

Abstract
Both gains and losses of DNA methylation are common in cancer, but the factors controlling this balance of methylation remain unclear. Triple-negative breast cancer (TNBC), a subtype that does not overexpress hormone receptors or HER2/NEU, is one of the most hypomethylated cancers observed. Here we discovered that the TET1 DNA demethylase is specifically overexpressed in about 40% of patients with TNBC, where it is associated with hypomethylation of up to 10% of queried CpG sites and a worse overall survival. Through bioinformatic analyses in both breast and ovarian cancer cell line panels, we uncovered an intricate network connecting TET1 to hypomethylation and activation of cancer-specific oncogenic pathways including PI3K, EGFR, and PDGF. TET1 expression correlated with sensitivity to drugs targeting the PI3K-mTOR pathway, and CRISPR-mediated deletion of TET1 in two independent TNBC cell lines resulted in reduced expression of PI3K pathway genes, upregulation of immune response genes, and substantially reduced cellular proliferation, suggesting dependence of oncogenic pathways on TET1 overexpression. Our work establishes TET1 as a potential oncogene that contributes to aberrant hypomethylation in cancer and suggests that TET1 could serve as a druggable target for therapeutic intervention.
Keywords: Cancer, Epigenetics, DNA methylation, Oncogenic signaling, Triple negative breast cancer
Introduction
Cancer cells display abnormal DNA methylation, where both gains and losses of methylation are observed. Aberrant methylation in CpG islands (CGIs) or surrounding CGIs (CGI shores) can lead to changes in expression of tumor suppressor genes and oncogenes (1)(2)(3). Triple negative breast cancer (TNBC) is a subtype of breast cancer that occurs in 10-20% of patients, and is defined by tumors that do not overexpress the estrogen, progesterone or HER2 receptors. These patients lack options for targeted therapy, as there are no obvious tumor-specific receptors or pathways to target. TNBC tumors have widespread genome-wide hypomethylation when compared to other breast cancer subtypes and normal breast controls (4)(5)(6). Furthermore, this hypomethylation is independently associated with a worse overall survival (7). How TNBCs become hypomethylated and why it is associated with a worse prognosis remains unknown.
The TET enzymes (TET1, TET2, TET3) are DNA demethylases that convert 5 methyl-cytosine (5mC) into 5 hydroxymethylcytosine (5hmC), which can then be further oxidized or converted to un-methylated cytosine (8)(9). In addition to its demethylase activity, TET1 acts as both a transcriptional co-activator and co-repressor, where its transcriptional activity can be dependent or independent of its demethylase activity (10)(11)(12). TET1 has a CXXC domain that binds to unmethylated CGIs and protects them from gaining aberrant methylation (12). We recently identified an isoform of TET1 (TET1ALT) that lacks targeting to CpG islands and that is overexpressed in some cancers (13). We now report that TET1 and TET1ALT mRNA are upregulated specifically in a subset of TNBC tumors that display genome-wide hypomethylation and activation of genes in the PI3K-Akt-mTOR pathway. Furthermore, this hypomethylation is mutually exclusive with activating PI3K mutations, suggesting that demethylation may be an alternate mechanism to activate this oncogenic pathway.
Materials and Methods
Cell culture
The cell lines Hs578T, MDA-MB-231, HCC1599 and HCC2218 were obtained from American Type Culture Collection (ATCC). Hs578T and MDA-MB-231 cells were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS. HCC2218 and HCC1599 cells were cultured in RPMI-1640 media with 10% FBS. All cell lines were routinely tested for mycoplasma contamination and cell line identification was validated in September 2017 by DNA fingerprinting.
CRISPR
To knockout TET1, we used the Lenti CRISPR V2 plasmid (Addgene 52961). CRISPR gRNAs were designed using http://crispr.mit.edu/ to target three different TET1 exons (Supplementary Table S1). Lentiviruses were generated using HEK293T cells and viral supernatant was collected at 48 and 72 hours and incubated on MDA-MB-231 or Hs578T cells for 7 hours in the presence of polybrene (6μg/mL, Millipore). We puromycin selected (1μg/mL) the cells for three days, followed by single cell cloning using serial dilution. After selection, cells were maintained in normal media supplemented with 0.25 μg/mL puromycin.
Western blot and slot blot analysis
A previously established protocol was followed (13). The following primary antibodies were used in this study: anti-TET1 (GT1462, Sigma), anti-β-actin (A5316, Sigma), anti-phospho 4EBP1-Thr37/46 (9459S, Cell Signaling) and anti-5hmC (catalog # 39769, Active Motif). For the DNA slot blot analysis, we followed the protocol established previously with the exception of using a slot blot apparatus instead of a dot blot (12).
Digital restriction enzyme analysis of methylation (DREAM)
Genome-wide DNA methylation assay was carried out using DREAM as described previously (14). The coverage threshold was set to greater than 19 reads per sample. TET1 knock out cell lines were compared to empty vector control single clone and data were filtered for sites that change methylation by greater than 10% with p<0.05. GEO accession number GSE100640. DREAM data for normal breast epithelium (NBE) were downloaded from GEO series GSE74910 (15). For NBE analysis parameters see supplementary methods.
RNA-seq
RNA was isolated using Qiagen’s RNeasy Plus Mini kit following manufacturer’s instructions from experiments done in biological triplicates. Strand-specific RNA libraries were generated from 1 μg of RNA using TruSeq stranded total RNA with Ribo-Zero Gold (Illumina). For details, see supplementary methods. The expression level and fold change of each treatment group was evaluated using Cuffdiff (16). For MDA-MB-231 cells, genes were considered differentially upregulated with FC>2 and downregulated with FC<0.5 and p<0.001. For Hs578T cells, genes were considered differentially upregulated with FC>1.5 and downregulated with FC<0.66 and p<0.05. GEO accession number GSE100483.
Proliferation and cell migration
20,000 cells were seeded per well in 24 well plates for proliferation assays. Biological triplicates were counted twice for a total of six counts per sample. Cells were counted every 24 hours for 120 hours and the data were plotted as the total number of cells vs the number of hours. For the wound healing assay, cells were seeded in 24-well plates and incubated to confluency. Wounds were scratched using a 200 μl sterile pipette tip. After scratching, detached cells were removed by washing three times with PBS and replenished with DMEM 10% FBS. The migration of the cells at the edge of the scratch was photographed at 0, 24, and 48 hours. The gap distance was quantitatively evaluated using ImageJ Wound Healing Tool (http://rsb.info.nih.gov/ij/download.html). To reduce variability, multiple wells of each cell line were evaluated.
Cell viability assay
The sensitivity of cells to the different drugs at the indicated time points was measured using the CellTiter-Blue Cell Viability assay (Promega) according to the manufacturer’s protocol. Briefly, in this assay, viable cells retain the ability to reduce resazurin into resorufin -a fluorescent end product. Cells treated in 96 well plates for the indicated time points, are then incubated with the redox dye for up to 3 hours. Fluorescence intensity which is proportional to the number of viable cells, is measured using GloMax multi detection system (Promega).
Bioinformatics
For details, see supplementary methods. DNA methylation data (Illumina HumanMethylation 27K and 450K array platform beta-values) and level 3 RNA-sequencing data (RSEM) were downloaded from The Cancer Genome Atlas (TCGA). We used publicly available TCGA data from deidentified patients; all studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) and were approved by the Temple University institutional review board. RNA-sequencing BAM files were downloaded from the Genomic Data Commons Portal. TET1 expression in ovarian and pancreatic cancer were downloaded using CBioPortal (RNA-seq, RSEM). DNA mutation data were downloaded using CBio Portal for the TCGA Provisional cohort of breast cancer patient samples.
Drug sensitivity data in breast and ovarian cancer cell lines were downloaded from the Genomics of Drug Sensitivity in Cancer database (cancerrxgene.org). Gene expression data (Affymetrix U133 array) for cancer cell lines were downloaded from the Cancer Cell Line Encyclopedia (17). IC50 values for 265 drugs were correlated to TET1 expression values in all breast and ovarian cancer cell lines available using Spearman analysis.
Survival data were downloaded from CBioPortal using the following studies: Breast cancer (METABRIC, Nature 2012 & Nature Communications 2016) (18)(19). Survival curves and all graphs unless otherwise noted were generated using GraphPad Prism 4.0.
Statistics
Calculations were done in GraphPad. The Student’s t-test was used to calculate significant p values unless otherwise stated. All p-values are two-sided. * p<0.05, ** p<0.01, *** p<0.001 denotes significance. Mann-Whitney test was used to test significance of normalized TET1 exon reads. Significance of the cell viability assay was measured by two-way ANOVA. Significance of clinical characteristics, including mutation levels, for each cluster was calculated using Fisher’s exact test. Significance of overlap for the datasets was calculated using the Chi-squared test. To statistically analyze the Spearman correlations, 1000 permutations were performed on the TET1 expression values and empirical p values were computed. Significance of survival curves were calculated using the log-rank test. Error bars are standard error of the mean (SEM) unless otherwise noted. The following graphs/statistical tests were generated using R software: principal component analysis, permutations, Spearman correlations, histogram distribution of R values, differential TCGA gene expression plot and Venn diagrams. All pathway analyses were performed using Consensus Path Database, and pathway analyses of methylation data were background corrected dependent on the assay performed (20).
Results
TET1 and TET1ALT are overexpressed in TNBC
DNA methylation can divide breast cancers into multiple groups, with TNBCs characterized as having the lowest levels of methylation compared to the other breast cancer subtypes (4)(5)(6). We used RNA-Seq data downloaded from The Cancer Genome Atlas (TCGA) to investigate whether expression of DNMTs or TETs could explain the methylation differences between TNBCs (N=100 patients) and hormone receptor positive breast cancers (HRBCs) (N=732 patients). Compared to normal breast (N=105), TET1 showed dramatic differences; it was significantly repressed in HRBCs (p<0.0001) and substantially overexpressed in TNBCs (p<0.0001), while unchanged in HER2 cases (p=0.65) (Fig. 1A). TET2 was downregulated in HRBCs and TNBCs while TET3 was upregulated in all subtypes (Supplementary Fig. S1A and S1B). The fact that TET3 is high in all patients is interesting but does not explain the differences in methylation between the subtypes. We focused on TET1 because it shows this dichotomy where it is high in TNBC but low in HRBC and we hypothesized that TET1 repression could contribute to the hypermethylation characteristic of HRBCs, while TET1 overexpression could potentially account for the TNBC-specific hypomethylation. Overexpression of TET1 in TNBC was confirmed in two independent datasets (METABRIC and GSE27447) (Supplementary Fig. S1C and S1D). Further, TET1 immunohistochemical staining from three representative breast cancer patient samples showed TET1 protein is expressed in tumor cells (Supplementary Fig. S1E, www.proteinatlas.org) (21). Since TET1 expression is variable within TNBC, we asked whether TET1 expression associates with survival. We analyzed survival data in the METABRIC cohort and found TNBC patients with high TET1 (N=64) had a significantly worse overall survival compared to all other TNBC patients (N=96, p=0.04, log-rank test) (Fig. 1B). Importantly, this was not observed with TET2 or TET3 expression (p=0.38 and 0.96, respectively) nor was TET1 associated with survival in HRBCs (p=0.4), suggesting this is a TET1-TNBC specific event (Supplementary Fig. S1F-H).
Figure 1. TET1 overexpression is associated with hypomethylation.
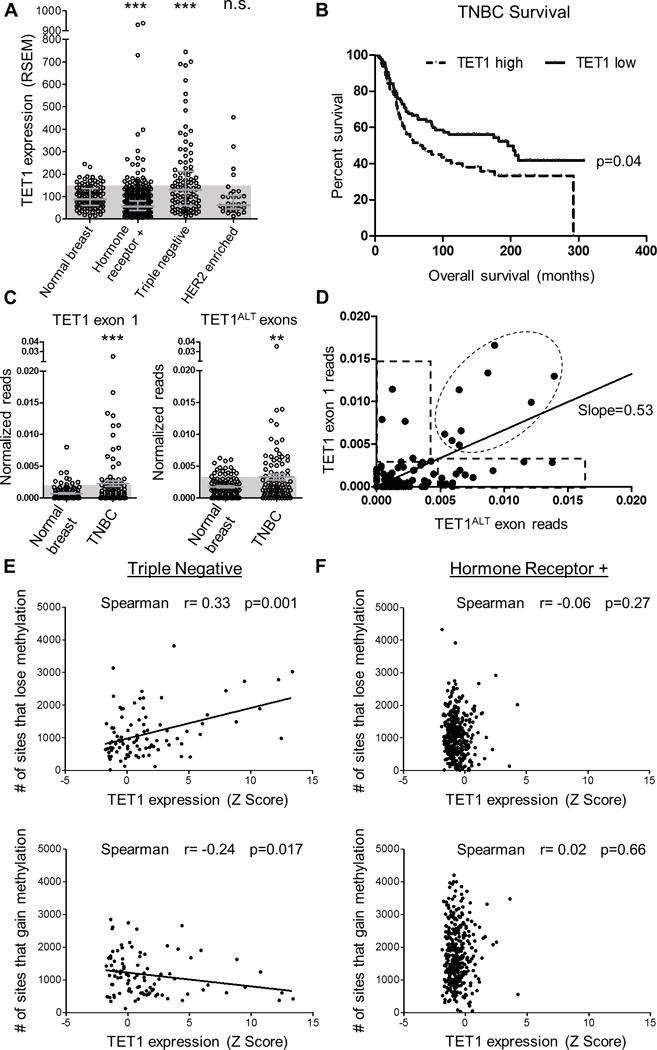
(A) TET1 expression (RNA-seq, RSEM values) for normal breast, HRBC, TNBC and HER2 enriched. Shaded blue boxes represent 1 standard deviation above the mean of normal. Error bars are median with interquartile range. T-test was used to measure significance. (B) Survival curve based on TET1 expression in TNBC. TET1 high is classified as patients with stdev>1 above the mean. (C) In vivo analysis of TCGA RNA-seq reads from normal breast and TNBCs. Normalized exon reads for TET1FL exon 1 (left) and TET1ALT exons (right). Y axis is reads from TET1ALT exon 1a + exon 1b or TET1 exon 1/exon length/chromosome 10 reads × 1e6. Mann-Whitney test was used for significance. D) TET1 exon 1 reads vs TET1ALT exon reads for TNBC patients. Expression was considered high if it was >2 standard deviations above the mean of normal for each isoform. (E) TNBC analysis and (F) HRBC analysis of # of sites that lose methylation (top) and # of sites that gain methylation (bottom) compared to normal breast vs TET1 expression Z score.
We recently identified an alternate, truncated isoform of TET1 (TET1ALT) that lacks the CXXC domain but retains the demethylase domain (13). To determine whether the TNBCs express the full length TET1 (TET1FL) or the truncated TET1, we analyzed RNA-seq read counts for normal breast (N=107) and for TNBCs (N=91) for exon 1 (only expressed in TET1FL, left) and TET1ALT exons (only expressed in TET1ALT, right) and found both isoforms are overexpressed in TNBC (Fig. 1C, Mann-Whitney test p=0.007 and p=0.02 respectively). Next, we plotted TET1ALT reads against TET1FL reads (Fig. 1D). We identified four groups of TNBC patients: Group 1 overexpress only TET1FL (N=5), Group 2 overexpress only TET1ALT (N=12), Group 3 overexpress both TET1FL and TET1ALT (N=10, note one patient with very high levels of both isoforms is not depicted) and Group 4 overexpress neither isoform (N=64). Thus, TET1ALT is the predominant isoform overexpressed in TNBC, where 22/27 of the TET1 overexpressing patients, overexpress TET1ALT. It is important to note that this analysis only quantifies reads for 1 exon per isoform, and is not representative of overall TET1 expression. Instead this analysis provides evidence that both isoforms are expressed in vivo. For subsequent analyses, RNA-seq RSEM values are used for TET1 expression as this takes all TET1 exons into account and is a more global view of TET1 transcript expression. In this analysis, 40 patients are classified as TET1 high and 55 as TET1 low.
TET1 expression correlates with hypomethylation in TNBC
To examine if TET1 expression is associated with DNA methylation, we analyzed RNA-seq and 27K methylation array data from the TCGA. We calculated the number of sites that gain or lose methylation (change> 20% compared to normal breast) for each patient and plotted it against TET1 expression values (normalized RNA-seq RSEM reads that quantify the whole TET1 transcript). TNBCs (N=95) displayed a significant correlation, with high TET1 patients having the most hypomethylation (top, p=0.001) and the least hypermethylation (bottom, p=0.01) (Fig. 1E). On the contrary, HRBCs (N=368) displayed no correlation between TET1 expression and DNA methylation (Fig. 1F), likely due to the relatively low TET1 expression in HRBCs. Next, we determined whether TET2 or TET3 expression was correlated with DNA methylation. TET2 displayed no correlation in TNBCs or HRBCs (Supplementary Fig. S1I and S1J), while TET3 was weakly correlated in TNBC, and not correlated in HRBCs (Supplementary Fig. S1K and S1L).
To identify potential TET1 target genes in breast cancer, we used the more comprehensive 450K arrays and computed Spearman correlations between TET1 expression and methylation values for 450,000 CpG sites for both TNBC (N=67) and HRBC (N=402). In this analysis, negative r values indicate sites that hypomethylate when TET1 is high, while positive r values correspond to sites that hypermethylate. We plotted histograms of the r values and found a predominance of negative values in TNBC, consistent with the hypothesis of TET1 mediated hypomethylation (Fig. 2A). To statistically analyze this observation, we performed 1000 permutations of the data (green distribution) and computed empirical p values to measure significance. If the correlation observed is due to chance, the permutated data will display a similar pattern to the real dataset. However, as expected, the distribution of the actual correlations (orange) showed a marked excess of negative r values compared to the permutated data. We used a cutoff of r<−0.3 to identify TET1 correlated probes, as these sites have a high likelihood of being significant (empirical p value <0.05) and due to the asymmetry of the data (more negatively correlated sites than positively correlated sites). We identified 42,559 probes, a striking number suggesting that TET1 could potentially regulate up to 10% of the methylome. In contrast, we found a narrow range of r values in HRBC, with none exceeding the low correlation of −0.2 (Supplementary Fig. S2A). This lack of correlation between TET1 expression and methylation in HRBC can be explained by the lack of diversity in TET1 expression among HRBCs, and is consistent with the uniform repression of the gene: 287/732 (39%) of HRBC patient’s downregulate TET1, while only 4% overexpress TET1.
Figure 2. Identification of putative TET1 targets in TNBC.
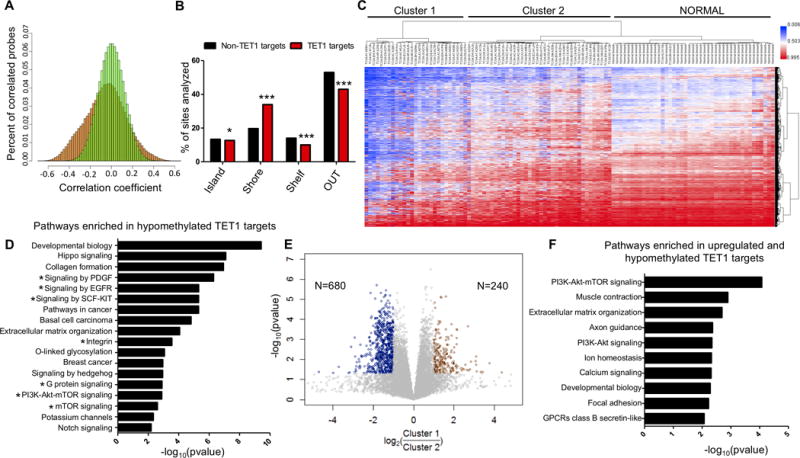
(A) Frequency distribution of Spearman correlation r values (TET1 expression vs DNA methylation) in TNBC patients. X axis (correlation coefficient) and Y axis (percent of correlated probes). 1000 permutations of the data (green) and real dataset (orange). (B) Location of putative TET1 targets and non TET1 targets relative to CpG islands. (C) Unsupervised cluster analysis of methylation in TNBC for the putative TET1 targets (N=17,521 sites), including 41 normal breast controls. (D) Pathway analysis of putative TET1 targets. X axis is −log10(pvalue). (E) Differential gene expression analysis between Cluster 1 and Cluster 2. Y axis is −log10(pvalue) and x axis is log2(Cluster 1/Cluster 2). Genes were considered upregulated if FC>2 or FC<0.5 and p<0.05 (t-test). (F) Pathway analysis of genes that are hypomethylated and upregulated in Cluster 1.
Given that hypomethylation might have the greatest biological impact on CpG sites showing high levels of methylation at baseline, we filtered the negatively correlated probes (r<−0.3) for sites that have an average methylation >40% in normal breast (putative TET1 targets). This strategy identified 17,521 CpG sites corresponding to 6,962 genes. Interestingly, compared to non-TET1 targets (r values between −0.05 to 0.05, and methylated >40% in normal breast), putative TET1 targets are significantly enriched for CpG island shores (p<0.001, Chi-squared test) (Fig. 2B), consistent with previous data linking TET1 to methylation of CpG island borders (12). Cluster analysis of the TNBC and normal breast cases using these probes revealed two main clusters, with Cluster 1 (N=27) having the highest levels of TET1 and the most hypomethylation (blue) (Fig. 2C, Table 1). As expected, normal breast clustered separately from TNBCs and patients in Cluster 2 (N=38) are slightly more methylated than normal breast. Next we asked if this pattern of methylation is specific to TNBC or if HRBC with high TET1 expression are also hypomethylated at these sites. We added HRBCs that overexpress TET1 (greater than 2 stdev above the mean of normal, N=8) and 16 randomly selected HRBC cases that do not overexpress TET1 to the methylation cluster in Fig. 2C (Supplementary Fig. S2B). 5/8 TET1 high HRBCs clustered in the hypomethylated Cluster 1. In contrast, all non-TET1 high patients were found in Cluster 2, with the exception of 2 patients that were found in the normal breast cluster. The HRBC patients found in Cluster 1 have even higher TET1 expression than the TNBCs found in Cluster 1, and in Cluster 2 the TNBCs and HRBCs have nearly identical TET1 expression levels (Supplementary Fig. S2C). These data suggest that the TET1 associated demethylator phenotype is likely not subtype specific but rather is dependent on TET1 expression levels. Nevertheless, the fact that TET1 is overexpressed in ~40% of TNBCs compared to only 4% of HRBCs is a likely explanation for the methylation differences between the two subtypes.
Table 1. Clinical features of TNBC patients in methylation Cluster 1 and Cluster 2, related to Figure 2C.
P values were calculated using Fisher’s exact test, except for age at diagnosis which was calculated using student’s t-test.
| Cluster 1 | Cluster 2 | p value | |
|---|---|---|---|
| Number of TNBC Patients | N=27 | N=38 | |
| Age at Diagnosis | 55 | 55 | n.s. |
| Stage I (%) | 22 | 14 | n.s. |
| Stage II (%) | 59 | 62 | n.s. |
| Stage III (%) | 15 | 24 | n.s. |
| Stage IV (%) | 4 | 0 | n.s. |
| Asian (%) | 11 | 9 | n.s. |
| White (%) | 70 | 66 | n.s. |
| Black (%) | 19 | 26 | n.s. |
| TET1 Z score expression (AVG) | 3.2 | 0.2 | 0.0002 |
| Mutation in PIK3CA or PTEN (%) | 0 | 21 | 0.01 |
| BRCA1 mutation (%) | 7.4 | 7.4 | n.s. |
| BRCA2 mutation (%) | 7.4 | 0 | n.s. |
| TP53 mutation (%) | 77.8 | 65.8 | n.s. |
To more clearly illustrate that the TNBC tumors with the highest TET1 expression have the greatest degree of hypomethylation, we used the putative TET1 targets identified above to plot TET1 expression vs the number of sites that lose methylation (Supplementary Fig. S2D, r=0.64, p<0.0001). Pathway analysis of these putative TET1 targets (N=6,962 genes) showed enrichment for many cancer relevant pathways, including a striking number of genes in the PI3K/mTOR pathway (115 genes enriched out of 299 total genes in the pathway) as well as Hippo signaling, pathways in cancer, and extracellular matrix organization (Fig. 2D). We decided to focus on the PI3K pathway because PI3K is targetable by drugs (22) and many of the top candidates also feed into this pathway (indicated with *), including EGFR, PDGF, KIT, G protein coupled receptors (GPCR), etc. For validation, we analyzed a larger cohort of patients (N=95) studied on the 27K array platform (27,000 CpG sites total) and obtained nearly identical results despite the lower number of sites (Supplementary Fig. S2E and S2F).
To address if these changes in methylation are leading to changes in gene expression, we performed a differential gene expression analysis between Cluster 1 and Cluster 2. We identified 240 genes that are upregulated and 680 genes downregulated in Cluster 1 (Fig. 2E). Pathway analysis of the upregulated genes revealed enrichment for the PI3K pathway, and the downregulated genes were enriched for immune system pathways (Supplementary Fig. S2G and S2H). Overlap of the genes upregulated in Cluster 1 with the genes identified as putative TET1 methylation targets above, revealed 119/240 (50%) of the genes upregulated in Cluster 1 are also hypomethylated in Cluster 1, (Chi-squared test p<0.0001, compared to overlap with genes downregulated in Cluster 1). Interestingly, the overlapping genes are enriched for the PI3K-Akt-mTOR pathway (Fig. 2F). Upon further analysis of these genes, we identified that 8/9 are upstream of the PI3K pathway, including tyrosine kinase receptors, GPCRs and integrin receptors which all activate PI3K. These genes are hypomethylated and upregulated in Cluster 1 patients. Gene expression and methylation levels for select target genes (e.g. KIT, ITGA10) can be found in Supplementary Fig. S2I and S2J.
Identification of putative TET1 targets in serous ovarian cancer
To determine if TET1 mediated hypomethylation is found in other cancer types, we downloaded methylation and gene expression data for all serous ovarian cystadenocarcinoma cases (N=304) from the TCGA. We examined serous ovarian cancers because this type has similar TET1 expression levels as TNBC, unlike pancreatic cancer for example where TET1 expression is very low (Supplementary Fig. S3A); in addition, promoter hypomethylation has been previously reported to activate oncogenes and associate with reduced overall survival in ovarian cancer (23). Further, serous ovarian cancer has been reported to have very similar molecular features to TNBC (4). Similar to TNBC, we identified putative TET1 targets using spearman correlation analysis (Supplementary Fig. S3B). Ovarian cancer patients with high TET1 levels show dramatic hypomethylation compared to TET1 low expressing Cluster 3 patients (Supplementary Fig. S3C and S3D). Further, the putative TET1 targets are enriched for genes in the GPCR pathway, a prominent activator of PI3K/Akt (Supplementary Fig. S3E). These data suggest TET1 mediated hypomethylation may be a more wide-spread mechanism utilized by cancer cells. Importantly, similar to TNBC, high expression of TET1 in ovarian cancer is associated with a worse overall survival (13).
TET1 is essential for growth in TNBC
Phosphoinositide 3-kinase (PI3K) regulates cell proliferation, survival and migration (22). Activating PI3K mutations are found in 41% of HRBCs, but only 7% of TNBCs (4). However, gene expression and proteomic data revealed that the PI3K pathway is more active in TN tumors compared to non-TN tumors, but it is unknown why (4). We were interested in the fact that TNBCs can be divided into TET1 high and low based on their methylation levels (Clusters 1 and 2 in Fig. 2C). When we compared these two groups, they had similar clinical characteristics but differed significantly in the rate of mutations affecting PI3K-AKT signaling (Table 1). 0% of Cluster 1 patients have PIK3CA or PTEN mutations, whereas 21% of Cluster 2 patients have mutations in the pathway (Fisher’s exact test, p=0.01). Thus, in TNBC, there is an inverse correlation between TET1 levels and PI3K-AKT pathway mutations, raising the possibility that the two molecular events are not co-selected because they are equivalent ways of activating the same oncogenic pathway. This is consistent with DNA methylation and gene expression studies in Cluster 1 (see above).
To identify whether TET1 is important for growth and maintenance of TNBC and to test whether there is a link between TET1 and PI3K, we generated CRISPR Cas9 TET1 KO cells in the TNBC cell line MDA-MB-231. Single cell clones were generated from pooled cells with three different sgRNAs targeting different exons of TET1: exon 6 (KO-1), exon 3 (KO-2) and exon 11 (KO-3). KO was confirmed by western blot analysis using a TET1 antibody (Fig. 3A) and reduced 5hmC levels were observed (Supplementary Fig. S4A and S4B). Of note, the largest effect of the TET1 KO was observed at 162 kDa, the size of TET1ALT, as it is the predominant isoform expressed in MDA-MB-231 cells. We have additional evidence by RNA-seq (Supplementary Fig. S4C) that the full-length isoform of TET1 is not expressed in this cell line, suggesting that the bands observed around 235 kDa are non-specific. Loss of TET1 resulted in a loss of phospho-4EBP1 (Thr37/46) (Fig. 3B) and significantly reduced cellular migration (Fig. 3C) and proliferation (Fig. 3D). Of note, CRISPR KO of TET1 in another TNBC cell line (Hs578T) also substantially reduced cellular proliferation (Fig. 3E and 3F).
Figure 3. TET1 is essential for growth in TNBC.
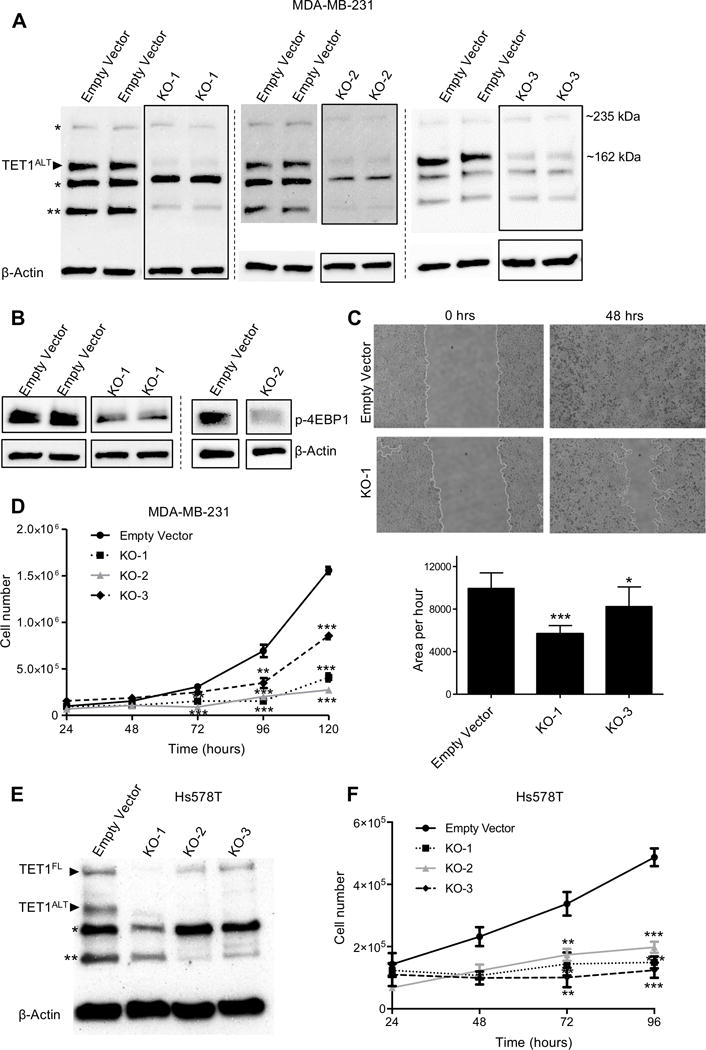
(A) Western blot (in duplicate) of MDA-MB-231 empty vector and TET1 knockout single clones. β-actin used as a loading control. (* denotes putative non-specific band, ** denotes another potential truncated isoform of TET1) (B) Western blot of phospho-4EBP1 (Thr37/46) in MDA-MB-231 TET1 KO cells. KO-1 is in duplicate, KO-2 is single experiment. (C) Representative images of wound-healing assays in empty vector and TET1 KO-1 cells at 0 and 48 hours (top) and quantification (bottom), experiment performed in triplicate. (D) Cell proliferation assay for MDA-MB-231 empty vector, KO-1, KO-2 and KO-3. Cells were counted every 24 hours (in triplicate, each sample counted twice). X axis is time, Y axis is total cell number. (E) Western blot of empty vector and TET1 knockout single clones in Hs578T cells. (F) Cell proliferation assay for Hs578T empty vector, KO-1, KO-2 and KO-3. X axis is time, Y axis is total cell number.
If TET1 is important for maintaining PI3K/mTOR activation, it is expected that cell lines with high TET1 will be more sensitive to inhibitors targeting this pathway. To determine potential corresponding vulnerabilities in a panel of 50 breast cancer cell lines, we computed Spearman correlations between TET1 expression and drug sensitivity (IC50) for 265 drugs in a public database (Genomics of Drug Sensitivity) (24). Multiple drugs showed high negative correlations with TET1 levels, including PI3K pathway targeting drugs (ERK, MAPK etc.), consistent with our hypothesis (Supplementary Table S2). These drugs constitute a potential entry towards targeted therapy for TNBC. Three representative drugs, XMD8-85 (targets ERK, r= −0.85), VX-680 (targets AURK, r= −0.86) and TGX-221 (targets PI3Kβ, r= −0.51) show strong negative correlations, with breast cancer cell lines with high TET1 having the lowest IC50 (Supplementary Fig. S4D). We validated two of the highly correlated drugs (VX-680 and Rapamycin, an mTOR inhibitor), and found they selectively killed the TET1 high expressing breast cancer cell line (HCC1599) over the TET1 low expressing line (HCC2218) (Supplementary Fig. S4E and S4F). Similar results were found in serous ovarian cancer, where of the top eight negatively correlated drugs (r<−0.3), four inhibitors directly affect the PI3K or mTOR pathway, including two IGF1R inhibitors (BMS-754807, Lisitinib), a receptor tyrosine kinase inhibitor (Axitinib) and an ERK/MAPK inhibitor (HG-6-64-1) (Supplementary Table S3).
Gene expression targets of TET1 in vitro
Next we performed RNA-seq in the TET1 KO cell lines generated above to investigate whether TET1 expression is important for maintaining oncogenic signaling pathways (including PI3K) in TNBC. In MDA-MB-231, compared to empty vector control, TET1 KO cells showed dramatic changes in expression as indicated by a principal component analysis (Fig. 4A). We identified both upregulated and downregulated genes, consistent with previous findings showing TET1 can act as both a transcriptional repressor and co-activator (10)(11)(12). We identified 566 genes that are downregulated (FC<0.5, p<0.001, t-test) in at least 2 of the 3 KO clones (Fig. 4B). Interestingly, we performed a pathway analysis and found several overlapping pathways (indicated by black circles) with the pathways identified as TET1 hypomethylated targets in Fig. 2D, including oncogenic pathways that feed into PI3K (EGFR, VEGF, Integrins, etc.) (Fig. 4C). Next, we analyzed genes that are upregulated upon loss of TET1 (FC>2, p<0.001, t-test) (Fig. 4D). We identified 343 genes upregulated in at least 2 of the 3 KO clones, and found they are enriched for pathways involved with immune system function (Fig. 4E). This agrees with the differential gene expression analysis in the TCGA datasets (immune system genes downregulated in Cluster 1). We verified the gene expression changes in another TNBC cell line (Hs578T) upon TET1 KO using RNA-seq and detected similarly enriched pathways, where immune genes are upregulated and PI3K, FGF and VEGF genes are downregulated. (Supplementary Fig. S5A-C).
Figure 4. Gene expression targets of TET1 in vitro.
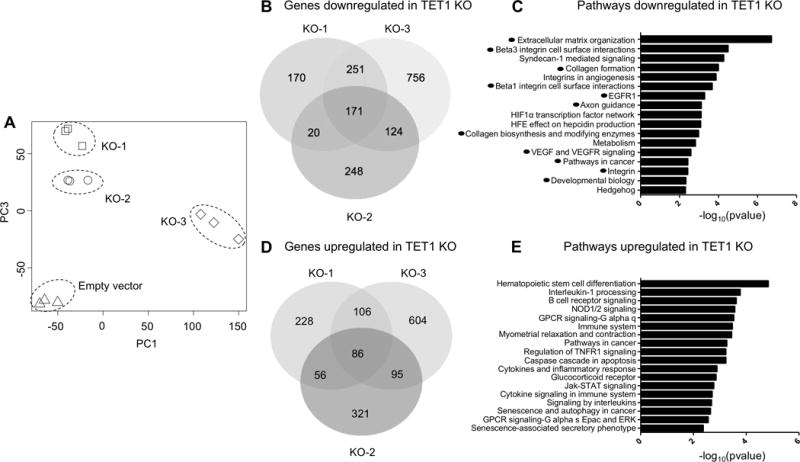
(A) Principal component analysis of RNA-seq data for empty vector, TET1 KO-1, KO-2 and KO-3 (in triplicate) in MDA-MB-231 cells. (B) Venn diagram overlap of genes downregulated in the TET1 KO cells. (C) Pathway analysis of genes downregulated in TET1 KO clones. X axis −log10(pvalue). (D) Venn diagram overlap of genes upregulated in the TET1 KO cells. (E) Pathway analysis of genes upregulated in TET1 KO clones.
Next, we overlapped the differentially expressed genes identified in the TCGA datasets in Fig. 2 with the differentially expressed genes in the MDA-MB-231 TET1 KO cells, and found 50 genes that are differentially expressed in both datasets (Chi-squared test p<0.0001, when compared to genes not differentially expressed in TET1 KO). This list includes several genes in the PI3K-Akt pathway including THBS3, ANGPT1, PIK3CG, MAP2, and IL7R. We validated this finding in Hs578T TET1 KO cells and found 157 genes that are differentially expressed in TET1 KO cells and TCGA datasets. THBS3 (a glycoprotein that activates ITGA/ITGB integrin receptors upstream of PI3K) is an example of a gene upregulated in Cluster 1 TCGA patients (and has promoter hypomethylation), and is downregulated in both MDA-MB-231 and Hs578T TET1 KO cells (Supplementary Fig. S5D and S5E). Interestingly, this protein has been implicated in facilitating tumor growth (25).
Methylation targets of TET1 in vitro
Next, we sought to investigate if the changes in expression are dependent or independent of changes in DNA methylation. We performed DREAM, a genome-wide DNA methylation assay, in the parental cell line MDA-MB-231, empty vector control pooled cells, empty vector single clones and in the TET1 KO cells. A principal component analysis of the data revealed that the parent line and pooled cells are very similar, however, there is a minor drift in methylation in the empty vector single clone cells, indicating minor cell to cell heterogeneity in methylation over time (Fig. 5A). By contrast, the methylome of the TET1 KO cells was dramatically different from all control cells. To analyze these changes in more detail, we first controlled for the methylation drift in the single clones by excluding sites that had a standard deviation of greater than 3% between all control cells (parental, pooled and single clone empty vector). This method identified the CpG sites that were the least variable in the controls (N=31,070 CpG sites). Since we were interested in methylation changes that correlated with changes in gene expression, we next filtered for CpG sites within promoter regions (+/- 2000 bp from the TSS), which left 15,221 CpG sites corresponding to 6,715 genes. We identified gene promoters that gain methylation >10% compared to empty vector single clones in each TET1 KO cell line. In this genomic compartment, the predominant change was a gain in methylation in TET1 KO cells (with two-fold more sites gaining methylation rather than losing), as expected. Overall, 212 promoters gained methylation in at least 2 of the 3 TET1 KO clones (Supplementary Fig. S6A). We further validated these changes in methylation upon TET1 KO in Hs578T cells, where we see even more dramatic gains in methylation at promoters (~five-fold more gains than loses) and an overall 109 promoters gaining methylation (Supplementary Fig. S6B)
Figure 5. Methylation targets of TET1 in vitro.
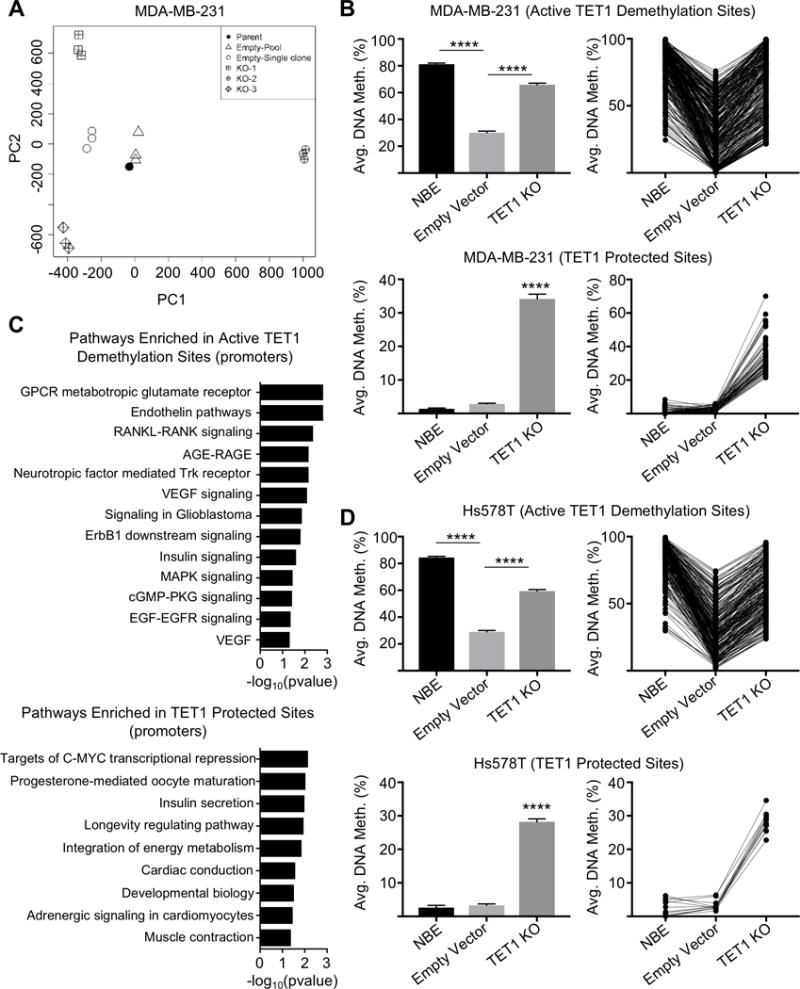
(A) Principal component analysis of DREAM methylation data for MDA-MB-231 parental cell line, empty vector pooled cells, empty vector single clone cells, and TET1 KO-1, KO-2, KO-3 single clone cells (all in triplicate except for parental cells). (B) Active TET1 demethylation sites in MDA-MB-231 cells (top) and TET1 protected sites (bottom). Includes average methylation values for NBE, empty vector control and TET1 KO cells. Y axis is average DNA methylation (%). (C) Pathway analysis of genes with hypermethylated promoters in TET1 KO clones that are at active TET1 demethylation sites (top) and TET1 protected sites (bottom). X axis −log10(pvalue). (D) Active TET1 demethylation sites in Hs578T cells (top) and TET1 protected sites (bottom).
Pathway analysis of these hypermethylated genes in MDA-MB-231 and Hs578T TET1 KO cell lines revealed enrichment for GPCR pathways, a prominent activator of the PI3K/mTOR pathway (Supplementary Fig. S6C). Next, we overlapped the genes that gained methylation upon loss of TET1 with the TET1 methylation targets identified in the TCGA datasets and found that 47% of the genes that gained methylation in MDA-MB-231 TET1 KO cells are also TET1 methylation targets identified in TNBC patient samples. Several of the overlapping genes are upstream regulators of PI3K, including FGF19, INSR, GHR and THBS4, of which methylation values from both datasets can be found in Supplementary Fig. S6D and S6E. A few of the methylation targets identified also lose expression in the TET1 KO cells and are overexpressed in Cluster 1 patients, including INSR (Supplementary Fig. S6F-I). This overlap was also observed in Hs578T cells where 46% of genes that gain methylation in TET1 KO are also TET1 methylation targets identified in TNBC patient samples.
Next, we wanted to parse out the dual roles of TET1 as both a protector of CGI methylation (at sites that are usually un-methylated in normal), from TET1’s role as an active DNA demethylase (targeting sites that are methylated). We computed the average methylation genome-wide of normal breast epithelium (NBE) and compared it to methylation of the cancer cell line MDA-MB-231 empty vector control cells and TET1 KO cells. First, we identified the active TET1 demethylation sites by filtering for sites that lose methylation in the cancer cell line (>20%) compared to NBE (N=5,105). Of these demethylated sites, 7% re-methylate upon loss of TET1 (>20%, p<0.05) (Fig. 5B, top) which is 2.5-fold more than sites that de-methylate upon loss of TET1. Of the re-methylated sites, 37% are hypomethylated in TET1 high expressing cluster 1 patients from Fig. 2C. We also identified that 0.5% of sites that are un-methylated in NBE and empty vector control cells, gain methylation in TET1 KO cells (TET1 protective sites) and 29% of these sites overlap with TET1 TCGA methylation targets (Fig. 5B, bottom). Pathway analysis at promoters of the active TET1 demethylation sites were enriched for cancer promoting pathways upstream of PI3K, including Trk, VEGF, and EGFR whereas the TET1 protective sites were enriched for pathways including developmental biology, longevity and energy metabolism (Fig. 5C). Of note, insulin secretion/signaling pathways were observed in both TET1 demethylated and protected sites and may not be specific to one such category.
We validated these finding in Hs578T TET1 KO cells where a striking 8.5-fold more genes re-methylate in TET1 KO cells as compared to de-methylate, revealing a role for active TET1 demethylation at sites normally methylated in NBE (Fig. 5D, top). Similar to MDA-MB-231 cells, 35% of the re-methylated sites overlap with TCGA TET1 targets. The active TET1 demethylation sites were enriched for RAC1 activity (p=0.002) also reported to activate PI3K/Akt signaling (26). An important difference was observed in the TET1 protection sites, as only 0.08% of sites un-methylated in NBE and Hst578T cells gain methylation in TET1 KO cells (Fig. 5D, bottom). This suggests a stronger role for TET1 mediated protection of methylation in MDA-MB-231 cells, however, active TET1 demethylation is found in both cell lines.
Discussion
In this study, we highlight the importance of TET1 in TNBC. We show that TET1 expression (but not TET2 or TET3) is associated with a worse overall survival in TNBC where high TET1 expression is highly correlated with DNA hypomethylation. In two TNBC cell lines, we show that deletion of TET1 leads to decreased proliferation and promoter hypermethylation in cancer promoting pathways. The differentially methylated genes in TNBC patient samples (putative TET1 targets) overlap with the differentially methylated genes upon TET1 KO in both TNBC cell lines. We also show a strong overlap between the differentially expressed genes in TCGA TET1 low vs. high TNBCs and those that change following TET1 KO in TNBC cell lines, illuminating roles for TET1 as both an activator and repressor of transcription.
Our attempt to better understand mechanisms leading to hypomethylation in cancer, shed light on how hypomethylation affects tumorigenesis. We identified a hypomethylator phenotype in breast cancer that is dependent on TET1 expression. Hypomethylator phenotypes have been described in other cancers, including acute-myeloid leukemia, where distinct demethylator phenotypes were found to associate with specific genetic backgrounds and clinical features, implicating hypomethylation as a clinically relevant, non-random phenomenon (36). Our results revealed a previously uncharacterized role for TET1 in TNBCs, where it acts as an oncogene leading to hypomethylation and activation of oncogenic signaling pathways. Approximately 42% of TNBCs overexpress TET1 and TET1ALT, where high levels of expression associate with a worse overall survival. On the contrary, remarkable downregulation of TET1 is found in HRBCs. Indeed, several studies have reported TET1 as a tumor suppressor in breast cancer, with one study finding that overexpression of TET1 led to reduced tumor volume in mice (27)(28). This contradictory evidence raises the interesting possibility of TET1 being both an oncogene and a tumor suppressor, a phenomenon that has been observed with other epigenetic regulators including EZH2 and DNMTs. EZH2 shows inactivating mutations in human myeloid leukemias and disruption of EZH2 in mice is enough to cause T-acute lymphoblastic leukemia, suggesting a tumor suppressor function (29). On the contrary, EZH2 shows activating mutations in lymphomas and is overexpressed in several cancers including melanoma, breast, and endometrial cancer where its expression is associated with disease progression (30). The opposing roles of TET1 add an additional layer of complexity in breast cancer and pose the question of whether TET1 should be inhibited or induced. For example, Vitamin C is a potent activator of the TET enzymes (31). Should TNBC patients avoid Vitamin C and HRBC patients take Vitamin C as part of their treatment regimen? More work is required to answer these questions, and future studies should parse out the varying roles of TET1 in different cellular contexts.
One possible explanation for the varying roles of TET1 is different interacting partners that are cell-type specific. For example, HIF1a and XBP1 have been shown to be activated in TNBC (32). XBP1 drives TNBC tumorigenicity by assembling a transcriptional complex with HIF1a that leads to the recruitment of RNA polymerase II at HIF1a target genes (32). Importantly, another study found in neuroblastoma cells that under hypoxic conditions, HIF1a induces TET1 expression, leading to hypomethylation and activation of HIF1a target genes (33). Therefore, TET1 upregulation may be an early response to hypoxic stress conditions, but as a consequence, leads to the demethylation of both hypoxic response genes and oncogenic signaling pathways, ultimately driving malignant transformation. Another possibility is that the opposing roles of TET1 may be isoform specific; full length TET1 may function as a tumor suppressor by protecting against CpG island hypermethylation and the truncated TET1ALT may function as an oncogene by mediating hypomethylation upon targeting by partner proteins. Indeed, TET1ALT, unlike full length TET1, lacks a DNA binding domain and must be recruited to DNA by interacting proteins and, both TET1 proteins lack sequence specificity and presumably rely solely on interacting proteins for target specificity.
Through the analysis of publicly available data from the TCGA, CCLE, and the genomics of drug sensitivity database, we provide evidence to suggest that a subset of TNBC and ovarian cancer patients may benefit from treatment with PI3K/Akt/mTOR inhibitors. So far, clinical results have been equivocal for using PI3K inhibitors in breast cancer and more focus on patient selection is needed to yield better results (22). We propose that TET1 high patients may be particularly vulnerable to this type of therapy. Through KO experiments, we confirmed that TET1 is important for maintaining activation of the PI3K/mTOR pathway as loss of TET1 resulted in decreased phosphorylation of 4EBP1 and decreased gene expression of upstream regulators of the PI3K pathway. In addition, we observed decreased cellular proliferation and migration in the TET1 KO cells, further evidence that TET1 may be playing a more oncogenic role.
It remains unclear whether the downregulation of the PI3K genes is due to methylation dependent or independent effects of TET1. We showed that select genes, such as the insulin receptor, have decreased expression and CGI shore promoter hypermethylation in TET1 KO cells. Interestingly, the insulin receptor is also upregulated and hypomethylated in TET1 high Cluster 1 patients. However, not all of the differentially expressed PI3K genes had promoter methylation data available through the DREAM assay. For example, ANGPT1, a gene involved in the PI3K pathway (34), is upregulated and hypomethylated in Cluster 1 patients and is downregulated upon TET1 KO. However, the ANGPT1 promoter is not covered in the DREAM assay so we are unable to say if the downregulation is dependent or independent of TET1’s demethylase activity. Thus, a limitation of our study includes the low number of CGI shores sites covered by DREAM, especially since TET1 methylation targets are enriched in CGI shores. In the future, a technique that covers more CpG sites outside of CGIs should be used to discern the methylation targets of TET1, such as whole genome bisulfite sequencing.
Another interesting finding is that TET1 may be a suppressor of the immune system. TNBC patients with high TET1 have decreased expression of immune pathway genes, and upon KO of TET1, immune genes are upregulated in both MDA-MB-231 and Hs578T TNBC cell lines. In contrast, TNBCs with low TET1 have upregulated immune mediators and therefore could be sensitive to checkpoint inhibitors. Indeed, early results from several phase 1 clinical trials have shown success in using PD-L1 inhibitors in TNBC, with one study reporting a 19% response rate in women with heavily pretreated TNBC (35). If our hypothesis is correct, TNBC patients with low TET1 will be more likely to respond to PD-L1 treatment, and TET1 expression may be a way to pre-screen TNBCs to determine likelihood of response to immune checkpoint therapy. Thus, for cancer patients with high TET1 expression, one might envision combining TET1 inhibitors with immune checkpoint inhibitors such as PD-1 or PD-L1 in the future.
Our work addresses a critical gap in knowledge of how and why methylation is prognostic in breast cancer, and sheds light on how this information can be used to stratify TNBC patients for targeted therapy. We also provide evidence that TET1 mediated hypomethylation occurs not only in TNBC, but also in serous ovarian cancer. Although the ovarian cancer analyses are preliminary with no in vitro experimental validation, the bioinformatic analyses suggest that an oncogenic role for TET1 may be a more widespread phenomena utilized by cancers cells to hijack signaling pathways. Of note, a recent report found overexpressing TET2 and TET3 had beneficial effects in ovarian cancer, which contradicts our findings for TET1(37). It is possible that low and high 5hmC levels are deleterious in cancer, as is known for 5mC. It is also likely that TET1, TET2 and TET3 have varying target CpG sites and these distinct targets may have different consequences for cancer, potentially explaining how TET2 suppresses growth while TET1 promotes it. Lastly, our work establishes TET1 as an oncogene that could serve as a novel druggable target for therapeutic intervention in TNBC, ovarian cancer and beyond.
Supplementary Material
Statement of significance.
This study addresses a critical gap in knowledge of how and why methylation is prognostic in breast cancer and shows how this information can be used to stratify TNBC patients for targeted therapy.
Acknowledgments
Financial support: C.R. Good received grant CA210424 and JP.J. Issa received grants CA158112 and CA100632 from the National Institutes of Health; S. Panjarian received grant PDF17479825 from the Susan G Komen Foundation. JP.J. Issa is an American Cancer Society Clinical Research professor supported by a generous gift from the Ellison Medical Foundation and F. M. Kirby Foundation.
Footnotes
Conflicts of interests: The authors declare no potential conflicts of interest.
References
- 1.Smith ZD, Meissner A. Nat Rev Genet. Vol. 14. Nature Publishing Group; 2013. DNA methylation: roles in mammalian development; pp. 204–20. [DOI] [PubMed] [Google Scholar]
- 2.Rao X, Evans J, Chae H, Pilrose J, Kim S, Yan P, et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene. 2013;32:4519–28. doi: 10.1038/onc.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2010;1:239–59. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefansson OA, Moran S, Gomez A, Sayols S, Arribas-Jorba C, Sandoval J, et al. Mol Oncol. Vol. 9. Elsevier, B.V; 2014. A DNA methylation-based definition of biologically distinct breast cancer subtypes; pp. 555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JS, Fackler MJ, Lee JH, Choi C, Park MH, Yoon JH, et al. Basal-like breast cancer displays distinct patterns of promoter methylation. Cancer Biol Ther. 2010;9:1017–24. doi: 10.4161/cbt.9.12.11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LGT, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3:75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (80-) 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PAC, Rappsilber J, et al. Nature. Vol. 473. Nature Publishing Group; 2011. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity; pp. 343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Nature [Internet] Vol. 473. Nature Publishing Group; 2011. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells; pp. 389–93. [cited 2014 Mar 20]; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3539771&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin C, Lu Y, Jelinek J, Liang S, Estecio MRH, Barton MC, et al. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014;42:6956–71. doi: 10.1093/nar/gku372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Good CR, Madzo J, Patel B, Maegawa S, Engel N, Jelinek J, et al. A novel isoform of TET1 that lacks a CXXC domain is overexpressed in cancer. Nucleic Acids Res. 2017:1–13. doi: 10.1093/nar/gkx435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jelinek J, Liang S, Lu Y, He R, Ramagli LS, Shpall EJ, et al. Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics. 2012;7:1368–78. doi: 10.4161/epi.22552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato T, Cesaroni M, Chung W, Panjarian S, Tran A, Madzo J, et al. Transcriptional selectivity of epigenetic therapy in cancer. Cancer Res. 2017;77:470–81. doi: 10.1158/0008-5472.CAN-16-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Nat Biotechnol. Vol. 28. Nature Publishing Group; 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation; pp. 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature [Internet] 2012;483:603–307. doi: 10.1038/nature11003. Available from: http://www.nature.com/doifinder/10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereira B, Chin S-F, Rueda OM, Vollan H-KM, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB - A database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:623–8. doi: 10.1093/nar/gkn698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science (80-) 2015;347:1260419–1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 22.Fruman DA, Rommel C. Nat Rev Drug Discov. Vol. 13. Nature Publishing Group; 2014. PI3K and cancer: lessons, challenges and opportunities; pp. 140–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W, Barger C, Link P, Mhawech-Fauceglia P, Miller A, Akers S, et al. DNA hypomethylation-mediated activation of Cancer/Testis Antigen 45 (CT45) genes is associated with disease progression and reduced survival in epithelial ovarian cancer. Epigenetics. 2015;10:00–00. doi: 10.1080/15592294.2015.1062206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–61. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalla-Torre CA, Yoshimoto M, Lee C-H, Joshua AM, de Toledo SR, Petrilli AS, et al. Effects of THBS3, SPARC and SPP1 expression on biological behavior and survival in patients with osteosarcoma. BMC Cancer. 2006;6:237. doi: 10.1186/1471-2407-6-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murga C, Zohar M, Teramoto H, Gutkind JS. Rac1 and rhog promote cell survival by the activation of pi3k and akt, independently of their ability to stimulate jnk and nf-κb. Oncogene. 2002;21:207–16. doi: 10.1038/sj.onc.1205036. [DOI] [PubMed] [Google Scholar]
- 27.Hsu C-H, Peng K-L, Kang M-L, Chen Y-R, Yang Y-C, Tsai C-H, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep The Authors. 2012;2:568–79. doi: 10.1016/j.celrep.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 28.Yang H, Liu Y, Bai F, Zhang J-Y, Ma S-H, Liu J, et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663–9. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hock H. A complex Polycomb issue: The two faces of EZH2 in cancer. Genes Dev. 2012;26:751–5. doi: 10.1101/gad.191163.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–6. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature Nature Publishing Group. 2014;508:103–7. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y, et al. TET1-Mediated Hydroxymethylation Facilitates Hypoxic Gene Induction in Neuroblastoma. Cell Rep The Authors. 2014;1:1–10. doi: 10.1016/j.celrep.2014.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT–TIE2 pathway in malignancy. Nat Rev Cancer Nature Publishing Group. 2010;10:575–85. doi: 10.1038/nrc2894. [DOI] [PubMed] [Google Scholar]
- 35.McArthur HL. Checkpoint inhibitors in breast cancer: Hype or promise? Clin Adv Hematol Oncol. 2016;14:392–5. [PubMed] [Google Scholar]
- 36.Kelly AD, Madzo J, Madireddi P, Kropf P, Good CR, Jelinek J, et al. Leukemia [Internet] Springer; US: 2018. Demethylator phenotypes in acute myeloid leukemia; pp. 1–11. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tucker DW, Getchell CR, McCarthy ET, Ohman AW, Sasamoto N, Xu S, et al. Epigenetic Reprogramming Strategies to Reverse Global Loss of 5-Hydroxymethylcytosine, a Prognostic Factor for Poor Survival in High-grade Serous Ovarian Cancer. Clin Cancer Res [Internet] 2018;24:1389–401. doi: 10.1158/1078-0432.CCR-17-1958. Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-17-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.