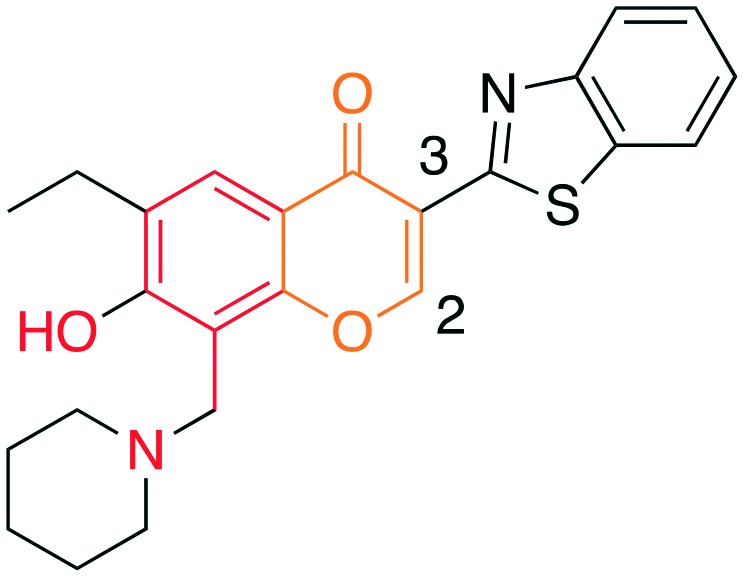
|
• In vitro inhibition of p27 ubiquitylation | Compound #25 | 7 |
| • Cell-based stabilization of p27 | ||||
| • Decrease viability | ||||
| • G2/M arrest | ||||
| 4 |
|
• Antiproliferation | 22d | 9 |
| 5 |
|
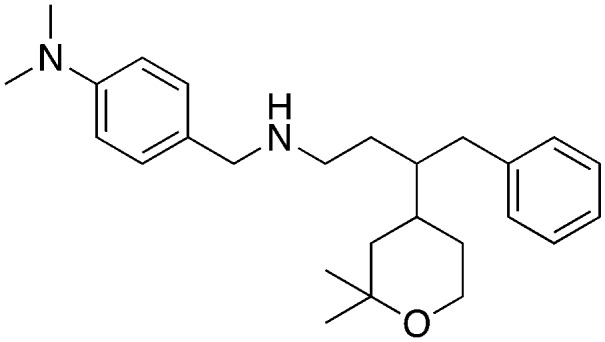
• In vitro inhibition of p27 ubiquitylation | Compound A | 8 |
| • Cell-based stabilization of p27 | ||||
| • Decrease viability | ||||
| • G1 arrest | ||||
| 6 |
|
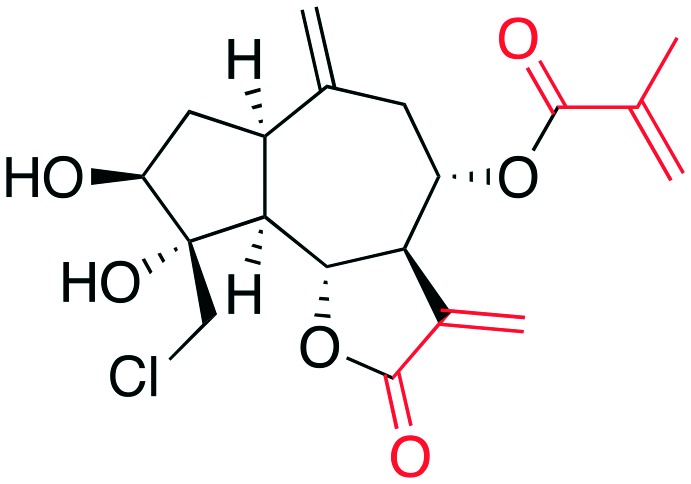
• In vitro inhibition of p27 ubiquitylation | Linichlorin A | 11 |
| • Cell-based stabilization of p27 | ||||
| • Antiproliferation | ||||
| 7 |

|
• In vitro inhibition of p27 ubiquitylation | Gentian violet | 11 |
| • Cell-based stabilization of p27 | ||||
| • Cell-based | ||||
| • Antiproliferation | ||||
| • G1 arrest | ||||
| 8 |
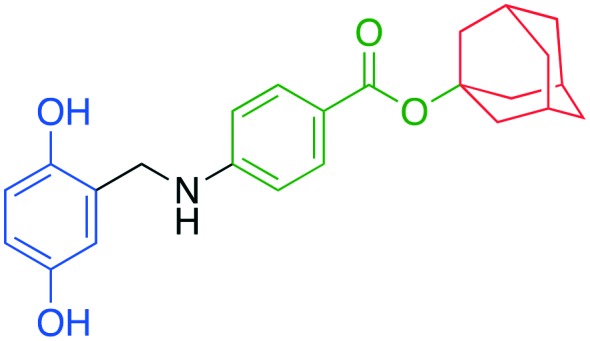
|
• In vitro inhibition of p27 ubiquitylation | NSC689857 | 10 |
| • Cell-based stabilization of p27 |
3D structure of SCFSkp2 inhibitor target sites
X-ray co-crystal structures of small molecule inhibitors bound to the SCFSkp2 are not yet available. Thus, computational or empirical approaches are required in order to either identify new agents or rationalize the activity of existing ones. Computational approaches to drugging the SCF complexes that use available crystallographic data have previously been described.27,28 The ; 2AST co-crystal structure of SCFSkp2 complex contains the Skp1/Skp2/Cks1 subunits together with a phosphorylated peptide representing the binding region of its major substrate, p27.29 The presence of a defined surface pocket of suitable size rendered by a suitable computational algorithm30 has previously been hypothesized as a necessary pre-requisite for accommodation of a drug-like small molecule for any target protein surface.27,28 Removing the p27 ligand reveals such a pocket at the p27–Skp2–Cks1 interface. This pocket was targeted using VLS to find compounds C1 and C2.6 Chan et al. identified two pockets at the Skp1–Skp2 interface by visual inspection of the structure, without the use of a computational pocket rendering algorithm.7 These sites were then targeted with VLS to find compound #25; however, the leading computational algorithm for objectively identifying druggable pockets fails to identify a pocket of suitable size at these sites under default parameters (Fig. 2). Using this same algorithm, a rather large pocket is evident at the Skp2–Cks1 interface, but its surface to volume ratio is larger than the acceptable range seen in known drug binding pockets.27,28
Fig. 2. SCFSkp2 druggable pockets. ICM-Skin and ribbon representation of PDB: ; 2AST. Skp1 (orange), Cks1 (purple) and phospo-p27 (green) are shown as mesh while Skp2 is represented as a yellow ribbon structure. The Skp2–Cks1–p27 interface pocket (cyan) is the target site for C1 and C2. It is not certain if linichlorin A and gentian violet also target this site. This pocket exists at an interface between Skp2 and Cks1 when phospo-p27 is removed. The Skp2–Cks1 interface pocket (red) has been previously noted to be an unusually large pocket and is the target site for 22d and NSC689857. It is also not certain if linichlorin A and gentian violet also target this site. The Skp2–Skp1 interface with no pocket is indicated with an arrow and compound #25 and compound A target this region.

Since no structural rationale has been disclosed for the majority of compounds in this review and since the absence of a pocket might not absolutely preclude binding of a drug, we performed a meta-analysis of docking studies to the proposed binding sites on SCFSkp2 that were either observed by probing the ; 2AST co-crystal structure (unpublished) or reported in the literature (compound #25).30 Only C1 and NSC689857 docked to their expected target sites with acceptable scores of –38.5 and –37.3, respectively using ICM-Dock, the docking algorithm that has shown the best accuracy in head to head comparisons of docking algorithms.31 ICM-Dock employs fully flexible 3D compound structures and rigid, energy-grid representations of the receptor, and has been described previousy.32 Compounds with an ICM-Dock score of ≤–32 are considered to have a higher probability of physiologic relevance. The reported docked poses of compound #25 and compound A could not be confirmed with ICM-Dock. The discrepancy is likely due to different docking parameters employed as well as to differences in performance between the HiPCDock algorithm used to identify compound #25 and ICM-Dock, although mutagenesis studies provide evidence of the binding site for compound #25. Placing compound #25 and compound A into their reported docked pose and calculating energy using the ICM-Dock implementation did not result in acceptable docking scores (≤–32, Fig. 3). The docking score of compound 22d to its target Cks1–Skp2 pocket was also unfavourable (Fig. 3). Although this is the expected binding region for this agent we could not generate a plausible binding mode. Nevertheless, compound 22d was shown to dissociate Cks1 and Skp2, therefore our understanding of this pocket is incomplete and would be greatly enhanced by further crystallographic data.9 The hypothesized binding sites for linichlorin A, gentian violet and NSC689857 on Skp2 were not identified,10 and after docking studies to the Skp2–Cks1 and Skp2–phospo-p27 pockets, neither linichlorin A nor gentian violet scored well in these studies (Fig. 3).
Fig. 3. ICM docking scores of SCFSkp2 inhibitors. The dotted line represents the putative threshold ICM docking score predictive of binding (≤–32). C1 and C2 were docked to their corresponding Skp2–P–p27 pocket. NSC689857 and compound A were docked to the Skp2–Skp1 interface with no originally identifiable pocket. 22d and NSC689857 were docked to the Skp2–Cks1 interface pocket. Both linichlorin A and gentian violet were docked to the Skp2–P–p27 and the Skp2–Cks1 pockets. Superscript 1 denotes docking to the Skp2–P–p27 pocket and superscript 2 denotes docking to the Skp2–Cks1 pocket.

These findings suggest that compound #25, 22d, linichlorin A, and gentian violet target an alternative form of SCFSkp2 not seen in the crystal structure, if they operate by direct binding to Skp2. This could be an alternative conformation of the current domains29 or one formed by elements missing in the current structure like the N-terminus of the Skp2 loops that were truncated in order to crystallize the protein, or the L16 loop of Skp1, or yet unknown binding domains, including oligomeric forms of Skp2, Cks1, p27 and/or all three.33
Bioactivity of Skp2-targeted inhibitors of p27 ubiquitylation and degradation
In vitro biochemical and cell-based assays are used to efficiently test and validate hit compounds for target binding, protein interactions, enzymatic function, and biological phenotypes. Below we consider these assays in decreasing order of target specificity and examine how the proposed inhibitors performed in each. A final list of assays tested is seen in Tables 2 and 3. It should be noted that none of these compounds have been tested with detergents in the biochemical assays used to demonstrate their Skp2 inhibitory activity. Detergents are useful to block compound aggregation, which is a common source of false positives in biochemical assays.34,35
Biochemical assays
Biochemical assays can be divided into those where only purified proteins are used versus those that employ cellular lysate/extract. The latter engenders more uncertainty about the target specificity of a compound. The purpose of the assay is to identify inhibitors that disrupt specific protein–protein interfaces. The only purely in vitro binding assays used in these investigations were ELISA-based assays that screened for 22d and NSC68985. Linichlorin A and gentian violet were monitored with an ELISA, but not with the same degree of transparency about the protein complexes being disrupted.
The ELISA system used to evaluate 22d used His-Skp1 and untagged Skp2 proteins that were co-expressed in SF9 insect cells. These proteins were purified as a His-Skp1/Skp2 hetero-dimeric complex. 96-well plates were coated with His-Cks1 that was expressed in and purified from E. coli cells. The purified His-Skp1/Skp2 complex was then added to each well in the presence of 22d precursor molecules. An anti-Skp2 antibody was used to detect the binding of the His-Skp1/Skp2 complex to His-Cks1. This assay format was used during both hit-finding and hit-optimization to identify 22d (and others).
The AlphaScreen system used to identify NSC689857 employs GST-Skp2/Skp1 and His-Cks1 proteins both expressed in and purified from E. coli. His-Cks1 and the inhibitor were incubated and added to 386-well microplates followed by the addition of GST-Skp2/Skp1. The plates were then treated with GSH-coated donor beads and nickel chelate acceptor beads designed to detect the Skp2–Cks1 complex. Upon incubation, laser excitation of the donor beads provided an emission from 520 nm to 620 nm. In the presence of NSC689857, the emission signal decreased due to the disruption of the Skp2–Cks1 complex.
Lastly, linichlorin A and gentian violet were both identified by an ELISA-based assay. Phospho-p27 peptides containing the target sequence for Skp1–Skp2–Cks1 were chemically synthesized, purified and covalent linked to 96-well plates. The 96-well plates were treated with inhibitor and insect cell lysate expressing Skp1 and fluorescent monomeric Azami Green (mAG) Skp2 and Cks1. Binding of the Skp1–mAGSkp2–Cks1 to phospho-p27 peptides was confirmed by spectrofluorometry detection of mAGSkp2, which indicated assembly of the SCF complex (Skp1–mAGSkp2–Cks1–phospho-p27). In the presence of linichlorin A or gentian violet the fluorescent signal by mAGSkp2 decreased; however, this assay does not rule out disruption of other interfaces within the complex, such as Skp2 to Cks1 or Cks1 to phopho-p27.
Binding assays
Pull-down/cell-based binding assays are specific for confirming target binding partners in the cellular milieu, but not necessarily the target interface.36 These assays have the advantage of being more biologically relevant, but maybe confounded by the presence of heterogeneous and undefined cell lysate. These types of assays were used in the testing of C1, C2, compound #25, and compound A. C1 and C2 were shown to target the Skp2-phospho-p27 interface; levels of p27 bound to HA-Skp2 were reduced in the presence of C1 and C2, whereas Cks1 binding was unaffected.6 Compound #25 used in the treatment of PC3 cells demonstrated an inhibition of in vivo binding of Skp2 to Skp1 in a dose-dependent manner.7 Compound A treatment of HeLa cells transfected with HA-Skp1 resulted in a decrease of detectable Skp2.8
Ubiquitylation assays
Ubiquitylation assays are conducted using in vitro translated rabbit reticulocyte lysate or purified protein components of E1, E2, and E3 proteins. Although, in cells this is a very reliable indicator of p27 ubiquitylation, this readout does not provide information about target specificity. All compounds mentioned except for 22d, which was not tested, inhibited ubiquitylation of p27, either in vitro or in mammalian cells. C1 and C2 were tested in vitro at 10 μM in the presence of the SCF complex proteins (His6-Skp1/Skp2, His6-Cul1/Roc1, His6-cyclin E/Cdk2 and His6-Cks1) previously expressed and purified from insect or E. coli cells.6 C1 and C2 met the first screening criteria of inhibiting ubiquitylation of p27 by at least 50%. Compound #25 was tested both in vitro at 10 μM and in HEK293T cells at 20 μM and both assays demonstrated inhibited Skp2-mediated p27 ubiquitylation.7 MM1.S myeloma cells treated with compound A at varying concentrations (10 μM, 20 μM, and 25 μM) resulted in a reduction of p27 ubiquitylation by 8%, 44%, and 51%, respectively.8 An in vitro system where the SCF complex was expressed and immunoprecipitated from HEK293T cells was used to detect changes in p27 ubiquitylation caused by linichlorin A and gentian violet.11 Treatment of linichlorin A at 3.2 μM and gentian violet at 0.4 μM reduced p27 ubiquitylation by 70–80%. Finally, in vitro testing of NSC689857 resulted in inhibition of p27 ubiquitylation at an IC50 of ∼30 μM.10
G1 cell cycle arrest
While the role of p27 in eukaryotic cells varies, its primary purpose is to prevent cell cycle progression from G1 phase to S phase. Halting this transition is the principal target-specific cellular phenotype expected of a successful Skp2–p27 inhibitor. This biology justifies the anticipated utility of Skp2 inhibitors in the treatment of cancer, and accordingly proposed inhibitors should be challenged in rapidly cycling, transformed cells. C1, C2, compound A, linichlorin A, and gentian violet were the only compounds reported to demonstrate G1 arrest in cultured cancer lines via flow cytometric analysis.
The effect of C1 and C2 were evaluated in melanoma, 501 Mel cells, at 10 μM for 16 hours.6 C1 and C2 both showed p27 protein induction and an increase in G1 phase cells and a decrease in S phase cells. The effects of C1 and C2 were additionally monitored in breast cancer cells, MCF-7 and T47D, at 5 μM for 16 hours. In contrast, MCF-7 cells responded to C1 with a significant reduction in G1 phase and an increase in G2/M phase. While T47D cells treated with C1 displayed an increase in G1 phase and a decrease in S phase, correlating with p27 protein induction. However, neither breast cancer cell displayed a significant change in cell cycle after treatment with C2. Finally, C1 and C2 were also used to treat endometrial carcinoma cells-1 (ECC-1) at 10 μM for 18 hours and both showed an increase in p27 and in cells at G0/G1.20 The effect of compound A on the cell cycle was investigated in myeloma, RPMI 8226 cells.8 The cells were treated with compound A at either 5 μM or 10 μM for 24 hours. The results showed an increase in G0/G1 phase with a very slow progression to S and G2/M phases. The cell cycle effects of both linichlorin A and gentian violet were investigated on mouse cancer, tsFT210 cells.11 Cells were treated at each compound's IC50 values (linichlorin A 1.6 μM; gentian violet 0.6 μM) for either 8 hours or 12 hours. The results showed a delayed in the initiation of S phase and significantly increase in p27 levels.
Cell cycle inhibition should lead to overall cell growth inhibition in cultured cells, but this phenotype is certainly not specific to the inhibition of the interaction between Skp2 and p27. Nevertheless, for compounds that meet biochemical criteria, such a phenotype is highly suggestive of an effective in vivo inhibitor. All compounds exhibited this property in cells, but only C1, C2, compound A, linichlorin A, and gentian violet were reported arrest cancer cells at G1 phase.
In vivo studies
A very useful property for any chemical probe is target-specific in vivo activity in animal studies. C1 and C2 were shown to stabilize p27 in a mouse model of endometrial hyperplasia and collapsed p53/pRB double-knockout prostate tumor organoids, which are exquisitely sensitive to p27 stabilization.20,37 Compound #25 exhibited non-target specific inhibition of both a lung adenocarcinoma xenograft and prostate adenocarcinoma xenograft in mice, showing an up regulation of p27, p21 and apoptosis, and a down regulation of the glycolysis regulators, Akt and Glut1 (glucose transporter 1).7 No additional in vivo data are reported for the other compounds mentioned.
Structure activity relationship (SAR) of SCFSkp2 inhibitors
The structures of the compounds described in the preceding text are identified in Tables 3 and 4. In Table 3, colored portions of the molecules represent known or suspected PAINS-like substructural features.26 The bold numbering will be used in conjunction with each compound's literature identifier to aid the discussion of structure activity relationship (SAR).
Table 4. SAR examples of reported inhibitors.
| Entry | Active | Inactive | Assay | Mode of inhibition |
| 1 |
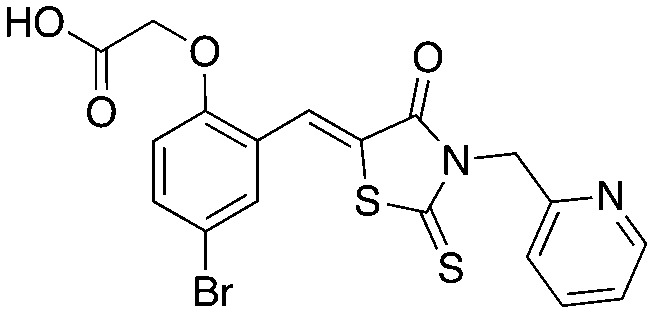 C1 C1 |
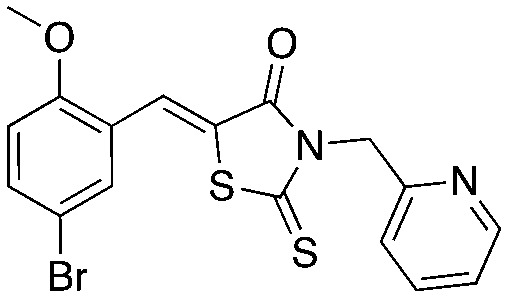 UM-C1 UM-C1 |
Ubiquitylation | Block phospho-p27 binding to Skp2–Cks1 |
 Neg-3 Neg-3 | ||||
 Neg-5 Neg-5 | ||||
| 2 |
 C2 C2 |
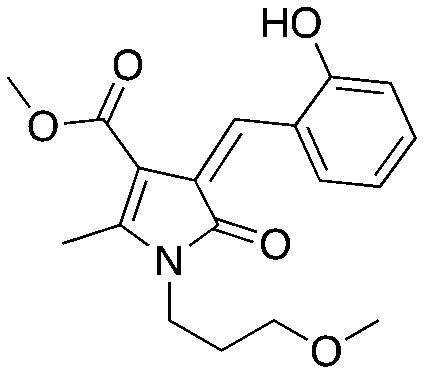 UM-C2a UM-C2a |
Ubiquitylation | Block phospho-p27 binding to Skp2–Cks1 |
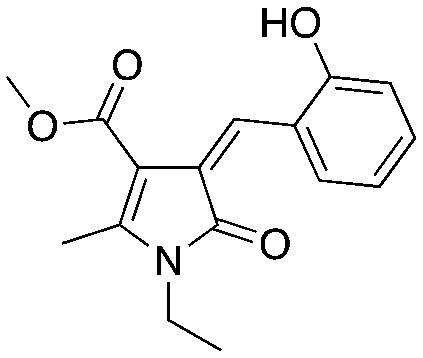 UM-C2b UM-C2b | ||||
| 3 |
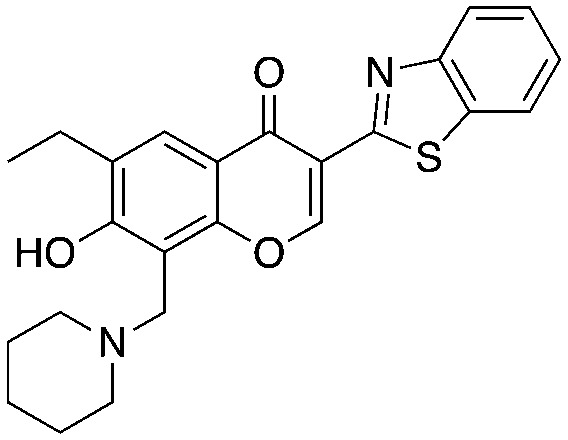 25 25 |
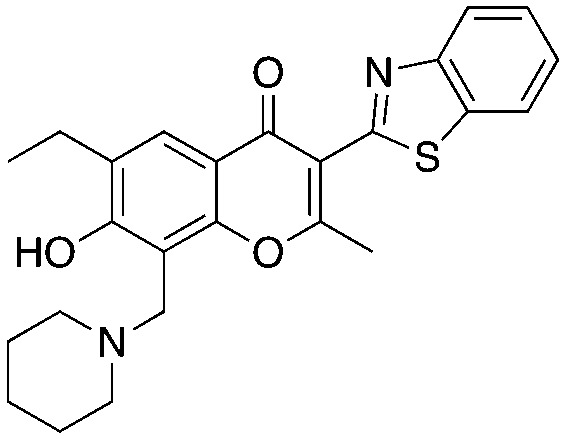 25-12 25-12 |
Ubiquitylation | Block Skp2–Skp1 association |
 25-5 25-5 |
 25-1 25-1 |
|||
| 4 |
 22d 22d |
 23a 23a |
Cks1–Skp2 interaction | Block Skp2–Cks1 interaction |
 22c 22c |
 4 4 |
|||
 1 1 | ||||
| 5 |
 Compound A Compound A |
 3h 3h |
HeLa cell proliferation | Block Skp2–Skp1 interaction |
| 8 |
 Q857 Q857 |
 3h 3h |
AlphaScreen PPI assay | Block Skp2–Cks1 interaction |
 NSC689857 NSC689857 | ||||
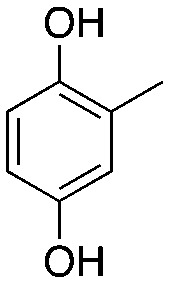 MHQ MHQ |
Compounds C1 and C2 (1, 2) were identified in a virtual screen that targeted the p27 binding pocket at the interface of Skp1 and Cks1. Compound C1 (1) is built upon the notorious electrophilic 5-benzylidene-rhodanine ring system (shown in blue), which is known to interfere with fluorescence-based assays. However, no fluorescence-based assays were used to evaluate the Skp2 inhibitory activity of 1 and 2 and the other benzylidene rhodanines, shown in Table 4 (UM-C1, Neg 3, Neg 5) tested did not inhibit the poly-ubiquitinylation of p27. The N-arylpyrrolin-2-one, C2 (2), contains two potentially electrophilic double bonds (Table 3, shown in red). The SAR in Table 4 demonstrates that this compound (2) also appears to require the 4-bromophenyl moiety for potency; however, it is unknown what other features are important for activity, or whether close analogs of the bromophenyl are tolerated. Similarly, the activity of C1 (1) depends on the acetate moiety as UM-C1 (Table 4) is inactive. In this case a structural rationale was provided from modeling where this group is proposed to form an electrostatic interaction with the side-chain of Cks1. The inactive analogs of 1 lack the functionality to perform this interaction.
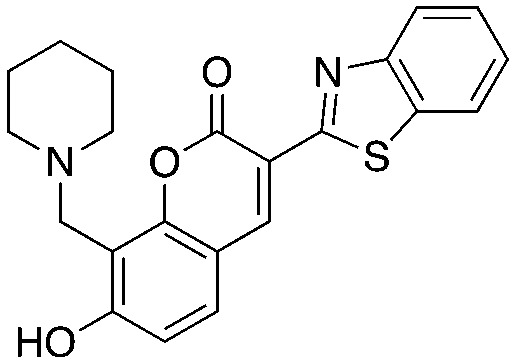
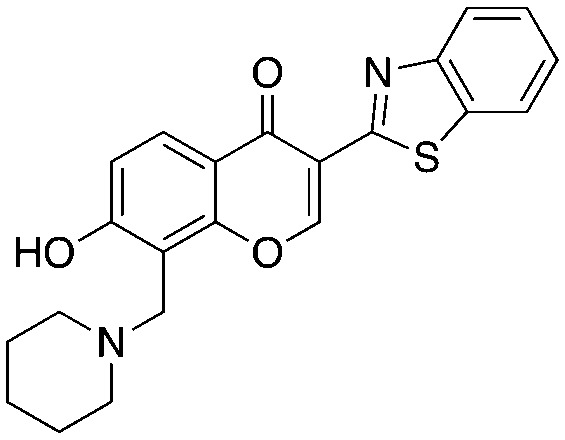
The chromone series exemplified by compound #25 (3) emerged from virtual screening against a pocket within the F-box binding region of Skp2 and exerts their effects by preventing the Skp1–Skp2 association. The chromenone scaffold at the center of compound #25 (3, orange) is also known to be electrophilic at carbon 2 (numbering shown) when activated at carbon 3 by an electron withdrawing group such as a carbonyl. Additionally, the mannich-type base of 3 (Table 3, red) is a well-known potential liability as this can eliminate under acidic conditions through a quinone methide intermediate. Nevertheless, this compound is stable enough to display serviceable pharmacokinetics when dosed at 40 and 80 mpk (ip), which at the higher dose translated to plasma and tumor Cmax values of 3.4 μM and 11.4 μM, respectively. One of the key SAR points was the requirement of the ethyl group (Table 4, compare 25 to 25-1). Modeling studies suggest that this is not critical for compound binding to Skp2, but is required to prevent Skp1–Skp2 from associating. Surprisingly, isochromone 25-5 is also active. The authors proposed that the binding mode is altered from that of 25 and that the piperidine group performs the same function as the ethyl group. The drawing of 25-5 in Table 4 is oriented to reflect this possibility. The C2 carbon of 25 cannot tolerate any substitution as shown by inactive analog 25-12.
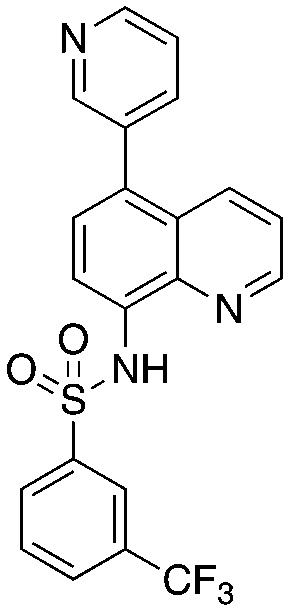
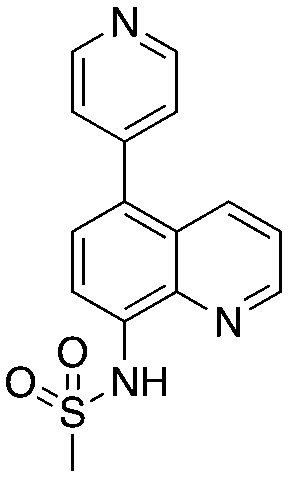
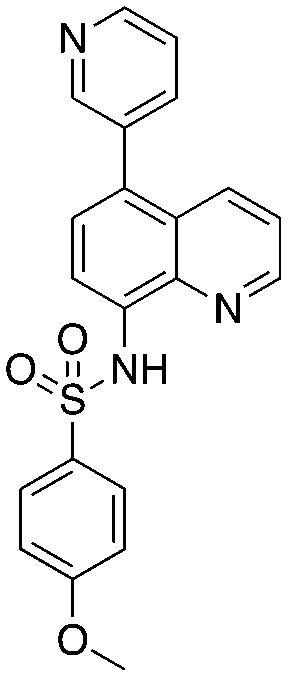
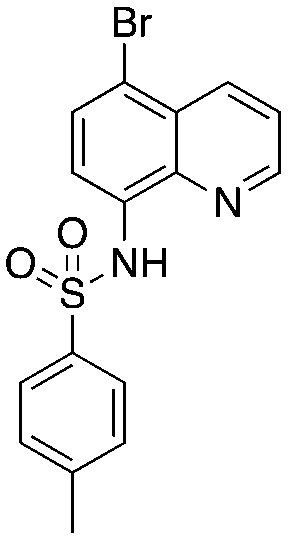
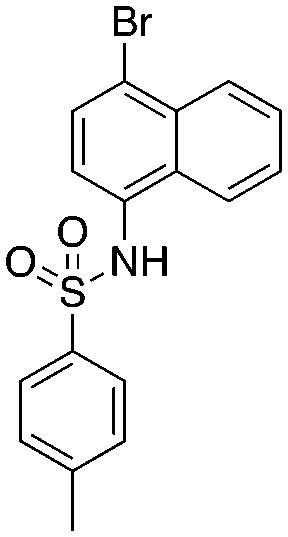
Quinoline-based compound 4 is the most potent of a series of inhibitors disclosed by Rigel Pharmaceuticals that block the Cks1–Skp2 interaction in an ELISA screen. The quinolone-based inhibitor 22d (4) was identified after a systematic SAR exploration of hit 1, which emerged from an HTS campaign to find agents that block the Cks1–Skp2 interaction. The SAR in this paper is extensive (compared to other Skp2 inhibitor papers) and will not be recapitulated here. Two key findings are of interest: the dependence of potency on both the bottom aryl ring (compare 22d, 22c, and 1 with 23a) and the quinolone nitrogen (compare 1 with 4). Surprisingly, the authors do not discuss any structure-based rationale for these (and other) SAR observations.
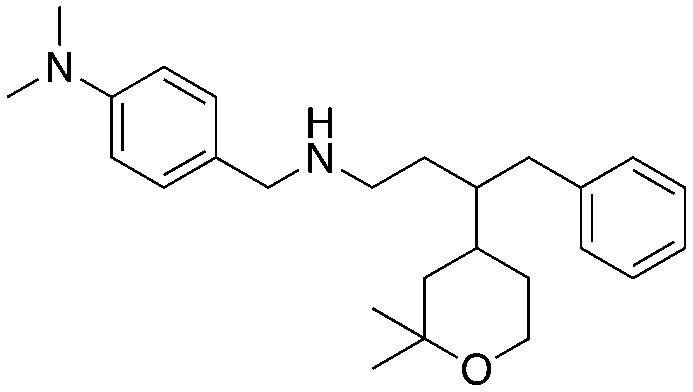
The biological activity of compound A (5) was disclosed by Celgene in 2008.8 While the synthesis and SAR for compound A (5) was published by researchers at the University of Newcastle.24 Intriguingly, the Newcastle researchers observed that the effects on HeLa cell proliferation correlated more with the lipophilicity of the analogs and not the potency in the ubiquitination assay.
Linichlorin A and gentian violet (6, 7) are two publically disclosed inhibitors from the RIKEN Institute in Japan. These compounds were able to displace a fluorescently labeled Skp2 molecule from a complex of Skp1–Skp2–Cks1 and immobilized phospho-p27 peptide despite the vastly different structures of 6 and 7, the former being a natural product with two electrophilic alkene systems (Table 3, red) and the latter a tri-arylmethane dye-like compound. No structural or modeling rationale was provided. Given the electrophilic centers on linichlorin A it can be inferred that it may operate as a covalent inhibitor. No SAR was reported for linichlorin A and gentian violet and given the vast difference in their structures none is inferable.
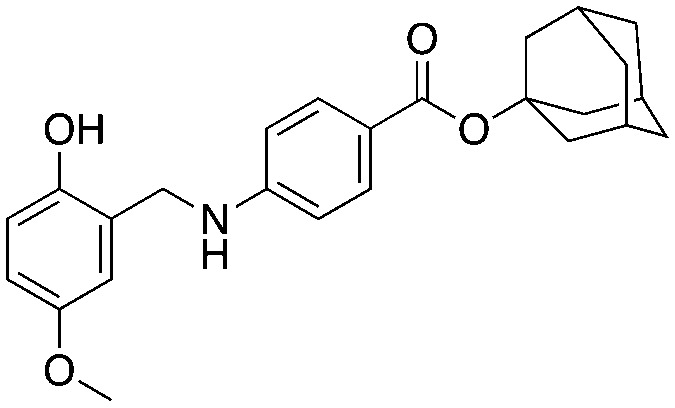
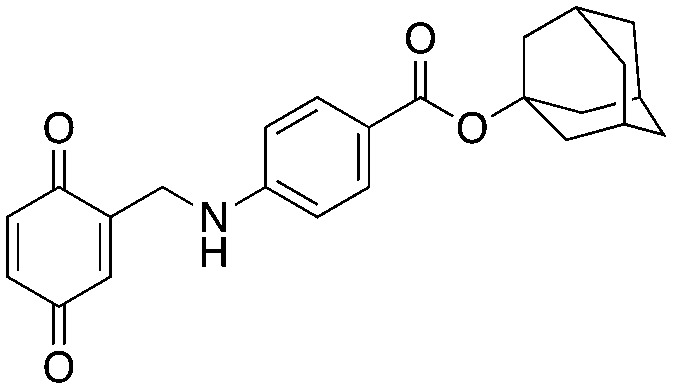
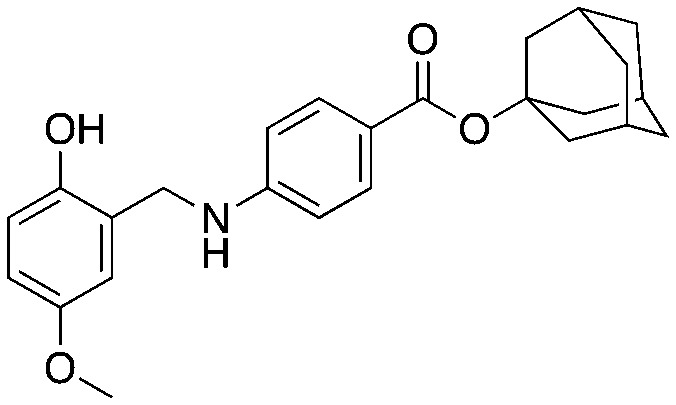
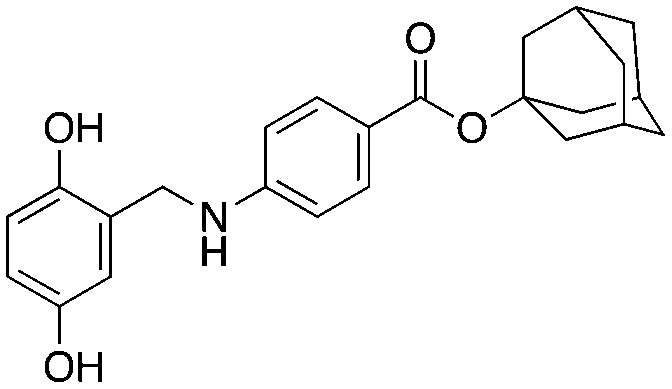
Compound NSC689857 (8) and related compounds contain the hydroquinone or benzoquinone moieties (blue) as well as adamantyl esters (red). The former are moieties considered worrying by medicinal chemists and the latter as something to be avoided as they are large lipophilic substructures that tend to have deleterious effects on aqueous solubility.38 The activity of this compound NSC689857 (and related hits) depends on the dihydroquinone (blue) and the benzoate (green) as methylation of the oxygens of the former or truncating the latter removes activity. Replacing the adamantyl group with a methyl significantly lowers the potency.
All of the compounds were shown to attenuate the ubiquitination of p27 in vitro and in cells and have effects on cell proliferation and in the cases of 1–3 display anti-tumor effects in animals. For compounds 1–5 and 8, some SAR is available whereby apparent key structural features have been identified. For most of the compounds reported here; however, the SAR reporting was limited by what analogs were commercially available. Selected SAR is summarized in Table 4 where examples of active and inactive compounds for 1–5 and 8 are shown.
Discussion
Skp2 is overexpressed and frequently observed in human cancers and thus Skp2 is thought to be an oncogene product. The oncogenic role of Skp2 was recognized when Skp2 knockout mice became resistant to tumorigenesis typically induced by the loss of the tumor suppressor proteins, p19ARF or PTEN.39 These results in turn validated Skp2 as a potential therapeutic target. Skp2 has been a high-profile cancer drug target for many years, so the absence of specifically-targeted chemical probes or drugs hints at the challenge of drugging this protein complex. Reported inhibitors of Skp2-mediated p27 ubiquitylation targeted each of the known functional interfaces of Skp2 independently: the p27 binding site, the Cks1 binding site, and the Skp1 binding site. None of the reported Skp2-targeted inhibitors of p27 degradation meet the full criteria of a validated hit, and there is significant lack of clarity as to the actual atomic site of action of every compound on Skp2. A wide range of intriguing p27-stabilizing bioactivity in orthogonal experiments are reported for these compounds, and weak SAR is apparent. However, none of these studies have concurrently investigated whether these inhibitors inhibit transcription of the p27 gene or translation of p27 mRNA, which is a potential confounding factor. Compound 22d has the best medicinal chemistry profile for a bioactive compound, but details of the bioactivity are scant.
These inhibitors promise greater specificity for the stabilization of specific SCF substrates, such as tumor suppressors like p27, over the broadly active proteasome inhibitors. For example, proteasome inhibitors broadly target the UPS and as a result down-regulate Skp2, accumulate p27, and trigger apoptosis; however, due to their broad target range their side effects are severe and include anemia, neutropenia, nausea, thrombocytopenia, peripheral neuropathy, diarrhea, and fever.40,41 In addition to such side effects, broadly active inhibitors also non-specifically degrade any protein including those that block the degradation of proteins involved in growth promoting or cancer promoting activities. In the case of these Skp2 inhibitors, having a more selective UPS inhibitor may decrease some of these unwanted off-target effects. However, even a “specific” Skp2 inhibitor is likely to have off-target effects, as most if not all licensed drugs do. For instance, the G1 arrest observed by these compounds may be a result of an off-target effect and not to the Skp2 on-target activity. Effects on cell growth are one of the most common off-target effects of chemical compounds, so showing that inhibitors of p27 degradation specifically act directly on Skp2 in cells is an important unmet challenge in the field. Indeed, very little cellular or animal toxicity data has been reported for any of the compounds reviewed here.
The extensive and diverse effort to find a validated Skp2 inhibitor without a clear success suggests that our knowledge of the structural biochemistry of the Skp2–p27 complex remains incomplete and highlights the need for novel modes of inquiry. Skp2 likely adopts alternative conformations or oligomeric states that strongly influence its druggability. In addition, its absence of a classical enzymatic active site disconnects the bioassays used for screening from compound optimization pathways by obscuring the target sites on Skp2 of compounds emerging from HTS. Studies used to evaluate C1, C2, compound #25, and NSC689857 used mutagenesis to support the targeted location, but at present, obtaining an experimental (X-ray, cryo-EM) structure of the complex of Skp2 with a compound is the next frontier in this field.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Institutes of Health award CA176502 to T. C. and a training supplement under that award to L. L.
References
- Skaar J. R., Pagan J. K., Pagano M. Nat. Rev. Drug Discovery. 2014;13(12):889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bross P. F., Kane R., Farrell A. T., Abraham S., Benson K., Brower M. E., Bradley S., Gobburu J. V., Goheer A., Lee S. L., Leighton J., Liang C. Y., Lostritto R. T., McGuinn W. D., Morse D. E., Rahman A., Rosario L. A., Verbois S. L., Williams G., Wang Y. C., Pazdur R. Clin. Cancer Res. 2004;10(12 Pt 1):3954–3964. doi: 10.1158/1078-0432.CCR-03-0781. [DOI] [PubMed] [Google Scholar]
- Kane R. C., Dagher R., Farrell A., Ko C. W., Sridhara R., Justice R., Pazdur R. Clin. Cancer Res. 2007;13(18 Pt 1):5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- Argyriou A. A., Iconomou G., Kalofonos H. P. Blood. 2008;112(5):1593–1599. doi: 10.1182/blood-2008-04-149385. [DOI] [PubMed] [Google Scholar]
- Orlowski R. Z., Kuhn D. J. Clin. Cancer Res. 2008;14(6):1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- Wu L., Grigoryan A. V., Li Y., Hao B., Pagano M., Cardozo T. J. Chem. Biol. 2012;19(12):1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. H., Morrow J. K., Li C. F., Gao Y., Jin G., Moten A., Stagg L. J., Ladbury J. E., Cai Z., Xu D., Logothetis C. J., Hung M. C., Zhang S., Lin H. K. Cell. 2013;154(3):556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Xie W., Kuhn D. J., Voorhees P. M., Lopez-Girona A., Mendy D., Corral L. G., Krenitsky V. P., Xu W., Moutouh-de Parseval L., Webb D. R., Mercurio F., Nakayama K. I., Nakayama K., Orlowski R. Z. Blood. 2008;111(9):4690–4699. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Sran A., Carroll D. C., Huang J., Tsvetkov L., Zhou X., Sheung J., McLaughlin J., Issakani S. D., Payan D. G., Shaw S. J. Bioorg. Med. Chem. Lett. 2015;25(22):5199–5202. doi: 10.1016/j.bmcl.2015.09.067. [DOI] [PubMed] [Google Scholar]
- Ungermannova D., Lee J., Zhang G., Dallmann H. G., McHenry C. S., Liu X. J. Biomol. Screening. 2013;18(8):910–920. doi: 10.1177/1087057113485789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi L. C., Watanabe N., Futamura Y., Sulaiman S. F., Darah I., Osada H. Cancer Sci. 2013;104(11):1461–1467. doi: 10.1111/cas.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Cai L., Yu Y., Meng Q., Cheng X., Zhao Y., Sui G., Zhang F. Acta Biochim.Acta Biochim. Biophys. Sin.Biophys. Sin. 2007;39(12):999–1007. doi: 10.1111/j.1745-7270.2007.00361.x. [DOI] [PubMed] [Google Scholar]
- Kitagawa K., Kotake Y., Kitagawa M. Cancer Sci. 2009;100(8):1374–1381. doi: 10.1111/j.1349-7006.2009.01196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossatz U., Dietrich N., Zender L., Buer J., Manns M. P., Malek N. P. Genes Dev. 2004;18(21):2602–2607. doi: 10.1101/gad.321004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K., Nagahama H., Minamishima Y. A., Matsumoto M., Nakamichi I., Kitagawa K., Shirane M., Tsunematsu R., Tsukiyama T., Ishida N., Kitagawa M., Hatakeyama S. EMBO J. 2000;19(9):2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K., Nagahama H., Minamishima Y. A., Miyake S., Ishida N., Hatakeyama S., Kitagawa M., Iemura S., Natsume T., Nakayama K. I. Dev. Cell. 2004;6(5):661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Frescas D., Pagano M. Nat. Rev. Cancer. 2008;8(6):438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu I. M., Hengst L., Slingerland J. M. Nat. Rev. Cancer. 2008;8(4):253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- Zhao H., Bauzon F., Fu H., Lu Z., Cui J., Nakayama K., Nakayama K. I., Locker J., Zhu L. Cancer Cell. 2013;24(5):645–659. doi: 10.1016/j.ccr.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlides S. C., Huang K. T., Reid D. A., Wu L., Blank S. V., Mittal K., Guo L., Rothenberg E., Rueda B., Cardozo T., Gold L. I. Endocrinology. 2013;154(11):4030–4045. doi: 10.1210/en.2013-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T., Pagano M., Wu L. and Gold L. I., Method of treating cancer with modulators of SCFSkp2, 2016, (US 14/055,501). [Google Scholar]
- Wen Y., Wang K., Yang K. Mol. Med. Rep. 2016;14(4):3917–3924. doi: 10.3892/mmr.2016.5676. [DOI] [PubMed] [Google Scholar]
- Grey W., Ivey A., Milne T. A., Haferlach T., Grimwade D., Uhlmann F., Voisset E., Yu V. Biochim. Biophys. Acta. 2018;1865(1):105–116. doi: 10.1016/j.bbamcr.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouksmith A. E., Evans E. L., Tweddle D. A., Miller D. C., Willmore E., Newell D. R., Golding B. T., Griffin R. J. Aust. J. Chem. 2015;68:660–679. [Google Scholar]
- Rico-Bautista E., Yang C. C., Lu L., Roth G. P., Wolf D. A. BMC Biol. 2010;8:153. doi: 10.1186/1741-7007-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B., Holloway G. A. J. Med. Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Cardozo T., Abagyan R. Methods Enzymol. 2005;399:634–653. doi: 10.1016/S0076-6879(05)99042-3. [DOI] [PubMed] [Google Scholar]
- Cardozo T., Pagano M. Nat. Rev. Mol. Cell Biol. 2004;5(9):739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Hao B., Zheng N., Schulman B. A., Wu G., Miller J. J., Pagano M., Pavletich N. P. Mol. Cell. 2005;20(1):9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- An J., Totrov M., Abagyan R. Mol. Cell. Proteomics. 2005;4(6):752–761. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- Bursulaya B. D., Totrov M., Abagyan R., 3rd Brooks C. L. J. Comput.-Aided Mol. Des. 2003;17(11):755–763. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- Neves M. A., Totrov M., Abagyan R. J. Comput.-Aided Mol. Des. 2012;26(6):675–686. doi: 10.1007/s10822-012-9547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra Dantu S., Nathubhai Kachariya N., Kumar A. Proteins. 2016;84(1):159–171. doi: 10.1002/prot.24963. [DOI] [PubMed] [Google Scholar]
- Bruns R. F., Watson I. A. J. Med. Chem. 2012;55(22):9763–9772. doi: 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- Thorne N., Auld D. S., Inglese J. Curr. Opin. Chem. Biol. 2010;14(3):315–324. doi: 10.1016/j.cbpa.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikis H. G., Guan K. L. Methods Mol. Biol. 2004;261:175–186. doi: 10.1385/1-59259-762-9:175. [DOI] [PubMed] [Google Scholar]
- Zhao H., Lu Z., Bauzon F., Fu H., Cui J., Locker J., Zhu L. Oncogene. 2017;36(1):60–70. doi: 10.1038/onc.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton J. L., Trush M. A., Penning T. M., Dryhurst G., Monks T. J. Chem. Res. Toxicol. 2000;13(3):135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- Lin H. K., Chen Z., Wang G., Nardella C., Lee S. W., Chan C. H., Yang W. L., Wang J., Egia A., Nakayama K. I., Cordon-Cardo C., Teruya-Feldstein J., Pandolfi P. P. Nature. 2010;464(7287):374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A. R., Ahmed M., Ahmed S. O., Al-Thari S., Khan A. S., Razack S., Platanias L. C., Al-Kuraya K. S., Uddin S. Leuk. Lymphoma. 2009;50(7):1204–1213. doi: 10.1080/10428190902951799. [DOI] [PubMed] [Google Scholar]
- Palumbo A., Gay F., Bringhen S., Falcone A., Pescosta N., Callea V., Caravita T., Morabito F., Magarotto V., Ruggeri M., Avonto I., Musto P., Cascavilla N., Bruno B., Boccadoro M. Ann. Oncol. 2008;19(6):1160–1165. doi: 10.1093/annonc/mdn018. [DOI] [PubMed] [Google Scholar]