

Abstract
In this report, we identify the relevant factors to increase production of medium chain n-alcohols through an expanded view of the reverse β-oxidation pathway. We began by creating a base strain capable of producing medium chain n-alcohols from glucose using a redox-balanced and growth-coupled metabolic engineering strategy. By dividing the heterologous enzymes in the pathway into different modules, we were able to identify and evaluate homologs of each enzyme within the pathway and identify several capable of enhancing medium chain alcohol titers and/or selectivity. In general, the identity of the trans-2-enoyl-CoA reductase (TER) and the direct overexpression of the thiolase (FadA) and β-hydroxy-acyl-CoA reductase (FadB) improved alcohol titer and the identity of the FadBA complex influenced the dominant chain length. Next, we linked the anaerobically induced VHb promoter from Vitreoscilla hemoglobin to each gene to remove the need for chemical inducers and ensure robust expression. The highest performing strain with the autoinduced reverse β-oxidation pathway produced n-alcohols at titers of 1.8 g/L with an apparent molar yield of 0.2 on glucose consumed in rich medium (52% of theoretical yield).
Keywords: Beta-reduction, reverse beta oxidation, fatty alcohol, medium chain alcohol, anaerobic, E. coli, oleochemical, acyl-CoA reductase
Graphical Abstract

1. Introduction
Increasing market demand for oleochemicals, which are typically derived from petrochemical and plant oil feedstocks, has led to the search for new sources of certain classes of these compounds. Medium chain (i.e. C6-C12), n-fatty alcohols have sparked particular interest due to their widespread use in industry, cosmetics, and consumer products. In combination with limited natural supply, this utility is reflected in higher prices relative to other classes of oleochemicals, such as fatty acids and biodiesel (Pfleger et al., 2015). The production of linear alcohols is thought to be so important that production is listed as one of the top ten challenges for catalysis (Haggin, 1993). Commercially, fatty alcohols are produced through the ethylene-based Ziegler process, although this process only produces a minority of alcohols in the medium-chain C6-C10 range (Falbe et al., 2013). While these alcohols can be derived from plant oils or other fatty acid sources that are rich in desired chain lengths, most plant oils are, unfortunately, dominated by longer chain fatty acids (≥ C16).
Metabolic engineering strategies for producing fatty alcohols often leverage a native fatty acid biosynthesis pathway. In this strategy, free fatty acids (FFAs) are formed by the hydrolysis of an acyl-ACP using a chain length selective thioesterase. The FFAs are subsequently reduced to a fatty alcohol directly (Akhtar et al., 2013) or through an acyl-CoA intermediate (Youngquist et al., 2013). Production of alcohols from this pathway requires a significant amount of ATP for malonyl-CoA production and FFA activation to an acyl-CoA (Lennen and Pfleger, 2012). Because of the high ATP requirement, these strategies require aerobic cultivation of the relevant microorganisms, leading to significant carbon-loss through respiration (Youngquist et al., 2012).
To address the loss of carbon as CO2, researchers have explored fermentative β-reduction pathways that use reversible thiolases for creating carbon-carbon bonds. These pathways have higher theoretical yields because ATP is not expended in the creation of malonyl-CoA or CoA activation of FFAs. One such pathway, the butanol fermentation pathway from Clostridium acetobutylicum, has been extended for butanol, hexanol, and some octanol production (Dekishima et al., 2011; Machado et al., 2012; Shen et al., 2011). Similar chemistry has been explored in the reversal of the β-oxidation pathway (Clomburg et al., 2012; Dellomonaco et al., 2011; Kim et al., 2015), which functionally reverses the catabolic β-oxidation pathway by replacing the irreversible, catabolic acyl-CoA dehydrogenase (fadE) with a trans-2-enoyl-CoA reductase (TER), for synthesizing a variety of fatty acid-derived products. While theoretically promising, yields and titers of strains targeting production of longer chain-length fatty alcohols (>C4) have remained low in published demonstrations (Kim et al., 2015; Machado et al., 2012).
In this study, we explored many of the variables available for comprising a β-reduction pathway in the synthetic biology workhorse, Escherichia coli. We used bioinformatic methods to identify diverse candidate strains and enzymes for the major steps in the pathway and tested each by compiling the full pathway in a series of exchangeable modules. We compared the capabilities of common E. coli, MG1655 and the recently sequenced strain LS5218 (Rand et al., 2017), which has been used as a host for producing bioplastics through use of β-oxidation enzymes. We implemented genomic changes, predicted by genome scale metabolic modeling, to couple cell growth to the production of fatty alcohols. We used the enzyme similarity tool (Gerlt et al., 2015) to identify diverse variants of the TER, thiolase (FadA), β-hydroxy-acyl-CoA reductase (FadB), and acyl-CoA reductase (ACR) enzymes and found that combinations of the enzymes altered the titer and chain-length selectivity of n-fatty alcohols. In general, the identity of the TER improved titer, and identity of the FadA and FadB influenced the dominant chain length. Finally, we found that using an anaerobically auto-induced promoter (Lara et al., 2017) to transcribe the genes comprising the β-reduction pathway improved product titer without the need for adding exogenous inducer at the appropriate time. Through our exploration, we identified a set of enzymes and promoters that achieved production of a mixture of medium- and long-chain fatty alcohols at ~1.8 g/L titers when cultured in LB plus 1% glucose - an apparent molar yield of 0.2 on glucose consumed (52% of theoretical yield). Given this success, the biggest remaining challenge is identifying strategies for controlling chain-length specificity of products made via a β-reduction pathway.
2. Materials and Methods
2.1. Chemicals, Reagents, and Media
Chemicals were purchased from either Sigma Aldrich (St. Louis, MO) or Fisher Scientific (Waltham, MA). Oligonucleotides and gene fragments were purchased from Integrated DNA Technologies (Coralville, IA) or Thermo Fisher Scientific (Waltham, MA). Enzymes were purchased from New England Biolabs (Ipswich, MA). DNA purification kits were purchased from Qiagen (Venlo, Netherlands). All cultures were started from single colonies grown on LB agar isolated from freezer stocks stored in 15% glycerol. Overnight cultures of strains were grown in LB media in a rotary shaker at 250 rpm at 37°C for cloning experiments and 30°C for precultures. When a selective pressure was necessary for plasmid retention, media was supplemented with the appropriate antibiotics (carbenicillin, 100 μg/mL; kanamycin, 50 μg/mL; chloramphenicol, 34 μg/mL).
2.2. Genome Scale Modeling
Flux balance analysis (FBA) was performed with the Cameo library for Python (Cardoso et al., 2018) using the iJO1366 model of E. coli metabolism (Orth et al., 2011). The model was based on a glucose uptake rate of 10 mmol gDW−1 hr−1. To model the pathway, heterologous reactions for the β-reduction pathway were added (Supplementary File). To prevent futile cycles in the model, reactions corresponding to thioesterases (e.g. FACOAE60), FadD (e.g. FACOAL60t2pp), and amino-acid degradation (e.g. THRD) were deleted (Supplementary Jupyter Notebook). For operation of the β-reduction pathway, FadE (e.g. ACOAD1f) was deleted. For the growth-coupled method, reactions corresponding to ethanol and mixed-acid fermentation (ACKr, ALCD2x, LDH_D, LDH_D2, ACALD, ACtex, FRD2, FRD3, POX, PYRt2rpp, LCARR, and ACALDtpp) were deleted and the oxygen exchange rate was set to 0.
2.3. CRISPR-Cas9 Recombineering
CRISPR/Cas9-assisted recombineering was performed with a protocol adapted from Li and co-workers (Li et al., 2015) using a two-plasmid system consisting of a cas9/λ-Red expression plasmid and a guide RNA (gRNA) plasmid. The expression plasmid, pMP11, was derived from the temperature-sensitive recombineering plasmid pKD46 (Datsenko and Wanner, 2000) by insertion of a copy of the Streptococcus pyogenes cas9 gene and an anhydrotetracycline-inducible gRNA targeting the pBR322 origin of replication. The gRNA plasmid encoded a constitutively expressed gRNA targeting the desired gene sequence, a pBR322 origin of replication, and either a kanamycin or chloramphenicol resistance marker. The S. pyogenes cas9 gene was obtained from the vector pwtCas9-bacteria (Addgene plasmid #44250). Desired guide RNA targeting regions were cloned into either pgRNA-kanR or pgRNA-cmR depending on whether kanamycin or chloramphenicol resistance was desired. These two vectors were derived from pgRNA-bacteria (Addgene plasmid #44251) by replacing the ampicillin resistance marker with the appropriate marker indicated above and replacing the gRNA targeting sequence with a null region containing two SapI restriction sites (Qi et al., 2013).
To create knockouts of specific genes (Table 1), gRNAs were designed using the gRNA designer from Atum (atum.bio). ssDNA oligos were designed with 30 base homology arms flanking the region to be knocked out and having the sequence of the lagging strand of DNA synthesis (Ellis et al., 2001; Mosberg et al., 2010). To make the knockout, 1mL of an overnight culture of a strain containing pMP11 was inoculated into 50mL SOB (Sambrook and Russell, 2001) and left to grow at 30°C for approximately one hour, or until it reached an OD600 of ~0.30, at which point the λ-Red genes were induced with 1% (w/v) arabinose. Once the culture reached an OD600 of 0.40-0.60, cells were made electrocompetent (Tu et al., 2016). The final washed pellet was resuspended in 500μL Milli-Q water and 50μL of the suspension was mixed with ~100ng pgRNA and 1μM ssDNA oligo; cells were then electroporated using a Bio-Rad MicroPulser. The transformed cells were allowed to recover in SOC for three hours in a 30°C shaker. Transformed cells were plated onto the appropriate dual antibiotic plate and incubated at 30°C for up to 24 hours to allow for substantial colony formation. Following colony PCR and sequencing to verify knockouts, correct strains were cured of the pgRNA plasmid by growing overnight in LB with carbenicillin and aTc (0.2ng/mL). pMP11 was cured out of strains by growing overnight at 42°C. Curing of plasmids was tested by streaking on agar plates with the appropriate antibiotics.
Table 1. Strains and Plasmids used in this study. Genbank files of all plasmids are available in the supplementary files.
| Strain/Plasmid | Relevant Genotype/Property | Source or Reference |
|---|---|---|
| Strains | ||
| Escherichia coli K-12 MG1655 | F- λ- ilvG- rfb-50 rph-1 | ECGSC |
| Escherichia coli LS5218 | ||
| Escherichia coli DH5α | Invitrogen | |
| Alcanivorax borkumensis | Source for abFadBA | ATCC 700651™ |
| Cupriavidus necator N-1 | Source for cnFadA2J2 and cnFadA4J3 | ATCC 43291™ |
| Marinobacter aquaeolei | Source for maFadBA | ATCC 700491™ |
| Pseudomonas oleovorans TF4-1L | Source for poFadBA | ATCC 29347™ |
| Pseudomonas putida KT2440 | Source for ppFadBA and ppTER | ATCC 47054™ |
| Salmonella enterica LT2 | Source for seFadBA | ATCC 700720™ |
| Vibrio fisheri ES114 | Source for vfFadBA | ATCC 700601™ |
| CM00 | MG1655 ΔaraBAD ΔfadR ΔfadE | This work |
| CM15 | MG1655 ΔaraBAD ΔfadR ΔfadE ΔldhA ΔackApta ΔadhE ΔpoxB ΔfrdABCD | This work |
| CM23 | MG1655 ΔaraBAD ΔFadRE ΔldhA ΔackApta ΔadhE ΔpoxB ΔfrdABCD ΔydiO ΔfadBA ΔfadIJ ΔfadD ΔatoC | This work |
| LSE | LS5218 ΔfadE | This work |
| CM16 | LS5218 ΔfadE ΔatoC | This work |
| CM20 | LS5218 ΔfadE ΔatoC ΔldhA ΔackApta ΔadhE ΔpoxB ΔfrdABCD | This work |
| CM24 | LS5218 ΔfadE ΔatoC ΔldhA ΔackApta ΔadhE ΔpoxB ΔfrdABCD ΔydiO ΔfadBA ΔfadIJ ΔfadD | This work |
| Plasmids | ||
| pMP11 | pKD46 with constitutively expressed Cas9 and an aTc gRNA targeting the ColE1 origin | This work |
| pgRNA | Constitutively expressed sgRNA targeting a desired gene | This work |
| pTRC99A | pBR322 Origin, AmpR, Trc Promoter | (Amann et al., 1988) |
| pTRC99A - vhTER | This work | |
| pTRC99A – vhTER - fdh | This work | |
| pTRC99A – tdTER - fdh | This work | |
| pTRC99A – lbbTER - fdh | This work | |
| pTRC99A – cmTER - fdh | This work | |
| pTRC99A – evcTER - fdh | This work | |
| pTRC99A – lboTER - fdh | This work | |
| pTRC99A – sdTER - fdh | This work | |
| pTRC99A – ppTER - fdh | This work | |
| pTRC99A – pVHb – tdTER – fdh | This Work | |
| pACYCtrc | pACYC Origin, CmR, Trc Promoter | (Youngquist et al., 2013) |
| pACYC – ecFadBA | This work | |
| pACYC – seFadBA | This work | |
| pACYC – ppFadBA | This work | |
| pACYC – poFadBA | This work | |
| pACYC – abFadBA | This work | |
| pACYC – maFadBA | This work | |
| pACYC – vfFadBA | This work | |
| pACYC – cnFadA2J2 | This work | |
| pACYC – cnFadA3J4 | This work | |
| pACYC – pVHb – seFadBA | This work | |
| pACYC – pVHb – seFadBA | This work | |
| pACYC – pVHb – seFadBA | This work | |
| pBTRK | pBBRl Origin, KmR, Trc Promoter | (Youngquist et al., 2013) |
| pBTRCk – maACR | (Youngquist et al., 2013) | |
| pBTRK – mcACR | This work | |
| pBTRK – rcACR | This work | |
| pBTRK – mhACR | This work | |
| pBTRK – mbACR | This work | |
| pBTRK – naACR | This work | |
| pBTRK – mtACR | This work | |
| pBTRK – caAdhE2 | This work | |
| pBTRK – raAdhE2 | This work | |
| pBTRK – aeAdhE2 | This work | |
| pBTRCk – pVHb – maACR | This work |
2.4. Plasmid Design & Construction
Variants of the TERs, ACRs, and AdhE2s were identified using Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST) (Gerlt et al., 2015) and visualized using Cytoscape (Shannon et al., 2003). TER variants were then cloned into the pTRC99A vector using Gibson assembly (Gibson et al., 2009) with a synthetic ribosome binding site (RBS) designed using the RBS Calculator version 1.1 (Espah Borujeni et al., 2014) for a translation initiation rate of approximately 50,000 a.u. The AdhE2 variants were cloned similarly into a pBTRK vector (Youngquist et al., 2013). The ACR homologs were cloned into the pBTRK vector with an N-terminal maltose binding protein fusions, as done previously (Youngquist et al., 2013). With the exception of maACR and ppTER, all TER, AdhE2, and ACR variants were codon optimized (Chin et al., 2014) and synthesized either by IDT (Coralville, IA) or Twist Bioscience (San Francisco, CA). The FadBA and FadIJ variants were designed and cloned into a pACYCtrc similar to the TER variants the genes were amplified directly from cell suspensions or purified genomic DNA to preserve the native operon. For all of the FadBA variants, the only synthetic RBS used was for the first gene in the operon to preserve the native translational coupling of the operon (Mutalik et al., 2013; Schümperli et al., 1982).
2.5. Cultivation for Alcohol Production
Cultures for alcohol fermentations were grown in 10mL of LB media supplemented with 1% glucose in 16 × 125mm Hungate Anaerobic tubes. The media was supplemented with appropriate antibiotics as previously described. The cultures were inoculated from an overnight culture (30°C) such that the starting optical density (OD) of the fermentation culture was 0.05. For microaerobic conditions, the tubes were sealed with ambient air in the tube’s headspace. Tubes were placed in test tube racks at 45-degree angles in a 30°C incubator and grown for approximately 2 hours until an OD of 0.2, at which point gene expression was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1mM. Fermentations were run for 48 hours after induction and then stored on ice for 1 hour to condense any vapors before analysis.
For the bioreactor experiments, 500mL of LB was added to 1L Infors Multifors bioreactors, sterilized, and supplemented with appropriate antibiotics. The aerobic growth phase was performed for 18 hours at 30°C, with the Rushton impellers at 500 rpm. The air flow rate was used to control the dissolved oxygen to 20% of the maximum dissolved oxygen and the pH was controlled by the addition of aqueous ammonia and 5M phosphoric acid. After the aerobic growth phase, 100mL of dodecane and 10mL of 50% (w/v) glucose feeding solution were added to each bioreactor. The feeding solution was adapted from Korz et al. (1995) by scaling the glucose concentration down to 50% (w/v) and scaling the additional minerals by the same factor. At the time glucose was added, the aeration was stopped and a 20mL/min overlay of N2 was started.
2.6. Extraction and GC-Analysis
Alcohols in the media were quantified by extraction into hexane and analysis on a GC-FID. Internal standards of each odd-chain-length alcohol from C3-C15 were added to the media to a concentration of 60mg/L and mixed by vortexing for 30 seconds. From each production vessel, 5mL of media was transferred to a centrifuge tube; in the case of a dodecane overlay, 5mL of media-dodecane emulsion were used. For small-scale fermentations (Hungate tubes and serum bottles), internal standards were added directly to the fermentation vessel. For bioreactor experiments, the internal standard was added to the 5mL sample after it was pulled from the reactor. The extraction was performed by adding 1mL of hexane to the centrifuge tube followed by 30s vortexing, 30s shaking, and 30s vortexing to allow the even-chain alcohols and odd-chain internal standards to reach equilibrium with the organic phase. The tubes were then centrifuged at 1000×g for 10min and the organic phase was pipetted from the top of the centrifuge and placed in a GC sample vial for analysis.
Analysis on the GC-FID was performed on a Stabilwax 60m column (Restek 10658) with a modified oven temperature program recommended from the manufacturer for alcohols: 45°C hold 10 min, ramp to 250 at 12°C/min, hold for 10 min. Peak areas were converted to concentrations in hexane by external standard curves. The concentrations from the organic (hexane) phase were then converted to the original concentration in the media by dividing organic concentration by the average concentration factor of the neighboring odd-chain alcohols. For example, if decanol has an organic concentration of 100mg/L, and the organic concentrations of nonanol and undecanol were, on average, 2× the known amount added, the reported concentration of decanol in the media would be 50mg/L. Data were analyzed for statistical significance in JMP (Version 13, SAS Institute Inc., Cary, NC) using a Tukey Honest Significant Difference test.
2.7. Western Blotting
Western blotting was performed as described in (Copeland et al., 2016). For the detection of maltose binding protein (MBP), the mouse anti-MBP monocolonal antibody from New England Biolabs (Ipswich, MA) was used.
3. Results & Discussion
3.1. Establishing an Alcohol Producing Strain
Our goal was to build a strain of E. coli capable of producing n-fatty alcohols from glucose anaerobically as a replacement for native mixed acid fermentation pathways of E. coli. Anaerobically, E. coli uses pyruvate formate lyase (Pfl) to generate acetyl-CoA and formate; this leaves one reducing equivalent in the formate molecule. To recapture this reducing power, a formate dehydrogenase (Fdh) from Candida boidinii can be expressed to generate NADH and CO2 (Figure 1a) (Shen et al., 2011). The elongation cycle of the reverse β-oxidation pathway is catalyzed by the β-oxidation complex (FadBA) and a trans-2-enoyl-CoA reductase (TER) (Figure 1b). Each elongation cycle requires one acetyl-CoA and two NADH to add two carbon units to a growing carbon chain. Addition of an alcohol forming acyl-CoA reductase (ACR) uses two NAD(P)H to form a terminal alcohol from the acyl-CoA. Addition of reactions (a) and (b) shows that this method is in redox balance at all chain lengths (Figure 1c). Consequently, this pathway should be able to be used as the sole fermentative pathway in E. coli. To implement the pathway, we designed a 3-plasmid system (Figure 1d), where each plasmid can be viewed as a “module” containing an enzyme(s) that can be swapped out with a plasmid containing a homologous enzyme.
Figure 1.
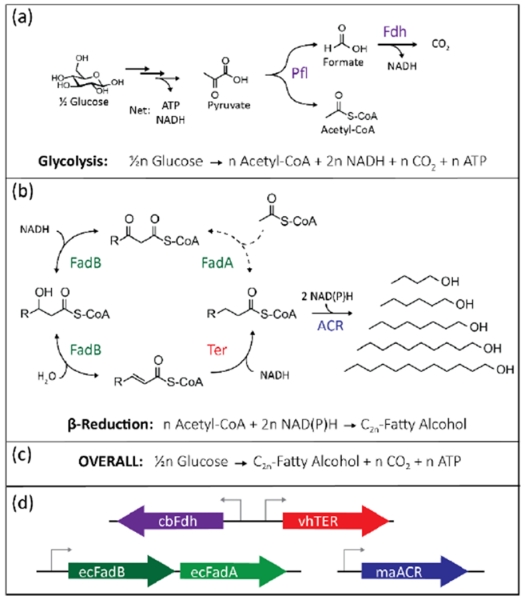
Design of a modular, redox balanced, β-reduction pathway. (a) Glycolysis yields one ATP and NADH. Under anaerobic conditions, acetyl-CoA is produced from pyruvate using pyruvate formate lyase (Pfl). The addition of a formate dehydrogenase (Fdh) recovers a reducing power from formate to create CO2, thus the entire glycolytic pathway yields two NADH, one CO2, and one ATP per acetyl-CoA produced. (b) Two-carbon acetyl-CoA are added to the chain in the β-reduction pathway by a thiolase (FadA) followed by a reduction, dehydration, and reduction (FadB), requiring two NADH. (c) By adding these reactions together, the pathway is redox balanced at every chain length of alcohol produced. (d) A modular system for expression of these genes, with the TER, FadBA, and ACR on separate plasmids allows each to be easily changed to a different variant.
With the modular system in mind, we built a genome-scale metabolic model of the β-reduction pathway based on the iJO1366 model of E. coli (Orth et al., 2011) and performed FBA with a basis glucose uptake rate of 10 mmol gDW−1 hr−1. For the addition of the reverse β-oxidation pathway to the model, the reactions corresponding to FadE had to be deleted, and reactions corresponding to the TER and ACR had to be added (Supplementary File). When we evaluated the model for anaerobic octanol production (as a representative medium-chain alcohol), no octanol production was predicted to occur at the maximum growth rate (0.234 hr−1) (Figure 2a). When all of the reactions corresponding to E. coli’s fermentative pathways were deleted, the model predicted an octanol production rate of 4 mmol gDW−1 hr−1 (a predicted carbon yield of 80% going to n-alcohol production) at a maximum specific growth rate of 0.153 hr−1 (Figure 2b). These results are similar to those reported previously for an genome scale metabolic model of E. coli producing alcohols from glycerol via the reverse β-oxidation pathway (Cintolesi et al., 2014).
Figure 2.
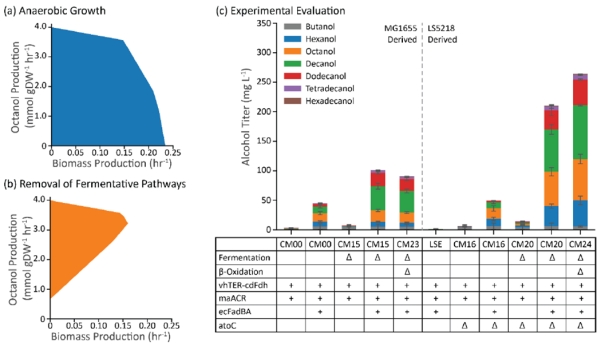
Strain design and evaluation. (a) The solution space for the E. coli genome scale model iJO1366 with the β-reduction pathway added and octanol as the model product. (b) Removal of all fermentative pathways from the E. coli model shows that alcohol production is growth coupled. (c) Implementation of this strategy in E. coli strains K12 MG1655 (left) and LS5218 (right) showed substantial differences in alcohol production. Error bars represent the standard deviation of triplicates performed on separate days.
We opted to implement this strategy in two strains of E. coli: K12 MG1655 and K12 LS5218 (Table 1). To satisfy the requirements for the operation of a β-reduction pathway, we deleted fadE in both strains of E. coli and overexpressed the β-oxidation FadBA complex. This was first implemented by inactivation of E. coli’s β-oxidation regulation gene, fadR, either by deletion of fadR in MG1655 (ΔfadE) or a natively inactive fadR in LS5218 (fadR*). These strains were titled CM00 and LSE, respectively. To build a full pathway for alcohol production, we also overexpressed a TER from Vibrio harveyi (vhTER) and an ACR from Marinobacter aquaeoli (maACR). After a 48-hour cultivation in Hungate tubes in LB media + 1% glucose, we observed only about 6 mg/L of total alcohols in CM00 and no significant alcohol production in LSE (Figure 2c). We suspected that the lack of alcohol production in strain LSE may be due to the constitutive activity of AtoC in LS5218, which activates the ato operon for short-chain fatty acid degradation; upon deletion of atoC in strain LSE (CM16), we observed alcohol production on the same order of magnitude as strain CM00.
After creating these two base strains for alcohol production, we further tested two possibilities to increase alcohol production: direct overexpression of the FadBA complex on a plasmid and the removal of native fermentative pathways (Figure 2c). Direct FadBA overexpression in strains CM00 and CM16 resulted in an order of magnitude increase in fatty alcohol titer over the course of the fermentation (45 mg/L and 50 mg/L, respectively). When we deleted the native fermentative pathways (for acetate, lactate, ethanol, and succinate production) to make strains CM15 and CM20, we observed little increase in alcohol production, however, when the fermentative pathway deletions were combined with FadBA overexpression from a plasmid, we noticed a 2-fold increase in alcohol titer in the MG1655-derived strain (101 mg/L) and a 4-fold increase in the LS5218-derived strain (210 mg/L). Finally, we created a strain with only plasmid-based FadBA overexpression. We deleted the chromosomal copies of fadBA and fadIJ from E. coli; from these strains, we noticed approximately the same alcohol production from the MG1655-derived strain (CM23) (91 mg/L) and a modest improvement in alcohol production from the LS5218-derived strain (CM24) (264 mg/L). Given the increased production in the LS5218 strains, the remainder of the research described in this report was performed in this strain background.
3.2. Non-Equivalence of Homologous β-Oxidation FadBA Complexes
After observing the substantial increase in fatty alcohol production from plasmid-based FadBA overexpression, we searched for diverse FadBA homologs (Figure 3a, Supplementary Figures 1 & 2) under the hypothesis that the native E. coli variants may not be optimal for operating a β-reduction pathway. To narrow the search, we only looked at proteobacteria with trifunctional β-oxidation enzyme complexes (Kazakov et al., 2009), similar to the FadBA complex in E. coli, that were organized in operons. From this search, we chose to sample operons from three groups of γ-proteobacteria: enteric bacteria (E. coli and Salmonella enterica), soil bacteria (Pseudomonas putida and Pseudomonas oleovorans), and marine bacteria (Alcanivorax burkhormensis, Marinobacter aquaeoli, and Vibrio fisherii). To expand beyond γ-proteobacteria, we also cloned two operons, fadA2J2 and fadA4J3, from the β-proteobacterium Cupravidus necator (not shown in Figure 3a).
Figure 3.
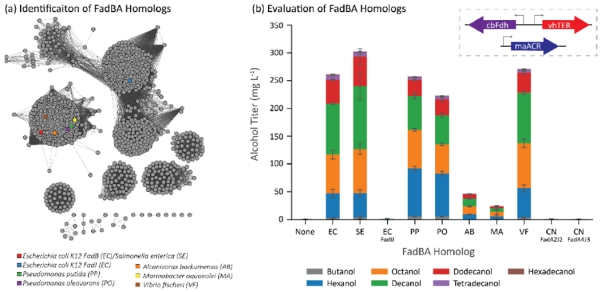
Identification and evaluation of FadBA homologs as components in a β-reduction pathway. (a) Sequence similarity map of FadBA homologs. Genes cluster into eight major families. The quantitative relationship between the sequences is shown in Supplementary Figure 1. We mainly tested variants from those close to E. coli FadBA to use their native operon structure. (b) FadBA homologs impacted the distribution of chain lengths in the fatty alcohols produced by the E. coli cells co-expressing vhTER, cbFdh, and maACR. Abbreviations on the x-axis refer to the first letter of the genus and species. Error bars represent the standard deviation of triplicates performed on separate days.
We tested the different FadBA homologs in strain CM24 (LS5218 ΔfadE ΔatoC ΔldhA ΔackApta ΔadhE ΔpoxB ΔfrdABCD ΔydiO ΔfadBA ΔfadIJ ΔfadD) containing the vhTER, cbFdh, and maACR (Figure 3b). We first tested the S. enterica FadBA (seFadBA) due to its similarity to E. coli’s FadBA (over 95% amino acid identity for both FadB and FadA) and its increased activity toward C4 chain-lengths in the catabolic direction (Iram and Cronan, 2006). With this system, we found moderately increased alcohol production to an average of 303 mg/L; while this difference was not statistically significant, we have found this trend to be repeatable across several experiments with different strains. We then attempted to overexpress E. coli’s anaerobic β-oxidation complex, FadIJ, to determine if the increase propensity of FadIJ to metabolize short- and medium- chain fatty acids would translate to shorter chain-length alcohol production (Campbell et al., 2003). Unexpectedly, this enzyme pair did not confer the ability to produce alcohols.
To expand on the expression of FadBA homologs from E. coli, we hypothesized that organisms that environmentally degrade fatty acids may have more efficient FadBA complexes that could be exploited in the reverse reactions. One example, Pseudomonas oleovorans is able to consume octane (Chakrabarty et al., 1973). Expression of FadBA complexes from Pseudomonas oleovorans and its near relative Pseudomonas putida produce significantly more hexanol and significantly less decanol when compared to the ecFadBA or seFadBA. We did not observe significant alcohol production from either strain expressing a FadBA complex from either marine oil-degrading bacteria (abFadBA and maFadBA), but we did notice approximately the same amount of alcohol production from the strain expressing the vfFadBA (271 mg/L).
3.3. Bio-Prospecting for Reductive Steps (TER and ACR)
Next, we furthered our search for homologous enzymes by comparing variants for catalyzing the irreversible reductive steps within the pathway: the terminal CoA reductase and the trans-2-enoyl-CoA reductase. Three types of acyl-CoA reductases (ACR) are commonly used for alcohol production: single domain aldehyde forming reductases (e.g. Acr1 from Acinetobacter calcoaceticus BD413 as in (Steen et al., 2010)) which are used in combination with an aldehyde reductase, a bi-functional alcohol forming acyl-CoA reductase, such as maACR from Marinobacter aquaeolei VT8 (Wahlen et al., 2009; Willis et al., 2011), and the alcohol dehydrogenase (AdhE) for butanol production from Clostridium acetobutylicum (caAdhE2). Sequence similarity searching for homologs to maACR showed two major groupings of ACR homologs (Figure 4a) with the pairwise amino acid identity between proteins in each grouping between 50 and 80% (Supplementary Figure 3a). The sequence similarity network based off caAdhE2 (Figure 4b) did not show individual grouping; three enzymes were chosen from this set with a pairwise amino acid identity between 32 and 37% (Supplementary Figure 3b).
Figure 4.
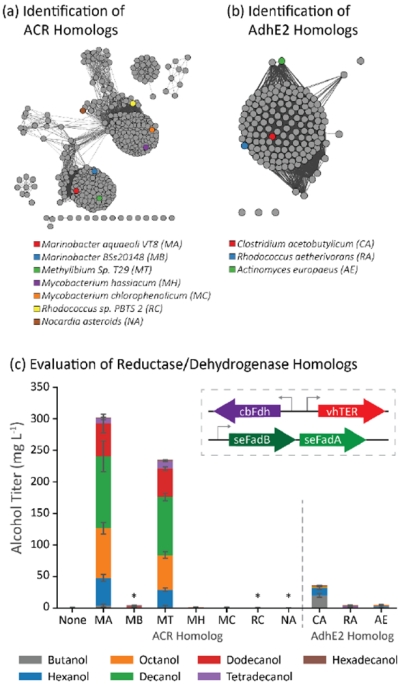
Identification and evaluation of ACR homologs as components in a β-reduction pathway. (a) The sequence similarity map of ACR homologs shows multiple groupings of sequence homologs. The quantitative relationship between the sequences is shown in Supplementary Figure 3a. (b) AdhE2 homologs identified were found in only a single grouping. The quantitative similarity between the AdhE2 homologs is shown in Supplementary Figure 3b (c) Most ACR homologs failed to generate alcohols in E. coli cells coexpressing vhTER, cbFdh, and seFadBA. Many of the nonproducing ACR variants failed to express soluble protein (labeled with *; see Supplementary Figure 4 for Western). Error bars represent the standard deviation of triplicates performed on separate days.
The ACR and AdhE2 homologs were evaluated in the background strain CM24 + pACYC − seFadBA + pTRC99a − vhTER − fdh (Figure 4c). We noticed the highest alcohol titer from maACR (303 mg/L) and similar results, with a lower titer, from the mtACR (235 mg/L). A western blot of the ACR homologs probed for 6x-histidine tag suggested that only maACR, mtACR, mhACR, and mcACR generated significant protein (Supplementary Figure 4). Not surprisingly, the expression of caAdhE2 in place of an ACR produced butanol as its major product and produced a total alcohol titer of 36 mg/L. Many of the homologs expressed in our test failed to produce alcohols.
Evaluation of multiple TER homologs was performed analogous to the ACR and AdhE homolog studies. The sequence similarity network for TER homologs (Figure 5a) was filtered for proteins with annotated reductase activity, from which seven candidates were chosen for further testing; the pairwise amino acid identities between each enzyme ranges from 30-60% (Supplementary Figure 5). The TER homologs were evaluated in strain CM24 with seFadBA and maACR overexpressed on separate plasmids and cbFdh overexpressed on the same plasmid (Figure 5b). The final alcohol titer from strains expressing evcTER (360 mg/L) and lboTER (325 mg/L) had about the same alcohol production as the control strain expressing the vhTER (280 mg/L). The strain expressing the tdTER, however, showed more than a 2-fold increase in total alcohol titer compared to the control (720 mg/L).
Figure 5.
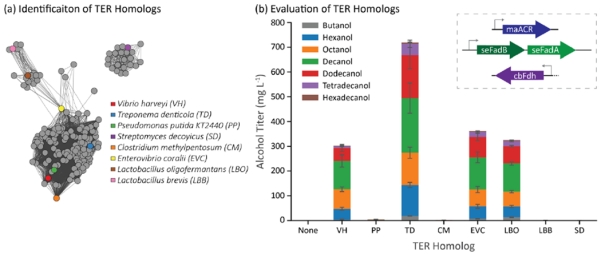
Identification and evaluation of TER homologs as components in a β-reduction pathway. (a) Sequence similarity map of TER homologs. Genes cluster into two major families with outliers. We tested variants from three clusters. The quantitative similarity can be seen in Supplementary Figure 5. (b) Several TER homologs failed to generate alcohols in E. coli cells co-expressing maACR, cbFdh, and seFadBA. The TER from Treponema denticola (TD) led to the highest alcohol titer. Error bars represent the standard deviation of triplicates performed on separate days.
3.4. Biphasic Fermentation and Anaerobic Auto-Induction
We combined the highest-titer-producers in the previous experiments (maACR, tdTER) with three different fadBA enzymes (seFadBA, ppFadBA, vfFadBA) and introduced a 20% (v/v) dodecane overlay (Figure 6a). Initially, we were unable to achieve proper mixing in a Hungate tube with a 20% dodecane overlay and found the cells settled at the bottom of the tube at the end of the cultivation. To improve mixing during cultivation, we moved our cultivations into serum bottles. Using the best alcohol producing strain from the previous set (seFadBA, tdTER, maACR), we achieved approximately 500 mg/L alcohol titers in vessels with new geometry. With the dodecane overlay added, we observed nearly 1.5 g/L of total alcohol production. We then cloned the anaerobically induced VHb promoter (mechanistic depiction Supplementary Figure 6) (Khosla et al., 1990; Khosla and Bailey, 1989) in front of all five β-reduction genes (tdTER, cbFdh, FadBA, maACR) and observed approximately 1.8 g/L of total alcohol production from the strain expressing vfFadBA (Figure 6b). This titer corresponded to an apparent molar yield of 0.2 on glucose, which represents 52% of the absolute theoretical yield and 65% of the theoretical yield from the growth-coupled genome scale metabolic model.
Figure 6.
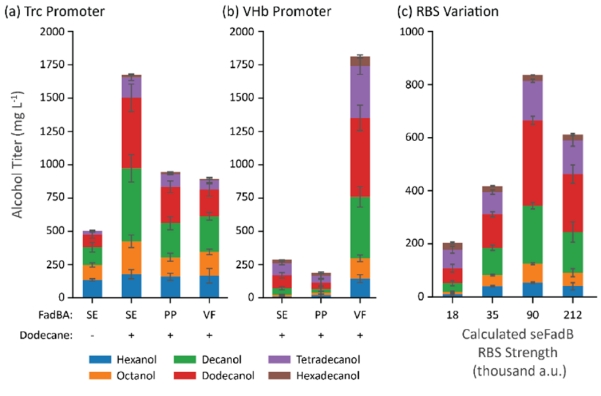
Combination of maACR, cbFdh, tdTER, and various FadBA enzymes in strain CM24 cultured in serum bottles with a 20% dodecane overlay. (a) Pathway genes expressed from the trc promoter and a no-dodecane control (left). (b) Changing the promoter to the VHb promoter strongly impacted production of fatty alcohols in both positive and negative directions for the same sets of genes. (c) Alcohol production in strains expressing tdTER, cbFdh, maACR, and seFadBA with different strength RBSs controlling FadB translation. Abbreviations: SE = S. enterica, PP = P. putida, VF = V. fischeri. Error bars represent the standard deviation of triplicates performed on separate days.
We were also interested to determine why the seFadBA and ppFadBA operons only produced alcohols at about 250 mg/L when transcribed from the anaerobic promoter. Because our earlier experiments had shown that increased expression of a FadBA complex can dramatically improve alcohol production (Figure 2c), we suspected that the artificial ribosome binding site (RBS) designed for the fadB gene may be causing suboptimal gene expression. To test this hypothesis, we built a small set of RBSs for the seFadB gene. The internal FadA RBS was not changed to preserve the natural translational coupling from the operon (Mutalik et al., 2013; Schümperli et al., 1982). From this experiment, observed a volcano-plot-like correlation between RBS strength before the FadBA gene and total alcohol production (Figure 6c), suggesting that relative enzyme expression caused the low alcohol production in the seFadBA and ppFadBA strains when expressed from the VHb promoter.
3.5. Scale-up and Fed-Batch
Given a highly productive combination of enzymes, we next tested its performance in a bioreactor containing higher cell densities (Figure 7). To do this, we grew strain CM24 containing VHb induced vfFadBA, tdTER, and maACR aerobically on LB media for 18 hours until it reached stationary phase (an OD of approximately 6). After the aerobic growth phase on LB, we added glucose (to 10 g/L), dodecane (to 20%), and trace minerals. To create an anaerobic environment, aeration was stopped and a nitrogen overlay was started. From this experiment, we found that the higher cell density culture consumed all the glucose within about 24 hours and generated alcohols with a titer of about 1.4 g/L. This corresponds to a molar yield of 0.15 on glucose, which is 41% of the maximum theoretical yield and 52% of the yield predicted by the genome scale metabolic model. The alcohol production in the two-stage bioreactor experiment is likely lower than the previous experiment for two reasons. First, more carbon from the LB should have been consumed during the aerobic growth stage, as evidenced in the higher OD, and would not be used for alcohol production. Second, the process in the bioreactor uses a low N2 flow (an open system) as compared to the completely closed system of the tubes, meaning that some of the alcohols may be lost due to evaporation in the bioreactor. Because of the separation between aerobic growth stage on LB and the alcohol production stage on glucose, we expect this yield (0.41) to be more reflective of the true alcohol yield on glucose as the carbon in the LB media had been consumed in the aerobic growth phase.
Figure 7.
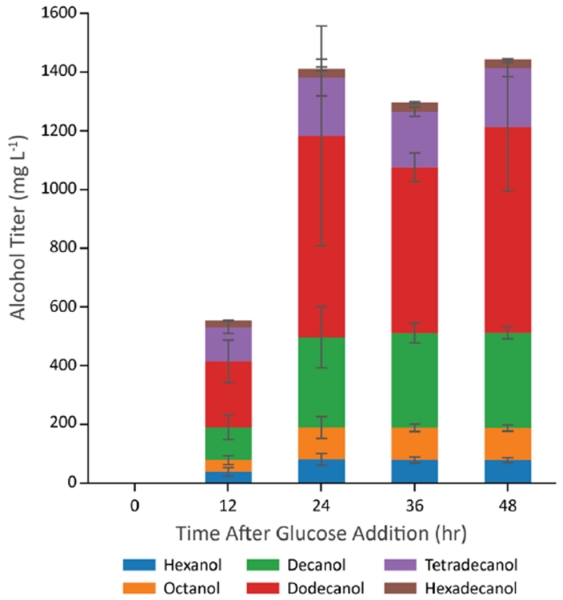
Bioreactor-scale production of alcohols at higher cell density. Cells were grown to stationary phase at which point (Time 0) glucose feeding solution was added to 10 g/L. Error bars represent the standard deviation of biological triplicates performed simultaneously.
4. Conclusions
We have evaluated several factors for alcohol production in E. coli via an engineered β-reduction pathway. Our initial comparison of E. coli strains K12 MG1655 and LS5218 suggests that strain LS5218 is a substantially superior strain for alcohol production through this pathway. The strong performance requires deletion of native fermentative pathways to couple redox balance to fatty alcohol production. We created sequence similarity networks of each of the four key enzymes in the β-reduction pathway and sampled variants from across the sets. From this exploration, we gained a better understanding of the effect of the sequence space on alcohol production.
We found that heterologous overexpression of FadBA enzymes from different organisms played two roles in the ability of cells to produce alcohols: first, overexpression dramatically increased total alcohol production from the pathway, and second, the source of the FadBA complex appears to influence the chain-length specificity of the pathway to an extent. We identified multiple new TER enzymes capable of driving the β-reduction pathway, suggesting that similar chemistry may be found broadly in nature. Lastly, we found that in situ product extraction through a biphasic fermentation significantly improved alcohol production, likely by removing product toxicity and/or product inhibition. This strategy allowed us to produce a range of fatty alcohols with our best-performing strain producing 1.8 g/L. Our work has demonstrated strains capable of crossing the 50% theoretical yield barrier and indicated that balancing expression of enzymes in the pathway will have a strong influence on titer and selectivity.
The work performed here leaves several opportunities for further improvement. First, it should be noted that our screening method requires genes that express functionally in the cell. Because of this, sub-optimally expressed genes may cause more efficient enzymes to be overlooked. Continued efforts in regularizing gene expression platforms, such as codon optimization (Boël et al., 2016) and consistent gene expression (Mutalik et al., 2013) will aid future work in screening enzyme variants in a more balanced manner. Furthermore, toxicity of medium-chain length products likely limits the maximum titer of fatty alcohols. Work to increase medium chain product resistance in E. coli (Royce et al., 2015) or selection of a host tolerant to high titers of fatty alcohols (Rutter and Rao, 2016) may improve the final titer and specific productivity of the alcohol fermentation process. In addition, work to optimize selectivity of the pathway to desired chain-lengths is highly desirable. Some work has been performed on this front for fatty acid production (Kim and Gonzalez, 2018), however improving chain-length selectivity without a highly selective thioesterase remains elusive. Finally, process optimization in terms of gene expression and media formulation may drastically increase productivity. This work would be best performed on an industrial high-throughput platform using statistical design of experiment strategies.
Supplementary Material
Highlights.
Anaerobic production of medium chain n-alcohols by a β-reduction pathway
Improved variants of TER, thiolase, and β-ketoreductases were identified
Inclusion of an anaerobic-responsive promoter increased production
Acknowledgements
This work was funded by a research grant to the University of Wisconsin-Madison from The Dow Chemical Company. MFP was supported by the NIH NHGRI Genomic Sciences Training Program (T32 HG002760). The authors are grateful to Chris Stowers and Devon Rosenfeld for helpful discussions. The authors are also grateful to Jonathan Greenhalgh for his help plotting the pairwise alignment data.
Abbreviations
- ACR
Acyl-CoA Reductase
- FBA
Flux Balance Analysis
- FFA
Free Fatty Acid
- TER
trans-2-enoyl-CoA reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar MK, Turner NJ, Jones PR. Carboxylic acid reductase is a versatile enzyme for the conversion of fatty acids into fuels and chemical commodities. Proc. Natl. Acad. Sci. U. S. A. 2013;110:87–92. doi: 10.1073/pnas.1216516110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann E, Ochs B, Abel KJ. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene. 1988;69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- Boël G, Letso R, Neely H, Price WN, Wong K-H, Su M, Luff J, Valecha M, Everett JK, Acton TB, Xiao R, Montelione GT, Aalberts DP, Hunt JF. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature. 2016;529:358–363. doi: 10.1038/nature16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JW, Morgan-Kiss RM, Cronan JE. A new Escherichia coli metabolic competency: Growth on fatty acids by a novel anaerobic b-oxidation pathway. Mol. Microbiol. 2003;47:793–805. doi: 10.1046/j.1365-2958.2003.03341.x. [DOI] [PubMed] [Google Scholar]
- Cardoso J, Jensen K, Lieven C, Hansen ASL, Galkina S, Beber ME, Özdemir E, Herrgard M, Redestig H, Sonnenschein N. Cameo: A Python Library for Computer Aided Metabolic Engineering and Optimization of Cell Factories. ACS Synth. Biol. 2018 doi: 10.1021/acssynbio.7b00423. acssynbio.7b00423. [DOI] [PubMed] [Google Scholar]
- Chakrabarty AM, Chou G, Gunsalus IC. Genetic Regulation of Octane Dissimilation Plasmid in Pseudomonas. Proc. Natl. Acad. Sci. 1973;70:1137–1140. doi: 10.1073/pnas.70.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JX, Chung BKS, Lee DY. Codon Optimization OnLine (COOL): A web-based multi-objective optimization platform for synthetic gene design. Bioinformatics. 2014;30:2210–2212. doi: 10.1093/bioinformatics/btu192. [DOI] [PubMed] [Google Scholar]
- Cintolesi A, Clomburg JM, Gonzalez R. In silico assessment of the metabolic capabilities of an engineered functional reversal of the β-oxidation cycle for the synthesis of longer-chain (C≥4) products. Metab. Eng. 2014;23:100–15. doi: 10.1016/j.ymben.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Clomburg JM, Vick JE, Blankschien MD, Rodríguez-Moyá M, Gonzalez R. A synthetic biology approach to engineer a functional reversal of the β-oxidation cycle. ACS Synth. Biol. 2012;1:541–54. doi: 10.1021/sb3000782. [DOI] [PubMed] [Google Scholar]
- Copeland MF, Politz MC, Johnson CB, Markley AL, Pfleger BF. A transcription activator–like effector (TALE) induction system mediated by proteolysis. Nat. Chem. Biol. 2016;12:254–260. doi: 10.1038/nchembio.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K. a, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekishima Y, Lan EI, Shen CR, Cho KM, Liao JC. Extending Carbon Chain Length of 1-Butanol Pathway for 1-Hexanol Synthesis from Glucose by Engineered Escherichia coli. J. Am. Chem. Soc. 2011;133:11399–11401. doi: 10.1021/ja203814d. [DOI] [PubMed] [Google Scholar]
- Dellomonaco C, Clomburg JM, Miller EN, Gonzalez R. Engineered reversal of the β-oxidation cycle for the synthesis of fuels and chemicals. Nature. 2011;476:355–9. doi: 10.1038/nature10333. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espah Borujeni A, Channarasappa AS, Salis HM. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014;42:2646–59. doi: 10.1093/nar/gkt1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falbe J, Bahrmann H, Lipps W, Mayer D, Frey GD. Ullmann’s Encyclopedia of Industrial Chemistry. 2013. Alcohols, Aliphatic. [DOI] [Google Scholar]
- Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, Whalen KL. Enzyme function initiative-enzyme similarity tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta - Proteins Proteomics. 2015;1854:1019–1037. doi: 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison C. a, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Haggin J. Chemists Seek Greater Recognition for Catalysis. Chem. Eng. News. 1993;71:23–27. doi: 10.1021/cen-v071n022.p023. [DOI] [Google Scholar]
- Iram SH, Cronan JE. The beta-oxidation systems of Escherichia coli and Salmonella enterica are not functionally equivalent. J. Bacteriol. 2006;188:599–608. doi: 10.1128/JB.188.2.599-608.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazakov AE, Rodionov DA, Alm E, Arkin AP, Dubchak I, Gelfand MS. Comparative genomics of regulation of fatty acid and branched-chain amino acid utilization in proteobacteria. J. Bacteriol. 2009;91:52–64. doi: 10.1128/JB.01175-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla C, Bailey JE. Characterization of the oxygen-dependent promoter of the Vitreoscilla hemoglobin gene in Escherichia coli. J. Bacteriol. 1989;171:5995–6004. doi: 10.1128/jb.171.11.5995-6004.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla C, Curtis JE, Bydalek P, Swartz JR, Bailey JE. Expression of Recombinant Proteins in Escherichia Coli Using an Oxygen-Responsive Promoter. Nat. Biotechnol. 1990 doi: 10.1038/nbt0690-554. [DOI] [PubMed] [Google Scholar]
- Kim S, Clomburg JM, Gonzalez R. Synthesis of medium-chain length (C6-C10) fuels and chemicals via β-oxidation reversal in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2015;42:465–475. doi: 10.1007/s10295-015-1589-6. [DOI] [PubMed] [Google Scholar]
- Kim S, Gonzalez R. Selective production of decanoic acid from iterative reversal of β-oxidation pathway. Biotechnol. Bioeng. 2018;115:1311–1320. doi: 10.1002/bit.26540. [DOI] [PubMed] [Google Scholar]
- Korz DJ, Rinas U, Hellmuth K, Sanders E. a, Deckwer WD. Simple fed-batch technique for high cell density cultivation of Escherichia coli. J. Biotechnol. 1995;39:59–65. doi: 10.1016/0168-1656(94)00143-z. [DOI] [PubMed] [Google Scholar]
- Lara AR, Jaén KE, Sigala J-C, Mühlmann M, Regestein L, Büchs J. Characterization of Endogenous and Reduced Promoters for Oxygen-Limited Processes Using Escherichia coli. ACS Synth. Biol. 2017;6:344–356. doi: 10.1021/acssynbio.6b00233. [DOI] [PubMed] [Google Scholar]
- Lennen RM, Pfleger BF. Engineering Escherichia coli to synthesize free fatty acids. Trends Biotechnol. 2012;30:659–67. doi: 10.1016/j.tibtech.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lin Z, Huang C, Zhang Y, Wang Z, Tang Y, Chen T, Zhao X. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab. Eng. 2015;31:13–21. doi: 10.1016/j.ymben.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Machado HB, Dekishima Y, Luo H, Lan EI, Liao JC. A selection platform for carbon chain elongation using the CoA-dependent pathway to produce linear higher alcohols. Metab. Eng. 2012;14:504–11. doi: 10.1016/j.ymben.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Mosberg JA, Lajoie MJ, Church GM. Lambda Red Recombineering in Escherichia coli Occurs Through a Fully Single-Stranded Intermediate. Genetics. 2010;186:791–799. doi: 10.1534/genetics.110.120782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutalik VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, Mai Q-A, Tran AB, Paull M, Keasling JD, Arkin AP, Endy D. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013;10:354–60. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- Orth JD, Conrad TM, Na J, Lerman J. a, Nam H, Feist AM, Palsson BØ. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Mol. Syst. Biol. 2011;7:1–9. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger BF, Gossing M, Nielsen J. Metabolic engineering strategies for microbial synthesis of oleochemicals. Metab. Eng. 2015;29:1–11. doi: 10.1016/j.ymben.2015.01.009. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JM, Gordon GC, Mehrer CR, Pfleger BF. Genome sequence and analysis of Escherichia coli production strain LS5218. Metab. Eng. Commun. 2017;5:78–83. doi: 10.1016/j.meteno.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royce LA, Yoon JM, Chen Y, Rickenbach E, Shanks JV, Jarboe LR. Evolution for exogenous octanoic acid tolerance improves carboxylic acid production and membrane integrity. Metab. Eng. 2015;29:180–188. doi: 10.1016/j.ymben.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Rutter CD, Rao CV. Production of 1-decanol by metabolically engineered Yarrowia lipolytica. Metab. Eng. 2016;38:139–147. doi: 10.1016/j.ymben.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning : a laboratory manual. Cold Spring Harbor Laboratory Press; 2001. (David W. [Google Scholar]
- Schümperli D, McKenney K, Sobieski DA, Rosenberg M. Translational coupling at an intercistronic boundary of the Escherichia coli galactose operon. Cell. 1982;30:865–71. doi: 10.1016/0092-8674(82)90291-4. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CR, Lan EI, Dekishima Y, Baez A, Cho KM, Liao JC. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl. Environ. Microbiol. 2011;77:2905–2915. doi: 10.1128/AEM.03034-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539–539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen EJ, Kang Y, Bokinsky G, Hu Z, Schirmer A, McClure A, Del Cardayre SB, Keasling JD. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature. 2010;463:559–562. doi: 10.1038/nature08721. [DOI] [PubMed] [Google Scholar]
- Tu Q, Yin J, Fu J, Herrmann J, Li Y, Yin Y, Stewart AF, Müller R, Zhang Y. Room temperature electrocompetent bacterial cells improve DNA transformation and recombineering efficiency. Sci. Rep. 2016;6:1–8. doi: 10.1038/srep24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlen BD, Oswald WS, Seefeldt LC, Barney BM. Purification, characterization, and potential bacterial Wax production role of an nadph-dependent fatty aldehyde reductase from Marinobacter aquaeolei VT8. Appl. Environ. Microbiol. 2009;75:2758–2764. doi: 10.1128/AEM.02578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis RM, Wahlen BD, Seefeldt LC, Barney BM. Characterization of a fatty acyl-CoA reductase from Marinobacter aquaeolei VT8: A bacterial enzyme catalyzing the reduction of fatty acyl-CoA to fatty alcohol. Biochemistry. 2011;50:10550–10558. doi: 10.1021/bi2008646. [DOI] [PubMed] [Google Scholar]
- Youngquist JT, Lennen RM, Ranatunga DR, Bothfeld WH, Ii WDM, Pfleger BF. Kinetic modeling of free fatty acid production in Escherichia coli based on continuous cultivation of a plasmid free strain. Biotechnol. Bioeng. 2012;109:1518–1527. doi: 10.1002/bit.24420. [DOI] [PubMed] [Google Scholar]
- Youngquist JT, Schumacher MH, Rose JP, Raines TC, Politz MC, Copeland MF, Pfleger BF. Production of medium chain length fatty alcohols from glucose in Escherichia coli. Metab. Eng. 2013;20:177–86. doi: 10.1016/j.ymben.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.