

Abstract
DNA damage repair capacity is required for cells to survive catastrophic DNA damage and proliferate under conditions of intra-tumoral stress. The ability of the minor histone protein H2AX to serve as a hub for the assembly of a productive DNA damage repair complex is a necessary step in preventing DNA damage-induced cell death. The Eyes Absent (EYA) proteins dephosphorylate the terminal tyrosine residue of H2AX thus permitting assembly of a productive DNA repair complex. Here we use genetic and chemical biology approaches to separately query the roles of host vascular endothelial cell and tumor cell EYA in tumor growth. Deletion of Eya3 in host endothelial cells significantly reduced tumor angiogenesis and limited tumor growth in xenografts. Deletion of Eya3 in tumor cells reduced tumor cell proliferation and tumor growth without affecting tumor angiogenesis. A chemical inhibitor of the EYA tyrosine phosphatase activity inhibited both tumor angiogenesis and tumor growth. Simultaneously targeting the tumor vasculature and tumor cells is an attractive therapeutic strategy since it could counter the development of the more aggressive phenotype known to emerge from conventional anti-angiogenic agents.
Keywords: anti-angiogenic, EYA, H2AX, targeted therapy, Benzarone, DNA damage
Introduction
Several mechanisms that differentiate between developmental and pathological angiogenesis have been recently delineated [1–5]. Among these the DNA damage repair (DDR) pathway has emerged as a potentially targetable mechanism permitting survival of endothelial cells in hostile microenvironments and thus promoting pathological angiogenesis. Notably, the H2AX-dependent DNA damage response is activated and promotes neovascularization specifically under hypoxic conditions [2]. Hypoxia-induced tumor angiogenesis was reduced in H2AX−/− mice [2] suggesting that H2AX is necessary for endothelial cells to survive and proliferate under conditions of oxygen deprivation. A critical step in the H2AX-dependent DDR pathway is phosphorylation at Serine-139 to form γ-H2AX, a commonly used marker of DNA damage and the formation of repair foci. H2AX is also constitutively phosphorylated on its terminal tyrosine-142 by the William-Beuren Syndrome Transcription Factor kinase [6]. Dephosphorylation of H2AX-Tyr142 is necessary for the assembly of productive DDR complexes via recruitment of the adaptor protein MDC1 [7]. The Eyes Absent (EYA) protein tyrosine phosphatases (PTP) dephosphorylate H2AX-Tyr142 [7–9], promoting DDR and cell survival under DNA-damaging hypoxic conditions [7]. Indeed, the PTP activity of EYA contributes to neovascularization in the oxygen-induced retinopathy model [10], a classic example of hypoxia-induced pathological angiogenesis.
The Eyes Absent proteins are expressed during embryonic development and their transcription is largely downregulated after development (reviewed in [11]). Elevated levels of EYA proteins in cancer cells are linked to increased tumor growth, metastasis [12–14] and resistance to DNA damaging therapeutics [15]. EYA1 promotes breast cancer cell proliferation [13] and medulloblastoma growth [16], EYA4 promotes the formation of malignant peripheral nerve sheath tumors (MPNST) [12], and over-expression of Eya2 in ovarian cancer cell lines promotes tumor growth in xenografts [17]. These observations have led to the suggestion that the EYA proteins are good targets for the development of cancer therapeutics [11, 18–20]. The ability of the EYA-PTP activity to promote tumor cell survival under DNA-damaging conditions, whether intrinsic (oxidative and replication stress in a growing tumor) or extrinsic (chemotherapy, radiation) provides an additional rationale for EYA as a therapeutic target.
Here we separately probed the contributions of host endothelial EYA3 and tumor cell EYA3 to tumor growth and vascularity using genetic manipulations. Further, the specific contribution of the tyrosine phosphatase activity of the EYA proteins was examined through the use of a small molecule inhibitor. The results suggest that targeting the EYA-PTP activity can simultaneously attenuate tumor angiogenesis and tumor growth.
Materials and Methods
Antibodies and Reagents
Anti-EYA3 (Abcam, Cambridge, MA), anti-Ki67 (Thermoscientific, Florence, KY), pan anti-actin C4 (Seven Hills Bioreagents, Cincinnati OH [21]), anti-endomucin (Santa Cruz, Dallas, TX), anti-CD31 (Dako Agilent, Santa Clara CA), anti-γ-H2AX (Novus Biological, Littleton CO), biotin-SP conjugated goat anti-mouse secondary antibody (Jackson Immuno-Research, West Grove PA), biotinylated goat anti-rabbit secondary antibody (Vector Labs, Burlingame CA). TUNEL assay kit (Promega, Madison, WI), Benzarone (2-Ethyl-1-benzofuran-3-yl)-(4-hydroxyphenyl)methanone, obtained from Toronto Research Chemicals, Canada), CCK-8 kit (Dojindo Molecular Technologies, Rockville, MD USA). Lewis Lung Carcinoma cells were obtained from ATCC (CRL-1642 LL/2 (LLC1), obtained January 2016), tested for mycoplasma contamination (ATCC Mycoplasma detection kit, 30-1012K), maintained in DMEM containing 1% vv−1 penicillin (100 IU ml−1), streptomycin 100 mg ml−1, 10% vv−1 Fetal Bovine Serum (FBS), and used within 10 passages. CRISPR/Cas9 was used to disrupt Eya3 expression by causing a double-strand break within the gene using the Eya3 CRSIPR/Cas9 KO system (Santa Cruz Biotechnology SC-420256, Dallas TX) consisting of a pool of 3 plasmids encoding the Cas9 nuclease and one of three target-specific guide RNAs:
Plasmid 1; GGTGCAGGCACCATAGCACT
Plasmid 2: TTGCAGATGAAGTGGGTATC
Plasmid 3: CAGTACCTGGTTTGAGATG
Formalin-fixed, paraffin-embedded de-identified lung cancer samples were obtained from University Hospital in Cincinnati, OH.
Animals
C57Bl/6 mice obtained from Jackson Laboratories (Bar Harbor, ME, USA). Conditional loss-of-function allele, Eya3flox was generated using conventional gene targeting as described previously [10]. To generate Eya3VEC-KO mice, Eya3flox mice were crossed with the endothelial cell specific tamoxifen-inducible Cre-recombinase Pdgfb-iCreER [10, 22]. 300 μg/day tamoxifen intra-peritoneally for three days induced Eya3 deletion. All mice were bred in the CCHMC facility (Animal Protocol IACUC2016-0057).
Xenografts
Animal experiments were performed in accordance with the recommendations of the Institutional Animal Care and Use Committee at Cincinnati Children’s Hospital Medical Center (IACUC2016-0019). 1 × 106 tumor cells mixed with 30% Matrigel in 100 μl PBS (1:1) were injected subcutaneously into the flank of C57BL/6 mice (male, 6-week old), and tumor size monitored be caliper measurements (Volume (mm3) = (length × width2)/2). At the indicated times mice were randomly distributed into appropriate groups. For inhibitor experiments mice were either administered vehicle (5% DMSO) or BZ in 5% DMSO (25 μg/g body weight) via intraperitoneal injection. Xenografts were performed at least twice.
Immunohistochemistry
Tumor tissue was fixed in 4% PFA, embedded in paraffin, 5 μm serial sections de-paraffinized and rehydrated in a gradient of alcohol and TBST. After antigen retrieval (10 mM sodium citrate pH 6.0), sections were incubated with 3% hydrogen peroxide (10 mins) and 5% goat serum in PBST (30 – 60 mins) as required, followed by anti-Eya3 (dilution: 1:500), anti-CD31 (1:50) or anti-γ-H2AX (1:500) at 4°C overnight, followed by 30 mins at room temperature in Alexa-fluor conjugated secondary antibody for immunofluorescence or biotinylated secondary antibody for immunohistochemistry. Images were taken using standard light or confocal microscopy and analyzed with ImageJ [23]. To detect cell death tumor sections were stained with DeadEnd™ Fluorometric TUNEL System (Promega) following the manufacturer’s protocol.
COMET Assays
Cells were maintained in either standard cell culture conditions or in a hypoxia chamber (0.1% O2) for 16 hours before being subject to the alkaline COMET assay [24]. Briefly, cells embedded in agarose on glass slides were maintained in lysis buffer (2.5M NaCl, 100 mM EDTA, 10 mM Tris, 1% sarkosyl, 1% Triton X-100, pH 10; 1 hour, 4°C), neutralized with Tris-buffered EDTA, incubated with alkaline electrophoresis buffer (300 mM NaOH, 1mM EDTA, pH 12.3) for 30 minutes to allow DNA to unwind, electrophoresed (40 mins, 25V), slide immersed in 70% ethanol for 5 minutes, dried at 40°C for 15 minutes, and stained with propidium iodide. For quantification comets were imaged with a fluorescent microscope and tail moments calculated using OPENCOMET [25] in ImageJ [23].
Multicellular tumor spheroids
Multicellular tumor spheroids (MCTS) were formed using the liquid overlay method [26], collected and fixed in 4% paraformaldehyde/PBS. When appropriate, spheroids were incubated with pimonidazole prior to fixation. For cryopreservation fixed MCTS were dehydrated with 30% sucrose and snap-frozen with Optimal Cutting Temperature media (Tissue Tek, Torrance, CA). 5 μm cryo-sections were blocked in PBS pH 7.4, 0.15% triton-X 100 vv−1, 10% FBS vv−1, and 2% BSA wv−1 in a humidified chamber, incubated with mouse anti-γ–H2AX (1:200), and with goat anti-mouse secondary antibody conjugated to an Alexa Flour 674 Azide. DNA was labeled with Hoechst dye. Sections were mounted using Fluorogel with DABCO (17985-04; Electron Microscopy Science, Hatfield, PA). Fluorescently stained MCTS sections were imaged on a Zeiss confocal microscope. Nuclei were counted as Hoechst-positive cells by using watershed separation and quantification with particle analysis in ImageJ software followed by manual counting of the number of γ–H2AX positive nuclei. LLC survival in two-dimensional culture was measured using the CCK-8 kit as previously described [27, 28] and following the manufacturer’s protocol.
Statistics
Results presented as the mean ± SD (standard deviation). Statistical analyses were performed using Graphpad PRISM version 5.0 for Mac OSX, www.graphpad.com. A t-test was used when two samples/conditions were compared and ANOVA for more than two groups.
Results
Endothelial cell EYA3 promotes tumor angiogenesis
RT-PCR analyses on human endothelial cells (HUVEC & HMVEC) show a transcript for only Eya3 of the four vertebrate Eya isoforms (Eya1, Eya2, Eya3, Eya4) [28]. Staining of serial sections from primary human lung adenocarcinoma and lung squamous cell carcinoma samples with antibodies to CD31 (endothelial cell (EC) marker) and EYA3 also showed the presence of EYA3 in tumor endothelial cells (Fig. 1a, b).
Figure 1. EYA3 is expressed in human tumor endothelial cells.
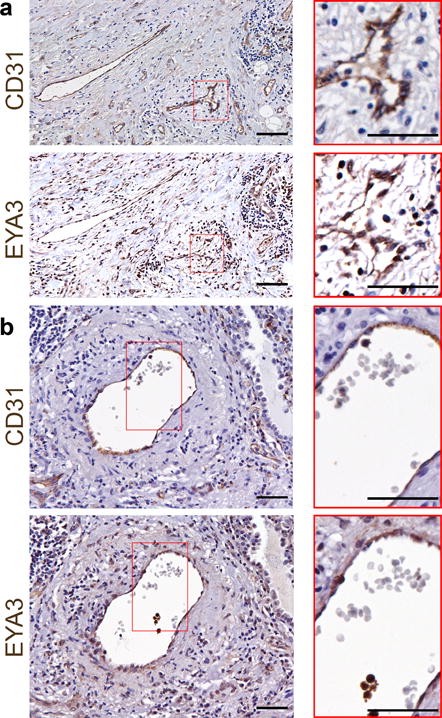
a) Immunohistochemistry shows endothelial and tumor cell EYA3 in human lung adenocarcinoma. Serial sections were stained with antibodies towards CD31 and EYA3. Magnified regions indicated by red boundaries; scale bars 50 μm.
b) Immunohistochemistry shows endothelial and tumor cell EYA3 in human lung squamous cell carcinoma. Serial sections were stained with antibodies towards CD31 and EYA3. Magnified regions indicated by red boundaries; scale bars 50 μm.
In order to investigate the endothelial cell autonomous contribution of EYA3 to tumor angiogenesis, we deleted Eya3 from ECs using the loxP recombination strategy and the vascular endothelial cell (VEC) specific tamoxifen-inducible Cre-recombinase Pdgfb-iCreER [22] (resulting mice are referred to as Eya3VEC-KO and have been previously described [10]). This targeting strategy results in over 80% reduction in EYA3 protein levels with no alterations in transcripts for other EYAs [10]. Sub-cutaneous transplantation of murine Lewis lung carcinoma (LLC) cells into littermates with Eya3VEC-KO and Eya3fl/fl genotypes was used. Once tumors were >100 mm3, all mice were administered tamoxifen. This led to VEC-specific deletion of Eya3 in the Eya3VEC-KO mice (Supplementary Fig. S1), while tamoxifen administered to all animals controlled for any effect of tamoxifen itself. In Eya3VEC-KO mice there was immediate flattening of the tumor growth curve that persisted for 12 days, after which the slope increased again. Despite this apparent recovery in tumor growth rate, tumors in Eya3VEC-KO mice were 50% smaller (Fig. 2a, b) than in control Eya3fl/fl mice at the end of the study. To determine whether tumor growth correlated with angiogenesis and cell proliferation, tumors were sectioned and stained with anti-CD31 antibody (specifically labels endothelial cells) and Ki-67 (Fig. 2c). Vessel density was quantified in 3 vascular hot-spot areas/section (Fig. 2d). Reduction in CD31-positive vasculature was apparent in LLC tumors from Eya3VEC-KO mice while no significant differences in tumor cell proliferation were measured (Fig. 2e). TdT-mediated dUTP nick-end labeling (TUNEL) positive apoptotic nuclei were also higher in tumors from Eya3VEC-KO mice (Fig. 2f), indicative of increased cell death with endothelial Eya3 deletion. Together these data suggest a role for host endothelial EYA3 in promoting vascularity and in the survival of tumor cells. We hypothesize that hypoxic conditions resulting from reduced tumor vascularity contribute to tumor cell apoptosis and slower tumor growth.
Figure 2. Endothelial EYA3 promotes tumor angiogenesis.
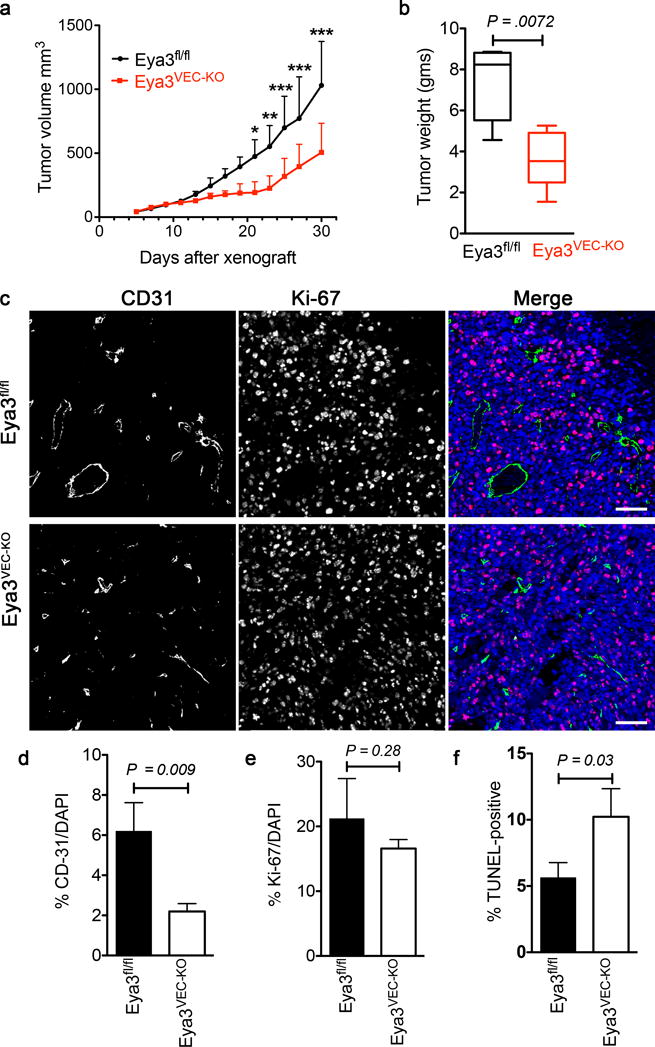
a) Tumor growth curve of Eya3fl/fl and Eya3VEC-KO mice bearing LLC tumors. Tamoxifen was administered to all mice on day 13 after the xenograft to induce loss of endothelial EYA3 in Eya3VEC-KO. Data represent mean and SD (n = 5 animals of each genotype, *P<0.05, ** P<0.01, *** P<0.001).
b) Weights of LLC tumors from Eya3fl/fl and Eya3VEC-KO mice. Data is represented as a box and whiskers plot where the box extends from the 25th to 75th percentile and the whiskers are drawn between the smallest and largest values. The median is indicated by a horizontal line in the box. n = 5 animals of each genotype.
c) LLC tumors from Eya3fl/fl and Eya3VEC-KO mice co-stained with the endothelial marker CD31 and the proliferation marker Ki-67. Sections were counter-stained with DAPI to show nuclei. Scale bar 100 μm.
d) Quantitation of CD31 positive area, in LLC tumors from Eya3fl/fl and Eya3VEC-KO mice. Four representative images were analyzed from each tumor. Data represents mean and SD; n = 3 tumors from animals of each genotype.
e) Quantitation of Ki-67 positive cells in LLC tumors from Eya3fl/fl and Eya3VEC-KO mice. Four representative images were analyzed from each tumor. Data represents mean and SD; n = 3 tumors from animals of each genotype.
f) Quantitation of TUNEL staining in LLC tumors from Eya3fl/fl and Eya3VEC-KO mice. Four representative images were analyzed from each tumor. Data represents mean and SD; n = 3 tumors from animals of each genotype.
Deletion of Eya3 in LLC cells limits growth of xenograft tumors
In order to specifically query the contribution of tumor cell EYA3 to tumor growth, we used the CRIPSR-Cas9 system to disrupt the Eya3 gene in LLC cells; the resulting cells will be referred to as EYA3-ko (Supplementary Fig. S2b). RT-PCR analysis shows high levels of Eya3 transcripts in LLC cells and low levels of Eya1. Eya2 and Eya4 were not detected (Supplementary Fig. S2a). Here we sought to determine the effect of loss of Eya3 (the major Eya homologue in LLC cells) on tumor growth. C57/Bl6 mice were used for xenografts in which one flank was injected with LLC cells and the other flank with the same number of Eya3-ko cells, allowing us to compare tumor characteristics in the same host animal. Tumor growth was monitored by caliper measurements (Fig. 3a). EYA3-ko tumors were over 50% smaller in size at the end of the study (Fig. 3a, b). Histological analyses revealed significant patches of necrosis in both large control and smaller EYA3-ko tumors. (Supplementary Fig. S2c). LLC tumors are fast-growing, hence the unevenly distributed necrosis in the large LLC tumors is likely to be a result of tumor cells outgrowing their nutrient supply. The degree of vascularity was similar in LLC and LLC-Eya3-ko tumors as measured by endomucin staining (Fig. 3c, d). In contrast, there were over 60% fewer Ki-67–positive cells and over 3.5-fold more apoptotic cells in the Eya3-ko tumors supporting a role for tumor cell EYA3 in cell survival and proliferation (Fig. 3e, f).
Figure 3. Loss of EYA3 in LLC tumor cells attenuates tumor growth.
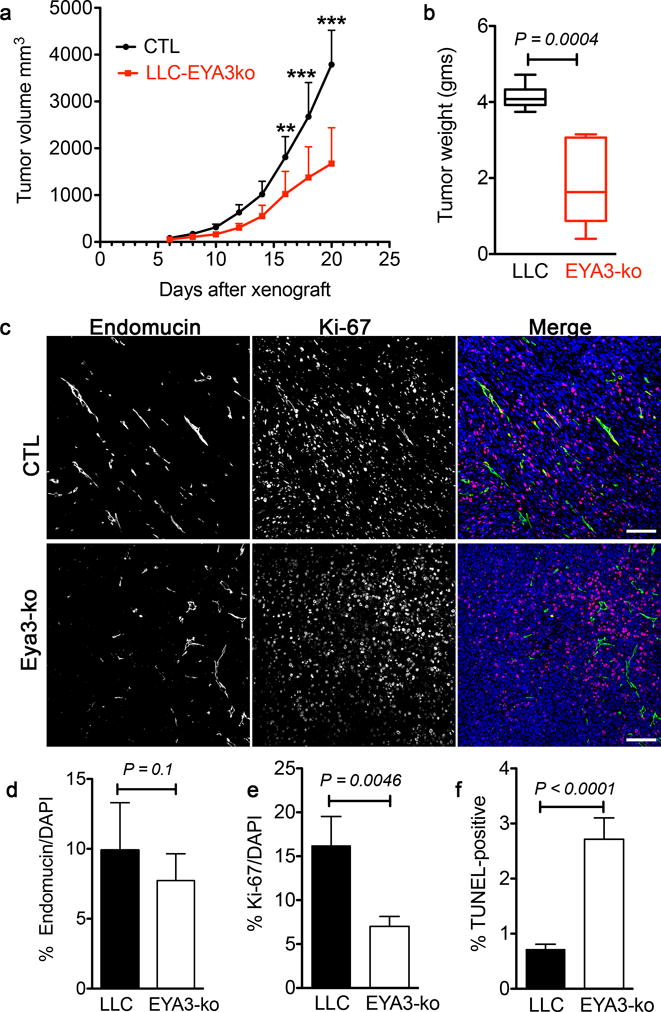
a) Tumor volume over time from xenografts performed with LLC and LLC-EYA3-ko cells in C57/Bl6 mice. Data represent mean and SD. Two-way ANOVA with Bonferroni’s post-test was used. n = 8 animals in each group, ** P<0.01, *** P<0.001.
b) Final tumor weights from xenografts performed with LLC and LLC-EYA3-ko cells in C57/Bl6 mice. Data represented as a box and whiskers plot where the box extends from the 25th to 75th percentile and the whiskers are drawn between the smallest and largest values. The median is indicated by a horizontal line in the box. A paired t-test was used comparing LLC (CTL) and EYA3-ko tumors in the same animal (n = 8 in each group).
c) LLC and EYA3-ko tumors co-stained with the endothelial marker Endomucin and the proliferation marker Ki-67. Sections were counter-stained with DAPI to show nuclei. Scale bar 100 μm.
d) Quantitation of Endomucin-positive area in LLC and EYA3-ko tumors. Four representative images were analyzed from each tumor. Data represents mean and SD (n = 5 tumors from each group).
e) Quantitation of Ki-67 index in LLC and EYA3-ko tumors. Four representative images were analyzed from each tumor. Data represents mean and SD (n = 5 tumors from each group).
f) Quantitation of TUNEL staining in LLC and EYA3-ko tumors. Four representative images were analyzed from each tumor. Data represents mean and SD (n = 4 tumors from animals of each group).
The PTP activity of EYA3 promotes tumor angiogenesis
The EYA family of proteins combine an unusual number of independent biochemical activities: the N-terminal domain has transactivation and threonine phosphatase activities [29] and the highly conserved C-terminal domain is a mechanistically novel protein tyrosine phosphatase (PTP) [30]. Previous studies have shown that the PTP activity of EYA3 contributes to retinal vascular development and to neovascularization in the oxygen-induced retinopathy model [10, 11, 14, 28]. To specifically query the contribution of the PTP activity to tumor angiogenesis and to tumor growth, we used the previously characterized EYA-PTP inhibitor Benzarone (BZ) [27, 28]. Cell proliferation measured using the CCK-8 cytotoxicity assay showed that BZ was anti-proliferative under standard culture conditions, with an IC50 for LLC cells of 1.2 μM (Supplementary Figure S3).
LLC xenografts in C57/Bl6 mice were used to establish whether BZ treatment inhibits tumor growth or vascularity. When tumors had reached >150 mm3, either 25 μg/g BZ or vehicle control was injected intra-peritoneally every other day. Tumor size was monitored using calipers. BZ treatment substantially reduced the rate of tumor growth (Fig. 4a, b), with over 65% tumor growth inhibition (%TGI = ((Vc-Vt)/(Vc-Vo)) × 100; Vc: median volume of vehicle-treated tumors at the end of the study, Vt: median volume of BZ-treated tumors at the end of the study, Vo: median volume of tumors at the start of treatment). Agents that lead to over 60% TGI in murine models have been shown to lead to clinical response in humans [31]. In a separate protocol, BZ (or vehicle) was administered when tumors had reached a median volume of 670 mm3. The experiment was terminated 4 days later, after two doses of BZ administration. There was an immediate change in the LLC tumor growth curve upon initiation of BZ-treatment (Fig. 4c), with 69% TGI measured at the end of the study. In contrast, there was no change in the growth rate of LLC-EYA3-ko tumors similarly treated with BZ (Fig. 4h). BZ treatment did not consistently induce necrosis; the level of necrosis in BZ treated tumors was either less or the same as that in controls (Supplementary Fig. S4).
Figure 4. Inhibition of EYA-PTP activity attenuates tumor growth, vascularization, and cell proliferation.
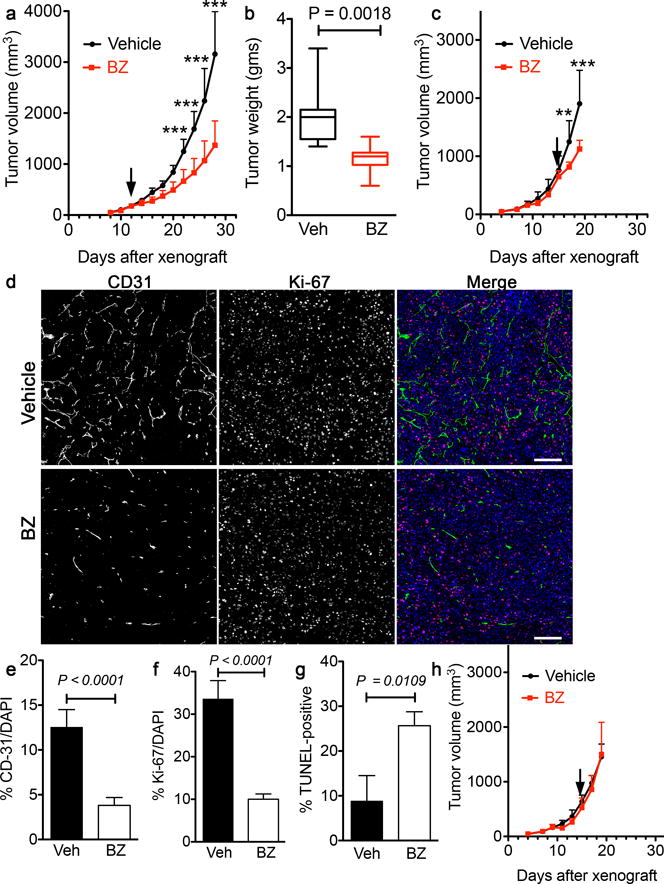
a) Tumor volume over time from xenografts performed with LLC cells in C57/Bl6 mice and treated with either BZ or vehicle starting when tumor volume was >100 mm3 (arrow). 25 μg/g body weight BZ or vehicle was delivered intra-peritoneally. Tumor growth was monitored by caliper measurements until the experiment was terminated. BZ treatment substantially reduced tumor size. Data represented as mean and SD; two-way ANOVA with a Bonferroni’s post-test was used. (n = 9 (vehicle), n = 8 (BZ), *** P<0.001).
b) Final tumor weights from xenografts performed with LLC cells in C57/Bl6 mice and treated with either BZ or vehicle. Data represented as a box and whiskers plot where the box extends from the 25th to 75th percentile and the whiskers are drawn between the smallest and largest values. The median is indicated by a horizontal line in the box. (n = 9 (vehicle), n = 8 (BZ), unpaired t-test).
c) Tumor volume over time from xenografts performed with LLC cells in C57/Bl6 mice and treated with either BZ or vehicle starting when tumor volume was >600 mm3 (arrow). 25 μg/g body weight or vehicle was delivered intra-peritoneally. Tumor growth was monitored by caliper measurements until the experiment was terminated. BZ treatment substantially reduced tumor size. Data represent mean and SD (n = 7 (vehicle), n = 6 (BZ), ** P<0.01, *** P<0.001).
d) Vehicle and BZ treated tumors co-stained with the endothelial marker CD31 and the proliferation marker Ki-67. Sections were counter-stained with DAPI to show nuclei. Scale 100 μm.
e) Quantitation of CD31-positive area in Vehicle and BZ treated tumors. Four representative images were analyzed from each tumor. Data represents mean and SD (n = 5 tumors in each group).
f) Quantitation of Ki-67 positive cells in LLC and EYA3-ko tumors. Four representative images were analyzed from each tumor. Data represents mean and SD (n = 4 tumors in each group).
g) Quantitation of TdT-mediated dUTP nick-end labeling (TUNEL) staining in LLC and EYA3-ko tumors. Four representative images were analyzed from each tumor. Data represents mean and SD (n = 3 tumors in each group).
h) Tumor volume over time from xenografts performed with LLC Eya3-ko cells in C57/Bl6 mice and treated with either BZ or vehicle starting when tumor volume was >600 mm3 (arrow). 25 μg/g body weight BZ or vehicle was delivered intra-peritoneally. BZ treatment had no effect on tumor size. Data represented as mean and SD (n = 7 (vehicle), n = 6 (BZ)).
To assess the effect of BZ on tumor vascularity and cell proliferation sections were co-stained for Ki-67 and the angiogenesis marker CD31 (Fig. 4d). Areas of high vascular density were chosen for quantification of both vascularity and proliferation as the ratio of CD31-positive or Ki-67 positive cells respectively. Tumor vascularity was reduced by over 65% in BZ-treated versus vehicle-treated LLC tumors (Fig. 4e). The percentage of tumor cells staining positive for the proliferation marker Ki-67 was also lower by over 50% in BZ-treated tumors (Fig. 4f). There was a substantial increase in cell death upon BZ treatment, reflected in nearly three-times as many TUNEL-positive cells (Fig. 4g).
EYA3 contributes to DNA Damage Repair, tumor cell survival, and tumor angiogenesis
The microenvironment within a tumor includes areas with varying oxygen levels ranging from anoxia to milder hypoxia. COMET assays show that LLC cells subject to acute hypoxic conditions undergo DNA damage in vitro (Fig. 5a). To mimic a 3D tumor environment, we used multicellular tumor spheroids. As has been previously described, LLC cells form spheroids within 48 hours of seeding and can grow to achieve a maximum diameter of ~1000 μm in 10–12 days [26]. Deletion of Eya3 results in a small (5%), but statistically significant, reduction in spheroid size measured 7 days after initial seeding. The divergence in size begins after spheroids reach 600 μm in diameter (Fig. 5b) and cells >100 μm from the spheroid surface are hypoxic (Fig. 5c). Ki-67 staining of spheroid sections revealed a gradient of proliferation with over 77% of cells within 100 μm of the surface staining positive for Ki67, while less than 55% of the cells in the hypoxic, non-necrotic center were Ki67-positive (Fig. 5d). Eya3 deletion led to a 26% reduction in the percentage of Ki-67 positive cells (Fig. 5d, e), suggesting that EYA3 contributes to cell proliferation in the 3D context. To examine the effect of Eya3 deletion on the DDR process, spheroid sections were stained for γ-H2AX. In spheroids between 600 – 700 μm in diameter, there is a distinct band of γ-H2AX positive cells present in the hypoxic, peri-necrotic area, suggesting that DDR processes are active in this zone (Fig. 5e). Loss of EYA3 led to over 50% reduction in γ-H2AX positive cells indicating defective DDR (Fig. 5e).
Figure 5. Multicellular tumor spheroids show reduced proliferation and γ-H2AX upon loss of EYA3.
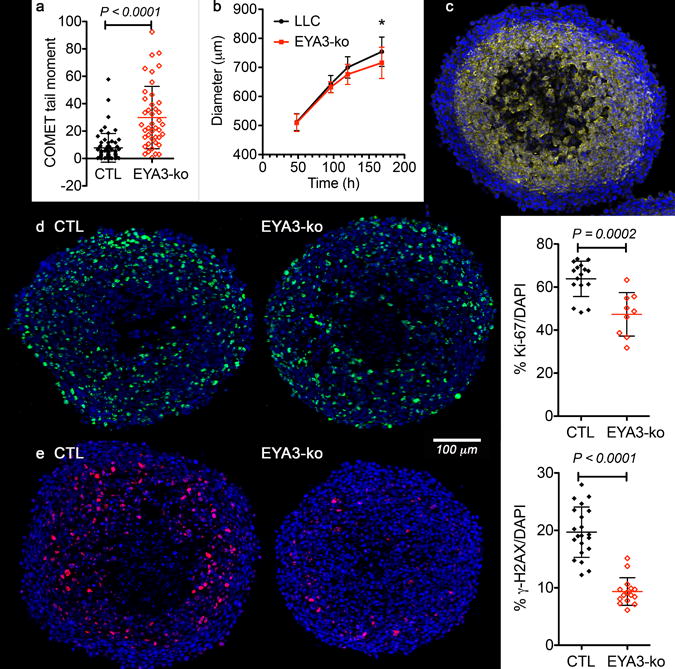
a) Hypoxia-induces DNA damage in LLC (CTL) and EYA3-ko cells. Alkaline COMET assays on cells maintained at 0.1% O2 for 16 hours shows increased DNA damage in EYA3-ko cells. Tail moments calculated using OPENCOMET. Graph shows mean and SD.
b) Growth curves for spheroids generated with LLC (black) or LLC-EYA3-ko (red) cells. The diameter and standard deviation of >15 spheroids representing two independent experiments are plotted over time. * P<0.05.
c) Hypoxia is present in spheroids >600 μm in diameter. Immuno-staining of an LLC spheroid (625 μm in diameter) section for the hypoxia marker pimonidazole (yellow), counter-stained with Hoechst 33342 (blue).
d) EYA3-ko spheroids have fewer proliferating cells. Representative images showing the proliferative marker Ki-67 (green, with blue Hoechst 33342 counterstain) in LLC and EYA3-ko spheroids 680 - 700 μm in diameter. Quantitation of the percentage of Ki-67 positive cells is shown as mean and SD.
e) EYA3-ko spheroids have fewer γ-H2AX positive cells. Representative images showing the DNA damage repair marker γ-H2AX (red, with blue Hoechst 33342 counterstain) in LLC and EYA3-ko spheroids 660 - 690 μm in diameter. Quantitation of the percentage of γ-H2AX positive cells is shown as mean and SD.
Tumor sections were similarly analyzed by co-staining for γ-H2AX and CD31. In control tumors patches of γ-H2AX positive tumor cells were found concentrated around vascular hotspots, and at lower frequency along the tumor surface (Fig. 6a). Eya3 deletion in tumor cells led to over 50% reduction in the percentage of γ-H2AX–positive cells (Fig. 6b, c).
Figure 6. Loss of EYA3 reduces the number of γ-H2AX positive cells in vivo.
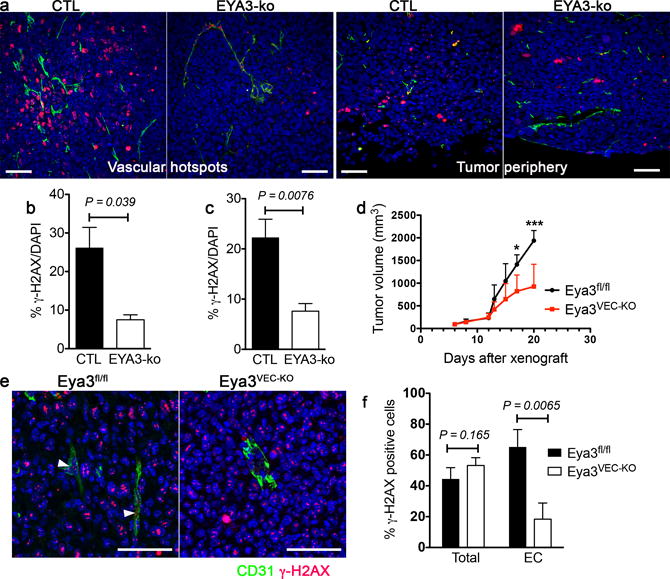
a) Representative sections from LLC (CTL) and EYA3-ko tumors in areas of maximal vascular density (vascular hotspots) and along the tumor periphery stained for the endothelial marker CD31 (green) and the DDR marker γ-H2AX (red); scale 50 μm.
b) Quantitation of the percentage of cells staining positive for γ-H2AX around areas of maximal vascular density. Three representative images were analyzed from each tumor. Data represents mean and SD (n = 3 tumors in each group).
c) Quantitation of the percentage of cells staining positive for γ-H2AX near the tumor surface. Three representative images were analyzed from each tumor. Data represents mean and SD (n = 3 tumors in each group).
d) Tumor growth curve of Eya3fl/fl and Eya3VEC-KO mice bearing LLC tumors. Tamoxifen was administered to all mice on day 12 after the xenograft to induce loss of endothelial EYA3 in Eya3VEC-KO. The experiment was terminated 8 days later. Data represent mean and SD (n = 4 for Eya3fl/fl and n = 5 for Eya3VEC-KO, * P<0.05, *** P<0.001).
e) γ-H2AX foci are present in both tumor and endothelial cells in tumor sections from Eya3fl/fl mice, but reduced in the vasculature of Eya3VEC-KO tumors. Sections stained for the endothelial marker CD31 (green) and the DDR marker γ-H2AX (red). White arrowheads indicate γ-H2AX foci-containing endothelial cells; scale 50 μm.
f) Quantitation of the percentage of cells staining positive for γ-H2AX in tumor and endothelial cells (EC). Three representative images were analyzed from each tumor were analyzed. Data represents mean and SD (n = 3 tumors in each group).
To evaluate the effect of Eya3 deletion in host endothelial cells, LLC xenografts were conducted in Eya3fl/fl and Eya3VEC-ko mice, with tamoxifen-induced Eya3 deletion initiated 12 days after tumor cell transplantation. As in Fig. 2a significant reduction in tumor growth was observed (Fig. 6d). Tumors were analyzed for the presence of γ-H2AX-positive cells on day 20. Upon host VEC-specific Eya3 deletion there was no change in the percentage of tumor cells staining positive for γ-H2AX (Fig. 6e, f). However, the percentage of endothelial (CD31-positive) cells that stained positive for γ-H2AX was substantially reduced (Fig. 6e, f). Together these analyses of tumor xenograft tissue are consistent with a defect in the γ-H2AX dependent DDR pathway upon Eya3 deletion in either tumor or endothelial cells.
Discussion
While defects in DNA repair pathways are commonly associated with cancer cells, residual DNA repair capacity is required for survival and proliferation under the conditions of oxidative stress present in tumors [32]. Evidence provided here in the context of tumor and endothelial cells, and in previous studies using other cell types [7, 10], support a role for the EYA-PTPs in promoting cell survival under DNA damaging conditions. Tumors with endothelial Eya3 deletion were smaller and significantly less vascular, with no change in the Ki67-index of tumor cells. More TUNEL-positive cells were seen in the tumor suggesting that reduced vascularity contributes to tumor cell death. An inverse correlation between neovascularization and spontaneous apoptosis in untreated tumors has been reported in numerous human cancers including lung adenocarcinoma [33] and colorectal cancer [34], consistent with an anti-apoptotic role for tumor angiogenesis. Indeed, angiogenesis inhibitors are known to sustain the dormancy of primary tumors [35] and micro-metastases [36] through increased apoptosis, while having no effect on the proliferation rate of tumor cells. Deletion of Eya3 in tumor cells reduced tumor size in xenografts but had no effect on vascularity, suggesting an endothelial cell autonomous role for EYA3 in tumor angiogenesis. The negative effect on the Ki-67 index and increase in apoptosis seen in LLC-Eya3-ko tumors is consistent with a role for EYA3 in cell proliferation and survival as previously reported in non-tumor contexts [7], and the documented correlation between levels of EYA proteins and tumor progression [13] [17].
Linking biochemical function with biological outcomes using simple knock-out studies is complicated for the multi-functional EYA proteins [11]. Here we have addressed this concern by the use of a validated inhibitor of the PTP activity, Benzarone [10, 27, 28]. The PTP activity of the EYA proteins utilizes a reaction mechanism distinct from that of the classical Cys-based PTPs [30, 37], and can be selectively targeted. BZ binds non-covalently to the EYA active site which is stereo-chemically conserved among all EYA proteins. Hence BZ inhibits all EYA family members (targeting both EYA3 and EYA1 in LLC cells). Treatment with BZ had a strong and immediate negative effect on tumor growth but did not result in tumor regression. Both tumor and stromal cells are exposed to BZ in our studies, consistent with the observed anti-angiogenic effect and reduction in tumor cell proliferation. The best characterized direct substrate for the EYA-PTP is the minor histone protein H2AX and dephosphorylation of H2AX-tyrosine 142 promotes the repair of DNA breaks and the survival of cells with DNA damage [7, 9]. There is independent evidence of a role for endothelial DDR [1, 2], and more specifically EYA3-associated DDR [10], in pathological angiogenesis. The data provided here extends those observations to the tumor context.
While the growth of solid tumors is limited without vascularization, tumor-associated vessels are inefficient and do not effectively oxygenate tumor tissue. Hence tumors have areas that are either chronically or acutely hypoxic with often high levels of reactive oxygen species. Such conditions, along with unresolved replicative stress in rapidly proliferating cells, can result in DNA damage. Previous investigations report that the level of constitutive H2AX phosphorylation in tumor cells varies between cell lines and is dependent on cell cycle status; S and G2M-phase cells showed higher levels of γ–H2AX [38]. Other contributors to cell-specific variability in constitutive γ–H2AX levels include genetic differences that affect DDR pathways and the response to oxidative stresses e.g. p53 mutational status [39–41], expression of oncogenes or tumor suppressors, and other defects in DDR pathway components. Furthermore several non-DDR associated γ-H2AX functions have been reported [42]. Numerous studies have examined the linkage between hypoxia, radiation sensitivity and γ-H2AX in tumors [43]. Here we note that there are distinct patches of γ-H2AX positive tumor cells around vascular hotspots, and that a γ-H2AX signal is also detected in endothelial cells in tumor sections. In the absence of EYA-PTP activity γ-H2AX containing repair foci formation is impaired tipping the balance towards apoptosis of DNA damaged cells, as evidenced here in both tumor cells and endothelial cells. Tumor hypoxia (resulting from conventional anti-angiogenic therapies) can promote a more aggressive phenotype (activation of stress responses, metabolic adaptations, tissue remodeling, escape of tumor cells), hence such simultaneous targeting of the tumor vasculature and tumor cells by EYA-PTP inhibition is an attractive therapeutic strategy.
BZ is an active metabolite of Benzbromarone, a uricosuric drug widely used in the treatment of gout. Isolated cases of hepatotoxicity have been reported in patients using these compounds for gout [44], but the risk-benefit assessments remain actively debated [45, 46]. In the context of gout treatment, Benzbromarone acts as a xanthine oxidase (XO) inhibitor. Hence it is possible that the effects seen in the present study are secondary to XO inhibition. However, treatment of LLC-Eya3-ko xenografts with BZ did not reduce tumor size (Fig. 4h), suggesting that the effectiveness of BZ in retarding tumor growth is EYA3-dependent. Despite this evidence of target specificity in the anti-tumor context, the ability of BZ to inhibit XO and thus reduce uric acid levels bears consideration. The association between uric acid levels and cancer is complicated [47]; while uric acid has anti-oxidant properties with potential benefit as a defense against cancer, elevated serum uric acid has primarily been associated with increased inflammatory stress and premature cancer mortality [48–50]. Furthermore, cancer patients are predisposed to hyperuricemia due to both tumor-related and treatment-related factors. Nevertheless, the proposal that therapeutic management of serum uric acid levels could facilitate the treatment of cancers associated with hyperuricemia [51] has been tempered by concern regarding the effect of XO inhibition in tumor cells (decreased XO levels are associated with many aggressive human cancer types [48–50]). XO inhibitors have never before been directly tested in tumor models. The data presented here shows that BZ, a XO and EYA-PTP inhibitor, retards tumor growth. This observation raises the possibility that the EYA-targeted effect of BZ combined with control of serum uric acid levels via XO inhibition could be useful in cancer management.
The LLC murine syngeneic model is highly tumorogenic, utilizes an immunocompetent host, and has been used extensively to evaluate the efficacy of chemotherapeutic agents [52, 53]. In the context of the present studies, it has the particular advantage of permitting the use of genetic manipulations to evaluate the role of host factors (EYA3 in host endothelial cells). Nevertheless, there are limitations. Responses evaluated in a completely murine background may not be directly extrapolated to human conditions, and the aggressive nature of LLC cells may not reflect human tumors. Ongoing studies are extending the results reported here to human tumor cell xenografts (both established cell-lines and patient-derived tissue) and orthotopic models.
In addition to the ability of EYA PTP inhibition to negatively regulate tumor angiogenesis and tumor cell survival/proliferation, impaired DDR could also sensitize cells to DNA damaging treatment strategies (radiation, chemotherapy) commonly used as first line treatments. Furthermore EYA-PTP activity promotes cell migration [14, 28], a process that contributes to both angiogenesis and metastasis. Hence in addition to the anti-angiogenic effect of EYA-PTP inhibition observed here, EYA-PTP inhibitors could be useful as anti-metastatic agents.
Supplementary Material
Acknowledgments
Funding sources: This work was supported by the National Institutes of Health (CA207068), the B+ Foundation, and Cancer Free Kids.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Okuno Y, Nakamura-Ishizu A, Otsu K, Suda T, Kubota Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat Medicine. 2012;18:1208–1216. doi: 10.1038/nm.2846. [DOI] [PubMed] [Google Scholar]
- 2.Economopoulou M, Langer HF, Celeste A, Orlova VV, Choi EY, Ma M, Vassilopoulos A, Callen E, Deng C, Bassing CH, Boehm M, Nussenzweig A, Chavakis T. Histone H2AX is integral to hypoxia-driven neovascularization. Nature Medicine. 2009;15:553–558. doi: 10.1038/nm.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alitalo K. A radical view of pathological vasculature. Cell Metab. 2012;16:287–288. doi: 10.1016/j.cmet.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Rankin EB, Giaccia AJ, Hammond EM. Bringing H2AX into the angiogenesis family. Cancer cell. 2009;15:459–461. doi: 10.1016/j.ccr.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen J, Frye M, Lee BL, Reinardy JL, McClung JM, Ding K, Kojima M, Xia H, Seidel C, Silva R, Lima E, Dong A, Hackett SF, Wang J, Howard BW, Vestweber D, Kontos CD, Peters KG, Campochiaro PA. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest. 2014;124:4564–4576. doi: 10.1172/JCI74527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, Patel DJ, Elledge SJ, Allis CD. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457:57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 9.Krishnan N, Jeong DG, Jung SK, Ryu SE, Xiao A, Allis CD, Kim SJ, Tonks NK. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. The Journal of biological chemistry. 2009;284:16066–16070. doi: 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Tadjuidje E, Pandey RN, Stefater JA, 3rd, Smith LE, Lang RA, Hegde RS. The Eyes Absent Proteins in Developmental and Pathological Angiogenesis. The American journal of pathology. 2016;186:568–578. doi: 10.1016/j.ajpath.2015.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tadjuidje E, Hegde RS. The Eyes Absent proteins in development and disease. Cell Mol Life Sci. 2013;70:1897–1913. doi: 10.1007/s00018-012-1144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, Hegde RS, Cripe TP, Cancelas JA, Collins MH, Ratner N. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010;29:368–379. doi: 10.1038/onc.2009.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu K, Li Z, Cai S, Tian L, Chen K, Wang J, Hu J, Sun Y, Li X, Ertel A, Pestell RG. EYA1 Phosphatase Function Is Essential to Drive Breast Cancer Cell Proliferation through Cyclin D1. Cancer research. 2013;73:4488–4499. doi: 10.1158/0008-5472.CAN-12-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandey RN, Rani R, Yeo EJ, Spencer M, Hu S, Lang RA, Hegde RS. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene. 2010;29:3715–3722. doi: 10.1038/onc.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robin TP, Smith A, McKinsey E, Reaves L, Jedlicka P, Ford HL. EWS/FLI1 regulates EYA3 in Ewing sarcoma via modulation of miRNA-708, resulting in increased cell survival and chemoresistance. Mol Cancer Res. 2012;10:1098–1108. doi: 10.1158/1541-7786.MCR-12-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisner A, Pazyra-Murphy MF, Durresi E, Zhou P, Zhao X, Chadwick EC, Xu PX, Hillman RT, Scott MP, Greenberg ME, Segal RA. The Eya1 phosphatase promotes Shh signaling during hindbrain development and oncogenesis. Dev Cell. 2015;33:22–35. doi: 10.1016/j.devcel.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Yang N, Huang J, Buckanovich RJ, Liang S, Barchetti A, Vezzani C, O’Brien-Jenkins A, Wang J, Ward MR, Courreges MC, Fracchioli S, Medina A, Katsaros D, Weber BL, Coukos G. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer research. 2005;65:925–932. [PubMed] [Google Scholar]
- 18.Blevins MA, Towers CG, Patrick AN, Zhao R, Ford HL. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert Opin Ther Targets. 2015;19:213–225. doi: 10.1517/14728222.2014.978860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong D, Liu Y, Liu Q, Han N, Zhang C, Pestell RG, Wu K, Wu G. The retinal determination gene network: from developmental regulator to cancer therapeutic target. Oncotarget. 2016 doi: 10.18632/oncotarget.9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Han N, Zhou S, Zhou R, Yuan X, Xu H, Zhang C, Yin T, Wu K. The DACH/EYA/SIX gene network and its role in tumor initiation and progression. Int J Cancer. 2016;138:1067–1075. doi: 10.1002/ijc.29560. [DOI] [PubMed] [Google Scholar]
- 21.Lessard JL. Two monoclonal antibodies to actin: one muscle selective and one generally reactive. Cell Motil Cytoskeleton. 1988;10:349–362. doi: 10.1002/cm.970100302. [DOI] [PubMed] [Google Scholar]
- 22.Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- 23.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 25.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV. OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2014;2:457–465. doi: 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riffle S, Pandey RN, Albert M, Hegde RS. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer. 2017;17:338. doi: 10.1186/s12885-017-3319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pandey RN, Wang TS, Tadjuidje E, McDonald MG, Rettie AE, Hegde RS. Structure-Activity Relationships of Benzbromarone Metabolites and Derivatives as EYA Inhibitory Anti-Angiogenic Agents. PLoS One. 2013;8:e84582. doi: 10.1371/journal.pone.0084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tadjuidje E, Wang TS, Pandey RN, Sumanas S, Lang RA, Hegde RS. The EYA Tyrosine Phosphatase Activity Is Pro-Angiogenic and Is Inhibited by Benzbromarone. PLoS One. 2012;7:e34806. doi: 10.1371/journal.pone.0034806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okabe Y, Sano T, Nagata S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature. 2009;460:520–524. doi: 10.1038/nature08138. [DOI] [PubMed] [Google Scholar]
- 30.Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, Hegde RS. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003;426:295–298. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- 31.Wong H, Choo EF, Alicke B, Ding X, La H, McNamara E, Theil FP, Tibbitts J, Friedman LS, Hop CE, Gould SE. Antitumor activity of targeted and cytotoxic agents in murine subcutaneous tumor models correlates with clinical response. Clin Cancer Res. 2012;18:3846–3855. doi: 10.1158/1078-0432.CCR-12-0738. [DOI] [PubMed] [Google Scholar]
- 32.Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014;30:326–339. doi: 10.1016/j.tig.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Chiba Y, Taniguchi T, Matsuyama K, Sasaki M, Kato Y, Tanaka H, Muraoka R, Tanigawa N. Tumor angiogenesis, apoptosis, and p53 oncogene in stage I lung adenocarcinoma. Surg Today. 1999;29:1148–1153. doi: 10.1007/BF02482263. [DOI] [PubMed] [Google Scholar]
- 34.Aotake T, Lu CD, Chiba Y, Muraoka R, Tanigawa N. Changes of angiogenesis and tumor cell apoptosis during colorectal carcinogenesis. Clin Cancer Res. 1999;5:135–142. [PubMed] [Google Scholar]
- 35.Parangi S, O’Reilly M, Christofori G, Holmgren L, Grosfeld J, Folkman J, Hanahan D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:2002–2007. doi: 10.1073/pnas.93.5.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- 37.Rayapureddi JP, Kattamuri C, Chan FH, Hegde RS. Characterization of a plant, tyrosine-specific phosphatase of the aspartyl class. Biochemistry. 2005;44:751–758. doi: 10.1021/bi0481794. [DOI] [PubMed] [Google Scholar]
- 38.Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol. 2006;29:495–501. [PubMed] [Google Scholar]
- 39.Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, Barelier S, Vasseur S, Spoto RP, Pebusque MJ, Dusetti NJ, Iovanna JL, Carrier A. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer research. 2009;69:219–226. doi: 10.1158/0008-5472.CAN-08-2320. [DOI] [PubMed] [Google Scholar]
- 40.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T, Kurose A, Huang X, Traganos F, Dai W, Darzynkiewicz Z. Extent of constitutive histone H2AX phosphorylation on Ser-139 varies in cells with different TP53 status. Cell Prolif. 2006;39:313–323. doi: 10.1111/j.1365-2184.2006.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turinetto V, Giachino C. Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015;43:2489–2498. doi: 10.1093/nar/gkv061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 44.Hautekeete ML, Henrion J, Naegels S, DeNeve A, Adler M, Deprez C, Devis G, Kloppel G. Severe hepatotoxicity related to benzarone: a report of three cases with two fatalities. Liver. 1995;15:25–29. doi: 10.1111/j.1600-0676.1995.tb00102.x. [DOI] [PubMed] [Google Scholar]
- 45.Stamp LK, Haslett J, Frampton C, White D, Gardner D, Stebbings S, Taylor G, Grainger R, Kumar R, Kumar S, Kain T, Porter D, Corkill M, Cathro A, Metcalfe S, Wyeth J, Dalbeth N. The safety and efficacy of benzbromarone in gout in Aotearoa New Zealand. Intern Med J. 2016;46:1075–1080. doi: 10.1111/imj.13173. [DOI] [PubMed] [Google Scholar]
- 46.Lee MH, Graham GG, Williams KM, Day RO. A benefit-risk assessment of benzbromarone in the treatment of gout. Was its withdrawal from the market in the best interest of patients? Drug safety: an international journal of medical toxicology and drug experience. 2008;31:643–665. doi: 10.2165/00002018-200831080-00002. [DOI] [PubMed] [Google Scholar]
- 47.Fini MA, Elias A, Johnson RJ, Wright RM. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin Transl Med. 2012;1:16. doi: 10.1186/2001-1326-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linder N, Butzow R, Lassus H, Lundin M, Lundin J. Decreased xanthine oxidoreductase (XOR) is associated with a worse prognosis in patients with serous ovarian carcinoma. Gynecol Oncol. 2012;124:311–318. doi: 10.1016/j.ygyno.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 49.Linder N, Haglund C, Lundin M, Nordling S, Ristimaki A, Kokkola A, Mrena J, Wiksten JP, Lundin J. Decreased xanthine oxidoreductase is a predictor of poor prognosis in early-stage gastric cancer. Journal of clinical pathology. 2006;59:965–971. doi: 10.1136/jcp.2005.032524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim AW, Batus M, Myint R, Fidler MJ, Basu S, Bonomi P, Faber LP, Wightman SC, Warren WH, McIntire M, Arvanitis LD, Gattuso P, Xu X, Liptay MJ. Prognostic value of xanthine oxidoreductase expression in patients with non-small cell lung cancer. Lung Cancer. 2011;71:186–190. doi: 10.1016/j.lungcan.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 51.Tsimberidou AM, Keating MJ. Hyperuricemic syndromes in cancer patients. Contrib Nephrol. 2005;147:47–60. doi: 10.1159/000082541. [DOI] [PubMed] [Google Scholar]
- 52.Perez-Soler R. The role of erlotinib (Tarceva, OSI 774) in the treatment of non-small cell lung cancer. Clin Cancer Res. 2004;10:4238s–4240s. doi: 10.1158/1078-0432.CCR-040017. [DOI] [PubMed] [Google Scholar]
- 53.Kellar A, Egan C, Morris D. Preclinical Murine Models for Lung Cancer: Clinical Trial Applications. Biomed Res Int. 2015;2015:621324. doi: 10.1155/2015/621324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.