

Abstract
Inhibition of the heat shock protein 90 (Hsp90) chaperone is a promising therapeutic strategy to target expression of the androgen receptor (AR) and other oncogenic drivers in prostate cancer cells. However, identification of clinically-relevant responses and predictive biomarkers is essential to maximize efficacy and treatment personalization. Here, we combined mass spectrometry (MS)-based proteomic analyses with a unique patient-derived explant (PDE) model that retains the complex microenvironment of primary prostate tumors. Independent discovery and validation cohorts of PDEs (n = 16 and 30, respectively) were cultured in the absence or presence of Hsp90 inhibitors AUY922 or 17-AAG. PDEs were analyzed by LC-MS/MS with a hyper-reaction monitoring data independent acquisition (HRM-DIA) workflow, and differentially expressed proteins identified using repeated measure analysis of variance (ANOVA; raw p value <0.01). Using gene set enrichment, we found striking conservation of the most significantly AUY922-altered gene pathways between the discovery and validation cohorts, indicating that our experimental and analysis workflows were robust. Eight proteins were selectively altered across both cohorts by the most potent inhibitor, AUY922, including TIMP1, SERPINA3 and CYP51A (adjusted p < 0.01). The AUY922-mediated decrease in secretory TIMP1 was validated by ELISA of the PDE culture medium. We next exploited the heterogeneous response of PDEs to 17-AAG in order to detect predictive biomarkers of response and identified PCBP3 as a marker with increased expression in PDEs that had no response or increased in proliferation. Also, 17-AAG treatment led to increased expression of DNAJA1 in PDEs that exhibited a cytostatic response, revealing potential drug resistance mechanisms. This selective regulation of DNAJA1 was validated by Western blot analysis. Our study establishes “proof-of-principle” that proteomic profiling of drug-treated PDEs represents an effective and clinically-relevant strategy for identification of biomarkers that associate with certain tumor-specific responses.
Keywords: Prostate cancer biomarkers, Patient cohorts, Cancer therapeutics, Tumor microenvironment, Biomarker: Diagnostic, Biomarker: Prognostic, ex vivo, Hsp90 inhibitors, patient-derived explants
Prostate cancer is the most commonly diagnosed cancer, and the second leading cause of cancer-related death, in men in the developed world (1). Despite intense research efforts, no curative therapies currently exist for men with advanced, metastatic prostate cancer. Consequently, there is an urgent need to develop new therapeutic approaches that will achieve more durable responses and thereby improve patient outcomes. A challenge that has constrained the clinical development of novel agents for prostate and other solid tumors is the difficulty in predicting and monitoring their clinical efficacy. Despite promising in vitro findings, preclinical efficacy of new therapeutics does not necessarily translate into clinical activity (2, 3), with only 5% of all potential anticancer compounds ever gaining regulatory approval (4). The reasons for this inefficiency of research translation are complex, but two clear problems have been identified: a lack of preclinical models that accurately predict activity of new agents, and a lack of robust biomarkers indicating an individual patient's response to an agent (5–7).
A class of agents that exemplifies these challenges is the heat shock protein 90 (Hsp90) 1 inhibitors. Targeting Hsp90 was considered a particularly attractive therapeutic strategy for prostate cancer as Hsp90 is commonly overexpressed in prostate cancer cells compared with normal prostate epithelium (8); therefore, prostate cancer cells are often selectively sensitive to targeting of Hsp90. Moreover, Hsp90 inhibition affords the opportunity to simultaneously degrade the androgen receptor (AR), the driver of prostate tumorigenesis, along with other oncogenic proteins that are Hsp90 clients (e.g. Her2, Akt, and Raf-1). However, despite robust preclinical data demonstrating anti-tumor activity of first-generation ansamycin-derived Hsp90 inhibitors (e.g. 17-AAG) in prostate cancer (9), poor clinical responses in prostate cancer trials initially cast doubt over this class of agent (10). This lack of efficacy has been attributed to poor solubility and pharmacokinetics, hepatotoxicity and multidrug resistance mechanisms that prevented adequate therapeutic doses from being achieved (11). Consequently, there has been considerable interest in developing new generation Hsp90 inhibitors such as AUY922, a synthetic resorcinylic isoxazole amide (12), that have improved clinical bioavailability and toxicity profiles. We previously reported on the efficacy of AUY922 in prostate cancer, showing that AUY922 is markedly more effective at killing prostate cancer cells in vitro and ex vivo than 17-AAG (13). Moreover, AUY922 maintained its efficacy even in prostate cancer cells containing constitutively active AR variants that are thought to drive advanced, castration-resistant prostate cancer.
Further complicating the clinical development of these agents is the lack of response markers to ensure the maximum benefit from a drug is obtained for an individual patient. Monitoring response markers will allow for optimal selection of agents, regimens, and patients for clinical trials. Protein markers of the AUY922 response have been investigated in a wide range of cancer cell line models using a priori approaches such as Western blot analysis (14–17). Global proteomic analysis was performed in Jurkat cells that highlighted 64 proteins (e.g. CDK1, CDK6, DNAJB1, SERPINH1, FKBP52, and mitochondrial chaperonin 10) to be markers of HSP90 inhibition by AUY922 (18). Nevertheless, there is substantial variation and a lack of conservation of results from these cell line studies emphasizing the need for a more clinically-relevant model system.
To circumvent the limitations of current cell line-based models of prostate cancer, we have developed a model of culturing human prostate cancer tissue ex vivo that retains the structure and stromal-epithelial interactions of the tumor microenvironment, has proliferative capacity, and most importantly takes into account the heterogeneous nature of the disease. Using this patient-derived explant model, we have showed that AUY922 but not 17-AAG, markedly inhibits cell proliferation and induces apoptosis in human prostate tumors, warranting further clinical investigation of this class of agents (13). When we assessed our patient-derived prostate explants (PDEs) for induction of the clinical pharmacodynamic biomarker of Hsp90 inhibition, Hsp70 (19), we saw equivalent induction of the biomarker with both 17-AAG and AUY922. This confirms that Hsp70 expression indicates target modulation but not anti-tumor activity of these inhibitors. These results are consistent with clinical studies wherein Hsp70 levels were not correlated with clinical response (20, 21), and highlight the urgent need for biomarkers that reflect biological activity rather than target modulation only.
The goal of the current study was to identify protein biomarkers associated with response or resistance to specific Hsp90 inhibitors in PDEs from clinical prostate tumors. Using HRM-DIA mass spectrometry, we report the identification of markers correlating with antiproliferative responses to the new-generation agent AUY922, and identify candidate predictive markers of 17-AAG responsiveness.
EXPERIMENTAL PROCEDURES
Reagents
The Hsp90 inhibitors 17-N-allylamino-17-demethoxygeldanamycin (17-AAG; National Cancer Institute, MD) and AUY922 (Novartis, now Vernalis, Winnersh, UK) were dissolved and diluted in dimethyl sulfoxide (DMSO). The effective dose of 500 nm for Hsp90 inhibitors, determined previously to induce Hsp70 and decrease levels of the androgen receptor and Akt in 8 independent tumors, was used in this study (13).
Patient-derived Explant (PDE) Culture of Prostate Tumors
Human ethical approval for this project was obtained from the Adelaide University Human Research Ethics Committee and the research ethics committees of the Royal Adelaide Hospital and St Andrew's Hospital. Fresh prostate cancer specimens were obtained with written informed consent through the Australian Prostate Cancer BioResource from men undergoing robotic radical prostatectomy at the Royal Adelaide Hospital and St Andrew's Hospital (Adelaide, South Australia). Tumors from two cohorts of patients were used for this study: a discovery cohort (n = 16) and a validation cohort (n = 30). Clinicopathological features of tumors used in each cohort are detailed in supplemental Table S1.
A single 6 mm core of tissue was obtained per patient. A longitudinal section of the entire core was taken for hematoxylin and eosin (H&E) analysis of tumor content. The remaining tissue was dissected into 1 mm3 pieces and cultured in triplicate on a presoaked gelatin sponge (Johnson and Johnson, New Brunswick, NJ) in 24-well plates containing 500 μl RPMI 1640 with 10% FBS, 1× antibiotic/antimycotic solution (Sigma, St Louis, MO), 0.01 mg/ml hydrocortisone, 0.01 mg/ml insulin (Sigma) and cultured for 48 h with 17-AAG, AUY922 (500 nm each) or DMSO vehicle alone as previously described (13). Tissues were cultured at 37 °C for 48 h, then were either formalin-fixed and paraffin-embedded or snap frozen in liquid nitrogen and stored at −80 °C until further analysis. Tissues containing ≥70% tumor content and ≥5% baseline Ki67 positivity, determined as outlined below, were included for proteomic analysis.
Immunohistochemical Staining and Microscopy
Paraffin-embedded tissues were sectioned (2 mm) on Ultraplus slides prior to hematoxylin and eosin (H&E) staining and immunohistochemical (IHC) detection of the proliferative marker, Ki67 (Agilent, M7240 antibody; 1:200 dilution, Santa Clara, CA). IHC staining was performed and tissues assessed for tumor content and Ki67 positivity in a blinded fashion as described previously (22).
Protein Preparation
Snap frozen PDEs were homogenized in 0.5 ml tubes containing 1.4 mm ceramic beads (Precellys® CK14 Lysing Kit, Bertin Instruments, Montigny-le-Bretonneux, France) and 100 μl 8 m Urea buffer (8 m Urea, 20 mm HEPES, 2.5 mm sodium pyrophosphate, 1 mm beta-glycerol phosphate, 1 mm sodium orthovanadate, 1 mm ethylenediaminetetraacetic acid, pH7.5) using the Precellys®24 tissue homogenizer (Bertin Instruments). Lysates from triplicate PDEs were combined and transferred to Eppendorf tubes, centrifuged at 10,000 rpm for 10 min, and supernatants stored at −80 °C. Total protein measurements were determined using the Bicinchoninic acid protein assay (Bio-Rad, Hercules, CA). 100 μg of protein extracts were denatured with 6 m urea in 25 mm Ammonium Bicarbonate, before reduction with 5 mm TCEP at 37 °C for 1h and alkylation with 32 mm iodoacetamide in the dark for 1 h. Alkylation was stopped by addition of 27 mm DTT. The samples were then diluted 1:10 with ammonium bicarbonate and digested with a 1:50 modified trypsin (Promega, Madison, WI) to protein weight at 37 °C for 18 h. Tryptic digests were slightly acidified with 10% TFA to pH 2–3, desalted with a C18 spin column (Thermo Fisher Scientific, Waltham, MA), and eluted with 0.1% TFA/40% ACN. Peptides were dried with a speed vacuum and resuspended in 2% ACN/0.1% FA before mass spectrometry analysis.
Mass Spectrometry Analysis
Samples were analyzed on an UltiMate 3000 RSLC nano LC system (Thermo Scientific) coupled to an LTQ-Orbitrap mass spectrometer (LTQ-Orbitrap, Thermo Scientific). Peptides for analysis were loaded via an Acclaim PepMap 100 trap column (100 μm × 2 cm, nanoViper, C18, 5 μm, 100Å, Thermo Scientific) and subsequent peptide separation was on an Acclaim PepMap RSLC analytical column (75 μm x 50 cm, nanoViper, C18, 2 μm, 100 Å, Thermo Scientific). For each liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, an estimated amount of 1 μg of peptides was loaded on the precolumn with microliter pickup. Peptides were eluted using a 2 h linear gradient of 80% acetonitrile/0.1% FA gradient flowing at 250 nL/min using mobile phase gradient of 2.5–42.5% acetonitrile. The eluting peptides were interrogated with an Orbitrap mass spectrometer. The HRM DIA method consisted of a survey scan (MS1) at 35,000 resolution (automatic gain control target 5e6 and maximum injection time of 120ms) from 400 to 1220 m/z followed by tandem MS/MS scans (MS2) through 19 overlapping DIA windows increasing from 30 to 222 Da. MS/MS scans were acquired at 35,000 resolution (automatic gain control target 3e6 and auto for injection time). Stepped collision energy was 22.5%, 25%, 27.5% and a 30 m/z isolation window. The spectra were recorded in profile type.
HRM-DIA Data Analysis
The DIA data were analyzed with Spectronaut 8, a mass spectrometer vendor-independent software from Biognosys (Schlieren, Switzerland). The default settings were used for the Spectronaut search. Retention time prediction type was set to dynamic iRT. Decoy generation was set to scrambled with no decoy limit. Interference correction on MS2 level was enabled. The false discovery rate (FDR) was set to 1% at peptide level. For generation of the spectral libraries, DDA measurements of each sample were performed. The DDA spectra were analyzed with the MaxQuant Version 1.5.2.8 analysis software using default settings. Enzyme specificity was set to Trypsin/P, minimal peptide length of 6, and up to 3 missed cleavages were allowed. Search criteria included carbamidomethylation of cysteine as a fixed modification, oxidation of methionine and acetyl (protein N terminus) as variable modifications. The mass tolerance for the precursor was 4.5 ppm and for the fragment ions was 20 ppm. The DDA files were searched against the human UniProt fasta database (v2015–08, 20,210 entries) and the Biognosys HRM calibration peptides. The identifications were filtered to satisfy FDR of 1% on peptide and protein level. The spectral library was generated in Spectronaut and normalized to iRT peptides (23). A peptide identification required at least 3 transitions in quantification. Quantification was based on the top 3 proteotypic peptide for each protein (24) and exported as an excel file with Spectronaut 8 software (23).
Statistical Rationale
Differentially expressed proteins between treatment groups were identified using repeated measure analysis of variance (ANOVA) with the Multi-Experiment Viewer analysis software (25). A raw p value <.01 and an F ratio >5 were used to define differential expression, and plots of local FDR estimates generated by LocalFDR from Anapuce R package. Based on analysis of the protein abundance data obtained from the initial cohort, an additional validation cohort of n = 30 patients was designed to have at least 95% power to detect an effect size of 1SD in a t test (2-sided alpha = 0.05) of within-sample difference between vehicle and AUY treatments.
Unpaired t test of PDE samples that responded compared with samples that had no response or showed poor response based on Ki67 proliferation was performed to determine differential proteins prior to 17-AAG treatment (raw p value <.05). A paired t test (raw p value <.01) prior and after treatment with 17-AAG was implemented with the Multi-Experiment Viewer software to determine differential proteins. All comparative tests were normalized with the Spectronaut software and exported for analysis (supplemental Fig. S1).
Functional Analysis
Functional annotation of the proteome was conducted using database for annotation, visualization, and integrated discovery (DAVID) software (26). Overrepresented functional categories among proteins enriched in each sample population were relative to a background of all identified proteins in study. Criteria for reported functional enrichment required a fold enrichment >1.5, FDR <5, and p value <.05. Experimentally verified and published protein-protein interactions from several resources including REACTOME (27, 28) and InnateDB (29) were assessed.
For gene set enrichment analyses, after voom transformation with quantile normalization of protein abundances (30), differential abundance analysis was performed with R limma package version 3.34.3 (31) using a pipeline like Law et al. (32). Protein uniprot IDs were matched to available gene IDs through Biomart query, and the gene set enrichment analysis was conducted using the camera method in R limma package (33) screening through the Molecular Signatures Database (MSigDB, version 6) (34). Pathways with Benjamini and Hochberg adjusted p values (35) less than 0.05 were accepted as statistically significant. Enrichment plots and heatmaps were generated by barcodeplot and heatmap.2 functions from limma and gplots R packages, respectively.
ELISA
TIMP1 levels in conditioned PDE media collected from the validation cohort (n = 25) were measured by the Human TIMP-1 Quantikine ELISA (R&D Systems, Minneapolis, MN) as per the manufacturer's instructions. Concentrations were extrapolated from simultaneously run standard curves. Differences between PDE samples exposed to either Hsp90 inhibitor and DMSO vehicle alone was assessed with repeat measure ANOVA with multiple comparisons using posthoc Tukey test (Prism7 software, Graphpad).
Western Blot Analysis
20 μg of remaining protein lysates from PDE samples prepared as described above were separated by 8% SDS-PAGE, transferred to PVDF membrane, and blocked for 1 h. Blots were incubated with mouse monoclonal antibody HDJ2/DNAJA1 (1:1,000, cat no. NA5–12748, Thermo Fisher Scientific) and α-tubulin (1:5000, cat no. T5168, Sigma) for 1 h and 30 min, respectively; followed by horseradish peroxidase-conjugated secondary antibody for 1 h and 30 min, respectively; developed with Western Lightening Plus ECL (Perkin Elmer); visualized with the ChemiDoc Touch Imaging System (BioRad); and quantitated using Image Lab (v5.2.1). Differences upon exposure to 17-AAG to control in two groups (Nonresponders n = 12 and Responders and Poor Responders n = 11) was assessed with one-tailed ratio paired t test (Prism7 software, Graphpad).
RESULTS
AUY922 is Significantly More Efficacious in PDE Tissues Than 17-AAG
Human PDEs cultured in the absence or presence of two Hsp90 inhibitors, 17-AAG (500 nm) or AUY922 (500 nm) for 48 h (Fig. 1A), were assessed for the biological response to these agents. Analysis was initially performed on a discovery cohort of 16 patients, and subsequently on a further validation cohort of 30 patients (supplemental Table S1). Parallel immunohistochemical analysis of the cohorts was performed to quantify treatment response by expression of the proliferative marker, Ki67. Overall, in both cohorts AUY922, but not 17-AAG, caused a statistically significant reduction in proliferation in the PDE tissues compared with the matched vehicle-treated control tissues (Fig. 1B), which confirmed our previously published findings (13).
Fig. 1.

Proliferative responses of explants cultured in the absence (DMSO) or presence of 17-AAG or AUY922. A, Workflow for drug treatment and MS analysis of patient-derived explant (PDE) cultures. B, Response of proliferation marker Ki67 to 17-AAG or AUY922. Mean and S.D. of Ki67 staining in PDEs are indicated. Statistical differences were calculated using repeat measure ANOVA with multiple comparisons using posthoc Tukey test.
The Proteome of PDEs Cultured with Specific Hsp90 Inhibitors Segregates Samples Based on Drug Exposure
HRM-DIA of PDE samples was performed to investigate alterations upon exposure to the Hsp90 inhibitors. From the combined proteome of an initial discovery cohort of 16 PDE samples cultured in the absence or presence of Hsp90 inhibitors, 4095 quantifiable proteins were identified. Proteins identified in PDE samples were functionally categorized to poly(A) RNA binding, membrane, mitochondrion, cell adhesion, ribosome, extracellular matrix and protein folding (adjusted p value < .01, proteins mapping to pathway >100, and hypergeometric fold enrichment distribution >2). Supplemental Tables S2A–S2B and S3 contain lists of identified proteins with peptides used for quantification and functional pathways, respectively. 625 differentially expressed proteins were identified between PDE samples exposed to vehicle (DMSO), either drug, or 17-AAG alone (supplemental Table S4). Principal component analysis of protein expression variability of these differential proteins segregated patient PDE samples based on Hsp90 inhibitor treatment (Fig. 2). The 328 proteins that significantly decreased upon exposure to both Hsp90 inhibitors (Fig. 3A) exhibited enrichment for processes involved in nucleoplasm, mRNA translation, RNA metabolism, and ribosome function (Table I), with AUY922 exhibiting a significant effect compared with 17-AAG (adj. p value <.0001). The 264 proteins that significantly increased upon exposure to Hsp90 inhibitors (Fig. 3B) were enriched for processes involving membrane rafts, muscle contraction, endoplasmic reticulum membrane and the TCA cycle. The most significant increase in expression was observed for heat shock 70 kDa proteins: HSPA6 (p value 9.14 × 10−12) and HSPA1A (p value 1.09 × 10−10), consistent with our previous results in prostate cancer PDE samples and their roles as markers of Hsp90 inhibition (13) (supplemental Table S4). The 33 proteins that showed a significant increase of expression with 17-AAG relative to AUY922 and control (Fig. 3C) were enriched for response to unfolded protein.
Fig. 2.

Principal component analysis with pearson correlation. Each sphere represents a patient-derived PDE sample whose location in “proteome space” is determined by protein expression variability of 625 differentially expressed proteins. Each axis represents a principal coordinate that captures a component of the variance in protein expression. PDE samples segregated based on drug exposure.
Fig. 3.

Differentially expressed proteins upon exposure to Hsp90 inhibitors. The mean (± S.D.) expression values for these proteins from 16 patients are presented. Statistical differences were calculated using repeat measure ANOVA with multiple comparisons using posthoc Tukey test (***adjusted p value ≤0.0001). A, 328 proteins with a significant decrease in expression upon treatment with both Hsp90 inhibitors. B, 264 proteins with a significant increase in expression upon treatment with both Hsp90 inhibitors. C, 33 proteins with a significant increase in expression upon treatment with 17-AAG compared with control and AUY922.
Table I. Functional analysis of differentially expressed proteins upon exposure to HSP90 inhibitors. Overrepresented functional categories among proteins enriched in each sample population (e.g., decreased and increased expression in presence of Hsp90 inhibitors, and increased expression in presence of 17-AAG) relative to all identified proteins using a permutation-based false discovery rate analysis (FDR).
| Functional term | p value | Fold enrichment | Adj. p value | FDR | |
|---|---|---|---|---|---|
| Decreased expression (+Hsp90 inhibitors) | Nucleoplasm | 1.13E-11 | 1.7 | 4.31E-09 | 1.56E-08 |
| Translational initiation | 7.51E-11 | 3.5 | 1.17E-07 | 1.25E-07 | |
| rRNA processing | 6.48E-09 | 3.1 | 5.07E-06 | 1.08E-05 | |
| Nuclear-transcribed mRNA catabolic process, nonsense-mediated decay | 7.84E-08 | 3.3 | 4.09E-05 | 1.31E-04 | |
| RNA binding | 2.60E-07 | 2.2 | 1.39E-04 | 3.78E-04 | |
| poly(A) RNA binding | 3.45E-07 | 1.6 | 9.24E-05 | 5.02E-04 | |
| Viral transcription | 6.21E-07 | 3.1 | 2.43E-04 | 1.04E-03 | |
| SRP-dependent cotranslational protein targeting to membrane | 3.38E-06 | 3.1 | 1.05E-03 | 0.01 | |
| Ribosome | 4.76E-05 | 2.5 | 4.54E-03 | 0.07 | |
| Translation | 1.02E-04 | 2.2 | 0.03 | 0.17 | |
| RNA transport | 1.99E-04 | 2.4 | 0.04 | 0.25 | |
| HIF-1 signaling pathway | 4.82E-04 | 3.3 | 0.05 | 0.60 | |
| Nucleolus | 1.07E-03 | 1.7 | 0.07 | 1.47 | |
| Cytosolic large ribosomal subunit | 1.88E-03 | 2.9 | 0.10 | 2.57 | |
| mRNA 3′-end processing | 1.94E-03 | 3.4 | 0.35 | 3.20 | |
| Nuclear speck | 2.08E-03 | 2.5 | 0.09 | 2.85 | |
| Cytosolic small ribosomal subunit | 2.44E-03 | 3.3 | 0.10 | 3.33 | |
| Formation of translation preinitiation complex | 3.16E-03 | 4.5 | 0.46 | 5.14 | |
| ATP-dependent RNA helicase activity | 3.49E-03 | 3.4 | 0.37 | 4.96 | |
| NuRD complex | 3.67E-03 | 6.9 | 0.13 | 4.96 | |
| Structural constituent of ribosome | 4.57E-03 | 1.9 | 0.34 | 6.44 | |
| cholesterol biosynthetic process | 4.95E-03 | 3.6 | 0.58 | 7.95 | |
| Translation initiation factor activity | 5.08E-03 | 3.0 | 0.32 | 7.14 | |
| Termination of RNA polymerase II transcription | 5.94E-03 | 2.9 | 0.61 | 9.47 | |
| Positive regulation of translation | 6.15E-03 | 3.5 | 0.58 | 9.78 | |
| Eukaryotic translation initiation factor 3 complex | 6.63E-03 | 4.7 | 0.21 | 8.81 | |
| Increased expression (+Hsp90 inhibitors) | Membrane raft | 5.22E-05 | 3.3 | 0.01 | 0.07 |
| Muscle contraction | 3.53E-04 | 3.9 | 0.44 | 0.59 | |
| Endoplasmic reticulum membrane | 7.28E-04 | 1.8 | 0.10 | 1.02 | |
| Tricarboxylic acid cycle | 1.10E-03 | 4.7 | 0.59 | 1.82 | |
| Extracellular matrix organization | 1.79E-03 | 2.4 | 0.62 | 2.96 | |
| Lipid particle | 2.38E-03 | 4.1 | 0.22 | 3.29 | |
| GTPase activity | 2.87E-03 | 2.2 | 0.74 | 4.01 | |
| Focal adhesion | 3.47E-03 | 1.7 | 0.22 | 4.76 | |
| GTP binding | 3.69E-03 | 1.9 | 0.58 | 5.13 | |
| Melanosome | 4.13E-03 | 2.5 | 0.22 | 5.66 | |
| Regulation of nitric-oxide synthase activity | 4.18E-03 | 6.9 | 0.82 | 6.79 | |
| Actin cytoskeleton | 4.34E-03 | 2.3 | 0.20 | 5.93 | |
| Guanyl nucleotide binding | 4.80E-03 | 10.1 | 0.52 | 6.62 | |
| Regulation of heart contraction | 4.86E-03 | 10.1 | 0.79 | 7.84 | |
| Regulation of cell death | 4.86E-03 | 10.1 | 0.79 | 7.84 | |
| Blood microparticle | 5.68E-03 | 2.3 | 0.23 | 7.69 | |
| response to cytokine | 5.95E-03 | 6.3 | 0.80 | 9.53 | |
| Increased expression (17-AAG) | Nucleus | 8.59E-04 | 1.9 | 0.09 | 0.96 |
| Cytosol | 9.36E-04 | 1.9 | 0.05 | 1.05 | |
| Response to unfolded protein | 9.48E-04 | 19.4 | 0.26 | 1.27 | |
| Cytoplasm | 1.88E-03 | 1.7 | 0.07 | 2.10 | |
| poly(A) RNA binding | 4.29E-03 | 2.3 | 0.34 | 4.62 |
Characterization of Hsp90 Inhibitor-induced Changes in an Independent Validation Cohort
To validate the identified proteomic changes in response to Hsp90 inhibition, we analyzed an additional validation cohort of 30 PDE tissues, which was powered based on the protein abundance data in the discovery cohort. From the combined proteome of the validation cohort, 5450 proteins were identified (supplemental Table S5A with peptides used for protein quantification in supplemental Table S5B). Of these proteins, 3770 were in common with the discovery cohort, equating to a marked 69.2% overlap between the two cohorts. Further analysis of the validation cohort identified 635 proteins as differentially expressed (DE) between PDE samples exposed to AUY922, 17-AAG, and vehicle (DMSO). Supplemental Tables S6 and S7 contains lists of DE proteins and functional pathways, respectively. Principal component analysis of each PDE sample's variability of expression based on 635 DE proteins in the validation cohort only segregated patient PDE samples exposed to AUY922, whereas control and treatment with 17-AAG samples were indistinguishable (Fig. 4). Overall, there were 100 DE proteins identified in both cohorts with consistent expression changes: 44 and 54 proteins exhibited decreased, and increased, expression in response to both drugs, respectively, and 2 increased in expression solely in response to 17-AAG (supplemental Tables S8A–S8G). Conservation of the proteomic response to specific Hsp90 inhibitors was further determined at a functional pathway level. Significantly enriched conserved pathways included a decrease in mRNA translation, ribosome function, and RNA metabolism whereas the tricarboxylic acid (TCA) cycle increased upon Hsp90 inhibition. The effects on ribosome function and RNA metabolism are consistent with a previous report (36). A response to unfolded protein was conserved specifically for 17-AAG (Fig. 5A) in both cohorts. The unfolded protein response (UPR) and Hsp90 chaperone system are closely related mechanisms for maintaining cellular homeostasis, and activation of the UPR has previously been reported for 17-AAG (37). The UPR can activate pro-survival signals or under persistent stress induces apoptotic cell death (38). In light of this, and our previous report that AUY922 but not 17-AAG induces apoptosis in prostate PDEs (13), it is likely that differential activation of the UPR by these agents reflects stimulation of pro-survival mechanisms by 17-AAG and apoptosis by AUY922.It is noted that an additional 34 DE proteins were identified in both cohorts that displayed inconsistency in protein expression direction change (supplemental Table S8H). Most of these proteins exhibited conflicting expression changes upon 17-AAG treatment, which is reflective of the heterogeneity in response to this drug as indicated by the contrasting grouping of 17-AAG-treated samples upon PCA (Figs. 2 and 4). Regardless, a marked conservation was observed at the functional pathway level despite the heterogeneity evident in these clinical samples.
Fig. 4.
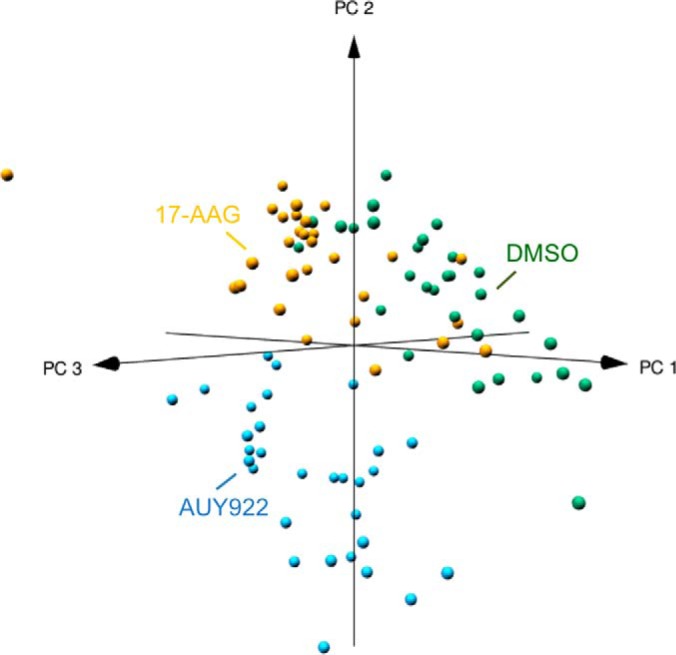
Principal component analysis with pearson correlation of PDE samples upon Hsp90 inhibitor treatment in Validation cohort (n = 30). Each sphere represents a location in “proteome space” determined by protein expression variability of 635 differentially expressed proteins. Each axis represents a principal coordinate that captures a component of the variance in protein expression. PDE samples exposed to AUY922 treatment segregate from PDE samples exposed to 17-AAG and DMSO.
Fig. 5.

Proteomic alterations in response to Hsp90 inhibitors in the Discovery and Validation cohorts exhibit concordance at the functional pathway level. A, Overrepresented functional categories in each sample population (e.g. decreased and increased expression in presence of Hsp90 inhibitors, and increased expression in presence of 17-AAG) relative to all identified proteins using a permutation-based false discovery rate analysis (FDR). Significantly altered and conserved pathways (-log10p value) > 2.2 are depicted for both cohorts. B, Barcode plots demonstrating significant alteration of the Ribosome pathway (KEGG) by AUY922 in both Discovery and Validation cohorts. C, Individual patient data on protein abundance for components of the Ribosome pathway for matched Control and AUY922-treated PDEs, in the Discovery and Validation cohorts.
Gene set enrichment analysis was performed for each agent alone compared with control and, given the substantial overlap in pathways altered by 17-AAG and AUY922, we focused on proteins selectively altered by AUY922 in the discovery and validation cohorts. Significant enrichment of key pathways was confirmed using the independent annotation methods REACTOME, KEGG, and GO (supplemental Table S9), and importantly, these were clearly conserved between the two cohorts. The most markedly enriched conserved pathways were involved in ribosomal function, gene translation, and RNA metabolism. Fig. 5B shows an example of one such function, Ribosome (KEGG), that was significantly enriched in discovery and validation cohorts, and when considered on an individual gene/protein level (Fig. 5C), despite the heterogeneity evident in these clinical samples, exhibits clear enrichment of the multiple genes annotated to this pathway.
A Signature of Proteins Selectively Altered by AUY922 in PDEs
To focus our list of proteins that were selectively altered by AUY922, a more stringent criterion for differential protein expression was applied (adjusted p value <.01). There were 12 proteins significantly altered by AUY922 in both cohorts. A group of 9 proteins significantly decreased upon AUY922 treatment compared with DMSO control (Fig. 6A and 6B), 2 proteins increased with AUY922 treatment (Fig. 6C) and 1 protein increased with both 17-AAG and AUY922 treatment (Fig. 6D). Of the 9 inhibited proteins, SERPINE1 significantly decreased with both Hsp90 inhibitors compared with DMSO but with greater potency by AUY922 (Fig. 6B). The remaining 8 proteins significantly decreased only in the presence of AUY922 and therefore represent selective response markers to this drug. Of the induced proteins, apoptosis inducer LGALS1 (39) showed a significant increase upon exposure to AUY922, whereas TMEM109 increased significantly in expression to both Hsp90 inhibitors but again with greater potency in the presence of AUY922 compared with 17-AAG (Fig. 6C The cognate Hsp70 family member, HSPA8 (40), showed a significant increase in expression upon treatment with both Hsp90 inhibitors and there was no statistical difference between AUY922 and 17-AAG (Fig. 6D). From the proteins that decreased in expression upon treatment with AUY922, three proteins are associated with the HIF pathway (i.e. SERPINE1, TFRC, and TIMP1); SERPINA3 (41), DDX21 (42), S100A2 (43), and ITGA2 (44) are associated with tumor progression; ACOT7 promotes hydrolysis of specific long chain fatty acids and is implicated in regulation of cell cycle progression (45) and CYP51A (46) is essential for cholesterol, sterol, and androgen production. Also, of note, SERPINE1 (decreased by both drugs), and both TIMP1 and SERPINA3 (decreased by AUY922 only) are all selective protease inhibitors that are associated with cancer progression and poor prognosis (47, 48) (41). The expression of these proteins in the Discovery cohort is reported in supplemental Fig. S2. TIMP1 was selected for validation as a selectively AUY922-modulated secretory protein (http://www.cbs.dtu.dk/services/SignalP/) (49). Conditioned media generated during culture of PDEs from the validation cohort (n = 25) were analyzed by ELISA and this confirmed that AUY922 significantly decreased the secretion of TIMP1 compared with DMSO vehicle and 17-AAG (Fig. 6E). Supplemental Fig. S3 shows the fold change for both inhibitors compared with DMSO vehicle for each individual PDE sample.
Fig. 6.

Validated proteins that alter expression upon treatment with Hsp90 inhibitors in both cohorts. A, 9 proteins with a significant decrease in expression upon treatment with AUY922. The mean (± S.D.) expression of these proteins in response to specific treatments in 30 PDEs from the validation cohort is presented. Statistical differences were calculated using repeat measure ANOVA with multiple comparisons using posthoc Tukey test (***adjusted p value ≤0.0001). B, Mean expression and 95% CI of 9 proteins that decrease in expression upon treatment with AUY922. t test corrected for multiple comparisons using Holm-Sidak method (***adjusted p value ≤0.0001). C, Mean expression and 95% CI of 2 proteins with a significant increase in expression upon treatment with AUY922. t test corrected for multiple comparisons using Holm-Sidak method (*adjusted p value ≤0.01, ***adjusted p value ≤0.0001). D, Mean expression and 95% CI of HSPA8 upon treatment with Hsp90 inhibitors. t test corrected for multiple comparisons using Holm-Sidak method (***adjusted p value ≤0.0001). E, ELISA analysis of conditioned explant media from PDEs from the validation cohort (n = 25). Statistical differences were calculated using repeat measure ANOVA with multiple comparisons using posthoc Tukey test (*adjusted p value ≤0.05).
PCBP3 is a Potential Predictive Biomarker for 17-AAG Response
Although treatment of PDEs with the second generation Hsp90 inhibitor AUY922 decreased cellular proliferation in the majority of PDE samples, responses to 17-AAG were more varied (Fig. 1). The latter response data provided us with the capability to determine whether our profiling platform could identify predictive biomarkers of response to a given drug. PDE samples were grouped into three categories based on proliferative response, as measured by Ki67 positivity, upon treatment with 17-AAG: (1) Responders showed a decrease in Ki67 positivity by -log2 (0.5), (2) Poor responders showed an increase in Ki67 positivity by +log2 (0.5), and (3) Non-responders showed positivity levels between the two cut-offs. From 40 PDE samples, 14 were grouped as “Responders” (Fig. 7A); 17 as “Nonresponders” (Fig. 7B); and 9 as “Poor Responders” (Fig. 7C). Statistical analysis comparing DE proteins in the 3 groups was performed first in the Discovery cohort and then in the Validation cohort. There were no differential proteins that validated in both cohorts. Responders were then compared with both Nonresponders and Poor Responders. In the Discovery cohort (n = 15) there were 202 DE proteins, and 474 DE proteins identified in the Validation cohort (n = 25) (unpaired t test, raw p value <0.05) (supplemental Table S10 and S11). Four proteins were validated to be over-expressed in Nonresponders and Poor Responders compared with Responders in both cohorts: UCHL3, S100P, PCBP3, WBP11 (Fig. 7D and 7E). PCBP3, a RNA binding protein that plays an important role in post-transcriptional control of gene expression (50), was the only protein passing statistical significance in both cohorts after p value adjustment.
Fig. 7.

Identification of predictive biomarkers of 17-AAG response. A, Ki67 levels for PDE samples that responded to 17-AAG treatment (decrease in Ki67 by -log2(0.5)). B, Ki67 levels for PDE samples unresponsive to 17-AAG treatment (no significant change in Ki67 positivity). C, Ki67 levels for PDE samples with a poor response to 17-AAG treatment (increase in Ki67 by +log2(0.5)). D, Mean expression and 95% CI of 4 proteins with significantly increased expression in discovery cohort “Nonresponder” and “Poor Responder” PDEs (n = 10) compared with “Responders” (n = 5). t test corrected for multiple comparisons using Holm-Sidak method (*adjusted p value ≤0.05, **adjusted p value ≤0.01). E, Mean expression and 95% CI of 4 proteins with significantly increased expression in validation cohort “Nonresponder” and “Poor Responder” PDEs (n = 16) compared with “Responders” (n = 9). t test corrected for multiple comparisons using Holm-Sidak method (*adjusted p value ≤0.05).
17-AAG Elicits Different Response Mechanisms in PDE Samples Grouped as Responders, Nonresponders, or Poor responders
To gain insight into potential mechanisms of sensitivity and resistance to 17-AAG, perturbations of the proteome were characterized before and after treatment with 17-AAG in the 3 response groups. DE proteins were identified in the Discovery cohort (n = 15) (supplemental Table S12) and the Validation cohort (n = 25) (supplemental Table S13) in the 3 response groups (paired t test, p value <0.05). Seventeen proteins increased in expression whereas 4 proteins decreased in expression in the Responders in both cohorts (Fig. 8A–8B). Functional pathway analysis of proteins with increased expression upon treatment with 17-AAG highlighted regulation of the neuron apoptotic pathway, suggesting a mechanism of sensitivity in the Responders (Table II). Next, to investigate mechanisms of resistance, differential proteins in PDE samples of Nonresponders and Poor responders were interrogated. Ten proteins increased in expression whereas 13 proteins decreased in expression in Nonresponders (Fig. 8C–8D). Proteins that increased expression upon treatment to 17-AAG demonstrated enrichment for negative regulation of protein localization to mitochondrion, response to DNA damage, and response to unfolded protein (Table II). The extracellular space and phosphoprotein binding were pathways associated with proteins that decreased in expression upon 17-AAG treatment in this group. Altogether, these pathways highlight potential resistance mechanisms associated with a cytostatic phenotype. Finally, nine proteins increased in expression in “Poor responders” (Fig. 8E) upon treatment with 17-AAG and were enriched for the axonogenesis and metabolic pathways (Table II), highlighting potential mechanisms that contribute to tumor growth in the presence of 17-AAG. Further validation of DNAJA1 expression was performed to confirm its potential role in resistance mechanisms to 17-AAG in Nonresponders. Western blot analysis confirmed a significant increase in expression of DNAJA1 in 10/12 Nonresponders (one tailed, ratio paired t test p value 0.001) (Fig. 9A–9B) whereas Responders and Poor responders showed no significant increase in expression upon exposure to 17-AAG (supplemental Fig S4, Fig. 9C).
Fig. 8.

Identification of potential mechanisms of sensitivity and resistance to 17-AAG treatment based on changes in protein expression in responders, poor responders, and non-responders. The plots indicate mean (± S.D.) of proteins that showed significant alteration upon treatment with 17-AAG (paired t test, *p value ≤0.01, **p value ≤0.001, ***p value ≤.0001). A, 17 proteins with increased expression upon 17-AAG treatment in validation cohort “Responder” PDE samples (n = 9). B, 4 proteins with decreased expression upon 17-AAG treatment in validation cohort “Responder” PDE samples (n = 9). C, 10 proteins with increased expression upon 17-AAG treatment in validation cohort “Non-responder” PDE samples (n = 12). D, 13 proteins with decreased expression upon 17-AAG treatment in validation cohort “Non-responder” PDE samples (n = 12). E, 9 proteins with increased expression upon 17-AAG treatment in Validation cohort “Poor responder” PDE samples (n = 4).
Table II. Functional analysis of differentially expressed proteins upon exposure to Hsp90 inhibitor 17-AAG in responder, poor responder, and nonresponder PDE samples.
| Functional term | p value | Proteins | Fold enrichment | FDR | |
|---|---|---|---|---|---|
| Increased expression in Responders (+17-AAG) | Regulation of neuron apoptotic process | 0.02 | PARK7, NPMI | 96.61 | 21.89 |
| Protein homodimerization activity | 0.04 | PARK7, AOC3, GSTMI, NPMI | 4.70 | 38.39 | |
| Protein heterodimerization activity | 0.05 | AOC3, NPMI, PPP2RIA | 8.05 | 41.76 | |
| Copper ion binding | 0.07 | PARK7, AOC3 | 24.15 | 59.08 | |
| Increased expression in Nonresponders (+17-AAG) | Negative regulation of establishment of protein localization to mitochondrion | 0.00 | DNAJA1, HSPH1 | 772.86 | 2.29 |
| DNA damage response, detection of DNA damage | 0.03 | DNAJA1, RPA3 | 59.45 | 26.05 | |
| Response to unfolded protein | 0.03 | DNAJA1, HSPH1 | 55.20 | 27.76 | |
| Drug binding | 0.03 | SIGMAR1, PYGL | 50.31 | 26.58 | |
| Pyridoxal phosphate binding | 0.04 | MARC2, PYGL | 46.84 | 28.25 | |
| Regulation of cellular response to heat | 0.07 | RPA3, HSPH1 | 25.34 | 50.86 | |
| Decreased expression in Non-responders (+17-AAG) | Extracellular space | 0.01 | QSOX2, SERPINB4, PLAT, KLK3, SFN | 4.76 | 11.14 |
| Phosphoprotein binding | 0.04 | PLAT, SFN | 41.17 | 31.65 | |
| Increased expression in Poor responders (+17-AAG) | Axonogenesis | 0.05 | FLOT1, CNP | 36.43 | 40.67 |
| Metabolic pathways | 0.09 | ALDH9A1, SUCLG1, MTR, PLA2G7 | 2.86 | 50.82 |
Fig. 9.

Western blot analysis of DNAJA1. Statistical differences were calculated using one-tailed ratio t test for paired PDE samples exposed to DMSO and 17-AAG. A, Log2(ratio 17-AAG/DMSO) for Non-responders (n = 12), Responders (n = 8), and Poor responders (n = 3). B, Log2(AUC) of PDE samples of Non-responders (n = 12) exposed to DMSO and 17-AAG. C, Log2(AUC) of PDE samples of Responders (n = 8) and Poor responders (n = 3) exposed to DMSO and 17-AAG.
DISCUSSION
The identification of informative biomarkers in the preclinical phase that can be integrated into clinical studies and decision making is increasingly being recognized as key to successful drug development and the most ethical way to minimize harm and maximize patient benefit (51). However, the clinical validation and implementation of biomarker discovery efforts has, to date, been limited. The rationale for this roadblock is multifactorial (52), but a major player is the short-comings of cell-based models. The majority of preclinical studies of prostate cancers are based on immortalized cell lines that are either grown in cultured dishes or xenografted onto mice (53). Although these models have provided valuable breakthroughs in our current understanding of prostate cancer, they represent a limited spectrum of disease and do not capture the unique and dynamic interplay of a heterogeneous population of cells within the tumor microenvironment. The tumor microenvironment encompasses the tumor epithelial cells and the surrounding stroma that encompasses fibroblasts, macrophages, lymphocytes, and components from the extracellular matrix (54). Stromal-epithelial signaling has been shown to be integral in prostate cancer metastasis and response to therapeutics (55, 56). Although prostatic cell lines have been subcutaneously xenografted onto the flanks of an immunocompromised murine host in an attempt to recapitulate stromal-epithelial signaling, the outcomes have not been predictive of clinical efficacy (57, 58). The use of primary human tumors for grafting is a promising technique to circumvent the drawbacks of cell-based xenograft models, but this methodology is technically challenging and aggressive tumors are needed to establish the model (59, 60). Despite these limitations, cell line models have remained the primary approach for preclinical assessment of therapeutic agents.
In this study we used our patient-derived explants as a more clinically relevant discovery model, capable of retaining inter-patient disease heterogeneity and the tumor microenvironment, to assess the efficacy of investigational agents (i.e. specific Hsp90 inhibitors) in prostate cancer. Notwithstanding the intra- and inter-tumoral heterogeneity inherent to primary tumors, our ex vivo model system was not only able to robustly identify signaling pathways that may be suitable for monitoring drug effectiveness, but also gave potential insight to mechanisms that lead to cytotoxic, cytostatic, and tumor progressive phenotypes induced by a drug. In addition, using posteriori data based on the Ki67 proliferation marker, potential predictive biomarkers have been identified that could be indicative of whether a patient responds to a given drug prior to treatment. Our results provide robust evidence that an ex vivo patient-derived explant model system in the cancer setting can be exploited to obtain a clinically translatable understanding of functional targets, variability and likelihood of patient response to clinical agents, and potential mechanisms of resistance. Although AUY922 has not yet been clinically evaluated in prostate cancer, the markers identified may be validated in other cancers that are being treated with Hsp90 inhibitors, and our explant-based approach to biomarker discovery will likely be applicable to other new investigational agents.
Extensive efforts have been invested into characterizing the proteome upon Hsp90 inhibition. Studies in yeast (61, 62), human cell lines of different origins (36, 63) including Jurkat cells (18) indicate that 58% (86/148 protein responses) are observed in one or more of the cell lines. These results capture a potential conserved signature that is inherent to Hsp90 inhibition, while also highlighting that some responses are specific to cell lines. We attempted to compare our results to studies performed in cancer cell lines. In the Discovery cohort, we found that AUY922 and 17-AAG share a highly conserved proteomic fingerprint of Hsp90 inhibition, and that most expression differences in these proteins are related to the potencies of the two drugs (Fig. 3). This is consistent with other comparative studies of AUY922 and 17-AAG in pancreatic, colorectal, and leukemia cancer cell lines (16, 18). Heat-shock cognate 70 protein, HSPA8, significantly increased upon treatment with both 17-AAG and AUY922 compared with control. HSPA8 regulates the heat shock response (HSR) and maintains homeostasis (64). The induction of the heat shock response is established as a bona fide component of the molecular signature of Hsp90 inhibition observed in numerous cancer cell line studies (18, 65–67) and is used to indicate target modulation in clinical trials (68, 69). AUY922 induces HSPA8 expression in breast cancer cell lines (17) whereas HSPA8 induction by 17-AAG has been observed in numerous cancer cell lines (18, 70–74), human ovarian cancer xenograft murine models (75), and patient tumor samples (68, 76). The Hsp70 proteins contribute to tumor cell survival though anti-apoptotic functions (77), and concurrent inhibition of both HSPA8 and inducible HSPA2 enhanced the clinical potency of 17-AAG in colon cancer (40).
Comparing data from both the Discovery and Validation cohorts using a stringent criterion for differential expression focused the list to 8 proteins that showed a selective and significant decrease with AUY922 treatment. Among the 8 proteins that were specifically decreased by AUY922 treatment in both cohorts were two components of the HIF-1 pathway, TFRC and TIMP1. Interestingly, hypoxia-related biomarkers are associated with poor prognosis in prostate cancer (78–81) and HIF-1 interacts with the AR to increase transcriptional activity and enhance PSA expression (82, 83). Consequently, although the impact of AUY922 on the HIF-1 pathway requires further characterization, treatment with this drug may present a novel strategy to target this pathway. TIMP1 is a secreted glycoprotein that inhibits matrix metalloproteinase (MMP) activity (84) and has been linked to (1) inhibition of tumor cell invasion by preventing extracellular matrix remodeling (85) and (2) promotion of tumor progression in a MMP-independent manner by inhibiting apoptosis and stimulating cancer cell growth (86, 87) in prostate cancer cell lines. Elevated TIMP1 levels in plasma predict worse survival outcome in metastatic CRPC patients to further support the pro-tumorigenic role of TIMP1 (88). Broad spectrum MMP inhibitors have been developed and tested in clinical trials resulting in disappointing outcomes, most likely because of the lack of understanding of the pro- and/or anti-tumor activities of distinct MMPs (89–91). The selective impact of AUY922 on several other proteins might also lead to therapeutic benefit. These proteins include ITGA2 and SERPINA3, both associated with tumor progression, and the cytochrome P450 enzyme, CYP51A1. Importantly, CYP51A1 first demethylates lanosterol as an early step in cholesterol biosynthesis, with cholesterol being metabolized by other enzymes including CYP17A1 to mediate androgen production. Abiraterone, a CYP17A1 antagonist, is commonly prescribed in patients to treat CRPC (92–94), and the identification of CYP51A1 as a AUY922 target identifies another potential strategy to target androgen biosynthesis.
A diverse proliferative response to 17-AAG was observed in the PDE samples, allowing us to identify protein biomarkers that predict response to this drug in individual patient tumors. PCPB3 was the only protein that was significantly overexpressed in non- and poor responders compared with responders in both cohorts. Interestingly, PCBP3 was identified, along with 27 other genes, as a predictive marker for clinical recurrence in early stage prostate cancer (95), and a paralog of PCBP3, PCBP1, is implicated as a regulator of cancer stem cells in DU145 and LNCAP prostate cell lines (96). Consequently, it will be important to characterize whether there is a direct role for this protein family in modulating response to specific drugs, and the underlying mechanisms. In addition, the heterogenous response to 17-AAG also enabled us to gain insights into potential drug resistance mechanisms. Here, a key protein identified to increase expression in the presence of 17-AAG in Nonresponders and was further validated was DNAJA1, which acts as a HSPA8 co-chaperone (97) and importantly, protects against apoptosis by inhibiting the stress-induced translocation of Bax from the cytosol to the mitochondria (98).
CONCLUSION
This study provides proof of concept for the utilization of MS-based proteomic profiling of patient-derived explant tissues for the identification of clinically-relevant response and predictive biomarkers to specific drugs and drug resistance mechanisms. Specifically, it provides this information in the context of certain Hsp90 inhibitors, which may help guide the more effective clinical use of these drugs.
Data Availability
The MS proteomic data have been deposited to the Mass spectrometry Interactive Virtual Environment (MassIVE) consortium (http://www.massive.ucsd.edu/ProteoSAFe/datasets.jsp) with data set identifier: MSV000082244.
Supplementary Material
Acknowledgments
We thank the Monash Biomedical Proteomics Facility for technical assistance.
Footnotes
* L.M.B., R.J.D., L.G.H., and M.M.C. acknowledge grant support from Cancer Australia/Prostate Cancer Foundation of Australia (ID 1050880 and 1085471). M.M.C. was supported by a Young Investigator Award (ID 0412) from the Prostate Cancer Foundation of Australia; L.M.B. is supported by a Future Fellowship from the Australian Research Council (FT130101004); and R.J.D. by a National Health and Medical Research Council Fellowship (1058540). This work was also supported by an EMBL Australia Group Leader award to D.J.L.
1 The abbreviations used are:
- Hsp90
- Heat shock protein 90
- 17-AAG
- 17-N-allylamino-17-demethoxygeldanamycin
- ADT
- Androgen deprivation therapy
- AGC
- Automatic gain control
- ANOVA
- Analysis of variance
- CRPC
- Castrate-resistant prostate cancer
- DIA
- Data Independent Acquisition
- FA
- Formic acid
- FBS
- Fetal bovine serum
- HCL
- Hydrochloric acid
- HIF-1
- Hypoxia-inducible factors −1
- HRM
- Hyper reaction monitoring
- iRT
- Retention-time-normalized
- IT
- Ion trap
- MS/MS
- Tandem mass spectrometry
- NH4OH
- Ammonium hydroxide
- PCA
- Principal component analysis
- PDE
- Patient-derived prostate cancer explant
- RPMI
- Roswell Park Memorial Institute.
REFERENCES
- 1. Jemal A., Siegel R., Xu J., and Ward E. (2010) Cancer statistics, 2010. Cancer J. Clin. 60, 277–300. doi: caac.20073 [pii] 10 3322/caac.20073 [DOI] [PubMed] [Google Scholar]
- 2. Johnson J. I., Decker S., Zaharevitz D., Rubinstein L. V., Venditti J. M., Schepartz S., Kalyandrug S., Christian M., Arbuck S., Hollingshead M., and Sausville E. A. (2001) Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 84, 1424–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Voskoglou-Nomikos T., Pater J. L., and Seymour L. (2003) Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 9, 4227–4239 [PubMed] [Google Scholar]
- 4. Kamb A., Wee S., and Lengauer C. (2007) Why is cancer drug discovery so difficult? Nat. Rev. Drug Discovery 6, 115–120 [DOI] [PubMed] [Google Scholar]
- 5. Scher H. I., Halabi S., Tannock I., Morris M., Sternberg C. N., Carducci M. A., Eisenberger M. A., Higano C., Bubley G. J., Dreicer R., Petrylak D., Kantoff P., Basch E., Kelly W. K., Figg W. D., Small E. J., Beer T. M., Wilding G., Martin A., and Hussain M. (2008) Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J. Clin. Oncol. 26, 1148–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tan D. S., Thomas G. V., Garrett M. D., Banerji U., de Bono J. S., Kaye S. B., and Workman P. (2009) Biomarker-driven early clinical trials in oncology: a paradigm shift in drug development. Cancer J. 15, 406–420 [DOI] [PubMed] [Google Scholar]
- 7. Adams D. J. (2012) The Valley of Death in anticancer drug development: a reassessment. Trends Pharmacol. Sci. 33, 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cardillo M. R., and Ippoliti F. (2006) IL-6, IL-10 and HSP-90 expression in tissue microarrays from human prostate cancer assessed by computer-assisted image analysis. Anticancer Res. 26, 3409–3416 [PubMed] [Google Scholar]
- 9. Solit D. B., Zheng F. F., Drobnjak M., Munster P. N., Higgins B., Verbel D., Heller G., Tong W., Cordon-Cardo C., Agus D. B., Scher H. I., and Rosen N. (2002) 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin. Cancer Res. 8, 986–993 [PubMed] [Google Scholar]
- 10. Heath E. I., Hillman D. W., Vaishampayan U., Sheng S., Sarkar F., Harper F., Gaskins M., Pitot H. C., Tan W., Ivy S. P., Pili R., Carducci M. A., Erlichman C., and Liu G. (2008) A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 14, 7940–7946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trepel J., Mollapour M., Giaccone G., and Neckers L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eccles S. A., Massey A., Raynaud F. I., Sharp S. Y., Box G., Valenti M., Patterson L., de Haven Brandon A., Gowan S., Boxall F., Aherne W., Rowlands M., Hayes A., Martins V., Urban F., Boxall K., Prodromou C., Pearl L., James K., Matthews T. P., Cheung K. M., Kalusa A., Jones K., McDonald E., Barril X., Brough P. A., Cansfield J. E., Dymock B., Drysdale M. J., Finch H., Howes R., Hubbard R. E., Surgenor A., Webb P., Wood M., Wright L., and Workman P. (2008) NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68, 2850–2860 [DOI] [PubMed] [Google Scholar]
- 13. Centenera M. M., Gillis J. L., Hanson A. R., Jindal S., Taylor R. A., Risbridger G. P., Sutherland P. D., Scher H. I., Raj G. V., Knudsen K. E., Yeadon T., Australian Prostate Cancer B., Tilley W. D., and Butler L. M. (2012) Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin. Cancer Res. 18, 3562–3570 [DOI] [PubMed] [Google Scholar]
- 14. Stingl L., Stuhmer T., Chatterjee M., Jensen M. R., Flentje M., and Djuzenova C. S. (2010) Novel HSP90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cell-cycle impairment, increased DNA damage and repair protraction. Br. J. Cancer 102, 1578–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garon E. B., Finn R. S., Hamidi H., Dering J., Pitts S., Kamranpour N., Desai A. J., Hosmer W., Ide S., Avsar E., Jensen M. R., Quadt C., Liu M., Dubinett S. M., and Slamon D. J. (2013) The HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growth. Mol. Cancer Ther. 12, 890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mayor-Lopez L., Tristante E., Carballo-Santana M., Carrasco-Garcia E., Grasso S., Garcia-Morales P., Saceda M., Lujan J., Garcia-Solano J., Carballo F., de Torre C., and Martinez-Lacaci I. (2014) Comparative study of 17-AAG and NVP-AUY922 in pancreatic and colorectal cancer cells: are there common determinants of sensitivity? Transl. Oncol. 7, 590–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jensen M. R., Schoepfer J., Radimerski T., Massey A., Guy C. T., Brueggen J., Quadt C., Buckler A., Cozens R., Drysdale M. J., Garcia-Echeverria C., and Chene P. (2008) NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 10, R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Voruganti S., Lacroix J. C., Rogers C. N., Rogers J., Matts R. L., and Hartson S. D. (2013) The anticancer drug AUY922 generates a proteomics fingerprint that is highly conserved among structurally diverse Hsp90 inhibitors. J. Proteome Res. 12, 3697–3706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dakappagari N., Neely L., Tangri S., Lundgren K., Hipolito L., Estrellado A., Burrows F., and Zhang H. (2010) An investigation into the potential use of serum Hsp70 as a novel tumour biomarker for Hsp90 inhibitors. Biomarkers 15, 31–38 [DOI] [PubMed] [Google Scholar]
- 20. Kummar S., Gutierrez M. E., Gardner E. R., Chen X., Figg W. D., Zajac-Kaye M., Chen M., Steinberg S. M., Muir C. A., Yancey M. A., Horneffer Y. R., Juwara L., Melillo G., Ivy S. P., Merino M., Neckers L., Steeg P. S., Conley B. A., Giaccone G., Doroshow J. H., and Murgo A. J. (2010) Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur. J. Cancer 46, 340–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramanathan R. K., Egorin M. J., Erlichman C., Remick S. C., Ramalingam S. S., Naret C., Holleran J. L., TenEyck C. J., Ivy S. P., and Belani C. P. (2010) Phase I pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. J. Clin. Oncol. 28, 1520–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Armstrong H. K., Koay Y. C., Irani S., Das R., Nassar Z. D., Australian Prostate Cancer B., Selth L. A., Centenera M. M., McAlpine S. R., and Butler L. M. (2016) A novel class of Hsp90 C-terminal modulators have pre-clinical efficacy in prostate tumor cells without induction of a heat shock response. Prostate 76, 1546–1559 [DOI] [PubMed] [Google Scholar]
- 23. Bruderer R., Bernhardt O. M., Gandhi T., Miladinovic S. M., Cheng L. Y., Messner S., Ehrenberger T., Zanotelli V., Butscheid Y., Escher C., Vitek O., Rinner O., and Reiter L. (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collins B. C., Hunter C. L., Liu Y., Schilling B., Rosenberger G., Bader S. L., Chan D. W., Gibson B. W., Gingras A. C., Held J. M., Hirayama-Kurogi M., Hou G., Krisp C., Larsen B., Lin L., Liu S., Molloy M. P., Moritz R. L., Ohtsuki S., Schlapbach R., Selevsek N., Thomas S. N., Tzeng S. C., Zhang H., and Aebersold R. (2017) Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 8, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saeed A. I., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., Thiagarajan M., Sturn A., Snuffin M., Rezantsev A., Popov D., Ryltsov A., Kostukovich E., Borisovsky I., Liu Z., Vinsavich A., Trush V., and Quackenbush J. (2003) TM4: a free, open-source system for microarray data management and analysis. BioTechniques 34, 374–378 [DOI] [PubMed] [Google Scholar]
- 26. Dennis G. Jr, Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., and Lempicki R. A. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 27. Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H and D'Eustachio P (2016) The reactome pathway knowledgebase. Nucleic Acids Res. 44, D481–D487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Milacic M., Haw R., Rothfels K., Wu G., Croft D., Hermjakob H., D'Eustachio P., and Stein L. (2012) Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers 4, 1180–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lynn D. J., Chan C., Naseer M., Yau M., Lo R., Sribnaia A., Ring G., Que J., Wee K., Winsor G. L., Laird M. R., Breuer K., Foroushani A. K., Brinkman F. S., and Hancock R. E. (2010) Curating the innate immunity interactome. BMC Syst. Biol. 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Law C. W., Chen Y., Shi W., and Smyth G. K. (2014) voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Law C. W., Alhamdoosh M., Su S., Smyth G. K., and Ritchie M. E. (2016) RNA-seq analysis is easy as 1–2-3 with limma, Glimma and edgeR. F1000Res 5, 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu D., and Smyth G. K. (2012) Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 40, e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liberzon A. (2014) A description of the Molecular Signatures Database (MSigDB) Web site. Methods Mol. Biol. 1150, 153–160 [DOI] [PubMed] [Google Scholar]
- 35. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statistical Soc. 57, 289–300 [Google Scholar]
- 36. Wu Z., Gholami A. M., and Kuster B. (2012) Systematic identification of the HSP90 candidate regulated proteome. Mol. Cell. Proteomics 11, M111 016675. doi: 10.1074/mcp.M111.016675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davenport E. L., Moore H. E., Dunlop A. S., Sharp S. Y., Workman P., Morgan G. J., and Davies F. E. (2007) Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 110, 2641–2649 [DOI] [PubMed] [Google Scholar]
- 38. Storm M., Sheng X., Arnoldussen Y. J., and Saatcioglu F. (2016) Prostate cancer and the unfolded protein response. Oncotarget 7, 54051–54066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He J., and Baum L. G. (2004) Presentation of galectin-1 by extracellular matrix triggers T cell death. J. Biol. Chem. 279, 4705–4712 [DOI] [PubMed] [Google Scholar]
- 40. Powers M. V., Clarke P. A., and Workman P. (2008) Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14, 250–262 [DOI] [PubMed] [Google Scholar]
- 41. Zhou J., Cheng Y., Tang L., Martinka M., and Kalia S. (2016) Up-regulation of SERPINA3 correlates with high mortality of melanoma patients and increased migration and invasion of cancer cells. Oncotarget doi: 10.18632/oncotarget.9409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y., Baysac K. C., Yee L. F., Saporita A. J., and Weber J. D. (2014) Elevated DDX21 regulates c-Jun activity and rRNA processing in human breast cancers. Breast Cancer Res. 16, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kwon Y. W., Chang I. H., Kim K. D., Kim Y. S., Myung S. C., Kim M. K., and Kim T. H. (2010) Significance of S100A2 and S100A4 expression in the progression of prostate adenocarcinoma. Korean J. Urol. 51, 456–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong K. F., Liu A. M., Hong W., Xu Z., and Luk J. M. (2016) Integrin alpha2beta1 inhibits MST1 kinase phosphorylation and activates Yes-associated protein oncogenic signaling in hepatocellular carcinoma. Oncotarget 7, 77683–77695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jung S. H., Lee H. C., Hwang H. J., Park H. A., Moon Y. A., Kim B. C., Lee H. M., Kim K. P., Kim Y. N., Lee B. L., Lee J. C., Ko Y. G., Park H. J., and Lee J. S. (2017) Acyl-CoA thioesterase 7 is involved in cell cycle progression via regulation of PKCzeta-p53-p21 signaling pathway. Cell Death Dis. 8, e2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stromstedt M., Rozman D., and Waterman M. R. (1996) The ubiquitously expressed human CYP51 encodes lanosterol 14 alpha-demethylase, a cytochrome P450 whose expression is regulated by oxysterols. Arch. Biochem. Biophys. 329, 73–81 [DOI] [PubMed] [Google Scholar]
- 47. Croucher D. R., Saunders D. N., Lobov S., and Ranson M. (2008) Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat. Rev. Cancer 8, 535–545 [DOI] [PubMed] [Google Scholar]
- 48. Jackson H. W., Defamie V., Waterhouse P., and Khokha R. (2017) TIMPs: versatile extracellular regulators in cancer. Nat. Rev. Cancer 17, 38–53 [DOI] [PubMed] [Google Scholar]
- 49. Nielsen H. (2017) Predicting secretory proteins with SignalP. Methods Mol. Biol. 1611, 59–73 [DOI] [PubMed] [Google Scholar]
- 50. Kang D. H., Song K. Y., Choi H. S., Law P. Y., Wei L. N., and Loh H. H. (2012) Novel dual-binding function of a poly (C)-binding protein 3, transcriptional factor which binds the double-strand and single-stranded DNA sequence. Gene 501, 33–38 [DOI] [PubMed] [Google Scholar]
- 51. Ferraldeschi R., Attard G., and de Bono J. S. (2013) Novel strategies to test biological hypotheses in early drug development for advanced prostate cancer. Clin. Chem. 59, 75–84 [DOI] [PubMed] [Google Scholar]
- 52. Adams DJ. (2012) The Valley of Death in anticancer drug development: a reassessment. Trends Pharmacol. Sci. 33, 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Centenera M. M., Raj G. V., Knudsen K. E., Tilley W. D., and Butler L. M. (2013) Ex vivo culture of human prostate tissue and drug development. Nat. Rev. Urol. 10, 483–487 [DOI] [PubMed] [Google Scholar]
- 54. Matrisian L. M., Cunha G. R., and Mohla S. (2001) Epithelial-stromal interactions and tumor progression: meeting summary and future directions. Cancer Res. 61, 3844–3846 [PubMed] [Google Scholar]
- 55. Swartz M. A., Iida N., Roberts E. W., Sangaletti S., Wong M. H., Yull F. E., Coussens L. M., and DeClerck Y. A. (2012) Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res. 72, 2473–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ricke E. A., Williams K., Lee Y. F., Couto S., Wang Y., Hayward S. W., Cunha G. R., and Ricke W. A. (2012) Androgen hormone action in prostatic carcinogenesis: stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis 33, 1391–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Johnson J. I., Decker S., Zaharevitz D., Rubinstein L. V., Venditti J. M., Schepartz S., Kalyandrug S., Christian M., Arbuck S., Hollingshead M., and Sausville E. A. (2001) Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 84, 1424–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Voskoglou-Nomikos T., Pater J. L., and Seymour L. (2003) Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 9, 4227–4239 [PubMed] [Google Scholar]
- 59. Toivanen R., Taylor R. A., Pook D. W., Ellem S. J., and Risbridger G. P. (2012) Breaking through a roadblock in prostate cancer research: an update on human model systems. J. Steroid Biochem. Mol. Biol. 131, 122–131 [DOI] [PubMed] [Google Scholar]
- 60. Lopez-Barcons LA. (2010) Human prostate cancer heterotransplants: a review on this experimental model. Asian J. Androl. 12, 509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McClellan A. J., Xia Y., Deutschbauer A. M., Davis R. W., Gerstein M., and Frydman J. (2007) Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131, 121–135 [DOI] [PubMed] [Google Scholar]
- 62. Zhao R., Davey M., Hsu Y. C., Kaplanek P., Tong A., Parsons A. B., Krogan N., Cagney G., Mai D., Greenblatt J., Boone C., Emili A., and Houry W. A. (2005) Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 120, 715–727 [DOI] [PubMed] [Google Scholar]
- 63. Hartson S. D., and Matts R. L. (2012) Approaches for defining the Hsp90-dependent proteome. Biochim. Biophys. Acta 1823, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Korfanty J., Stokowy T., Widlak P., Gogler-Piglowska A., Handschuh L., Podkowinski J., Vydra N., Naumowicz A., Toma-Jonik A., and Widlak W. (2014) Crosstalk between HSF1 and HSF2 during the heat shock response in mouse testes. Int. J. Biochem. Cell Biol. 57, 76–83 [DOI] [PubMed] [Google Scholar]
- 65. Zou J., Guo Y., Guettouche T., Smith D. F., and Voellmy R. (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480 [DOI] [PubMed] [Google Scholar]
- 66. Kim H. R., Kang H. S., and Kim H. D. (1999) Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life 48, 429–433 [DOI] [PubMed] [Google Scholar]
- 67. Clarke P. A., Hostein I., Banerji U., Stefano F. D., Maloney A., Walton M., Judson I., and Workman P. (2000) Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the hsp90 molecular chaperone. Oncogene 19, 4125–4133 [DOI] [PubMed] [Google Scholar]
- 68. Banerji U., O'Donnell A., Scurr M., Pacey S., Stapleton S., Asad Y., Simmons L., Maloney A., Raynaud F., Campbell M., Walton M., Lakhani S., Kaye S., Workman P., and Judson I. (2005) Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 23, 4152–4161 [DOI] [PubMed] [Google Scholar]
- 69. Maloney A., Clarke P. A., and Workman P. (2003) Genes and proteins governing the cellular sensitivity to HSP90 inhibitors: a mechanistic perspective. Curr. Cancer Drug Targets 3, 331–341 [DOI] [PubMed] [Google Scholar]
- 70. Ruckova E., Muller P., Nenutil R., and Vojtesek B. (2012) Alterations of the Hsp70/Hsp90 chaperone and the HOP/CHIP co-chaperone system in cancer. Cell. Mol. Biol. Lett. 17, 446–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Born E. J., Hartman S. V., and Holstein S. A. (2013) Targeting HSP90 and monoclonal protein trafficking modulates the unfolded protein response, chaperone regulation and apoptosis in myeloma cells. Blood Cancer J. 3, e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim S. H., Kang J. G., Kim C. S., Ihm S. H., Choi M. G., Yoo H. J., and Lee S. J. (2015) The effect of 17-allylamino-17-demethoxygeldanamycin alone or in combination with paclitaxel on anaplastic thyroid carcinoma cells. Endocrine 48, 886–893 [DOI] [PubMed] [Google Scholar]
- 73. Maloney A., Clarke P. A., Naaby-Hansen S., Stein R., Koopman J. O., Akpan A., Yang A., Zvelebil M., Cramer R., Stimson L., Aherne W., Banerji U., Judson I., Sharp S., Powers M., deBilly E., Salmons J., Walton M., Burlingame A., Waterfield M., and Workman P. (2007) Gene and protein expression profiling of human ovarian cancer cells treated with the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 67, 3239–3253 [DOI] [PubMed] [Google Scholar]
- 74. Hostein I., Robertson D., DiStefano F., Workman P., and Clarke P. A. (2001) Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 61, 4003–4009 [PubMed] [Google Scholar]
- 75. Banerji U., Walton M., Raynaud F., Grimshaw R., Kelland L., Valenti M., Judson I., and Workman P. (2005) Pharmacokinetic-pharmacodynamic relationships for the heat shock protein 90 molecular chaperone inhibitor 17-allylamino, 17-demethoxygeldanamycin in human ovarian cancer xenograft models. Clin. Cancer Res. 11, 7023–7032 [DOI] [PubMed] [Google Scholar]
- 76. Goetz M. P., Toft D., Reid J., Ames M., Stensgard B., Safgren S., Adjei A. A., Sloan J., Atherton P., Vasile V., Salazaar S., Adjei A., Croghan G., and Erlichman C. (2005) Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J. Clin. Oncol. 23, 1078–1087 [DOI] [PubMed] [Google Scholar]
- 77. Mosser D. D., and Morimoto R. I. (2004) Molecular chaperones and the stress of oncogenesis. Oncogene 23, 2907–2918 [DOI] [PubMed] [Google Scholar]
- 78. Ranasinghe W. K., Xiao L., Kovac S., Chang M., Michiels C., Bolton D., Shulkes A., Baldwin G. S., and Patel O. (2013) The role of hypoxia-inducible factor 1alpha in determining the properties of castrate-resistant prostate cancers. PLoS ONE 8, e54251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ragnum H. B., Vlatkovic L., Lie A. K., Axcrona K., Julin C. H., Frikstad K. M., Hole K. H., Seierstad T., and Lyng H. (2015) The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br. J. Cancer 112, 382–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Milosevic M., Warde P., Menard C., Chung P., Toi A., Ishkanian A., McLean M., Pintilie M., Sykes J., Gospodarowicz M., Catton C., Hill R. P., and Bristow R. (2012) Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin. Cancer Res. 18, 2108–2114 [DOI] [PubMed] [Google Scholar]
- 81. Vergis R., Corbishley C. M., Norman A. R., Bartlett J., Jhavar S., Borre M., Heeboll S., Horwich A., Huddart R., Khoo V., Eeles R., Cooper C., Sydes M., Dearnaley D., and Parker C. (2008) Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol. 9, 342–351 [DOI] [PubMed] [Google Scholar]
- 82. Horii K., Suzuki Y., Kondo Y., Akimoto M., Nishimura T., Yamabe Y., Sakaue M., Sano T., Kitagawa T., Himeno S., Imura N., and Hara S. (2007) Androgen-dependent gene expression of prostate-specific antigen is enhanced synergistically by hypoxia in human prostate cancer cells. Mol. Cancer Res. 5, 383–391 [DOI] [PubMed] [Google Scholar]
- 83. Mitani T., Yamaji R., Higashimura Y., Harada N., Nakano Y., and Inui H. (2011) Hypoxia enhances transcriptional activity of androgen receptor through hypoxia-inducible factor-1alpha in a low androgen environment. J. Steroid Biochem. Mol. Biol. 123, 58–64 [DOI] [PubMed] [Google Scholar]
- 84. Gong Y., Scott E., Lu R., Xu Y., Oh W. K., and Yu Q. (2013) TIMP-1 promotes accumulation of cancer associated fibroblasts and cancer progression. PLoS ONE 8, e77366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Deng X., He G., Levine A., Cao Y., and Mullins C. (2008) Adenovirus-mediated expression of TIMP-1 and TIMP-2 in bone inhibits osteolytic degradation by human prostate cancer. Int. J. Cancer 122, 209–218 [DOI] [PubMed] [Google Scholar]
- 86. Hayakawa T., Yamashita K., Tanzawa K., Uchijima E., and Iwata K. (1992) Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range of cells. A possible new growth factor in serum. FEBS Lett. 298, 29–32 [DOI] [PubMed] [Google Scholar]
- 87. Gong Y., Chippada-Venkata U. D., Galsky M. D., Huang J., and Oh W. K. (2015) Elevated circulating tissue inhibitor of metalloproteinase 1 (TIMP-1) levels are associated with neuroendocrine differentiation in castration resistant prostate cancer. Prostate 75, 616–627 [DOI] [PubMed] [Google Scholar]
- 88. Oh W. K., Vargas R., Jacobus S., Leitzel K., Regan M. M., Hamer P., Pierce K., Brown-Shimer S., Carney W., Ali S. M., Kantoff P. W., and Lipton A. (2011) Elevated plasma tissue inhibitor of metalloproteinase-1 levels predict decreased survival in castration-resistant prostate cancer patients. Cancer 117, 517–525 [DOI] [PubMed] [Google Scholar]
- 89. Bourboulia D, and Stetler-Stevenson WG. (2010) Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 20, 161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fu X., Parks W. C., and Heinecke J. W. (2008) Activation and silencing of matrix metalloproteinases. Semin. Cell Dev. Biol. 19, 2–13 [DOI] [PubMed] [Google Scholar]
- 91. Huang M., Narita S., Tsuchiya N., Ma Z., Numakura K., Obara T., Tsuruta H., Saito M., Inoue T., Horikawa Y., Satoh S., and Habuchi T. (2011) Overexpression of Fn14 promotes androgen-independent prostate cancer progression through MMP-9 and correlates with poor treatment outcome. Carcinogenesis 32, 1589–1596 [DOI] [PubMed] [Google Scholar]
- 92. Attard G., Reid A. H., Auchus R. J., Hughes B. A., Cassidy A. M., Thompson E., Oommen N. B., Folkerd E., Dowsett M., Arlt W and de Bono JS (2012) Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab. 97, 507–516 [DOI] [PubMed] [Google Scholar]
- 93. de Bono J. S., Logothetis C. J., Molina A., Fizazi K., North S., Chu L., Chi K. N., Jones R. J., Goodman O. B. Jr, Saad F., Staffurth J. N., Mainwaring P., Harland S., Flaig T. W., Hutson T. E., Cheng T., Patterson H., Hainsworth J. D., Ryan C. J., Sternberg C. N., Ellard S. L., Flechon A., Saleh M., Scholz M., Efstathiou E., Zivi A., Bianchini D., Loriot Y., Chieffo N., Kheoh T., Haqq C. M., Scher HI and C-A- Investigators (2011) Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 364, 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pezaro C. J., Mukherji D., and De Bono J. S. (2012) Abiraterone acetate: redefining hormone treatment for advanced prostate cancer. Drug Discov. Today 17, 221–226 [DOI] [PubMed] [Google Scholar]
- 95. Shahabi A., Lewinger J. P., Ren J., April C., Sherrod A. E., Hacia J. G., Daneshmand S., Gill I., Pinski J. K., Fan J. B., and Stern M. C. (2016) Novel gene expression signature predictive of clinical recurrence after radical prostatectomy in early stage prostate cancer patients. Prostate 76, 1239–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chen Q., Cai Z. K., Chen Y. B., Gu M., Zheng D. C., Zhou J., and Wang Z. (2015) Poly r(C) binding protein-1 is central to maintenance of cancer stem cells in prostate cancer cells. Cell Physiol. Biochem. 35, 1052–1061 [DOI] [PubMed] [Google Scholar]
- 97. Terada K, and Mori M. (2000) Human DnaJ homologs dj2 and dj3, and bag-1 are positive cochaperones of hsc70. J. Biol. Chem. 275, 24728–24734 [DOI] [PubMed] [Google Scholar]
- 98. Gotoh T., Terada K., Oyadomari S., and Mori M. (2004) hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 11, 390–402 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomic data have been deposited to the Mass spectrometry Interactive Virtual Environment (MassIVE) consortium (http://www.massive.ucsd.edu/ProteoSAFe/datasets.jsp) with data set identifier: MSV000082244.