

Abstract
Inhibition of ataxia-telangiectasia mutated (ATM) during radiotherapy of glioblastoma multiforme (GBM) may improve tumor control by short-circuiting the response to radiation-induced DNA damage. A major impediment for clinical implementation is that current inhibitors have limited CNS bioavailability, thus, the goal was to identify ATM inhibitors (ATMi) with improved CNS penetration. Drug screens and refinement of lead compounds identified AZ31 and AZ32. The compounds were then tested in vivo for efficacy and impact on tumor and healthy brain. Both AZ31 and AZ32 blocked the DNA damage response (DDR) and radiosensitized GBM cells in vitro. AZ32, with enhanced blood-brain barrier (BBB) penetration, was highly efficient in vivo as radiosensitizer in syngeneic and human, orthotopic mouse glioma model compared with AZ31. Furthermore, human glioma cell lines expressing mutant p53 or having checkpoint-defective mutations were particularly sensitive to ATMi radiosensitization. The mechanism for this p53 effect involves a propensity to undergo mitotic catastrophe relative to cells with wild-type p53. In vivo, apoptosis was >6-fold higher in tumor relative to healthy brain after exposure to AZ32 and low dose radiation. AZ32 is the first ATMi with oral bioavailability shown to radiosensitize glioma and improve survival in orthotopic mouse models. These findings support the development of a clinical-grade, BBB-penetrating ATMi for the treatment of GBM. Importantly, since many GBMs have defective p53 signalling, the use of an ATMi concurrent with standard radiotherapy is expected to be cancer-specific, increase the therapeutic ratio, and maintain full therapeutic effect at lower radiation doses.
Keywords: Blood-brain barrier, DNA damage response, p53
Introduction
Glioblastoma multiforme (GBM) is a highly lethal brain cancer presented as one of two subtypes with distinct clinical histories and molecular profiles (1). Significant characteristics of GBM include aggressive growth, diffuse infiltration, and resistance to undergo apoptosis. In older individuals, the most common form is the primary subtype, which arises with no prior symptoms or evidence of progression from a lower grade tumor. Secondary GBM occurs in younger patients by progression from a lower grade tumor. In addition, brain metastasis occurs frequently in many advanced cancers, including lung and breast, among others. The Cancer Genome Atlas (TCGA) has molecularly characterized GBM and defined more precisely key gene alterations in GBM (2). Primary GBMs have frequent EGFR mutations/ amplifications and mutations in CDKN2A and PTEN. On the other hand, secondary GBMs are usually associated with mutations in PDGFR, IDH1/2, and p53 (3). The frequency of p53 mutation in GBM varies depending on subtype with secondary GBM having the most (4, 5). However, it is now generally recognized that most if not all GBMs have defective p53 signaling and/or compromised cell cycle checkpoints (2). Together, these attributes account for GBM’s poor prognosis and resistance toward standard-of-care treatment, and a median survival of only about a year. Surgery followed by radio- and chemotherapy combinations has improved outcomes, but in most cases, not all cancer tissue can be removed and the disease progresses.
Ataxia telangiectasia mutated (ATM) protein is a critical kinase that responds to DNA double strand breaks (DSB) and reactive oxygen species (ROS) induced by cellular exposures to ionizing radiation (IR) and certain intrinsic stimuli (6, 7). ATM is an ATP-dependent phosphatidylinositol 3-kinase-related kinase (PIKK) serine/threonine protein kinase related to ATR, DNA-PKcs, mTOR, SMG1 and the non-enzymatic TRRAP within this enzyme family (6). ATM is auto-phosphorylated when recruited to sites of DSBs by the DNA end-tethering MRE11–RAD50–NBS1 (MRN) complex (8, 9), which in turn results in phosphorylation of H2AX at serine-139 (γ-H2AX) thereby initiating the assembly of DNA repair components at DSBs and amplifying the DDR (10, 11). Many hundreds of proteins are phosphorylated directly or indirectly by ATM (12, 13), including p53 at serine-15, MDM2 at serine-394 and CHK2 at threonine-68, which, in turn, also phosphorylates p53 on serine-20 (14–16), illustrating the intricacy of the DDR and ATM’s central role as a master regulator leading to p53/p21-mediated cell cycle arrest, and, ultimately, a decision between DNA repair and survival, or apoptotic end points.
It is logical to understand that blocking the function of ATM affects the ability of tumor cells to respond to DNA damage and ROS, making this a highly attractive pharmacological target to potentially radiosensitize tumors. The ATM inhibitors, KU-55933, CP466722 and KU-59403, were shown to sensitize tumor cells to radiation and certain chemotherapeutic agents that cause DSBs, such as irinotecan and doxorubicin (17–19). KU-60019 (20), an improved analog of KU-55933, was reported to radiosensitize human glioma cells in vitro and significantly reduced migration, invasion, and growth, and, when infused directly into mouse orthotopic gliomas prior to repeated doses of radiation, significantly increased the survival of mice compared to radiation or drug alone treatments (21). However, except for the broad spectrum PI3K/mTOR inhibitor (NVP-BEZ235) that also inhibits ATM and DNA-PKcs (22), there are no reports, to our knowledge, of small-molecule ATM inhibitors that significantly cross the blood-brain barrier (BBB) during systemic treatment and demonstrate effective radiosensitization of gliomas in vivo.
Herein, we show that a novel, orally bioavailable, mouse BBB-permeable ATMi significantly extends the survival of mice harboring intra-cranial tumors treated with ATMi and radiation. Furthermore, building on our previous study that p53 mutant/cell cycle defective gliomas were more sensitive to ATMi radiosensitization than matched p53 wild-type counterparts (21), we define that this selectivity is attributed to the increased tendency of p53 mutant cells to undergo mitotic catastrophe, which results in a >6-fold apoptosis differential selective for treated tumor over healthy brain.
Materials and Methods
Reagents
AZ31, AZ32, and KU-60019 (20) were dissolved in DMSO to concentrations specified. For in vivo studies, AZ31 was prepared in 10% DMSO + 90% Captisol (30%w/v) to 10 mg/ml, and AZ32 in hydroxypropyl-methyl cellulose (0.5%w/v)/0.1%w/v polysorbate-80 to 20 mg/ml. Both compounds were administered by oral gavage 1 hr prior to radiation.
Plasmids and viruses
Lentiviruses were generated in HEK293T cells (23). Lentivirus expressing H2B-mCherry has been described (23). The virus expressing the fusion EGFP-Centrin2 was constructed from pEGFP-Centrin 2 (provided by E. Nigg; Addgene plasmid #41147) and pWPXLd (23). Lentivirus shp53 pLKO.1 puro (provided by R. Weinberg; Addgene plasmid #19119) was used to knock down p53 with pBabe-puro (provided by H. Land, J. Morgenstern, and R. Weinberg; Addgene plasmid #1764) as empty vector control.
Cell culture
Certified malignant glioma LN18, T98G, Hs683, SW1088 (anaplastic astrocytoma), SW1783 (anaplastic astrocytoma), U118MG, U138MG, M059J, A172, U87MG, H4, CF5STTG1 cells were obtained from the American Type Culture Collection (ATCC). AstraZeneca purchases cell lines from reputable providers such as ATCC and the European Collection of Authenticated Cell Cultures (ECACC) to ensure cell line authenticity. The AstraZeneca Global Cell Bank performs comprehensive quality checks on all cell lines including species-specific Short Tandem Repeat (STR) profiling against a minimum of 9 published markers per species. Human glioma U1242, U87/luc-DsRed-p53(281G), and cell derivatives expressing reporter genes were previously described (21). Mouse glioma GL261 cells were infected with Fluc-DsRed2 lentivirus (21) and sorted prior to cell injections. Similarly, certified NCI-H2228 non-small lung cancer cells were obtained from the ATCC. These cells were also modified to express luciferase (NCI-H2228-Luc) suitable for BLI. Cells were acquired and modified between 2009 and 2016. Cells were grown in complete Dulbecco’s Modified Eagles Medium (Gibco) supplemented with 10% FBS and penicillin-streptomycin at 37°C and 5% CO2. Cultures were maintained for no longer than 2 month and routinely tested negative for mycoplasma. Radiosurvival (CFA) experiments were carried out as described (20, 21).
Antibodies
Primary antibodies include p-ATM Ser1981 (Epitomics), ATM (Genetex), p-KAP-1 Ser824 (Bethyl), KAP-1 (CalBiochem), p-p53 Ser15 (Cell Signaling), anti-p53 (Calbiochem or Santa Cruz DO-1), anti-p(T68)-CHK2 (Cell Signaling), anti-CHK2 (Cell Signaling), cleaved caspase 3 rabbit mAb (5A1E), anti-α-tubulin (Cell Signaling), and anti-GAPDH (EMD Millipore). Secondary antibodies for westerns were anti-rabbit Dylight-800 (Cell Signaling) and anti-mouse Alexa Fluor-680 (Invitrogen). Secondary antibodies for immunofluorescence were AlexaFluor-488, AlexaFluor-568, and AlexaFluor-647 anti-IgG (Invitrogen).
Western blotting
Western blotting of cell extracts were done as described (20).
Mouse surgery and irradiation
All procedures were carried out in accordance with protocol AM10197 approved by the VCU (Richmond, VA) Institutional Animal Care and Use Committee (IACUC). Partial (head only) irradiation was performed using an MDS Nordion Gammacell 40 irradiator with a Cs-137 source at a dose rate of 1.05 Gy/min. For conformal micro-irradiation of mice a SARRP (Gulmay) was utilized. Mouse brain tumors were irradiated with 5×5-mm field either laterally or from the top of the head on the side of the tumor.
Microscopy
Cells were grown on chamber slides and processed for immunocytochemistry as described (24, 25). Brain tissues were processed as described (21). Cell nuclei were counterstained with DAPI and mounted in VECTASHIELD mounting medium (Vector Laboratories). Imaging was performed on a Zeiss LSM 710 laser scanning confocal microscope and images were analyzed using Zeiss Zen software.
Live cell imaging
U87/H2B-mCherry/Centrin2-EGFP and U87/shp53/H2B-mCherry/Centrin2-EGFP cells were seeded on 4-chamber, glass-bottom CELLview tissue culture dishes (Grenier Bio-One) and allowed to grow for 48–72 hrs. Time-laps videos were captured using a Zeiss Cell Observer SD spinning disk confocal microscope as described (26). AZ32 was added 1 hr before irradiation (5 Gy). Images were taken every 7 min beginning 2 hrs after irradiation for a total of 16 hrs. Aberrant mitoses were identified visually by morphological abnormalities in chromatin and/or centrosomes (27). DNA was visualized by DAPI stain or by expression of a fluorescent histone H2B-mCherry fusion protein. Centrosomes were fluorescently labelled with antibodies against α-tubulin or visualized with an EGFP-Centrin2 fusion protein.
Statistics
Statistical analyses were calculated using ANOVA (IBM SPSS Statistics 22) or GraphPad Prism.
Results
Chemical synthesis and characterization of novel bioavailable ATM kinase inhibitors
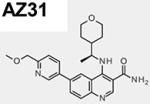
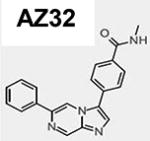
The chemical synthesis of AZ31 (6-[6-(methoxymethyl)-3-pyridinyl]-4-([(1R)-1- (tetrahydro-2H-pyran-4-yl)ethyl]amino)-3-quinolinecarboxamide; compound 72; ATM cell IC50 = 0.046 µM) was recently reported (28, 29). AZ32 (compound 8), is a specific inhibitor of the ATM kinase that possesses good BBB penetration in mouse (29). The structures and pharmacological properties of AZ31 and AZ32 are summarized in Table 1. Adequate oral exposure in mouse was observed with a dose of 50 mg/kg BID (twice daily) of AZ31 giving unbound plasma exposures in excess of the ATM cell IC50 for approximately 24 hr whereas 100 mg/kg barely made it above the IC50 at 0.046 µM in the brain and only for 2–3 hr (Fig. S1A). On the other hand, AZ32 showed moderate potency against ATM in cell (IC50 = 0.31 µM) and adequate selectivity over ATR, while also having high cell permeability. Following a single oral dose of AZ32 (200 mg/kg) in mice, the free brain concentrations of AZ32 was in excess of the cellular IC50 for approximately 22 hr (Fig. S1B). Altogether, AZ32 has enhanced BBB penetration at 8.7-fold and improved brain coverage over AZ31 but with reduced ATM selectivity (Table 1).
Table 1.
AZ31 and AZ32 chemical structures and biophysical properties. The AZ31 biophysical properties were reported recently28.
| Properties |
|
|
|---|---|---|
| ATM enzyme IC50 (µM) | <0.0012 | <0.0062 |
| pS1981ATM IC50 (µM) | 0.046 | 0.31 |
| Mol weight / Polar surface area | 421 / 99 | 328.4 / 54 |
| LogD | 2.5 | 2.94 |
| Solubility (µM) | >750 | 24 |
| Kinase selectivity -biochemical PIKK enzyme activity (µM) | ATR > 30 | ATR > 4.6 |
| DNA-PK < 30 | DNA-PK < 4.5 | |
| mTOR < 30 | mTOR < 4.6 | |
| PI3Kα = 30 | PI3Kα = 4.6 | |
| hERG (IC50 µM) | 17.6 | |
| Free Brain:Plasma (AUC) ratio (corrected for brain / plasma binding) from mouse brain homogenate after 200 mg/kg dose = Kpuu) | 0.03 | 0.26 |
Characterization of AZ31 in vitro and in vivo with glioma cells and orthotopic tumor models
Molecular and radiobiological characterization of AZ31’s ability to inhibit the DDR and radiosensitize glioma cells was determined by radiosurvival using colony-forming ability (CFA) of human glioma T98G cells. AZ31 radiosensitized cells to a similar extent as KU-60019 (Fig. S2A). As expected, AZ31 also effectively blocked phosphorylation of a panel of ATM targets including p53-S15, KAP1-S824, and ATM auto-phosphorylation at S1981 (Fig. S2B). AZ31 was at least 10-fold more potent compared to KU-60019 (compare lanes 2–6).
To determine whether AZ31 would be effective in vivo as a glioma radiosensitizer, we utilized the well-established syngeneic orthotopic mouse glioma model, GL261/C57bl6 (30), which shows morbidity within 3 weeks after intracranial cell injection. Initial experiments demonstrated that GL261 cells expressing mutant p53 (p53-153P) were radiosensitized by KU-60019 (p = 0.0001) when administered intra-tumorally by CED immediately prior to radiation, as described (21) (Fig. S2C). However, when administered orally during a prolonged low radiation dose treatment schedule, AZ31 was unable to significantly radiosensitize tumors using the same model (Fig. S2D). Thus, AZ31 was unable to radiosensitize GL261 tumors when administered orally, which is likely a reflection of poor brain coverage (see Table 1). Altogether, AZ31 is a potent radiosensitizer in vitro but low BBB permeability renders it unsuitable for systemic treatment of mouse brain tumors.
Characterization of AZ32 and demonstration that a BBB-penetrating ATMi improves the survival of mice with glioma
Similar to AZ31, AZ32 showed radiosensitizing and DDR blocking ability in vitro. A panel of human glioma cell lines with different p53 status, including LN18, U1242, and T98G (all with mutant p53), as well as DBTRG and U87MG (with wild-type p53) (21, 31), were all radiosensitized in vitro (Fig. 1A). However, all glioma cell lines with mutant p53 showed greater DER (>2) than cell lines with normal p53 (Supplementary Table 1).
Figure 1.

Radiosensitization of glioma cell lines by AZ32 in vitro. A, Human glioma cell lines with mutant p53 are radiosensitized by AZ32. B, AZ32 blocks the DDR in mouse glioma cells. Mouse glioma GL261 cells were irradiated (5 Gy) or not, exposed to KU-60019 (3 µM) or AZ32 (0.3, 1, or 3 µM) or not, followed by western blotting with anti-pp53 (S15), -pKAP1 (S824), and GAPDH or KAP1 (loading controls) antibodies. C, Radiosurvival colony-forming assay. Mouse GL261 cells were diluted, plated on tissue culture dishes, and exposed to AZ32 (3 µM) or not, and irradiated (2, 4 Gy) or not. Approximately 2 weeks later colonies (>50 cells) were counted after staining with crystal violet.
AZ32 also blocked the DDR and radiosensitized mouse GL261 glioma cells. Similar to KU-60019, AZ32 blocked the phosphorylation of p53 and KAP-1 after radiation (Fig. 1B). In a radiosurvival experiment, GL261 cells were compromised by AZ32 and became more radiosensitive (Fig. 1C). In vivo, AZ32 performed very well and significantly (p = 0.019) prolonged the survival of GL261/C57bl6 mice growing intra-cranial tumors when treated with whole-head irradiation from a 137-Cs source (4×2.5 Gy, Fig. 2A). Compared to radiation alone, more than half of the mice were apparently cured (no sign of tumor by BLI).
Figure 2.

Radiosensitization of glioma by AZ32 in vivo. A, AZ32 radiosensitizes orthotopic GL261 glioma. GL261/luc-red cells were injected intracranially into C57bl6 mice followed by BLI to confirm tumor growth prior to treatment. AZ32 (200 mg/kg; p.o. QD) was administered on day 11 - 15 1 hr prior to radiation (2.5 Gy) on day 12 - 15. B, AZ32 radiosensitizes human orthotopic p53 mutant glioma. Human glioma U87/281G cells expressing luciferase and DsRed were injected intracranially. AZ32 (200 mg/kg; p.o. QD) was administered on days 12–15 and 1 hr later irradiated by whole-head (137-Cs) with 2.5 Gy to a total dose of 4×2.5 Gy. C, AZ32 radiosensitizes orthotopic mouse GL261 glioma using SARRP. Injection of GL261/luc-red cells was followed by AZ32 (200 mg/kg; p.o. QD) and 2×5 Gy SARRP (5×5-mm lateral field). D, Brain NCI-H2228 non-small cell lung cancer brain mets are radiosensitized by AZ32. Nude mice were injected with NCI-H2228-Luc cells and randomized into groups to receive vehicle, radiation alone, AZ32 (50 mg/kg BID) alone or AZ32 in combination with radiation.
Previously, we demonstrated greater ATMi radiosensitization with mutant p53-281G glioma cells compared with parental wild type cells (21). To extend the AZ32 study to human glioma xenografts, we injected U87-281G cells, expressing mutant p53, intra-cranially into nude mice resulting in a significant (p = 0.0015) increase in survival of mice in the AZ32 and 4×2.5 Gy group relative to mice receiving radiation alone (Fig. 2B).
During these initial survival experiments, we noticed that the irradiated mice also treated with AZ32 developed difficulties eating and drinking resulting in weight loss. This effect was more pronounced with C57bl6 than with nude mice and was radiation dose-dependent, occurring consistently after a total dose of ~10 Gy to the head of the mouse. We suspected this might be caused by mucositis, ie, inflammation and ulceration occurring in the mouth, due to radiation. Therefore, we decided to apply conformal radiation (5×5-mm lateral field) using the Small-Animal-Radiation-Research-Platform (SARRP) rather than whole-head irradiation to potentially reduce mucositis. The more precise radiation delivery by SARRP can easily be seen by the whitening of the fur of treated mice observed post-irradiation (Fig. S3).
Again, C57bl6 mice growing GL261 tumors treated with AZ32 and 2×5 Gy (SARRP) resulted in a significant (p = 0.0001) increase in survival of the AZ32 and radiation group with almost half of the mice surviving for more than 120 days and showing no sign of tumor by BLI. Importantly, we did not notice any radiation toxicity in this experiment suggesting that avoiding the salivary glands was critical for reducing normal tissue toxicity. This result also demonstrated that as few as two fractions were sufficient to significantly improve survival.
To determine whether AZ32 would also be efficacious for treating metastatic brain cancer, we utilized the non-small lung cancer brain metastasis model, NCI-H2228 (BM), which grew into intra-cranial tumors based on BLI and brain histology (Fig. S4A, B). Both tumor growth (BLI) (Fig. S4C), and mouse survival (Fig. 2D) showed that mice treated with AZ32 in combination with radiation survived much longer than mice in the radiation alone or AZ32 alone groups. These data suggest that AZ32 is able to radiosensitize intracranial NCI-H2228 (BM) tumors and improve survival. Altogether, AZ32 administered orally and concurrently with radiation prolongs the survival of orthotopic tumor-bearing, immune-competent mice, as well as human glioma xenografts demonstrating excellent efficacy of AZ32 as a radiosensitizer for primary and metastatic glioma.
Differential response of p53 mutant versus wild-type glioma cells to an ATMi and radiation
To assess the potency and possible differential radiosurvival response to AZ31 and the impact of p53 status, we utilized a panel of human glioma cell lines with varying p53 mutant/checkpoint-defective mutations and normal p53. Cells were exposed to AZ31 or not followed by radiation and the radiation dose-modification-ratio (DMR) determined (Table 2). Cell lines were ranked in respect to DMR, p53 status, and whether they demonstrated normal or defective p21-induction after radiation (Fig. S5), and listed in order of descending DMR (Table 2). Human glioma cell lines with p53 mutations and deficiencies in p21 induction were more responsive to AZ31 radiosensitization than cells with normal p53 signalling. Thus, AZ31 blocks the DDR and radiosensitizes human glioma cells in vitro, correlating with impaired p53/cell cycle signaling, in line with similar results using AZ32 (Fig. 1, Supplementary Table 1).
Table 2.
Mutant p53 GBM cells are more sensitive to ATM kinase inhibition in combination with radiation. Human GBM cell lines with varying p53 and p21 status were treated with AZ31 (1 µM) followed by radiation (4 Gy). Average (n=3) of representative results in Supplementary Fig. S5 is shown. Cells were seeded in 384-well plates dosed with AZ31 an hour before irradiation. Total cell counts were measured by Saponin and live cells measured by Sytox green at day 0 and day 5 after irradiation. DMR (fold radiosensitization achieved by AZ31 calculated after irradiation with 4 Gy). Red fields represent cell lines with mutant p53/defective p53 signalling and green fields represent cell lines with wild type p53/proficient p53 signalling, respectively. Yellow fields represent exceptions and intermediate responders.

|
Increased mitotic catastrophe in glioma cells following treatment with AZ32 and radiation
It is well-established that an ATMi abrogates the radiation-induced G2/M checkpoint (32, 33). A major effect of an ATMi on radiation-induced checkpoints is the failure of treated cells to arrest in G2/M and prematurely permit cells to enter mitosis prior to repairing DNA damage (34). Our previous work demonstrated that human glioma U87 (p53 wild-type) and U87-281G (p53 mutant) cells have intact radiation-induced G2/M checkpoints, whereas the G1/S checkpoint is only impaired in the latter cells (21). To determine whether human glioma cells treated with AZ32 and radiation would overcome the G2/M checkpoint arrest and result in elevated chromosome aberrations, we examined treated U1242 (p53-R175H) (21) cells by ICC staining with α- and γ-tubulin antibodies as a means to assay for abnormal centrosome numbers (>2) and lagging chromosomes (Fig. 3). Many of the cells showed elevated, abnormal numbers of centrosomes after treatment with AZ32 and radiation but this was rare in untreated cells (Fig. 3A). Furthermore, we found a >2-fold increase over radiation alone in the number of lagging chromosomes and inter-nuclear bridges after treatment with AZ32 and radiation, suggesting increased mitotic failure (Fig. 3B).
Figure 3.

Increased mitotic aberrations in glioma cells exposed to AZ32 and radiation. A, U1242 cells show increased centrosomes and anaphase bridges relative to untreated cells. Human glioma U1242 cells were treated with AZ32 (3 µM) and radiation (2 Gy) or left untreated. At 48 hrs the cells were fixed and processed for ICC using anti- γ -tubulin (centrosomes) and - α -tubulin (microtubules). Cells were counterstained with DAPI to visualize nuclei. B, Quantification of inter-nuclear (anaphase)-bridges. Treatment of U1242 cells with AZ32 and IR produced 2.4-fold greater levels of bridges relative to IR alone. C, p53 knockdown sensitizes U87 cells to AZ32 radiation. p53 knockdown (~85%) in U87 cells after radiation. U87 cells were infected with either shp53 pLKO.1 puro (Addgene plasmid #19119) or pBabe-puro and selected for puromycin resistance. Resistant cells were treated with +/− AZ32 (3 µM) and +/− radiation (3 Gy) followed by western blotting with anti-p53 antibody. GAPDH was used as loading control. D, p53 knockdown results in increased AZ32 radiosensitization in vitro. E, Experimental outline. U87 cell expressing H2B-Cherry and EGFP-Centrin2 were infected with shp53 pLKO.1 puro or pBabe-puro, selected with puromycin and sorted by FACS to obtain mCherry+/EGFP+ cells. Cell populations were treated with AZ32 (3 µM) added at 0.5 hr prior to radiation (5 Gy) and subsequently monitored by time-lapse video for 16 hrs. F, Enhanced mitotic catastrophe in U87 cells with p53 knockdown. Representative images of mitosis and mitotic catastrophe after exposure of U87 parental (puro) and U87/shp53 cells to AZ32 and radiation. The table quantifies aberrant mitoses showing 4.2-fold (46/11) greater levels in shp53 cells relative to parental cells.
In an attempt to further investigate whether the increased response of p53 mutant glioma cells to ATMi radiosensitization is related to p53 function, we knocked down p53 in U87 glioma cells to create an isogenic pair only differing in p53 status. Western blotting showed that we achieved 85–90% reduced levels of p53 after radiation in the U87/shp53 cells relative to parental U87/puro cells (Fig. 3C). Most importantly, the U87/shp53 cells were much more sensitive (2–4 fold) to AZ32 radiosensitization relative to parental cells (Fig. 3D). This result recapitulates the finding we reported previously that U87 cells expressing p53-281G mutant is more sensitive compared to parental U87 cells isogenic with respect to p53 (21). Hence, both over-expression of mutant p53 and knockdown of p53 in human U87 cells give results suggesting that p53 status is central to determining the response to ATMi and radiation in glioma cells.
To determine whether p53 status directly influenced the extent of aberrant mitoses after treatment with AZ32 and radiation, we infected the two isogenically matched cell populations with lentiviruses expressing H2B-mCherry and EGFP-Centrin2 and followed mitotic cells by live cell imaging (Fig. 3E). Indeed, the shp53 knockdown cells had ~4-fold higher levels of mitotic aberrations leading to mitotic catastrophe (disappearance of cells in mitosis/cytokinesis with >2 sets of chromosomes) relative to the matched parental cells (Fig. 3F, Video 1 - 4).
To see whether the mitotic catastrophe response could be recapitulated in genetically matched p53 knockout and mutant knock-in cells, we examined human HCT116 colon carcinoma (p53 wild-type (+/+), null (−/−), and mutant knock-in R248W/-) cells and treated these with AZ32 with or without radiation (Fig. S6). As expected, the untreated p53−/− cells had a ~2-fold elevated mitotic index relative to p53+/+ cells. Likewise, irradiated p53+/+ cells had no mitotic cells indicating an intact G2/M checkpoint. On the other hand, p53−/− cells had ~2-fold reduced number of mitotic cells relative to control and they were all aberrant post-IR. The addition of AZ32 before IR treatment of p53−/− cells resulted in almost a doubling of aberrant mitoses and polyploidy cells which was not seen with p53+/+ cells (Fig. S6A). Similar results seen with p53 −/− cells were also observed with p53R248W/- cells. All combined, AZ32 in combination with radiation causes mitotic catastrophe at much greater levels in p53 null and R248W mutant colon carcinoma cells relative to wild-type which explains the enhanced response of p53 mutant cells to ATMi and radiation.
However, paradoxically HCT116 p53+/+ cells were more sensitive to AZ32 and radiation compared to mutant p53 cells by CFA (Fig. S6B). Thus, despite a higher rate of mitotic failure and catastrophe in p53 null and mutant cells, functional p53 is required for enhanced killing of colon carcinoma cells. In response to AZ32 and radiation, GBM cells respond in an opposite manner to colon carcinoma cells with respect to p53 status, ie, p53 null/mutant cells are more sensitive whereas p53 wild-type cells are more resistant (see Fig. 3D). This observation is in line with previous studies showing that malignant gliomas, and GBM in particular, are under extensive replicative stress and have gross, aberrant constitutive DDR (35), which might explain the enhanced response of p53 mutant cells to ATMi and radiation.
Mitotic catastrophe can be defined as triple-positive staining with γ-H2AX (DNA damage), p-histone H3 (mitosis), and cleaved caspase 3 (apoptosis) antibodies (36). To determine whether we could catch and quantify this cell population, we examined mouse GL261 glioma cells after treatment with AZ32 with or without radiation (Fig. 4A). At 24 hr, we were able to detect triple+ cells at 4% of all cells after treatment with AZ32 and radiation whereas untreated cells had no triple+ cells. Given the fact this is a snapshot of cells going through the cell cycle with triple+ cells likely disappearing relatively quickly, we believe that a propensity for and elevated levels of mitotic catastrophe accounts for the greater killing of p53 mutant/knockdown glioma cells to ATMi and radiation than p53 wild-type cells.
Figure 4.

Combination of AZ32 and radiation induces mitotic catastrophe in mouse GL261 glioma cells. A, Mitotic catastrophe (triple-stained+) is 4-fold higher in AZ32+IR treated mouse glioma cells than IR or AZ32 alone. Cells were grown on chamber slides and treated as indicated; AZ32 (3 µM), radiation (2 Gy). At 24 hr, the cells were fixed and incubated with the indicated antibodies (anti- γ -H2AX, -p-(S10)-histone H3, and -cleaved caspase 3). Mitotic catastrophe is quantified in the table showing 4-fold greater levels in AZ32+IR over IR alone. B, Apoptosis (cleaved caspase 3+) is significantly higher in tumor relative to brain after exposure to AZ32 and radiation. C57bl6 mice were injected intra-cranially with GL261/luc-red cells. Approximately 3 weeks later AZ32 was administered (200 mg/kg; p.o.) or not followed by SARRP irradiation (1 Gy; 5×5 mm) to the tumor side (approximate radiation field is indicated in red). The mice were euthanized 2 and 4 hr post-irradiation, respectively, fixed, imbedded in paraffin, and sectioned. Sections were exposed to anti-cleaved caspase 3 antibody and HRP secondary followed by DAB (brown) and hematoxylin staining (blue) counter staining. Coronal sections with tumors are shown (top) of untreated (left) and AZ32 + IR at 4 hr (right) with close-up (20X) images from areas indicated in red (inset). C, Quantification of cleaved caspase 3+ cells in treated brain and tumor by folds (left) and numbers (right). The area below the tumor were examined and scored as treated (AZ32 + radiation) brain whereas the left hemisphere was considered to be AZ32 alone (no radiation). p < 0.05*, p < 0.01**, p < 0.001***.
Increased levels of apoptosis in tumor relative to healthy brain in GL261/C57bl6 mice treated with AZ32 and low dose of conformal irradiation
Next, we wanted to determine whether AZ32 and radiation inflict greater damage to tumor than healthy brain, similar to the p53 mutant/knockdown glioma cell experiments in vitro. C57bl6 mice growing GL261/luc-red tumors were treated with AZ32 followed by low dose conformal radiation to the tumor in the right hemisphere using the SARRP. Brain sections were prepared and stained with anti-cleaved caspase 3 antibody to quantify apoptotic cells (Fig. 4B). We chose areas both within the irradiated tumor as well as irradiated (below the tumor) and non-irradiated (left hemisphere) healthy brain, and scored for cleaved caspase 3+ cells. On average, there were >2-fold more cells in each field in the tumor compared to brain (Fig. 4C). We also noted ~4-fold more cleaved caspase 3+ cells in untreated tumor compared to brain, probably reflecting apoptosis due to hypoxia and other pathophysiological alterations. Most importantly, the level and rate of apoptosis 4 hours after treatment with AZ32 and low dose radiation was >6-fold in the tumor compared to healthy brain parenchyma (Fig. 4C). Thus, mouse glioma with mutant p53 is more sensitive to ATMi radiosensitization and results in more apoptosis than in equally treated healthy brain.
Discussion
Despite recent advances, survival rates for patients diagnosed with GBM remain dismal. Following surgery, radiotherapy is an essential part of first-line treatment for GBM, but side-effects and inherent neurotoxicity remain significant and problematic issues. In addition, malignant gliomas are highly aggressive and resistant to radiation and chemotherapy, likely because glioma-derived cancer stem cells are able to preferentially activate DNA damage checkpoints and repair mechanisms to promote radioresistance (37). Therefore, inactivating proteins involved in the DDR by blocking the ATM kinase could be a promising new strategy for improving the efficacy of radiotherapy for GBM.
A next-generation ATMi, AZ32, was identified in a screen as a low molecular weight compound with enhanced BBB penetration. We found that ATM inhibition by AZ32 is dose-dependent and sustained even several hours after radiation. AZ32 significantly reduced the survival of human and mouse glioma cells in the low radiation dose range used in the clinic. Furthermore, human glioma cell lines with mutant p53 and/or defective cell cycle checkpoints were more sensitive to ATMi radiosensitization than p53 wild-type cells. Previous results from our group also demonstrated this phenomenon, but these studies were performed with cells in which mutant p53 was overexpressed (21). Thus, it was unclear if effective ATMi radiosensitization was due entirely to p53 status or if overexpression was a confounding variable. Therefore, we selected glioma cells expressing shRNA against p53 and compared their response and found that the radiosurvival in the absence of an ATMi did not differ significantly between shp53 and wild-type U87 glioma cells. However, radiosensitization by ATMi was enhanced in cells in which p53 was knocked down, and time-lapse imaging revealed that the ATMi preferentially increased the rate of mitotic catastrophe in shp53 cells following radiation.
Forced overexpression of mutant p53 or knockdown of p53 in human glioma cells and the comparison of responses between isogeneic cell pairs have provided consistent results in our hands with the overall conclusion that cells with defective p53 signaling are more sensitive to ATMi radiosensitization than glioma cells with normal p53. In further support of this result, we performed a set of experiments on a panel of human glioma cell lines and determined their response to ATMi radiosensitization. Cell lines with mutant p53 or defective cell cycle checkpoints were consistently more sensitive than cells with normal p53. In addition, to see whether this p53-dependent response was of general nature among tumor cells, we tested isogenic colon carcinoma HCT116 cells expressing wild-type p53, the p53 R248W gain-of-function mutant, and p53 null cells (38). Both mutant and null cells had increased mitotic aberrations and mitotic catastrophe after ATM inhibition and radiation. However, contrary to expectation, aberrant mitosis did not translate to reduced radiosurvival of mutant or null p53 colon carcinoma cells relative to p53 wild-type.
These observations are in stark contrast to our findings with glioma cells. We suspect that this difference may be due to the fact that basal levels of replicative stress and ROS are significantly higher in gliomas than in cancers derived from epithelial tissues (35). It may be that wild-type p53 induces apoptosis in response to replicative stress in HCT116 cells, but that such mechanism is inactive in glioma. Indeed, previous reports indicate that gliomas in general are often highly resistant to death by apoptosis (39). One possibility is that the reduced responsiveness of HCT116 cells with defective p53 to ATMi is due to that ATM signaling is already partially impaired in these cells. In fact, it was shown that MEFs expressing R248W were unable to efficiently recruit ATM to sites of DSBs due to defects in the MRE11-NBS1-RAD50 complex (40), in agreement with finding that MRE11 and DNA end-resection at DSBs was reported to be defective in HCT116 cells (41). Altogether, it appears that the response to AZ32 and radiation is opposite in glioma and colon carcinoma for survival in vitro, ie, p53 is required in the latter but not in the former, despite that both cell types show extensive mitotic catastrophe when p53 function is perturbed.
A similar observation regarding an inverse relationship between functional p53 and apoptosis was made using genetically engineered mouse models of breast cancer with mutant or null p53 breast carcinoma responding better to doxorubicin than matched p53 wild-type tumors (42). Along the same line, p53 mutant glioma cells may similarly rely on signaling through p38MAPK/MK2 for G2/M arrest when ATM activity is abrogated, as in mouse tumor models of lung and breast (42, 43). Thus, an ATMi might override radiation-induced G2/M arrest in cells with defective or absent p53 and reduce survival through mitotic catastrophe more effectively than in cells with normal p53. This discrepancy likely reflects a fundamental difference in genetic wiring between different types of cancers (44).
The current findings lend support to the idea that ATM is necessary for p53-independent G2/M arrest. This might involve an increased dependence on PLK1 as it was reported that nuclear signaling through p38 and MK2 is required for proper mitosis through an interaction and co-localization with p38 and MK2 at spindle poles during prophase and metaphase followed by PLK1 phosphorylation at serine-326 by MK2 (45). PLK-1 phosphorylation then permits mitotic progression by facilitating nuclear translocation of CDC25C (46). PLK1 is also necessary for the recovery from G2/M arrest and its expression is elevated in cells lacking wild-type p53 (38, 47).
In conclusion, orthotopic mouse and human gliomas were effectively radiosensitized by AZ32, a next-generation BBB penetrating ATMi, administered orally, whereas AZ31 was relatively ineffective. Our findings suggest that cells and tumors with defective p53 signaling are more sensitive to ATMi and low dose radiation than tumors with functional p53 signaling. In support of this, we have shown that combined ATM kinase inhibition and low dose radiation is selectively toxic to glioma with mutant p53 by inducing mitotic catastrophe and apoptosis, thus revealing a cancer-specific Achilles’ heel. We propose that targeting GBM with a BBB-penetrating small molecule inhibitor of ATM kinase in combination with standard radiotherapy would provide cancer-specificity and broadening of the therapeutic window in a significant subset of GBM patients.
We propose the following model for how an ATMi radiosensitization strategy might be cancer-specific for the treatment of GBM (Fig. S7). It was determined some time ago that ATM is required for p53-dependent radiation-induced neuronal apoptosis in the mouse (48). The negative effects of radiation on the brain are known to be dose-dependent (49), and thus could be minimized by using lower radiation doses. Considering that neurons and other terminally differentiated cells are not dividing and therefore cannot undergo cell cycle arrest, apoptosis would be p53-dependent but cell cycle-independent. On the other hand, proliferating cells of the CNS, for example neural stem and progenitor cells at different stages of neurogenesis, would have a p53-dependent G1/S checkpoint at least partially active (via ATR) in the absence of ATM or in the presence of an ATMi (50). Therefore, these cells would undergo cell cycle arrest and not apoptosis and thus be largely spared. Irradiation of a tumor with a p53 mutation while exposed to an ATMi would trigger mitotic catastrophe and subsequent apoptosis resulting in a more effective and cancer-specific therapy. Thus, transient brain exposure to an ATMi might not be unmanageably toxic and, in fact, could perhaps protect against neuronal cell death in an adjuvant setting.
Supplementary Material
Acknowledgments
Services and products in support of the research project were generated by the VCU Microscopy and Flow Cytometry Shared Resources and the Massey Cancer Center Mouse Model Core (CMMC) Facility, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30CA016059. We thank B. Vogelstein for the HCT116 cell panel.
Grant Support
Supported by R01NS064593, R21CA194789, R21CA156995, and funds provided by AstraZeneca (to K. Valerie).
Footnotes
Disclosure of Potential Conflicts of Interest
K. Valerie has received research funding from AstraZeneca. No other potential conflicts of interest were disclosed.
References
- 1.Reifenberger G, Wirsching HG, Knobbe-Thomsen CB, Weller M. Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol. 2017;14(7):434–52. doi: 10.1038/nrclinonc.2016.204. [DOI] [PubMed] [Google Scholar]
- 2.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009;100(12):2235–41. doi: 10.1111/j.1349-7006.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455(7216):1129–33. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews Molecular cell biology. 2013;14(4):197–210. [PubMed] [Google Scholar]
- 7.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9(10):759–69. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 8.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 9.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304(5667):93–6. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 10.Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22(37):5792–812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- 11.Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends in biochemical sciences. 2011 doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 13.Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang SY, Chen DJ, et al. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal. 2010;3(151):rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- 14.Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71(4):587–97. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 15.Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11(24):3471–81. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 17.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24):9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 18.Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008;68(18):7466–74. doi: 10.1158/0008-5472.CAN-08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batey MA, Zhao Y, Kyle S, Richardson C, Slade A, Martin NM, et al. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Molecular cancer therapeutics. 2013;12(6):959–67. doi: 10.1158/1535-7163.MCT-12-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8(10):2894–902. doi: 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biddlestone-Thorpe L, Sajjad M, Rosenberg E, Beckta JM, Valerie NC, Tokarz M, et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(12):3189–200. doi: 10.1158/1078-0432.CCR-12-3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gil del Alcazar CR, Hardebeck MC, Mukherjee B, Tomimatsu N, Gao X, Yan J, et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(5):1235–48. doi: 10.1158/1078-0432.CCR-13-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckta JM, Dever SM, Gnawali N, Khalil A, Sule A, Golding SE, et al. Mutation of the BRCA1 SQ-cluster results in aberrant mitosis, reduced homologous recombination, and a compensatory increase in non-homologous end joining. Oncotarget. 2015;6(29):27674–87. doi: 10.18632/oncotarget.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, Valerie K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol Ther. 2009;8(8):730–8. doi: 10.4161/cbt.8.8.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. 2007;67(3):1046–53. doi: 10.1158/0008-5472.CAN-06-2371. [DOI] [PubMed] [Google Scholar]
- 26.Beckta JM, Henderson SC, Valerie K. Two- and three-dimensional live cell imaging of DNA damage response proteins. J Vis Exp. 2012;67:e4251. doi: 10.3791/4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rello-Varona S, Kepp O, Vitale I, Michaud M, Senovilla L, Jemaa M, et al. An automated fluorescence videomicroscopy assay for the detection of mitotic catastrophe. Cell Death Dis. 2010;1:e25. doi: 10.1038/cddis.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Degorce SL, Barlaam B, Cadogan E, Dishington A, Ducray R, Glossop SC, et al. Discovery of Novel 3-Quinoline Carboxamides as Potent, Selective, and Orally Bioavailable Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase. Journal of medicinal chemistry. 2016;59(13):6281–92. doi: 10.1021/acs.jmedchem.6b00519. [DOI] [PubMed] [Google Scholar]
- 29.Pike KG. Discovery of AZD0156: The First Potent and Selective Inhibitor of ATM Kinase for Clinical Evaluation. In: Samuel Chackalamannil DPRaSEW., editor. Comprehensive Medicinal Chemistry III Volume 8. Oxford: Elsevier; 2017. pp. 161–77. [Google Scholar]
- 30.Szatmari T, Lumniczky K, Desaknai S, Trajcevski S, Hidvegi EJ, Hamada H, et al. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006;97(6):546–53. doi: 10.1111/j.1349-7006.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golding SE, Rosenberg E, Adams BR, Wignarajah S, Beckta JM, O'Connor MJ, et al. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle. 2012;11(6):1167–73. doi: 10.4161/cc.11.6.19576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22(4):1049–59. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carruthers R, Ahmed SU, Strathdee K, Gomez-Roman N, Amoah-Buahin E, Watts C, et al. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol Oncol. 2015;9(1):192–203. doi: 10.1016/j.molonc.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 35.Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 2010;29(36):5095–102. doi: 10.1038/onc.2010.249. [DOI] [PubMed] [Google Scholar]
- 36.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11(2):175–89. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 38.Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr, Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(10):3964–9. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinbach JP, Weller M. Apoptosis in Gliomas: Molecular Mechanisms and Therapeutic Implications. Journal of neuro-oncology. 2004;70(2):247–56. doi: 10.1007/s11060-004-2753-4. [DOI] [PubMed] [Google Scholar]
- 40.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nature cell biology. 2007;9(5):573–80. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- 41.Takemura H, Rao VA, Sordet O, Furuta T, Miao ZH, Meng L, et al. Defective Mre11-dependent activation of Chk2 by ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. The Journal of biological chemistry. 2006;281(41):30814–23. doi: 10.1074/jbc.M603747200. [DOI] [PubMed] [Google Scholar]
- 42.Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell. 2012;21(6):793–806. doi: 10.1016/j.ccr.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, et al. A Reversible Gene-Targeting Strategy Identifies Synthetic Lethal Interactions between MK2 and p53 in the DNA Damage Response In Vivo. Cell Rep. 2013;5(4):868–77. doi: 10.1016/j.celrep.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson JG, Lozano G. The mutant p53 mouse as a pre-clinical model. Oncogene. 2013;32(37):4325–30. doi: 10.1038/onc.2012.610. [DOI] [PubMed] [Google Scholar]
- 45.Tang J, Yang X, Liu X. Phosphorylation of Plk1 at Ser326 regulates its functions during mitotic progression. Oncogene. 2008;27(52):6635–45. doi: 10.1038/onc.2008.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toyoshima-Morimoto F, Taniguchi E, Nishida E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO reports. 2002;3(4):341–8. doi: 10.1093/embo-reports/kvf069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455(7209):119–23. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 48.Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280(5366):1089–91. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- 49.Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8(9):955–62. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- 50.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15(17):2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.