

Abstract
Radiotherapy (RT) used at immunogenic doses leads to accumulation of cytosolic double-stranded DNA (dsDNA) in cancer cells, which activates type I IFN (IFN-I) via the cGAS/STING pathway. Cancer cell-derived IFN-I is required to recruit BATF3-dependent dendritic cells (DCs) to poorly immunogenic tumors and trigger antitumor T-cell responses in combination with immune checkpoint blockade. We have previously demonstrated that the exonuclease TREX1 regulates radiation immunogenicity by degrading cytosolic dsDNA. Tumor-derived DNA can also activate cGAS/STING-mediated production of IFN-I by DCs infiltrating immunogenic tumors. However, how DNA from cancer cells is transferred to the cytoplasm of DCs remains unclear. Here, we showed that tumor-derived exosomes (TEX) produced by irradiated mouse breast cancer cells (RT-TEX) transfer dsDNA to DCs and stimulate DC upregulation of costimulatory molecules and STING-dependent activation of IFN-I. In vivo, RT-TEX elicited tumor-specific CD8+ T-cell responses and protected mice from tumor development significantly better than TEX from untreated cancer cells in a prophylactic vaccination experiment. We demonstrated that the IFN-stimulatory dsDNA cargo of RT-TEX is regulated by TREX1 expression in the parent cells. Overall, these results identify RT-TEX as a mechanism whereby IFN-stimulatory dsDNA is transferred from irradiated cancer cells to DCs. We have previously shown that the expression of TREX1 is dependent on the RT dose size. Thus, these data have important implications for the use of RT with immunotherapy.
Keywords: Cytosolic DNA, radiotherapy, STING, tumor-specific T cells, vaccination
INTRODUCTION
It is well-recognized that harnessing the power of the immune system with immune checkpoint blocking antibodies (ICBs) leads to the successful elimination of cancer in a subset of patients across different malignancies. However, for the majority of patients, additional interventions are required to elicit sufficient antitumor T cells and achieve responses to ICBs (1). Among them, tumor-targeted radiation therapy (RT) has shown synergy with ICB in pre-clinical studies and promising results in patients, and is currently under investigation in several trials (2,3). Priming of antitumor T cells by RT is due, at least in part, to its ability to induce an immunogenic cell death (ICD) of the cancer cells (4,5). ICD is a type of regulated cell death associated with the exposure and release of danger signals that activate DCs to uptake and cross-present tumor antigens to T cells (6). One of the critical signals for the spontaneous activation of antitumor T cells against immunogenic tumors, as well as RT-induced T-cell priming, is dsDNA (7,8). Cytosolic tumor-derived dsDNA is sensed by cyclic GMP-AMP synthase (cGAS) in DCs to generate cGAMP required for the activation of the adaptor STING (stimulator of interferon genes), resulting in the production of IFNβ and induction of several interferon-stimulated genes (ISGs) (7). DC phagocytosis of the cancer cells with the blockade of the CD47-signal regulatory protein alpha (SIRPα) axis was shown to result in the release of DNA into the DC cytosol (9). However, the mechanisms whereby dsDNA derived from the cancer cells reaches the cytosol of DCs in the absence of CD47-SIRPα remain incompletely understood.
Exosomes are membrane microvesicles (30–100 nm) secreted from all cell types, which provide a sophisticated means of local and distal intercellular communication (10). Tumor-derived exosomes (TEX) have been shown to shuttle a variety of bioactive molecules between cells, including proteins, miRNAs, and mRNAs (11–15). Importantly, TEX also carry dsDNA, which is present in cancer cell-derived exosomes in variable but larger quantities than in exosomes derived from normal cells (16). Overall, the molecular profiles of exosomes reflect their cell of origin. For instance, TEX carry tumor antigens that can be transferred to DCs and elicit tumor-specific immune responses (14). However, for the most part, TEX have been shown to promote immunosuppressive and pro-tumorigenic effects, a dichotomy that may depend on the molecular context of the parent and recipient cells (13,17,18). Notably, it has been shown that treatment-induced molecular changes in cancer cells can lead to the production of TEX with similarly altered cargo that can be transferred to recipient cells (19–21). Thus, we hypothesized that key molecular changes that occur in cancer cells treated with radiation may be detected in the TEX that they produce and result in enhanced TEX immunogenicity.
Here, we demonstrated that TEX released from TSA mouse breast carcinoma cells irradiated with 8Gy X 3 (RT-TEX), an optimally immunogenic dose and fractionation for this tumor (22,23), have an altered molecular composition compared to TEX from untreated cells (UT-TEX). We have previously shown that 8 Gy X 3 RT induces the accumulation of dsDNA in the cytosol of TSA cells that activates IFN-I pathway via cGAS/STING (23). Consistent with our hypothesis, only RT-TEX carried dsDNA capable of inducing IFN-I production by DCs in a STING-dependent fashion. IFN-stimulatory dsDNA carried by RT-TEX was regulated by the expression of the three-prime repair exonuclease 1 (TREX1) in the parent cells, and RT-TEX elicited significantly better protective antitumor immunity than UT-TEX when used to vaccinate mice. Overall, these results provide a mechanism whereby dsDNA derived from irradiated cancer cells reaches the cytoplasm of DCs and highlight the importance of TREX1 as a regulator of RT immunogenicity (24).
METHODS
Mice
Six-week-old BALB/c and C57BL/6 female mice, and B6(Cg)-Tmem173tm1.2Camb/J (STING-deficient) mice were purchased from Jackson Laboratories. All mice were maintained under pathogen-free conditions in the animal facility at Weill Cornell Medicine (New York, NY). All experiments were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine.
Tumor cell culture
TSA is a BALB/C mouse–derived mammary carcinoma (provided by P.L. Lollini, University of Bologna, Bologna, Italy) (25). A20 is a BALB/C mouse–derived B-cell leukemia/lymphoma (provided by G. Inghirami, Weill Cornell Medicine, New York, NY, USA) (26). B16.Flt3L cells, genetically engineered to stably secrete Flt3-ligand (27), were a gift from B. Reizis (NYU School of Medicine, New York, NY USA). TSA cell derivatives containing a doxycycline-inducible Trex1 gene (TSAKI Trex1), TSA cells expressing a doxycycline-inducible shRNA non-silencing construct (TSAshNS), and shRNA constructs targeting cGAS (TSAshcGAS ) and STING (TSAshSTING) were previously described (23). Cells were authenticated by morphology, phenotype, and growth, and routinely screened for Mycoplasma (LookOut® Mycoplasma PCR Detection kit, Sigma-Aldrich). Cells were cultured in DMEM (Invitrogen Corporation) supplemented with L-glutamine (2 mmol/L), penicillin (100 U/mL), streptomycin (100 μg/mL), 2-mercapthoethanol (2.5 × 105 mol/L), and 10% FBS (Life technologies). For B16.Flt3L culture, non-essential amino acids (Invitrogen Corporation) and sodium pyruvate (1 mmol/L; Invitrogen Corporation) were added. Cells were cultured for the minimum time required to achieve sufficient expansion, approximately one week or 3–5 passages for the preparation of exosome stocks in various experiments.
Exosome Isolation and Purification
TSA cells were cultured in complete medium as described above. To induce the expression of TREX1 or to induce the knockdown of cGAS and STING, 7×105 TSAKI Trex1, TSAshNS, TSAshcGAS, and TSAshSTING cells were plated in T75 flask in media containing doxycycline (4 μg/mL) 4 days prior to RT, as previously described (23). Cells either received Sham RT or 8 Gy on 3 consecutive days using the Small Animal Radiation Research Platform (SARRP Xstrahl Ltd). Following the last radiation exposure, culture medium was replaced with DMEM containing antibiotic solutions as outlined above and 10% exosome-depleted FBS (Exo-FBS, System Biosciences, Inc.). Supernatants from sham- or RT-treated cells were collected 48 hours later, and exosomes were isolated by sequential centrifugation as previously described (28). Briefly, cell supernatant was sequentially centrifuged at 2000 x g for 20 minutes at 4°C and 10,000 x g for 30 minutes at 4°C to remove cells and cellular debris. The supernatant was then centrifuged at 100,000 x g for 70 minutes at 4°C, and the pellet containing exosomes was collected, washed in cold PBS (Invitrogen), and centrifuged again at 100,000 x g for 70 minutes at 4°C. The washed exosomes were further purified by a sucrose step gradient as described (29). Total exosomal protein was measured by the Bradford Protein Assay (Bio-Rad). The yield was 0.4– 0.6 μg TEX/106 TSA cells. To confirm the morphological appearance of purified TEX by transmission electron microscopy (TEM), 5 μL of TEX in PBS were placed onto glow-discharged (Bench Top Turbo, Denton Vacuum, Morristown, NJ) home-made carbon coated 400 mesh Cu/Rh grid (Ted Pella Inc., Redding, CA). After staining with 1% uranyl acetate in distill water (Polysciences, Inc, Warrington, PA), the grids were examined under Philips CM-12 electron microscope and photographed with a Gatan (4k ൲2.7k) digital camera.
Mass spectrometry
TEX protein samples were prepared in biological triplicate for each group (0Gy TEX and RT-TEX), with 400 μg of exosomal protein per sample. Briefly, sucrose-purified TEX were lysed in SDS detergent containing buffer and prepared for mass spectrometric analysis using Filter Assisted Sample Preparation procedure (30). The tryptic peptides were eluted, desalted, and aliquots of the peptide mixtures were injected onto an Acclaim PepMap 100 (2 cm x 75 μm ID) trap column in line with an EASY Spray column (50 cm x 75 μm ID; PepMap RSLC C18, 2 μm) with the autosampler of an EASY nLC 1000 interfaced to a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific). Peptides were gradient eluted using the following gradient (solvent A: 2% acetonitrile in 0.5% acetic acid and solvent B: 90% acetonitrile in 0.5% acetic acid): 5–40% in 60 minutes, 40–100% in 10 minutes, and followed by 100% for 20 minutes. High resolution full MS spectra were acquired with a resolution of 70,000, an AGC target of 1e6, with a maximum ion time of 120 ms, and scan range of 400 to 1500 m/z. Following each full MS scan, 20 high-resolution HCD MS/MS spectra were acquired. The MS/MS spectra were collected at a resolution of 17,500, an AGC target of 5e4, maximum ion time of 120 ms, one microscan, 2.0 m/z isolation window, fixed first mass of 150 m/z, dynamic exclusion of 30 s and a Normalized Collision Energy (NCE) of 27.
Peptide identification, protein grouping, and protein quantitation was performed using the label-free quantitation (LFQ) algorithm in the MaxQuant software suite version 1.5.0.12 searched against a Uniprot Mus musculus database (31,32). For the first search, the peptide mass tolerance was set to 20 ppm, and for the main search, peptide mass tolerance was set to 4.5 ppm. Trypsin-specific cleavage was selected with two missed cleavages. Both peptide spectral match and protein FDR were set to 1% for identification. Carbamidomethylation of cysteine was added as a static modification. Oxidation of methionine and acetylation of N-termini were allowed as variable modifications. Protein quantitation was performed using unique and razor peptides. The data set was filtered to include proteins identified with two or more unique and/or razor peptides and proteins that were detected in all three replicates of at least one treatment. A two-sided Student’s t-test was performed, correcting for multiple testing by controlling for FDR at 5% using a permutation method. Proteins with a q value <0.05 were considered significant. 684 proteins met the filter requirements, and of those, 158 were defined as significant by a q value <0.05. Proteomics data have been uploaded to the MassIVE (accession number MSV000081341) and ProteomeXchange (accession number PXD007069) repositories.
Pathway analysis
QIAGEN’s Ingenuity Pathway Analysis software (IPA; Ingenuity Systems, Redwood City, CA, USA) was used to determine upstream regulators and impact on canonical pathways (Fisher’s exact test, p values <0.05). A pathway or upstream regulator was determined activated or inhibited for |z-scores| ≥ 2 (positive z-score: activation; negative z-score: inhibition).
TEX vaccination and tumor challenge
BALB/c mice were vaccinated s.c. near the base of the tail with TEX (20 μg/mouse, n=6/group). Mice received boost vaccinations 7 and 14 days later. PBS was used for control mice. Seven days after the last immunization, mice were challenged s.c. with 1 × 105 TSA tumor cells. Tumor growth was monitored 2–3 times weekly for 4 weeks. Perpendicular tumor diameters were measured with a Vernier caliper, and tumor volumes were calculated as length × width2 × 0.52. Mice were sacrificed 20 days after tumor inoculation for analysis of the immune infiltrate.
Analysis of tumor-infiltrating CD8+ T cells
Tumors were enzymatically digested using Miltenyi’s Mouse Tumor Dissociation Kit on a gentleMACS Octo Dissociator (Miltenyi Biotec). Following dissociation, red blood cell lysis was performed, and the cell suspension was filtered through a 40 μM strainer to remove large debris. Cells were blocked with CD16/32 antibody to Fc gamma III and II receptors (Thermo Fisher Scientific, Clone 93) and were then stained for flow cytometry analysis using Fixable Viability Dye eFluor® 506, (Thermo Fisher Scientific), APC-Cy7–conjugated anti-CD45 (Affymetrix – eBioscience; Clone I3/2.3), VioBlue-conjugated anti-CD8a (Miltenyi Biotec; Clone 53–6.7), and R-PE–labeled Pro5® MHC class I pentamers linked to the MuLV env gp70 423–431 (AH1 peptide, SPSYVYHQF; ProImmune Ltd.). Control pentamers were loaded with irrelevant MCMV IE1 168–176 peptide (YPHFMPTNL). After staining, cells were fixed with 4% paraformaldehyde, and samples analyzed using a MACSQuant® Analyzer 10 (Miltenyi Biotec), and FlowJo software (version 8.8.7). Cell density was calculated by dividing the total number of cells staining positive for a given marker for the tumor volume.
Analysis of tumor-reactive CD8+ T cells from spleen
T cells were isolated from the spleen of TSA tumor-bearing mice using a Pan T-Cell Isolation Kit II (Miltenyi Biotec), following manufacturer’s protocol. 1 × 106 T cells were then cultured with Mouse T-Activator CD3/CD28 Dynabeads® (ThermoFisher Scientific) in 1 mL T-cell medium supplemented with human rIL2 (10 U/mL; provided by the Biological Resources Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute) for 4 days in a 24-well tissue culture plate. Beads were removed by magnetic separation, and activated T cells were then cultured with 1 × 104 irradiated TSA or A20 lymphoma cells at effector to target ratio 20:1 for 4 hours. GolgiPlug™ containing Brefeldin A (1uL per 1mL culture media, BD Biosciences) was added for 8 hours to inhibit intracellular protein transport. T cells were then washed and stained with Fixable Viability Dye eFluor® 506 (Thermo Fisher Scientific). Cells were then stained for surface markers with APCcy7- conjugated anti-CD45 (Affymetrix – eBioscience; Clone I3/2.3) and Vio-Blue-conjugated anti-CD8a (Miltenyi Biotec, Clone 53–6.7). Intracellular staining was then performed for IFNγ using an Intracellular Fixation & Permeabilization Buffer Set (Thermo Fisher Scientific) according to manufacturer’s protocol followed by staining with FITC-conjugated anti-IFNγ (Thermo Fisher Scientific; Clone XMG1.2). Cells were analyzed as described above.
TEX labeling and uptake kinetics by DC in vivo
Purified TEX were fluorescently labeled with PKH67 membrane dye (Sigma-Aldrich), following the manufacturer’s protocol. Labeled exosomes were washed in 30 mL of exosome-free media and collected by ultracentrifugation at 100,000 x g for 70 minutes at 4°C. Absence of dye contamination was verified using a PBS control. 10 μg of labeled TEX were injected s.c. near the base of the tail. The draining inguinal lymph node was harvested 24, 48, or 72 hours post-injection for flow cytometric analysis (n=3 mice/group at each time point). Cell suspensions were stained with BV421-conjugated anti-CD11c (BioLegend; Clone N418), and analyzed as described above.
Isolation of primary DCs
To improve the yield of DCs, C57BL/6 female mice were injected s.c. with 7 X 105 B16.Flt3L, a cell line genetically engineered to stably secrete fms-related tyrosine kinase 3 ligand (Flt3L) (27,33). Spleens were harvested 14 days later and processed as described (34). Briefly, to prepare single cell suspensions, spleens were minced in RPMI, and the obtained mixture was filtered using a Falcon 40 μM strainer (Corning). Following red blood cell lysis by ACK buffer, the cell suspension was filtered again. After washing in PBS, cells were blocked with CD16/32 antibody to Fc gamma III and II receptors (Thermo Fisher Scientific; Clone 93), followed by. incubation with CD11c microbeads ultrapure (Milteny Biotec) and loaded onto a MACS column (Milteny Biotec) for the positive selection, as outlined in the kit protocol.
DC culture with TEX
Primary CD11c+ DCs were cultured with purified TEX (30 μg TEX per 1 X 106 DCs) or with TLR3 agonist Poly:ICLC (0.025 mg/mL; Sigma Aldrich) for 48 hours in medium supplemented with exosome-depleted FBS. Cells were collected for analysis by flow cytometry and quantitative PCR with reverse transcription (qRT-PCR), as described below. Cell culture supernatants were collected for measurement of secreted IFNβ using High Sensitivity Mouse IFNβ ELISA Kit (PBL Assay Science).
For phenotypic analysis, 2 X 105 DCs were washed and stained with Fixable Viability Dye (eFluor® 506, Thermo Fisher Scientific), FITC–anti-CD11c (Thermo Fisher Scientific; Clone N418), PE–anti-CD40 (Thermo Fisher Scientific; Clone 1C10), PE-Cy7–anti-CD80 (Thermo Fisher Scientific; Clone 16–10A1), and Pacific Blue–anti-CD86 (Thermo Fisher Scientific; Clone GL1), acquired by a MACSQuant® Analyzer (Miltenyi Biotec), and analyzed with FlowJo software (version 8.8.7).
Quantitative PCR with reverse transcription (qRT-PCR)
Total RNA from DCs was isolated using RNeasy Micro Kit (Qiagen). Real-time PCR was performed using the Applied Biosystems® 7500 real-time PCR cycler (Thermo Fisher). One microgram of RNA was used for cDNA synthesis, performed with SuperScript® IV VILO™ Master Mix (Thermo Fisher Scientific), followed by real-time RT-PCR with DyNAmo Flash SYBR Green qPCR Kit (Thermo Fisher Scientific), according to manufacturer’s protocol. Samples were normalized to housekeeping genes, and expression on untreated cells was assigned a relative value of 1.0. The PrimePCR™ SYBR® Green assay primers (BioRad) used in this study were UniqueAssay ID: qMmuCED0050444 for mouse IFNβ1, qMmuCID0023356 for mouse Mx1, qMmuCED0046382 for mouse Ifnar1, and qMmuCED0027497 for mouse Gapdh. Data were analyzed with the 7500 Dx Instrument’s Sequence Detection software (Thermo Fisher). To calculate the relative gene expression, the 2(−ΔΔCt) method was used. Experiments were performed in biological triplicate, and statistical significance was assessed using unpaired, two-tailed t-test.
Quantification of exosomal dsDNA
dsDNA was quantified from 5 ug of TEX, with or without DNase I pre-treatment (0.15 units/μL, Sigma-Aldrich) using the SpectraMax® Quant™ AccuBlue™ Pico dsDNA Assay Kit (Molecular Devices) as described (23). The samples were prepared according to manufacturer’s protocol and analyzed using the FlexStation 3 multi-mode microplate reader.
Statistics
Data are presented as mean ± standard error of the mean (s.e.m.). For comparisons with only two groups, p values were calculated using unpaired Student’s two-tailed t-tests. The two-way Analysis of Variance (ANOVA) test was used for tumor growth curve analyses. The s.e.m. are indicated by errors bars for each group of data. Differences were considered significant at p values below 0.05. All data were analyzed using GraphPad Prism software (GraphPad version 7).
RESULTS
Exosomes secreted by irradiated cancer cells show an altered proteomic profile
TEX can carry multiple tumor antigens and deliver them to recipient DCs for presentation to T cells (14). However, TEX have limited immunogenicity, which can be increased by modification of the parent cells, for example, by heat stress (35), suggesting that increased adjuvanticity of cancer cells can be transferred to the exosomes they secrete.
We have shown that RT can increase tumor immunogenicity and induce the generation of tumor-specific CD8+ T cells that mediate the regression of the irradiated tumors, as well as synchronous non-irradiated tumors in synergy with ICB (22). Because the pro-immunogenic effects of RT are dependent on the dose and fractionation (23), for these experiments, we used an optimal RT regimen of 8 Gy X 3 to irradiate the mouse breast cancer cells TSA and test whether this treatment altered the molecular composition of the secreted TEX. TEX were isolated from the supernatants of untreated and RT-treated TSA cells using a two-step procedure to achieve a high purity and their morphology verified by electron microscopy (Fig. 1A). Next, the proteomic profile of RT-TEX was compared to UT-TEX using label-free quantitation mass spectrometry (LFQ-MS). This analysis revealed significant differences between the two groups, with RT-TEX containing 114 proteins not detected in UT-TEX (Supplementary Table S1 and Supplementary Excel Sheet 1; entire data set available at ProteomeXchange ID: PXD007069). 27.6% of the shared proteins were found at significantly different levels in the two groups (q<0.05). To understand the biological context of the differences between UT-TEX and RT-TEX, Ingenuity Pathway Analysis (IPA) tool was used to compare all the proteins identified in each TEX group. Whereas 153 pathways were shared between the two groups, 10 pathways were unique to RT-TEX and 15 pathways were unique to UT-TEX (Fig. 1B). Interferon signaling was one of the 10 pathways unique to RT-TEX (Table 1), suggesting that RT-TEX cargo reflects the cellular environment of irradiated TSA cells, which display cancer cell intrinsic IFN-I pathway activation (23). Finally, among the 153 shared pathways, 21 were predicted to be activated in RT-TEX compared to UT-TEX, whereas only 3 pathways were predicted to be inhibited (Supplementary Table S2).
Figure 1. Isolation and proteomic profiling of TEX.
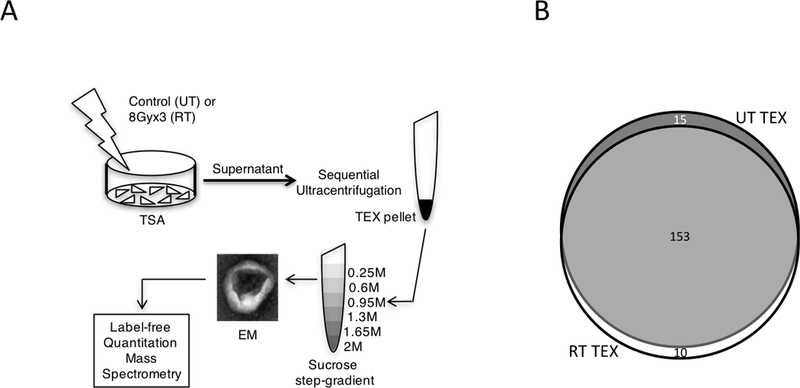
(A) Schema of TEX isolation method. TSA mouse breast cancer cells were left untreated (UT) or irradiated with 8 Gy X 3 (RT) and supernatants were collected after 48 hours (h) for TEX purification as indicated. Purified TEX were analyzed by transmission electron microscopy (EM) and analyzed by LFQ-MS. (B) Venn diagram illustrating the number of shared and unique pathways represented in each TEX group. White: RT-TEX; Dark grey: UT-TEX; Light gray: Overlap.
Table 1.
Pathways unique to UT-TEX and RT-TEX
| UT-TEX | p-value | RT-TEX | p-value |
|---|---|---|---|
| CTLA4 Signaling in Cytotoxic T Lymphocytes | 0.0267 | Folate Polyglutamylation | 0.0089 |
| Role of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis | 0.0271 | Methylglyoxal Degradation III | 0.0121 |
| Non-Small Cell Lung Cancer Signaling | 0.0321 | Pentose Phosphate Pathway (Non-oxidative Branch) | 0.0131 |
| Cytotoxic T Lymphocyte-mediated Apoptosis of Target Cells | 0.0365 | Interferon Signaling | 0.0242 |
| IL3 Signaling | 0.0423 | Cardiac beta-adrenergic Signaling | 0.0243 |
| Oncostatin M Signaling | 0.0426 | Folate Transformations I | 0.0297 |
| Aryl Hydrocarbon Receptor Signaling | 0.0431 | Prostanoid Biosynthesis | 0.0297 |
| nNOS Signaling in Skeletal Muscle Cells | 0.0454 | Neuroprotective Role of THOP1 in Alzheimer’s Disease | 0.0342 |
| Fatty Acid Biosynthesis Initiation II | 0.0454 | Antioxidant Action of Vitamin C | 0.0404 |
| Palmitate Biosynthesis I (Animals) | 0.0454 | CD28 Signaling in T Helper Cells | 0.0468 |
| Glycine Biosynthesis I | 0.0454 | ||
| S-methyl-5’-thioadenosine Degradation II | 0.0454 | ||
| Nitric Oxide Signaling in the Cardiovascular System | 0.0462 | ||
| Ephrin A Signaling | 0.0490 | ||
| HIF1a Signaling | 0.0496 |
STING-dependent activation of dendritic cells by exosomes from irradiated cancer cells
IFN-I is essential for activation of DCs and priming of antitumor T cells (36,37). Because immunogenic RT induces IFN-I production by cancer cells (23), and the proteomics analysis indicated that IFN-I pathway was selectively upregulated in RT-TEX, we next tested the effects of RT-TEX on DC activation. To this end, primary CD11c+ DCs were isolated from the spleens of mice and cultured ex vivo with TEX for 48hrs. DCs cultured with RT-TEX or with the powerful TLR3 agonist poly:ICLC showed significantly increased cell surface expression of co-stimulatory molecules CD40, CD80, and CD86 (Fig. 2A and B). RT-TEX and poly:ICLC also induced expression of Ifnβ, Mx1, and Ifnar1 genes and secretion of IFNβ by DCs (Fig. 2C and D). In contrast, UT-TEX were unable to induce DC activation and IFNβ production (Fig. 2A-D). Thus, RT-TEX exhibited adjuvant properties that were not seen in UT-TEX.
Figure 2. RT-TEX induce activation and IFN-I production by primary DCs.
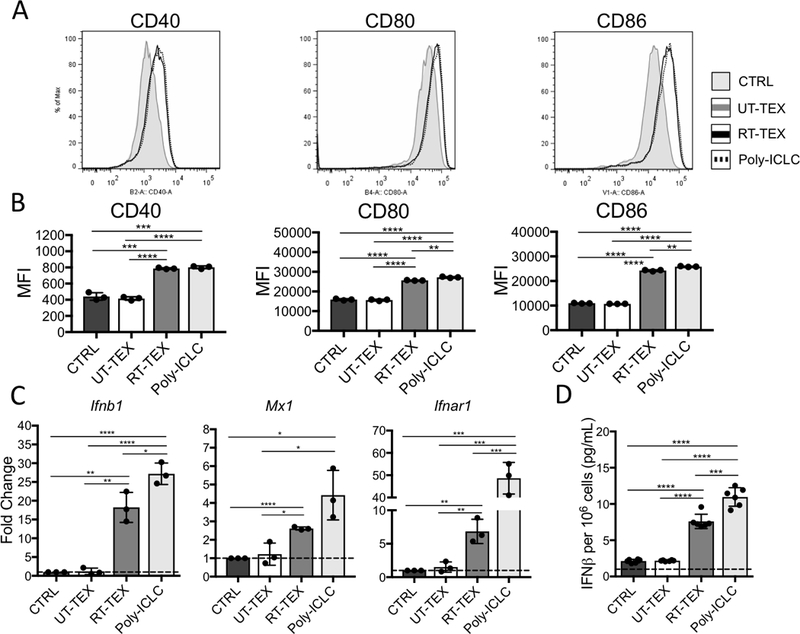
1 × 106 CD11c+ DCs isolated from spleens were cultured for 48h with TEX (30 μg) or TLR3 agonist poly:ICLC (0.025 mg/mL) and analyzed for (A, B) expression of co-stimulatory molecules and (C, D) IFN-I pathway activation. (A) Histograms showing co-stimulatory molecule expression on DCs cultured with PBS (CTRL: shaded gray), UT-TEX (gray line), RT-TEX (black line), and poly:ICLC (black broken line). (B) Mean fluorescence intensity (MFI)±s.e.m. of samples in each group (n=3/group). MFI was calculated after subtracting background. (C) Gene expression evaluated by qRT-PCR. (D) IFNβ measured by ELISA in supernatant of DCs. Results are representative of two independent experiments. Using a Student’s two-tailed t-test, *p<0.05; **p<0.005; ***p<0.0005; ****p<0.00005.
The STING pathway has been implicated in induction of IFN-I production by DCs exposed to irradiated cancer cells (8). To determine whether RT-TEX activate DCs through this pathway, TEX were cocultured with primary DCs isolated from STING-deficient (STING–/–) mice. STING–/– DCs were able to upregulate Ifnb1, Mx1, and Ifnar1 and produce IFNβ when stimulated with poly:ICLC. However, they failed to respond to RT-TEX, indicating that STING is an essential mediator of the adjuvant activity of RT-TEX (Fig. 3).
Figure 3. IFN-I pathway activation by RT-TEX is STING-dependent.
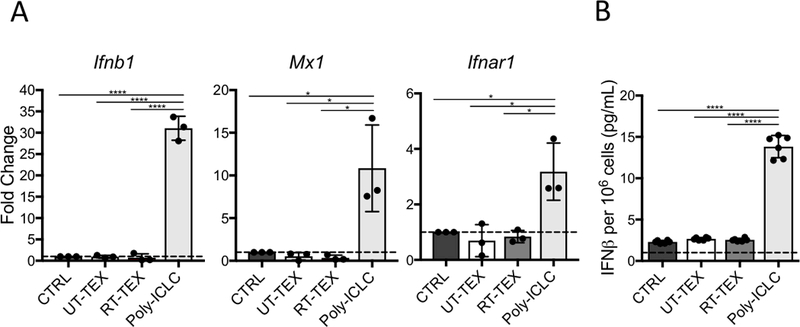
1×106 CD11c+ DCs isolated from spleens of STING-deficient mice were cultured for 48h with TEX (30 μg) or TLR3 agonist poly:ICLC (0.025 mg/mL) and analyzed for IFN-I pathway activation. (A) Gene expression evaluated by qRT-PCR. (B) IFNβ measured by ELISA in supernatant of DCs. Results are representative of two independent experiments. Using a Student’s two-tailed t-test, *p<0.05; ****p<0.00005.
TREX1-sensitive dsDNA carried by exosomes causes IFN-I activation in recipient DCs
Cancer cell-derived DNA has been shown to be responsible for the STING-mediated activation of IFN-I in DCs (7,8). DsDNA can be present inside exosomes as well as associated with their outer membrane (16). To determine the amount and location of dsDNA carried by TEX, dsDNA was measured before and after treatment with DNase I. This revealed that a large portion of dsDNA in RT-TEX is external, surface-associated DNA, but RT-TEX also contain significantly more internal dsDNA compared to UT-TEX (Supplementary Fig. S1A). To determine if the external DNA was responsible for the induction of IFN-I in recipient DCs, RT-TEX were treated with DNase I before culture with DCs. No significant decreases in the ability of DNase-treated RT-TEX to induce IFN-I pathway activation or IFNβ secretion by DCs were seen compared to untreated RT-TEX (Supplementary Fig. S1B and C), demonstrating that external DNA did not contribute to the IFN-stimulatory effects of RT-TEX.
We have shown that 8 Gy X 3 RT induces the accumulation of dsDNA in the cytosol of the irradiated TSA cells, where it activates cGAS leading to downstream STING-mediated IFN-I induction. The IFN-stimulatory cytosolic dsDNA is degraded by TREX1, which is induced by single RT doses above 12 Gy (23). Thus, to test if the IFN-stimulatory dsDNA cargo of RT-TEX originates from the cytosolic fraction that is sensitive to TREX1, we used TSA cells transduced with a doxycycline-inducible Trex1 cDNA (TSAKI Trex1). Doxycycline treatment of these cells, which we have demonstrated abrogates the cytoplasmic accumulation of dsDNA induced by RT (23), decreased markedly the internal dsDNA cargo of RT-TEX (Fig. 4A), and abrogated RT-TEX’s ability to induce IFNβ secretion by DCs and Ifnb1, Mx1, Ifnar1 gene expression (Fig. 4B and C).
Figure 4. IFN-stimulatory dsDNA in RT-TEX is regulated by Trex1 expression in parent cells.
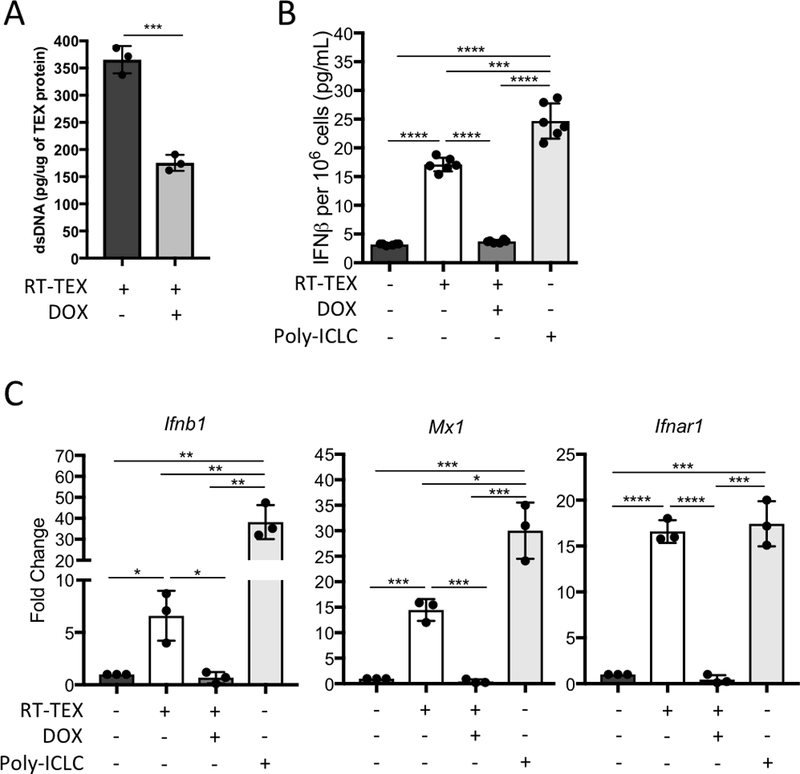
(A) Quantification of internal dsDNA carried by TEX secreted by irradiated TSAKI Trex1 cells treated or not with doxycycline (DOX), as indicated, to induce Trex1. (B, C) 1 × 106 CD11c+ DCs were cultured for 48h with TEX (30 μg) derived from TSAKI Trex1 cells treated or not with doxycycline (DOX; 4 μg/mL), as indicated, or with TLR3 agonist poly:ICLC (0.025 mg/mL) and analyzed for IFN-I pathway activation. (B) IFNβ measured by ELISA in supernatant of DCs. (C) Gene expression evaluated by qRT-PCR. Using a Student’s two-tailed t-test, *p<0.05; **p<0.005; ***p<0.0005; ****p<0.00005.
Because TREX1 removes the substrate for cGAS activation, we next asked if the abrogated IFN-stimulatory activity of RT-TEX produced by doxycycline-treated TSAKI Trex1 cells was entirely due to loss of dsDNA in the TEX cargo, or could it be explained by loss of cGAMP or other factors activated downstream of STING. To this end, RT-TEX were isolated from TSA cells with shRNA-mediated downregulation of cGAS or STING, and tested for the ability to activate DCs. RT-TEX derived from cGAS- or STING-deficient TSA cells were equally effective as RT-TEX from control TSA cells expressing a non-silencing construct at inducing the expression of Ifnar1 in recipient DCs and only slightly less effective at inducing Ifnb1 (Supplementary Fig. S2). Overall, these data demonstrate that dsDNA is responsible for the increased adjuvanticity of RT-TEX and is regulated by TREX1 in the parent cells.
Vaccination with RT-TEX induces protective antitumor immunity
Next, we tested if the increased adjuvanticity of RT-TEX results in improved induction of antitumor immune responses in vivo compared to UT-TEX. First, the ability of TEX injected subcutaneously to reach the DCs in the draining lymph node was evaluated. To this end, we used a well-established method to monitor uptake of PKH67-labelled exosomes in vivo (38). At 24hrs. post-injection, both UT-TEX and RT-TEX were taken up by DCs, but the percentage of DCs that acquired TEX, as indicated by PKH67-positivity, was slightly higher in mice injected with RT-TEX than with UT-TEX (mean 14% versus 9.5%, p<0.05; Fig. 5A and B). The percentage of positive DCs decreased similarly in both groups at later time points.
Figure 5. Uptake of TEX by DCs in vivo.
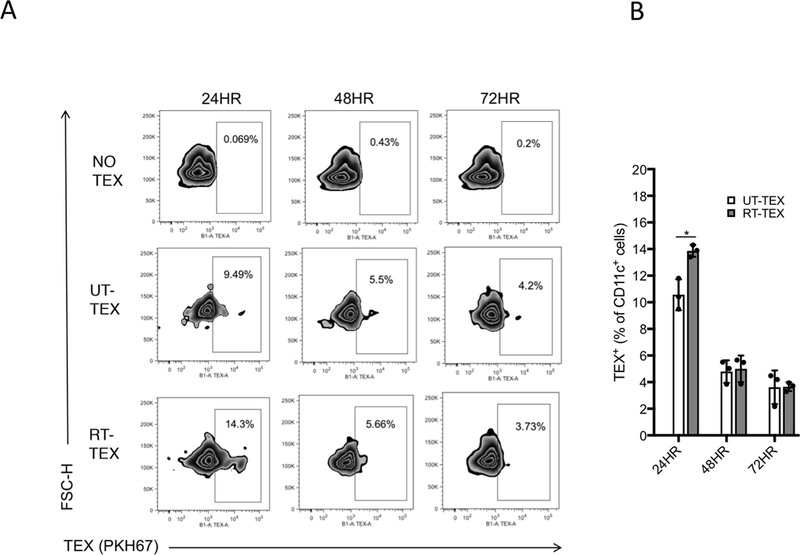
PKH67-labelled TEX (10 μg) were injected s.c. at the base of the tail, and the draining lymph nodes (DLN) were collected 24, 48, and 72h after injection (n=3/group per time point). DLN cells were stained with anti-CD11c to identify DCs. (A) Representative flow cytometry plots gated on CD11c+ DCs. Boxes encase PKH67+ DCs. (B) Mean percentage±s.e.m. of TEX+ CD11c+ cells at different times post-injection. Using a Student’s two-tailed t-test, *p<0.05.
Having confirmed that TEX injected subcutaneously could reach the DCs in the draining lymph nodes where T-cell priming occurs, mice were vaccinated three times with UT-TEX or RT-TEX, followed by challenge with live TSA cancer cells (Fig. 6A). All mice vaccinated with UT-TEX developed tumors, although tumor growth was slightly but significantly slower compared to control non-vaccinated mice (Fig. 6B and C). In contrast, 2/6 mice vaccinated with RT-TEX did not develop tumors, and the tumors in the remaining mice grew with a significantly larger delay compared to both control and UT-TEX vaccinated mice (Fig. 6B and C). The tumors with delayed growth contained on average more CD8+ T cells compared to untreated and UT-TEX-vaccinated mice, and a larger number among them were specific for an endogenous antigen that is immunodominant in TSA tumor (25) (Fig. 6D and E).
Figure 6. Vaccination with RT TEX induces protective antitumor immunity.
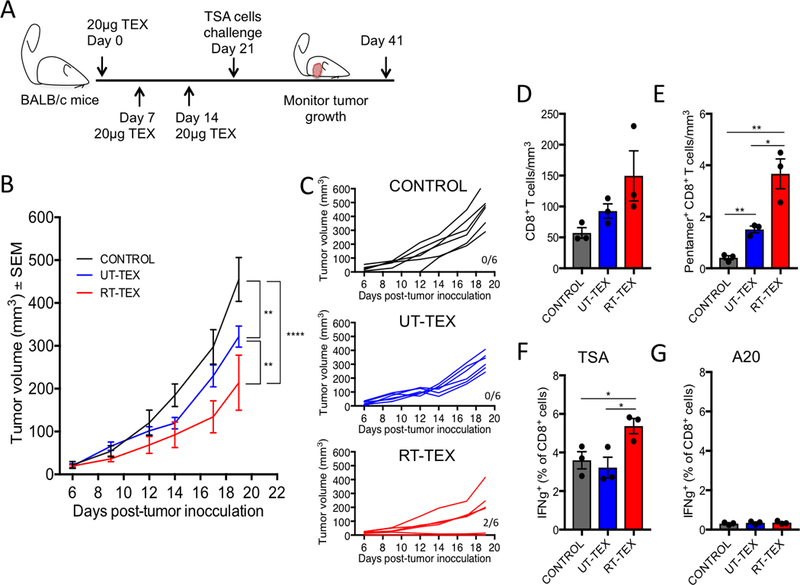
Mice were vaccinated s.c. with 20 μg TEX and received boost vaccinations 7 and 14 days later (20 μg/boost), followed by challenge with a tumorigenic inoculum of 1 × 105 TSA cells. (A) Experimental schema depicting vaccination and challenge schedule. (B) Tumor growth in each group post-challenge (n=6/group). (C) Individual mouse tumor growth curves. Numbers indicate mice without tumor/total mice. (D, E) Tumors infiltrating T cells analyzed at day 20 post challenge by flow cytometry. (D) Density of CD8+ T cells in each treatment group (n=3/group). (E) Density of tumor-specific CD8+ T cells as determined by Ld/AH1 pentamer staining. (F, G) Spleen T cells harvested at day 20 post challenge and tested for IFNγ production by intracellular staining after in vitro activation and incubation with (F) 1 × 104 of either TSA cells or (G) the irrelevant target A20 lymphoma cells. All data are representative of two independent experiments. Using a Student’s two-tailed t-test, *p<0.05; **p<0.005.
To assess for systemic tumor-specific T-cell responses, T cells isolated from spleen were activated for 4 days in vitro and then incubated with irradiated TSA cells or the unrelated A20 lymphoma cells for 3hrs., followed by analysis of IFNγ production by intracellular staining. A significant increase in TSA tumor-specific IFNγ+ CD8+ T cells was evident in mice vaccinated with RT-TEX compared to control and UT-TEX vaccinated mice, whereas no response was seen against A20 cells (Fig. 6F and G). Overall, these results indicated that RT-TEX were a more powerful vaccine than UT-TEX and elicited tumor-specific CD8+ T cells.
DISCUSSION
Exosomes can carry a variety of tumor antigens and are under investigation in the clinic as promising cell-free cancer vaccines (39). Exosomes secreted by cancer cells efficiently deliver tumor antigens to DCs (14), but their low/absent adjuvanticity and potential for immunosuppressive and pro-tumorigenic effects have hampered their use as vaccines (13,17,18). Because radiation can increase cancer cells’ adjuvanticity, we hypothesized that TEX produced by irradiated cancer cells would carry a cargo different from TEX produced by untreated cancer cells, and display increased immunogenicity. Proteomics analysis and vaccination experiments supported and validated both of these hypotheses.
Several proteins were found uniquely in RT-TEX, and others were present at different levels indicating that different pathways were represented and/or the represented pathways differed in their predicted functional orientation (activated versus inhibited) between RT-TEX and UT-TEX. We found that RT-TEX were capable of activating DCs, whereas UT-TEX were not. Thus, we further investigated the mechanisms underlying the increased adjuvanticity of RT-TEX. Depending on their nature, DAMPs are sensed by surface, endosomal, or cytoplasmic receptors that are shared with pathogen associated molecular patterns (PAMPs) and converge into activation of the IFN-I pathway in DCs (40). Various DAMPs are generated during ICD induced by cytotoxic treatments, including RT (6). However, evidence indicates that in in vivo irradiated tumor dsDNA, which induces IFN-I via the cGAS/STING pathway, is the critical signal for priming of antitumor T cells (8,23). We have shown that dsDNA accumulates in the cytosol of mouse and human cancer cells upon treatment with radiation and stimulates IFN-I release by the cancer cells via the cGAS/STING pathway. The released IFN-I signals to the cancer cells in an autocrine fashion, and to host DCs, increasing their recruitment to the tumor and inducing their activation (23). Here, we demonstrated that irradiated cancer cells could also signal by releasing TEX that carry the tumor dsDNA to the cytosol of DCs, leading to DCs activation and priming of antitumor T cells. Thus, RT-TEX provide a new mechanism whereby RT increases tumor DNA-mediated IFN-I activation in DCs.
Xu et al. (9) demonstrated that mitochondrial DNA from MC38 cells, which are phagocytosed by DCs when the “do not eat me” signal provided by CD47 is blocked, gains access to the DC cytosol and stimulates cGAS/STING. Thus, it is likely that different pathways mediate the transfer of tumor-derived DNA to DCs. TEX might have a more important role in delivering tumor-derived DNA to DCs in the draining lymph nodes, whereas tumor cell phagocytosis by DCs may underlie priming of spontaneous T-cell responses to immunogenic tumors that are infiltrated by sufficient DCs (7). The work of Xu et al. (9) also highlights the importance of other mechanisms, such as CD47/SIRPα, in regulating the access of the phagocytosed DNA to the cytosolic compartment. It remains to be ascertained if access to the DC cytosol of dsDNA contained in UT-TEX and RT-TEX is regulated by the same or similar mechanisms. Further experimentation will be required to determine the relative contribution of these different mechanisms of tumor DNA delivery to DCs for the radiation-induced in situ vaccination.
Variable but conspicuous amounts of dsDNA have been found in TEX produced by a number of cancer cells that were not treated with radiation (16). Similarly, UT-TEX contained some dsDNA but did not stimulate DC activation and IFN-I release. TREX1 upregulation in the parent cells reduced by approximately 50% but did not completely eliminate dsDNA carried by RT-TEX. However, the ability of RT-TEX to stimulate DCs was completely abrogated. Thus, it is possible that the IFN-stimulatory dsDNA selectively present in RT-TEX and sensitive to the levels of TREX1 in the parent cells has characteristics distinct from the dsDNA present in TEX derived from untreated cancer cells. Thakur et al. (16) performed an extensive characterization of dsDNA present within TEX and found no bias for gene-encoding versus intergenic regions and no enrichment in specific regions compared to genomic DNA. Interestingly, they also found that dsDNA within TEX was methylated similar to genomic DNA. Thus, multiple factors may regulate the IFN-I stimulatory function of dsDNA contained within RT-TEX, including abundance, structure/epigenetic modifications, and efficiency of delivery to recipient cell cytosol. In relation to the latter, it is intriguing to consider if RT-TEX but not UT-TEX contain protein(s) or other factors (miRNA, mRNA) that regulate the access of the dsDNA to the DC cytosol, where it can stimulate cGAS/STING pathway. Interestingly, exosomes derived from breast cancer cells treated with the topoisomerase I inhibitor topotecan were reported to produce exosomes that carry DNA capable to activate DCs in a STING-dependent way (41), suggesting that treatment-induced DNA damage may be coupled with the activation of innate immunity via a common pathway.
RT increases the expression of several antigens and expands the antigenic repertoire of cancer cells (42,43). It is possible that among the more than hundred proteins selectively identified in RT-TEX, proteins encoding antigenic peptides that could be delivered to DCs are present. Thus, TEX could contribute to mediate antigenic spread, which has been reported to occur following RT (42,44). Interferon signaling was one of the 10 pathways present in RT-TEX but not UT-TEX. The main proteins identified in RT-TEX from this pathway were IFITM2, IFITM3, OAS1, and PSMB8. Although it cannot be excluded that each of these proteins could perform functions in the recipient DCs that may contribute to their activation and IFN-I production, the fact that RT-TEX produced by TSAshcGAS and TSAshSTING cells were able to induce Ifnb1 and Ifnar1 expression in DCs indicates that they are not required. However, becuase TEX are found in the peripheral blood, the proteomic signature of IFN-I pathway activation could be a biomarker of radiation-induced IFN-I activation in the tumor and possibly be used to predict which patients may respond to combinations of radiation and ICB (23,45).
Finally, the finding that TREX1 expression in the parent cell determined the ability of RT-TEX to activate the STING pathway in recipient DCs has important implications for the choice of radiation doses and fractionation in the clinic, especially in trials testing radiation with immunotherapy. We have shown that there is a threshold for single radiation doses above which, TREX1 is induced and dsDNA is cleared from the cancer cell cytosol, abrogating cancer cell-intrinsic activation of IFN-I and subsequent RT-induced abscopal responses (23). Data presented here strongly suggest that this threshold, which ranges between 12 to 18 Gy in different cancer cells tested, will also affect the induction of IFN-I pathway in DCs, by modulating the dsDNA content of RT-TEX. Interestingly, Deng et al. showed that a single dose of 20 Gy did not elicit IFN-I production by MC38 cancer cells, but it did elicit IFN-I production by DCs infiltrating MC38 tumors via the STING pathway (8). Consistent with this report, we found that 20 Gy RT induced Trex1 expression, resulting in degradation of most dsDNA and precluding IFN-I activation in MC38 cells (23). Thus, it is possible that the residual dsDNA in MC38 cancer cells was sufficient to activate DCs when delivered to DC cytosol, possibly by RT-TEX. Because no other radiation doses were tested in this study, it cannot be ruled out that radiation doses of 8 to 10 Gy, which induce optimal accumulation of dsDNA in the cytosol of MC38 cells (23), would have induced a stronger IFN-I response in DCs. In support of this notion, the intratumoral delivery of a STING agonist was required to achieve optimal T cell-priming in mice treated with 20 Gy RT, suggesting a suboptimal DC activation (8).
In conclusion, we showed that RT-TEX act as messengers of the molecular changes that took place in irradiated cancer cells. Together with tumor antigens, RT-TEX carried adjuvants that activated DCs and could prime protective antitumor T-cell responses. These findings may lead to new vaccination strategies and provide a rationale for testing, if circulating TEX in peripheral blood can be used as biomarkers of immunogenic changes induced by RT in the tumor.
Supplementary Material
ACKNOWLEDGMENTS
We thank the laboratories of Dr. Robert J. Schneider at NYU School of Medicine and Dr. John P. Moore at Weill Cornell Medicine for the use of their ultracentrifuge machines. We thank Dr. Philippe Colin at Weill Cornell Medicine for technical assistance with the ultracentrifuge. We also thank Dr. Boris Reizis and his staff for providing the B16.Flt3-L cell line, and the NYU Langone School of Medicine DART Microscopy Lab Alice Liang and Kristen Dancel-Manning for their assistance with electron microscopy.
Grant support: This work was supported by NIH R01 CA201246 (to S.D.), and a grant from The Chemotherapy Foundation (to S.D.). Additional funding was provided by the Breast Cancer Research Foundation award BCRF-16–054 (to S.C.F. and S.D.), and S10 RR027619 (to S.C.F.). J.M.D. was supported, in part, by the Vittorio Defendi Fellowship in Pathobiology from NYU School of Medicine Department of Pathology. The NYU proteomics laboratory is partially supported by NYU School of Medicine and the Laura and Isaac Perlmutter Cancer Center support grant P30CA016087 from the National Cancer Institute.
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists, but S.C.F has received speaker compensation from Bristol-Myer Squibb, Sanofi, Regeneron, Varian, Elekta and Janssen, S.D. has received honorarium from Lytix Biopharma, Astrazeneca, StemImmune, and AbbVie for advisory services.
Authors Contributions
Conception and design: S.D. and J.M.D.
Acquisition of data: J.M.D., J.R.C., B.M.U., Y.S., C.V.-B, S.S.
Analysis and interpretation of data: J.M.D., S.D., K.A.P., N.-P.R., J.R.C., B.M.U.
Writing and/or review of manuscript: S.D., J.M.D., C.V.-B., S.C.F.
Approval of the manuscript: All authors.
REFERENCES
- 1.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161(2):205–14 doi 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demaria S, Coleman CN, Formenti SC. Radiotherapy: Changing the Game in Immunotherapy. Trends Cancer 2016;2(6):286–94 doi 10.1016/j.trecan.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vacchelli E, Bloy N, Aranda F, Buque A, Cremer I, Demaria S, et al. Trial Watch: Immunotherapy plus radiation therapy for oncological indications. Oncoimmunology 2016;5(9):e1214790 doi 10.1080/2162402X.2016.1214790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007;13(9):1050–9 doi 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 5.Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol 2015;25(1):11–7 doi 10.1016/j.semradonc.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17(2):97–111 doi 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 7.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41(5):830–42 doi 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014;41(5):843–52 doi 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 2017;47(2):363–73 e5 doi 10.1016/j.immuni.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ludwig AK, Giebel B. Exosomes: small vesicles participating in intercellular communication. The international journal of biochemistry & cell biology 2012;44(1):11–5 doi 10.1016/j.biocel.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Rabinowits G, Gercel-Taylor C, Day JM, Taylor DD, Kloecker GH. Exosomal microRNA: a diagnostic marker for lung cancer. Clinical lung cancer 2009;10(1):42–6 doi 10.3816/CLC.2009.n.006. [DOI] [PubMed] [Google Scholar]
- 12.Record M, Subra C, Silvente-Poirot S, Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochemical pharmacology 2011;81(10):1171–82 doi 10.1016/j.bcp.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 13.Whiteside TL. Exosomes carrying immunoinhibitory proteins and their role in cancer. Clin Exp Immunol 2017. doi 10.1111/cei.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 2001;7(3):297–303 doi 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- 15.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009;9(8):581–93 doi 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 16.Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014;24(6):766–9 doi 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunigelis KE, Graner MW. The Dichotomy of Tumor Exosomes (TEX) in Cancer Immunity: Is It All in the ConTEXt? Vaccines (Basel) 2015;3(4):1019–51 doi 10.3390/vaccines3041019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012;18(6):883–91 doi 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z. Induction of heat shock proteins in B-cell exosomes. J Cell Sci 2005;118(Pt 16):3631–8 doi 10.1242/jcs.02494. [DOI] [PubMed] [Google Scholar]
- 20.Eldh M, Ekstrom K, Valadi H, Sjostrand M, Olsson B, Jernas M, et al. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS One 2010;5(12):e15353 doi 10.1371/journal.pone.0015353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res 2005;65(12):5238–47 doi 10.1158/0008-5472.CAN-04-3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 2009;15(17):5379–88 doi 10.1158/1078-0432.CCR-09-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618 doi 10.1038/ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamazaki T, Galluzzi L. TREX1 Cuts Down on Cancer Immunogenicity. Trends Cell Biol 2017;27(8):543–5 doi 10.1016/j.tcb.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Rosato A, Dalla Santa S, Zoso A, Giacomelli S, Milan G, Macino B, et al. The cytotoxic T-lymphocyte response against a poorly immunogenic mammary adenocarcinoma is focused on a single immunodominant class I epitope derived from the gp70 Env product of an endogenous retrovirus. Cancer Res 2003;63(9):2158–63. [PubMed] [Google Scholar]
- 26.Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol 1979;122:549–54. [PubMed] [Google Scholar]
- 27.Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, Dranoff G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res 2000;60(12):3239–46. [PubMed] [Google Scholar]
- 28.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006;Chapter 3:Unit 3 22 doi 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Gonzalez R, Gauthier SA, Kumar A, Saito M, Saito M, Levy E. A Method for Isolation of Extracellular Vesicles and Characterization of Exosomes from Brain Extracellular Space. Methods Mol Biol 2017;1545:139–51 doi 10.1007/978-1-4939-6728-5_10. [DOI] [PubMed] [Google Scholar]
- 30.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009;6(5):359–62 doi 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 31.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26(12):1367–72 doi 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 32.Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 2010;32(2):279–89 doi 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 33.Arora P, Porcelli SA. An Efficient and High Yield Method for Isolation of Mouse Dendritic Cell Subsets. J Vis Exp 2016(110):e53824 doi 10.3791/53824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stagg AJ, Burke F, Hill S, Knight SC. Isolation of mouse spleen dendritic cells. Methods Mol Med 2001;64:9–22 doi 10.1385/1-59259-150-7:9. [DOI] [PubMed] [Google Scholar]
- 35.Dai S, Wan T, Wang B, Zhou X, Xiu F, Chen T, et al. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin Cancer Res 2005;11(20):7554–63 doi 10.1158/1078-0432.CCR-05-0810. [DOI] [PubMed] [Google Scholar]
- 36.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011;208(10):1989–2003 doi 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011;208(10):2005–16 doi 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527(7578):329–35 doi 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pitt JM, Andre F, Amigorena S, Soria JC, Eggermont A, Kroemer G, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest 2016;126(4):1224–32 doi 10.1172/JCI81137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007;81(1):1–5. [DOI] [PubMed] [Google Scholar]
- 41.Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, et al. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J Immunol 2017;198(4):1649–59 doi 10.4049/jimmunol.1601694. [DOI] [PubMed] [Google Scholar]
- 42.Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 2006;203(5):1259–71 doi 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res 2004;64(21):7985–94 doi 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 44.Gulley JL, Arlen PM, Bastian N, Morin N, Marte J, Beetham P, et al. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin Cancer Res 2005;11:3353–62. [DOI] [PubMed] [Google Scholar]
- 45.Vanpouille-Box C, Formenti SC, Demaria S. Toward Precision Radiotherapy for Use with Immune Checkpoint Blockers. Clin Cancer Res 2018;24(2):259–65 doi 10.1158/1078-0432.CCR-16-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.