

Abstract
Clear Cell Renal Cell Carcinoma (CC-RCC) is a devastating disease with limited therapeutic options available for advance stages. The objective of this study was to investigate HMG-CoA Reductase inhibitors, also known as statins, as potential therapeutics for CC-RCC. Importantly, treatment with statins was found to be synthetically lethal with the loss of the von Hippel-Lindau (VHL) tumor suppressor gene, which occurs in 90% of CC-RCC driving the disease. This effect has been confirmed in three different CC-RCC cell lines with three different lipophilic statins. Inhibition of mevalonate synthesis by statins causes a profound cytostatic effect at nanomolar concentrations and becomes cytotoxic at low micromolar concentrations in VHL-deficient CC-RCC. The synthetic lethal effect can be fully rescued by both mevalonate and geranylgeranylpyrophosphate, but not squalene, indicating that the effect is due to disruption of small GTPase isoprenylation and not the inhibition of cholesterol synthesis. Inhibition of Rho and Rho kinase (ROCK) signaling contributes to the synthetic lethality effect, and overactivation of Hypoxia Inducible Factor signaling resulting from VHL loss is required. Finally, statin treatment is able to inhibit both tumor initiation and progression of subcutaneous 786-OT1-based CC-RCC tumors in mice. Thus, statins represent potential therapeutics for the treatment of VHL-deficient CC-RCC.
Keywords: Statins, HMG-CoA Inhibitor, Clear Cell Renal Cell Carcinoma, Synthetic Lethality, VHL, HIF, small GTPases, ROCK
Introduction
CC-RCC is a life-threatening condition, especially in its metastatic manifestation. It is resistant to both radiation and chemotherapy(1), and although a recently introduced programmed death-1 inhibitor based immunotherapy shows promise with 25% of metastatic CC-RCC patients responding, the median overall survival remains at 2 years(2). In addition, toxicity to normal tissues is a limiting factor for current treatments. Thus, it is of primary importance to identify new therapeutics and their target pathways to successfully treat CC-RCC. Identified in 1993 as the tumor-suppressor gene affected in von Hippel-Lindau disease(3), the VHL gene is lost in 80-90% of CC-RCC(4). The goal of this study is to therapeutically target this large group of VHL-deficient CC-RCCs using a synthetic lethality approach.
Previously we conducted a chemical library screen and identified Y-27632 and inhibition of its target(ROCK1) being synthetically lethal with VHL loss(5). While multiple ROCK inhibitors have shown success in topical treatments for glaucoma(6), systemic treatments were conducted only with two ROCK inhibitors. Fasudil was approved in Japan for treatment of cerebral vasospasm complicating intracranial hemorrhage(7); and AT13148 is currently in a phase I clinical trial (NCT01585701) for solid tumors other than CC-RCC(8). Thus, systemic use of ROCK inhibitors requires further investigation to determine a therapeutic window for cancer patients. On the other hand, HMG-CoA Reductase inhibitors, also known as statins, can inhibit Rho/ROCK signaling in human patients(9), and their pharmacokinetics and doses are well established, including maximum tolerated doses(10).
Rho GTPases are upstream activators of ROCK(11). Rho/ROCK inhibition by statins occurs due to reduced synthesis of mevalonate and geranylgeranyl pyrophosphate (GGPP), in turn leading to inhibition of protein isoprenylation(9). This disrupts the intracellular trafficking of small GTPases like Rho, Ras, Rap1a and Rac and their recruitment to the cell membrane required for their activity(12). Although statins are not specific toward Rho, they are safe and taken by hypercholesterolemia patients at up to 1mpk (80 mg daily)(13). In addition, Lovastatin was evaluated as an anti-cancer agent for gastric carcinoma(14), anaplastic astrocytoma(15), and glioblastoma multiforme(15) in phase I and II clinical trials, and the maximum tolerated doses were established as high as 20-35mpk daily for 7 days, with monthly repeats, resulting in responses in 3 out of 18 patients(15). Accordingly, it has become clear that biomarkers are needed to stratify the patients into responders and non-responders. In addition, studies addressing the mechanism of statins’ anti-cancer action are largely absent. Since our studies show that statins trigger synthetic lethality with VHL deficiency, VHL can be used as a biomarker for tumor sensitivity. Furthermore, the effect is dependent on the overactivation of HIFs upon VHL loss, making HIF expression a second potential biomarker. Statin treatment selectively inhibits cell proliferation and induces cell death. Our studies also reveal that this effect occurs due to the disruption of GTPase isoprenylation and partially through the inhibition of Rho/ROCK1 signaling. Statin treatment is effective at inhibiting tumor initiation and tumor growth of established tumors in vivo, confirming their potential as therapeutics for treating VHL-deficient CC-RCC.
Materials and Methods
Cell culture and chemical treatments
All cell lines used in this study were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Caisson Labs #25-500, North Logan, UT) + 10% Fetal Bovine Serum (FBS; Omega Scientific #FB-12, Tarzana, CA) + 1% Penicillin/Streptomycin (Caisson Labs #25-512) in 5%CO2, 21%O2 at +37°C. The cell lines used in this study were a gift from Dr. Giaccia (Stanford). The identities of RCC4, RCC10, and 786-O were confirmed via STR analysis through the University of Arizona Genetics Core.786-OT1 cells are a sub-line of 786-O described in (5). Simvastatin, Pravastatin, mevalonate, GGPP, squalene (Sigma-Aldrich, St. Louis, MO), Fluvastatin, Lovastatin (Selleck Chemicals, Houston, TX), and Arachidonic acid (MP Biomedicals, Santa Ana, CA). Fluvastatin and Pravastatin were diluted in Dimethyl Sulfoxide (DMSO) and serially diluted for each experiment. Simvastatin and Lovastatin were dissolved in ethanol and activated in 0.1N NaOH by incubation at 50°C for 2 hours, followed by neutralization with 1N HCl, and dilution to 20mM in DMSO. The vehicle control was subjected to the same process and is approximately 20% ethanol and 80% DMSO.
Clonogenic assay
Clonogenic assays were performed using 300 cells/plate as previously described(5). For rescue experiments Fluvastatin (0.6125 or 1.25μM) and metabolite (GGPP [10 and 20μM], or mevalonate [500, 1000, and 2000μM], or squalene [10 and 100μM]) were dosed together and the clonogenic assays analyzed 7 days after treatment.
Live/Dead cell viability assay
The Live/Dead cell viability assays were performed by plating 300 cells per well into a 96-well plate, allowing them to attach overnight. The following day statins. On the 6th day Calcein AM fluorescent dye from Thermo Fisher (1:1000) and propidium iodide (PI) from Sigma-Aldrich (1:250) were added to each well, incubated for 10 minutes at 37°C, and then images were obtained on a Nikon TI-E at 4×. The live cells (Calcein-positive) and dead cells (PI-positive) were counted per field. Treatments were normalized to vehicle-controls for proliferation calculations. Normalized % viability was calculated by dividing the number of dead PI-positive cells by the total number of cells (Calcein-positive + PI-positive), subtracting the result from 1, and then multiplying by 100%. Treatments were conducted in quadruplicate and each experiment was repeated three times.
Western blot analysis
After treatments, cells were lysed and western blot was conducted as previously described(5). Proteins were visualized using primary antibodies recognizing HIF1α, VHL (BD Biosciences, #610959, #564183, San Jose, CA), HIF2α (Novus Biological, #NB100-122, Littleton, CO and abcam, #ab199, Cambridge, MA), α-tubulin (Fitzgerald, #10R-842, North Acton, MA), β-actin (Sigma, #A5441, St. Louis, MO), Phospho-LIMK1 (Thr508)/LIMK2 (Thr505), LIMK1, Rap1a(Cell Signaling, #3841S, #3842S, #4938S, Danvers, MA), unprenylated Rap1a (Santa Cruz Biotechnology, #SC-1482, Dallas, TX), LDHA, CAIX (GeneTex, #GTX101416, #GTX70020, Irvine, CA); and Horseradish peroxidase conjugated Goat anti-Rabbit IgG and Goat anti-Mouse IgG secondary antibodies (Thermo Scientific, #31460, #31430). Blots were imaged using a ChemiDoc XRS+ (BioRad, Hercules, CA).
Cholesterol Detection Assay
The Cholesterol Cell-Based Detection Assay Kit (Cayman Chemical, #10009779, Ann Arbor, Michigan) was performed following the manufacture’s protocol. Cells were treated 24hr before fixation. U-18666A, at 1.25μM, served as a positive control and came with the kit.
Lentiviral constructs and virus production
HEK 293T cells were transfected with lentiviral plasmids (pLKO.1shARNT: 5′AAATAAACCATCTGACTTCTC3′ (target sequence, OpenBiosystems, Huntsville, AL)) or pLKO.1shScr: 5′CCTAAGGTTAAGTCGCCCTCGC3′ (target sequence, Addgene, Cambidge, MA, #1864), along with packaging plasmids, pVSVG and ΔR8.2. Virus collection and infection were conducted as previously described(5).
Cell cycle analysis
50,000 cells were seeded per well of a 6-well plate and treated the following day with vehicle (DMSO) or Fluvastatin for 6 days. BrdU analysis was performed using the FITC BrdU Flow Kit (BD Biosciences, San Jose, CA, #559619) following the manufacturer’s protocol.
In vivo experiments
All in vivo experiments were conducted in accordance with and approval of the UCI IACUC. 25 RAG1 (B6.129S7-Rag1tm1Mom/J, Jackson Labs) mice (11–20 weeks old) were injected subcutaneously into the right flank with 5×106 786-OT1 cells resuspended in 50μl of 50%PBS/50%matrigel (BD Bioscience # 354248) mixture. See details in the “Results” section. Kaplan Meier curves were derived using GraphPad software and statistical significance calculated based on a Mantel-Cox test. Tumor volume was calculated using the formula: V=(a)(b2/2), where “a” is the shorter measurement of length/width. Statistical analysis was performed using a one-way ANOVA between the two groups per day.
Tumor Sample Processing
Tumor samples were fixed in formalin overnight and stored in 70% ethanol until processing on a Leica Tissue Processor (TP1020) following the manufacturer’s protocol. Samples were then embedded in paraffin on a Leica EG 1150 embedding/cooling station and sectioned on a Leica RM2255.
Ki67 Staining
Samples were baked at 65°C overnight and then deparaffinized and rehydrated. Antigen retrieval was performed in 10mM sodium citrate, samples were blocked in goat serum. Samples were then incubated overnight in Ki67 primary antibody (Genetex, #GTX16667) at 4°C, followed by incubation for 1 h in secondary antibody (Goat Anti-Rabbit IgG H&L [Alexa Fluor® 488], Abcam, Cambridge, UK, #ab150077) at room temperature. Cell nuclei were visualized with DAPI using Vectashield with DAPI (VWR, Radnor, PA, #101098-044). Images were obtained on a Nikon TI-E.
TUNEL Assay
TUNEL analysis was conducted using the DeadEnd Fluorometric Tunel System (Promega, Madison, WI, #G3250) following the manufacturer’s protocol for paraffin-embedded tissue sections. The positive control was prepared by treating 786-O cells with 2μM Staurosporine for 4 hours. The cells were then trypsinized, washed with PBS, and spun down onto slides using a cytospin centrifuge.
Growth curves and statistical analysis
Dose response and cell growth curves were generated using GraphPad Prism. IC50 values were calculated by transforming the X axis using X=Log(X), normalizing the transformed data to the vehicle control with 0 as 0%, and then fitting the normalized transformed data with a nonlinear trend line either using a normalized response (“log(inhibitor) vs. normalized response”) or a variable slope (“log(inhibitor) vs. normalized response – variable slope”). The correct nonlinear trendline was selected using GraphPad’s comparison of fits, which directly compares both fit lines statistically using an extra sum-of-squares F test. The IC50 values for each experiment were then calculated from the best-fit values. Statistical analysis was conducted in Minitab 16 using a paired t-test or ANOVA between cell lines with a p-value of less than 0.05 considered statistically significant. All error bars represent the SEMs. The number of biological replicates is indicated in each figure legend.
Results
Treatment with statins selectively targets VHL-deficient CC-RCCs of multiple genetic backgrounds
Since statins inhibit Rho/ROCK signaling(9) and we recently showed that ROCK1 inhibition is synthetically lethal with VHL-loss in CC-RCC(5) we decided to test if statin treatment would be synthetically lethal with VHL-loss. Isogenic cell line pairs were generated from the parental VHL-deficient CC-RCC cell lines by re-expressing the full-length wild-type VHL cDNA(16). VHL is a substrate-recognition subunit of an E3 ubiquitin ligase complex that targets the α subunits of Hypoxia Inducible Factors 1 and 2 (HIF1 and HIF2) for proteasomal degradation in the presence of oxygen (in normoxia)(17). Accordingly, VHL loss causes overexpression of HIF1α and HIF2α in RCC4 and RCC10 cells and HIF2α in 786-O cells, and VHL reintroduction causes a decrease in HIF expression (Supplementary Fig. 1). We conducted clonogenic assays and showed that both Simvastatin (Fig. 1A-C, Supplementary Fig. 2A-C) and Fluvastatin (Fig. 1D-F, Supplementary Fig. 2D-I) treatments are synthetically lethal with VHL loss. Both RCC4 (Fig. 1A, D) and RCC10 (Fig. 1B, E) showed sensitivity to statin treatment and a nearly 15-fold difference in IC50 values over respective RCCVHL cell lines. 786-O (Fig. 1C, F) showed a 5-fold difference in IC50 values over 786-OVHL.
Figure 1. Simvastatin and Fluvastatin treatment causes synthetic lethality with VHL loss in multiple CC-RCC cell lines.
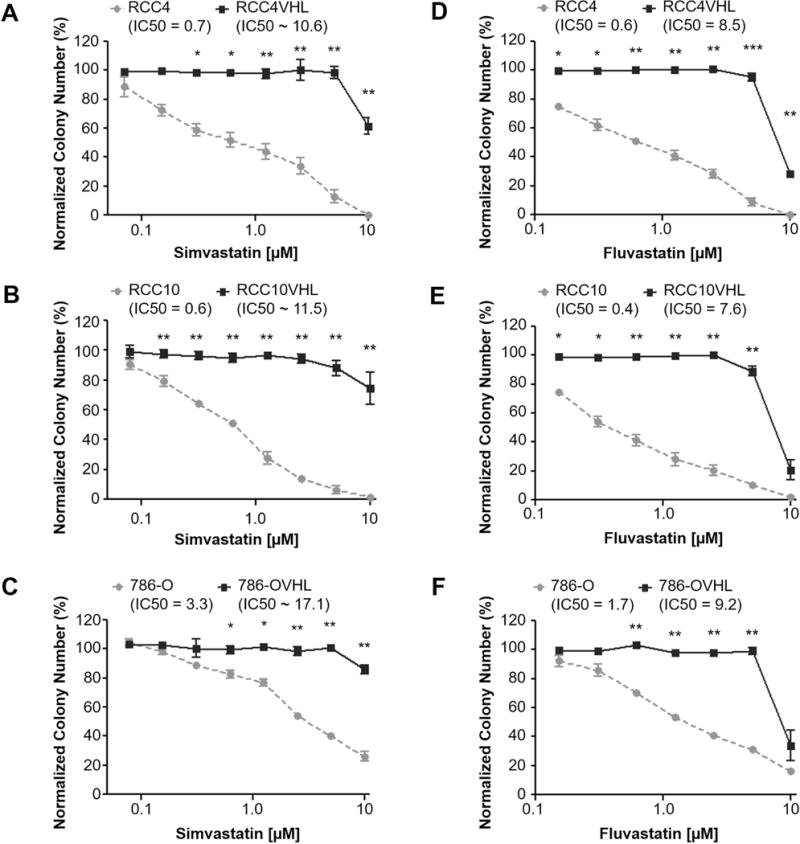
In a clonogenic assay activated Simvastatin (a-c) and Fluvastatin (d-f) show selective toxicity towards VHL-deficient (a, d) RCC4, (b, e) RCC10, and (c, f) 786-O while sparing their VHL-expressing isogenic cell line pairs. The cells were plated and allowed to adhere overnight, followed by addition of statins. Colonies were fixed and stained after 7 days. Data for Simvastatin and Fluvastatin treatments were normalized to an 80%DMSO/20%Ethanol and DMSO vehicle controls, respectively. Each dose within each experiment was tested in duplicate, and each experiment was repeated three times for each isogenic cell line pair. IC50s are indicated where “~” corresponds to IC50s extrapolated based on a best fit line of the data. Statistical analysis in (a-f) was performed using a paired t-test between the matched cell lines at each dose (* p < 0.05, ** p < 0.01, *** p < 0.001), SEMs are shown.
We also tested the effect of Lovastatin and Pravastatin, on the colony forming ability of the RCC4±VHL isogenic cell lines. RCC4 (Supplementary Fig. 3A) showed sensitivity to Lovastatin treatment and a 9-fold difference in IC50 values over RCC4VHL. Since treatment with Pravastatin up to 80μM did not reduce the colony forming ability of both RCC4 and RCC4VHL (Supplementary Fig. 3B), we assessed the inhibitory effect of each statin on isoprenylation of Rap1a, which depends on the mevalonate pathway. Unprenylated Rap1a was detected by western blot(18) after treatment with all statins but Pravastatin (Supplementary Fig. 3C), which is consistent with the lack of the effect of Pravastatin on colony forming ability of RCC4 cells. Unlike lipophilic statins Simvastatin, Fluvastatin, and Lovastatin, Pravastatin is hydrophilic and requires a liver-specific transporter OATP1B1(19) to be delivered inside the cells; thus it is likely not delivered to CC-RCC cells. Together, these data indicate that treatment with multiple lipophilic statins is synthetically lethal with VHL loss in several CC-RCC genetic backgrounds.
Treatment with statins is cytostatic and cytotoxic in VHL-deficient CC-RCC
We next sought to determine if the effect of statins on colony forming ability of VHL-deficient CC-RCC was caused by cell death, inhibition of proliferation, or both. We treated RCC4±VHL cells with Simvastatin at doses ranging from ~600nM to 10μM. We found that Simvastatin decreases RCC4 cell proliferation starting at nanomolar doses and increases RCC4 cell death starting at low micromolar doses based on Calcein-based Live/Dead assay (Fig. 2A-C). We confirmed these effects in the 786-O±VHL cells (Supplementary Fig. 4). Thus, statin treatment is predominantly cytostatic in VHL-deficient CC-RCC, but becomes cytotoxic as the concentration increases.
Figure 2. Statin treatment is cytostatic and cytotoxic in VHL-deficient CC-RCC.
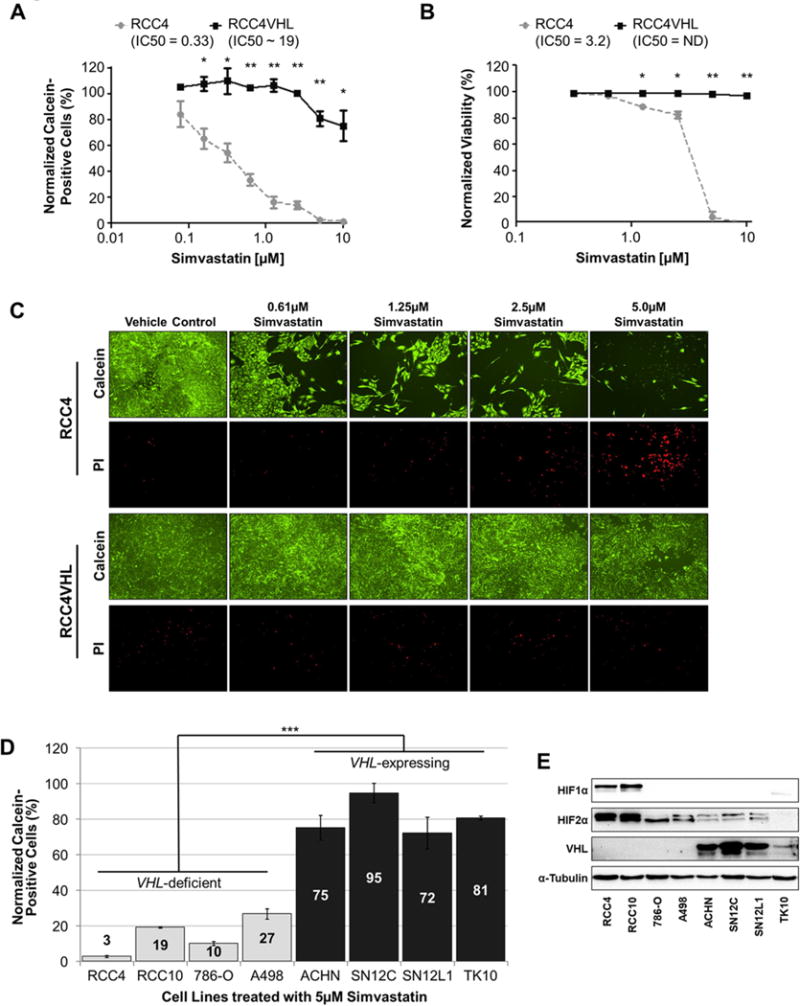
LIVE/DEAD assay measuring live cell numbers via Calcein staining (a) and dead cell numbers via PI staining (b) reveals that Simvastatin treatment inhibits RCC4 cell proliferation at nanomolar and micromolar doses, and triggers cell death at higher micromolar doses. RCC4±VHL cells were treated with Simvastatin or vehicle control (80%DMSO/20%Ethanol) for 6 days. Calcein-positive cells in (a) were normalized to the vehicle control. Statistical analysis in (a-b) was performed using a paired t-test between the matched cell lines at each dose (* p < 0.05, ** p < 0.01), SEMs are shown. (c) Representative images of Live/Dead assay. (d) VHL-deficient CC-RCC are more sensitive to Simvastatin treatment than renal cancer cell lines endogenously expressing VHL. Cell lines were treated with 5 μM Simvastatin for 6 days and the live cell number was assessed by Calcein staining. The results were normalized to vehicle-treated cells. Statistical significance was determined using a one-way ANOVA followed by Tukey’s post-hoc analysis (*** p < 0.001), SEMs are shown. The experiment was conducted in duplicate and repeated two times. (e) Western blot confirming VHL expression in cell lines used in (d), HIF1α and HIF2α expression is also shown. α-tubulin serves as a loading control.
Since each of the RCCVHL cell lines used above was genetically modified to overexpress VHL, we then asked if endogenous VHL expression could protect against the cytostatic effects of Simvastatin treatment. We compared the sensitivity of four VHL-deficient and four VHL-expressing kidney cancer cell lines to 5μM Simvastatin by Calcein-based assay. As expected, the VHL-deficient RCC4, RCC10, 786-O, and A498 were all more sensitive to Simvastatin treatment in comparison to the VHL-expressing ACHN, SN12C, SN12L1, and TK10 (Fig. 2D). VHL expression and HIF expression in these cell lines was confirmed by western blot (Fig. 2E). Thus, endogenous VHL expression is sufficient to protect cells against Simvastatin treatment.
Synthetic lethality depends on statins’ blocking effect on small GTPase isoprenylation
Next, we sought to confirm that the synthetic lethal interaction between statins and VHL loss was due to the inhibition of HMG-CoA reductase (see Supplementary Fig. 5 for schematic). Since HMG-CoA reductase catalyzes generation of mevalonate, we performed experiments with exogenous mevalonate to see if we could rescue cell proliferation of Fluvastatin-treated RCC4. As expected, 500μM and 1000μM/2000μM mevalonate treatments were able to partially and fully rescue the colony forming ability of Fluvastatin-treated RCC4 (Fig. 3A). The effectiveness of treatment by statins and rescue by mevalonate was assessed by their effect on Rap1a isoprenylation at 24 hours, which was blocked by Fluvastatin and rescued by mevalonate (Fig. 3C). Since 500μM mevalonate was able to fully restore Rap1a isoprenylation at 24 hours, but provided just the partial rescue of colony forming ability at 10 days, we assume that mevalonate stability and/or metabolic rate over the prolonged period of time contributes to the partial rescue. Furthermore, the addition of mevalonate to the RCC4±VHL cells treated with high doses of Simvastatin (10 and 20μM) resulted in a partial rescue of proliferation and a complete rescue of cell death in the Live/Dead assay (Supplementary Fig. 6). BrdU cell cycle analysis revealed that statin treatment selectively decreases S phase progression and increases apoptotic/debris cells in RCC4 cells, but not in RCC4VHL cells (Supplementary Fig. 7).
Figure 3. The effect of statins on GTPase isoprenylation is important for synthetic lethality with VHL loss.
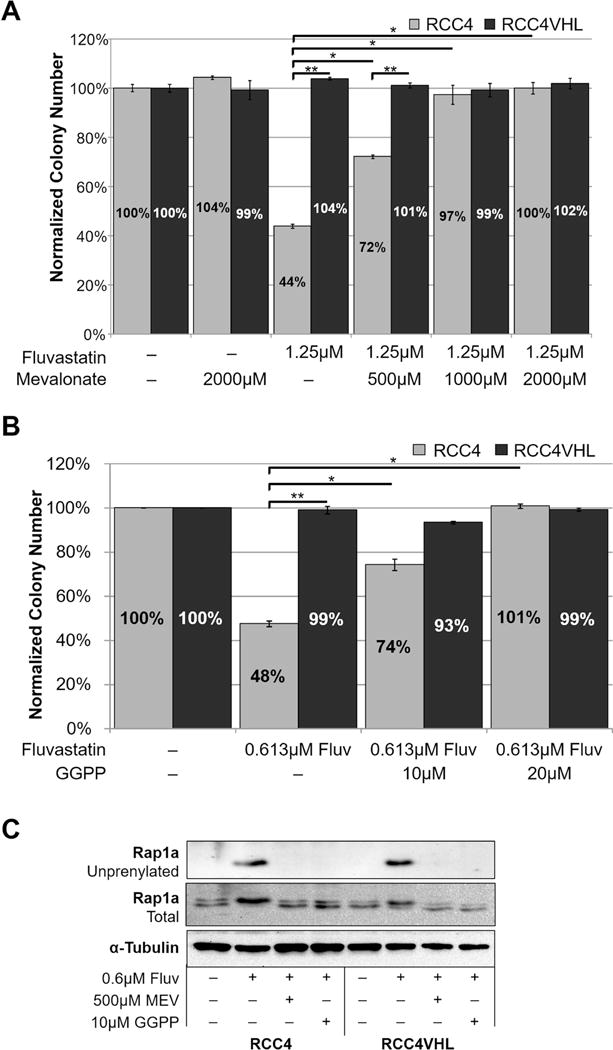
(a–b) Addition of 1000μM mevalonate (a) or 20μM GGPP (b) rescues the effect of seven-day treatment with Fluvastatin on colony forming ability of RCC4 cells. Each chemical was added at the time of Fluvastatin treatment and remained in the medium throughout the experiment. Statistical analysis in (a-b) was performed using a paired t-test comparing treatments, and comparing RCC4 to RCC4VHL (* p < 0.05, ** p < 0.01), SEMs are shown. In (a-b) each dose of Fluvastatin or vehicle control (DMSO) within each experiment was tested in duplicate, and the experiment was repeated three times for each isogenic cell line pair. (c) Western blot showing that the addition of mevalonate or GGPP rescues the effect of Fluvastatin on GTPase isoprenylation by blocking the appearance of unprenylated Rap1a. α-tubulin serves as a loading control.
Since the mevalonate pathway has multiple downstream metabolic products, we next sought to elucidate which arm of the pathway is involved in the synthetic lethal effect. It is important to note that in our experiments we used medium containing cholesterol from the serum. Since we are seeing the difference in RCC and RCCVHL colony forming ability in that medium, it suggests that this arm of the mevalonate pathway is not important for the synthetic lethal effect. In addition, to rescue cholesterol synthesis in Fluvastatin-treated RCC4±VHL cells, we added up to 100μM squalene (cholesterol precursor) and conducted clonogenic assays. The inability of squalene to rescue colony numbers (Supplementary Fig. 8) confirmed that cholesterol synthesis does not contribute to the synthetic lethal effect.
Previous studies have identified the inhibitory effect of statins on small GTPases, including the Rho GTPase and Rap1a GTPase(9,18,20,21) (used as a readout of statin inhibitory action on the mevalonate pathway in (Fig. 3C and Supplementary Fig. 2B). One of the arms of the mevalonate pathway generates GGPP, which is used as a substrate for isoprenylation of small GTPases by GGTase, directing GTPases’ membrane localization (see Supplementary Fig. 5 for schematic). In order to rescue isoprenylation of small GTPases in Fluvastatin-treated RCC4±VHL cells, we added 10μM or 20μM GGPP and conducted clonogenic assays. GGPP treatment led to a partial (10μM) and full (20μM) rescue of colony forming ability (Fig. 3B). Similar to mevalonate, 10μM GGPP treatment fully rescued the Rap1a isoprenylation at 24 hours as assessed by western blot (Fig. 3C). Together, these results indicate that the synthetic lethal effect between statin treatment and VHL loss is through inhibition of HMG-CoA reductase and the resulting effect of inhibiting GTPase isoprenylation.
The inhibitory effect of statins on the Rho/ROCK pathway is important for synthetic lethality with VHL loss
Previously we reported that ROCK1 inhibition results in a synthetic lethal interaction with VHL loss in CC-RCC(5). Thus, we hypothesized that inhibition of Rho GTPase isoprenylation and subsequent inhibition of ROCK by statins, is responsible for the synthetic lethality with VHL loss. First, we assessed whether treatment with statins causes Rho/ROCK pathway inhibition. We treated the RCC4±VHL cells with Fluvastatin for 24 hours and observed a decrease in phosphorylation of LIMK1 at Thr508 and LIMK2 at Thr505 (Fig. 4A). This effect was also replicated in the RCC10±VHL and the 786-O±VHL (Supplementary Fig. 9).
Figure 4. The inhibitory effect of statins on the Rho/ROCK pathway contributes to synthetic lethality with VHL loss.
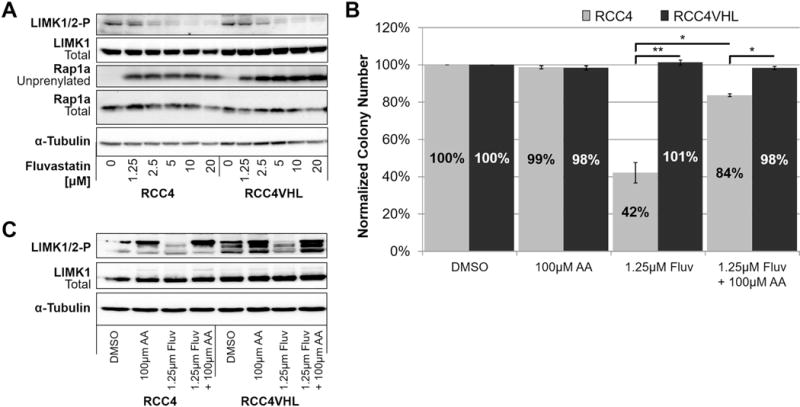
(a) Western blot showing that 24h treatment with Fluvastatin is sufficient to inhibit phosphorylation of LIMK1 (Thr508) and LIMK2 (Thr505) (ROCK substrates) in RCC4±VHL. Unprenylated Rap1a is used as a readout for the disruption of GTPase isoprenylation by treatment with Fluvastatin. (b) The effect of Fluvastatin on RCC4 colony forming ability can be partially rescued by administration of 100μM Arachidonic Acid (AA) (ROCK activator). Each dose of Fluvastatin or vehicle control (DMSO) within each experiment was tested in duplicate, and the experiment was repeated three times. Statistical analysis was performed using a paired t-test between the matched cell lines at each dose (* p < 0.05, ** p < 0.01), SEMs are shown. (c) Western blot showing that 24h treatment with Fluvastatin inhibits phosphorylation of LIMK1/2, and co-treatment with AA rescues phosphorylation of LIMK1/2 in the RCC4±VHL. (a-c) α-tubulin serves as a loading control.
Second, in order to rescue ROCK pathway activity in Fluvastatin-treated RCC4±VHL cells, we added 100μM Arachidonic acid (AA), which binds and activates ROCK by releasing it from its own autoinhibition(22,23), and conducted clonogenic assays. AA was able to partially rescue the colony forming ability of Fluvastatin-treated RCC4 cells (Fig. 4B). AA treatment activated ROCK signaling as judged by increased phospho-LIMK1/2 (Fig. 4C). Together, these results indicate that inhibition of the Rho/ROCK pathway by statins contributes to synthetic lethality with VHL loss in CC-RCC.
The synthetic lethal interaction between statin treatment and VHL loss is dependent on the activation of HIFs
Since mutation or deletion of VHL results in the stabilization and activation of HIF1α and HIF2α(24–26), we conducted three experiments to test if the synthetic lethal effect of statin treatment with VHL loss is dependent on overactivation of the HIF pathway. In the first experiment, we knocked down the Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT), which heterodimerizes with both HIF1α and HIF2α and is required for their activity(27). We then treated shARNT- and shScramble-transduced CC-RCC cells with Fluvastatin and conducted clonogenic assays. shARNT-transduced RCC4, RCC10, and 786-O cells were protected from Fluvastatin treatment in comparison to respective shScramble-transduced control cells (Fig. 5A). ARNT knockdown led to reduced expression of HIF target gene LDHA as confirmed by western blot (Fig. 5B). The protection of the shARNT-transduced CC-RCC cell lines from Fluvastatin treatment mimics VHL reintroduction, indicating that the synthetic lethal effect is dependent on HIF signaling.
Figure 5. HIF activation sensitizes CC-RCC to Fluvastatin.
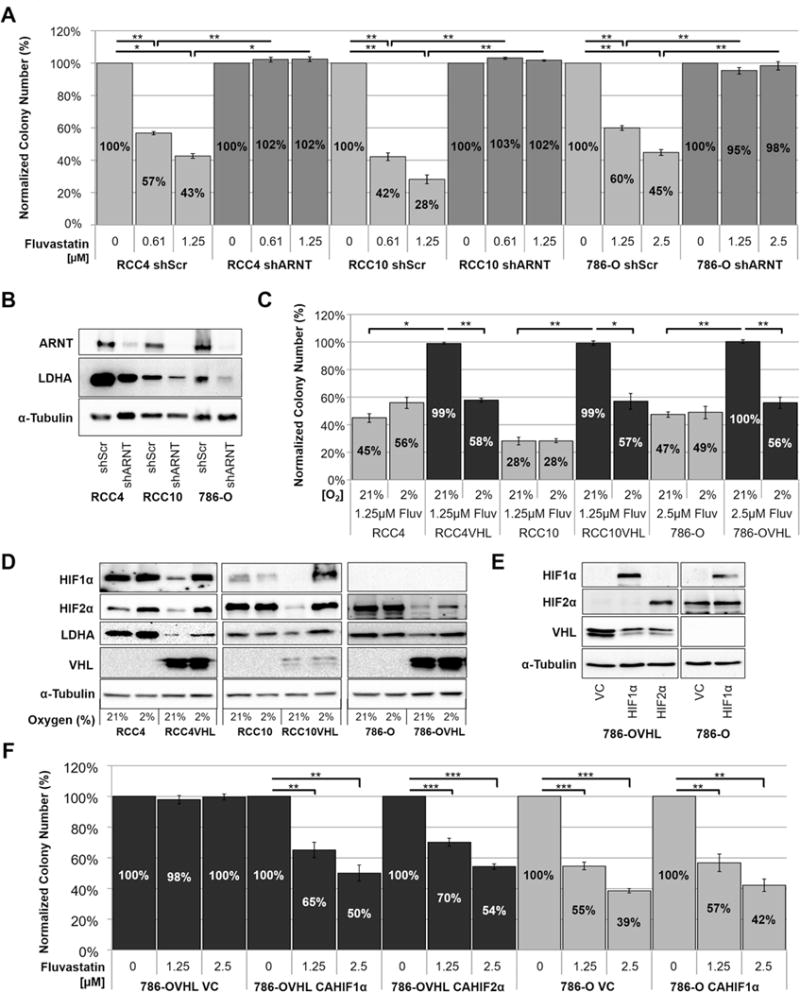
(a) Clonogenic assay showing that CC-RCC cells transduced with shARNT are protected against Fluvastatin treatment and their colony forming ability is comparable to RCCVHL cells. (b) Western blot confirming the downregulation of ARNT in shRNA-transduced cells, accompanied by downregulation of HIF’s downstream target LDHA. Cells transduced with scramble shRNA (shScr) serve as controls. (c) RCC±VHL cells were treated with 1.25μM Fluvastatin, plated for clonogenic assays and replicate plates were subjected to either normoxia (21% O2) or hypoxia (2% O2) for the duration of the experiment. Colony numbers were normalized to the DMSO vehicle control. RCC4VHL, RCC10VHL and 786-OVHL cells were sensitized to Fluvastatin treatment in hypoxia. (d) Western blot showing the induction of HIF1α and HIF2α and their downstream target LDHA in hypoxia (2% O2, 24h). (e) Western blot confirming the overexpression of constitutively active (CA) non-degradable hemagglutinin-tagged HIF1α or HIF2α (CA-HA-HIF1α and CA-HA-HIF2α). Cells transduced with vector-control (VC) serve as controls. Western blots shown are from the same gel. (f) Clonogenic assay showing that overexpression of CAHIF1α or CAHIF2α sensitizes the indicated cell lines to Fluvastatin treatment. Each assay in (a, c, and f) was performed in duplicate and repeated three times, and statistical analysis was performed using a paired t-test (* p < 0.05, ** p < 0.01 and *** p < 0.001), SEMs are shown. α-tubulin serves as a loading control in (b, d, and e).
In the second experiment, we treated RCC4±VHL, RCC10±VHL, and 786-O±VHL cells with Fluvastatin and subjected them to clonogenic assays in normoxia (21% oxygen) or hypoxia (2% oxygen). Each treatment was then normalized to the DMSO vehicle control. RCCVHL cell lines were sensitized to Fluvastatin treatment in hypoxia having decreased colony-forming ability in hypoxia in comparison to normoxia (Fig. 5C). Activation of HIF1α and HIF2α and induction of their downstream target in hypoxia were confirmed by western blot (Fig. 5D). The sensitization of the RCCVHL cells to Fluvastatin treatment in hypoxia mimics VHL loss, indicating that the synthetic lethal effect is dependent on HIF signaling.
In the third experiment, we used 786-OVHL cell line expressing a non-degradable constitutively active hemagglutinin-tagged HIF1α (CA-HA-HIF1α) or HIF2α (CA-HA-HIF2α) (Fig. 5E), which sensitized 786-OVHL cells to Fluvastatin treatment in comparison to vector-control cells (Fig. 5F). Expression of CA-HA-HIF1α in 786-O cells did not further sensitize them to Fluvastatin treatment in comparison to the vector-control cells (Fig. 5F). Together, these results confirm that the synthetic lethal effect between statins and VHL loss is dependent on the stabilization and overactivation of either HIF1α or HIF2α signaling.
Fluvastatin delays tumor initiation and inhibits tumor growth in vivo.
There are reports suggesting that statins reduce the risk of developing cancer(28–30), while other studies suggest that statins could serve as cancer therapeutics(21,31). Accordingly, we decided to test if statins delay CC-RCC tumor initiation and also inhibit tumor growth. For in vivo studies, we used 786-OT1 cells, which were established from a 786-O-based tumor and are characterized by fast tumor growth kinetics in vivo as previously described(5). 786-OT1 showed similar sensitivity to Fluvastatin in vitro as the parental 786-O cell line (Supplementary Fig. 10). 786-OT1 cells were injected subcutaneously into the right flank of 25 RAG1 mice. The mice were then randomized into three groups and two out of three groups were treated daily with (1% DMSO in PBS) vehicle-control (n=8) or 10mpk Fluvastatin (n=8) via intraperitoneal (ip) injection. The third group was left untreated (n=9) until the tumors reached approximately 300mm3. Mice were examined daily and palpable tumors were recorded. Treatment with 10mpk Fluvastatin inhibited tumor initiation as shown by Kaplan Meier curves (Fig. 6A). Once tumors had formed, tumor size was measured biweekly by calipers; tumor volume increased rapidly in the vehicle-treated control group, while tumor volume in the 10mpk Fluvastatin-treated group increased at a significantly slower rate (Fig. 6B). When tumors reached approximately 300mm3 in the third group (at day 54), we began treatments with 15mpk Fluvastatin daily for 21 days. Tumor size was measured triweekly and while tumor size constantly increased in the control group, treatment with 15mpk Fluvastatin resulted in a significant regression in tumor size (Fig. 6C). Both treatments resulted in a reduction of tumor weight and size (Fig. 6D-E). We also confirmed that 10mpk and 15mpk Fluvastatin treatments resulted in increased unprenylated Rap1a in tumor samples by western blot indicative of effective Fluvastatin delivery to tumor tissue (Fig. 6F). The group treated with 10mpk did not show a statistically significant difference in both proliferation (Ki67 staining) and apoptosis (TUNEL assay), which is consistent with the primary tumor growth curves in Figure 6, where at the time of sacrifice the slope of the curves for PBS-treated and 10mpk statin-treated groups becomes similar. As expected, Ki67 staining of the tumor tissue revealed that 15mpk Fluvastatin treatment resulted in decreased tumor cell proliferation in vivo at the time of sacrifice (Fig. 6G, Supplementary Fig. 11A). At the same time, TUNEL staining revealed that statin treatment slightly increased apoptotic cell death in vivo (Fig. 6H, Supplementary Fig. 11B). In summary, in vivo treatment with Fluvastatin successfully inhibited tumor initiation and decreased tumor growth in established tumors.
Figure 6. Fluvastatin prevents tumor initiation and inhibits tumor growth in vivo.
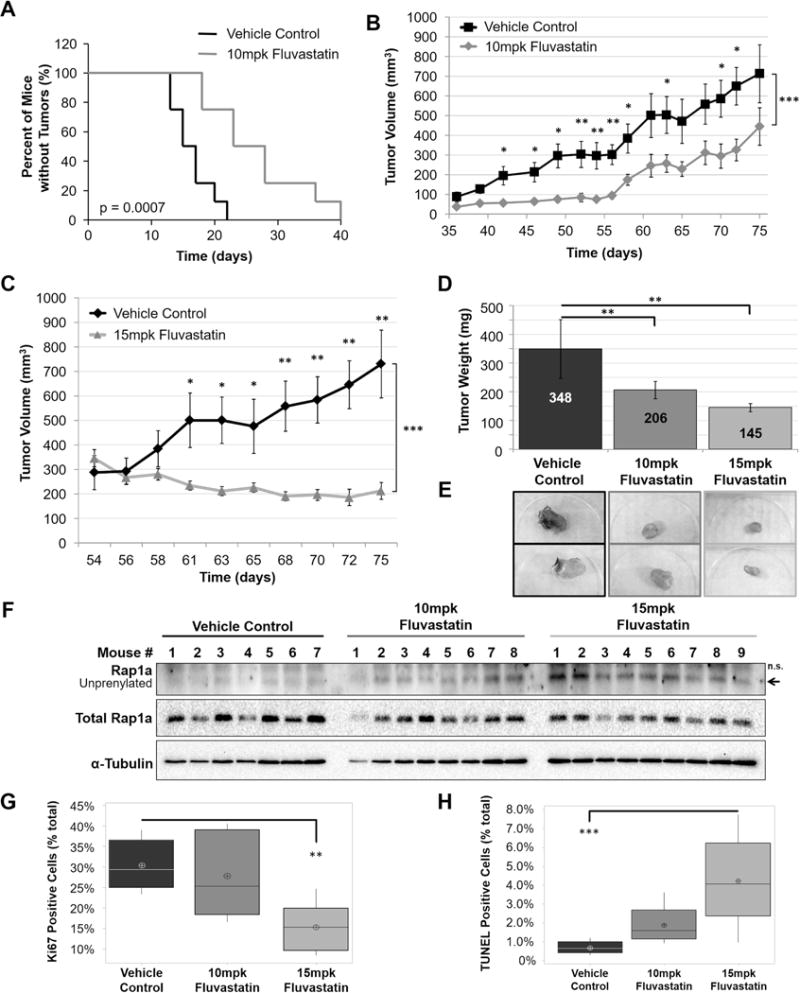
(a) 5 × 106 786-OT1 cells were injected subcutaneously into 25 RAG1 mice. The mice were then randomized into three groups: vehicle control (n=8), 10mpk Fluvastatin (n=8), and no treatment (n=9). Treatment was administrated immediately for the vehicle control and 10mpk Fluvastatin groups. 10mpk Fluvastatin treatment inhibited tumor initiation (a) as shown by Kaplan Meier analysis and inhibited tumor growth (b). When the “vehicle control” and “no treatment” tumors reached approximately 300mm3, 15mpk Fluvastatin treatment was initiated for the “no treatment” group, which inhibited tumor growth (c). (d) Administration of Fluvastatin to both groups (10mpk and 15mpk) resulted in reduced tumor weight in comparison to vehicle-control group at sacrifice. (e) Representative images of tumors. (f) Representative western blot of two showing that 10mpk and 15mpk Fluvastatin treatments resulted in appearance of unprenylated Rap1a in the tumor samples in comparison to the vehicle control, indicating that the drug was effectively delivered to the tumors. Administration of 15mpk Fluvastatin resulted in (g) decreased proliferation measured by Ki67 staining and (h) increased apoptotic cell death measured by TUNEL assay in comparison to vehicle-control group at sacrifice. Statistical analysis in (a) was conducted using Log-rank (Mantel-Cox) Test (p = 0.0007) and Gehan-Breslow-Wilcoxon Test (p = 0.0015). Statistical analysis in (b-c) was conducted using a paired t-test between doses and a two-way ANOVA comparing the response of each treatment group over time. Statistical analysis in (d, g, h) was conducted using a one-way ANOVA comparing treatments. (* p < 0.05, ** p < 0.01, *** p < 0.001) and Dunnett post-hoc with vehicle set as the control group. SEMs are shown.
Discussion
In this study, we have shown that Fluvastatin may serve as a potential therapeutic to target VHL-deficient CC-RCC. We have further determined that the therapeutic effect depends on blocking GTPase isoprenylation, including blocking of the Rho/ROCK pathway. Importantly, overactivation of the HIF pathway triggered by VHL loss contributes to CC-RCC sensitivity to statins. Treatment of VHL-deficient CC-RCC with statins in vitro inhibits proliferation and induces cell death. Accordingly, in vivo Fluvastatin is effective at both preventing tumor initiation and at inhibiting tumor growth of established xenografts, confirming its therapeutic potential.
Multiple synthetic lethal interactions have been identified in VHL-deficient CC-RCC, including stimulation of autophagy(32), inhibition of Glut1(26), CDK6(33), MET(33), MEK1(33), protein translation(34,35), and inhibition of ROCK1(5). Interestingly, with the exception of MET inhibition, statins target each of these synthetic lethality partners of VHL. Statin treatment has been shown to increase autophagy through inhibition of Rheb and Ras(36), inhibit glucose uptake and glucose metabolism in cancer cells(37), inhibit CDKs(31,38) (including CDK6(31)), inhibit the MEK pathway(39) through inhibition of Ras, and finally inhibit the ROCK pathway through inhibition of Rho(20,21). Since our studies show only partial reliance on Rho/ROCK pathway inhibition for statins’ selective targeting of VHL-deficient CC-RCC, statins’ inhibitory effect on other synthetic lethality targets likely contributes to the observed therapeutic effect. Furthermore, statins inhibitory effect on all of these synthetic lethality targets, except for the inhibition of glucose uptake, can be rescued by GGPP(21,36,38,39), indicating that inhibition of small GTPase isoprenylation is important.
The majority of patients with CC-RCC lose the function of VHL(17), which results in constitutive activation of HIFs, which is vital to the pathogenesis of the disease. Our finding that the synthetic lethal interaction is dependent on HIF overactivation suggests that other cancer types with overactivation of the HIF pathway might be sensitive to statin treatment. Thus, the stratification based on HIF activity and the status of small GTPase signaling needs to be tested to predict tumor’s sensitivity to statins.
In this study, we chose Fluvastatin for our in vivo studies because it has been shown to be the only statin that is able to achieve micromolar peak plasma concentrations (Cmax) with oral doses that are currently approved for treating hypercholesterolemia. This is due to Fluvastatin’s saturable first-pass metabolism(40), which results in greater than proportional increases in Cmaxv as the dose is increased in patients. Thus, Fluvastatin at hypercholesterolemia doses reaches Cmax concentrations that are greater than the IC50s determined in our study (Fig. 1D-F). Corsine and coauthors(10) showed that one 40mg oral dose has a Cmax of 448ng/ml (1.09μM) in patients. Another study by Siekmeier and coauthors(40) found that an 80mg dose has an average Cmax of 1024.7 (2.49μM). These studies make Fluvastatin a statin of choice for evaluation as CC-RCC therapeutic.
In addition, the doses of statins used in anti-cancer clinical trials far exceed the doses used for hypercholesterolemia, and although they have side-effects, are tolerated by patients. Lovastatin was evaluated as an anti-cancer agent for gastric carcinoma(14), anaplastic astrocytoma(15), and glioblastoma multiforme(15) in phase I and II clinical trials, and the maximum tolerated doses were established as high as 20-35mpk/day daily for 7 days, with monthly repeats(15). Hartman and coauthors reported that rhesus monkeys tolerated well 48/84/108mpk/day doses of Fluvastatin for 26 weeks(41). López-Aguilar and coauthors established a maximum tolerated dose for Fluvastatin in pediatric patients at 8mpk/day(42). The dose limiting side effect of statin treatment is apoptosis of skeletal muscle cells, which is preventable by the co-administration of ubiquinone(43). Accordingly, a maximum tolerated dose for Lovastatin can be increased from 25mpk/day to at least 45mpk/day by co-administration of ubiquinone (maximum tolerated dose was not reached in the study)(43). Thus, statin dosing and regimen for treatment of CC-RCC need to be carefully evaluated in patients.
There is another study reporting on the effect of statins on kidney cancer proliferation, migration, and tumor growth(44). The study did not concentrate on VHL-deficient CC-RCC and included several types of VHL-positive and -negative kidney cancers. Simvastatin was found to block tumor cell proliferation via the inhibition of the AKT/mTOR, ERK, and JAK2/STAT3 pathways at μM doses(44), and block tumor growth of A498-based xenografts. Our data show that A498 cells are the least sensitive to statin treatment among the VHL-deficient cell lines tested (Fig. 2D) and the reported Simvastatin Cmax concentrations in the blood(10) are far lower than for Fluvastatin used in our study. At the same time, we provide additional mechanisms of action for statins in CC-RCC via VHL loss and the resulting overactivation of the HIF pathway and inhibition of small GTPases, including Rho GTPase. Importantly, Rho GTPase was reported to regulate AKT signaling in melanoma(45), making our findings consistent with the ones discussed above. We also provide data on tumor initiation in addition to tumor progression.
Although statin administration led to reduction of tumor growth in both 786-OT1-based and A498-based(44) xenografts, additional evaluation of statins in vivo would be valuable. Although CC-RCCs tend to lose HIF1α during tumor progression(46) and multiple CC-RCC tumors and cell lines are expressing HIF2α only, including 786-O and A498, the efficacy of statins on HIF1α/HIF2α tumors in vivo would strengthen our conclusions. Furthermore, two autochthonous CC-RCC mouse models with the intact immune system have recently been developed(47,48), which could be used as future testing platforms for statins.
While a number of epidemiological studies have been conducted on the ability of statins to reduce the risk of CC-RCC, there is conflicting literature on the subject. Although, there are studies showing that people on statins have a lower risk of CC-RCC development(28), and that CC-RCC patients on statins have a better overall survival and lower risk of progression after surgery(29,30), there are two studies which found no correlation of statin intake and CC-RCC recurrence-free and progression-free survival(49,50). This discrepancy may be explained by the absence of stratification of CC-RCC patients by VHL status, difference in pharmacokinetics (Cmax achieved) for different statins, and intake of lipophilic vs hydrophilic statins, targeting both hepatic and non-hepatic tissues vs mainly hepatic tissue, respectively. Taking into consideration the above factors, a more careful epidemiologic analysis should be conducted to draw the conclusions.
In addition, we see a conceptual difference between tumors arising in patients already on statins (those tumors should be statin-resistant), and VHL-deficient statin-naïve tumors (those tumors should be statin-sensitive and respond to statin therapy). Accordingly, we propose that VHL-deficient CC-RCC patients, who were never on statins before, may benefit from lipophilic statin intake; and that VHL-deficient CC-RCC tumor patients, who are on hydrophilic statins, may benefit from switching to lipophilic statins. Furthermore, patients with VHL disease, lacking one copy of VHL at birth, may benefit from taking lipophilic statins to prevent initiation of CC-RCC, hemangioblastoma, and pheochromocytoma(17).
In conclusion, statin treatment is synthetically lethal with VHL loss in CC-RCC, and Fluvastatin could serve as a viable therapy for the disease. Treatment with Fluvastatin has a profound effect on VHL-deficient CC-RCC cells inhibiting proliferation, inducing cell death, and inhibiting both tumor initiation and growth. It is expected that patient stratification by the HIF and small GTPase signaling status will predict the response to lipophilic statin therapy; it is also expected that the reanalysis of the existing epidemiologic data on the CC-RCC initiation taking into account the administrated statin’s Cmax, the lipophilic vs hydrophilic statins, and VHL status of the tumor, will generate valuable data. Further studies are needed to evaluate statins as single agent CC-RCC therapeutics or combined with currently approved treatments.
Supplementary Material
Acknowledgments
Funding: This work was supported by an NCI/NIH R03 to O. V. Razorenova (R03 CA202563-01), an ACS RSG-170151-01-CDD to O. V. Razorenova, an NIH T32 (2T32CA009054-36A1) to J. M. Thompson, and an NIH ICTS (UL1 TR001414) to L. J. Nelson.
Abbreviations
- AA
Arachidonic acid
- ARNT
Aryl Hydrocarbon Receptor Nuclear Translocator
- CC-RCC
Clear Cell Renal Cell Carcinoma
- Cmax
maximum blood concentration
- DMSO
Dimethyl Sulfoxide
- GGPP
geranylgeranyl pyrophosphate
- HIF
Hypoxia Inducible Factors
- ROCK
Rho kinase
- siRNA
small interfering RNA
- Statin
HMG-CoA Reductase Inhibitor
- VHL
von Hippel-Lindau
Footnotes
Conflict of Interest: The authors have no conflicts of interest to disclose.
References
- 1.Motzer RJ. Renal cell carcinoma: progress against an elusive tumor. Semin Oncol. 2000;27:113–4. [PubMed] [Google Scholar]
- 2.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015:1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Latif F, Tory K, Gnarra J, Yao M, Duh F-M, Lou Orcutt M, et al. Identification of the von Hippel-Lindau Disease Tumor Suppressor Gene. Source Sci New Ser. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 4.Nickerson ML, Jaeger E, Shi Y, Durocher Ja, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–34. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson JM, Nguyen QH, Singh M, Pavesic MW, Nesterenko I, Nelson LJ, et al. Rho-associated kinase 1 inhibition is synthetically lethal with von Hippel-Lindau deficiency in clear cell renal cell carcinoma. Oncogene Nature Publishing Group. 2016;36:1080–9. doi: 10.1038/onc.2016.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Araie M. Phase 1 Clinical Trials of a Selective Rho Kinase Inhibitor, K-115. JAMA Ophthalmol. 2014;131:1288–95. doi: 10.1001/jamaophthalmol.2013.323. [DOI] [PubMed] [Google Scholar]
- 7.Dong M, Yan BP, Liao JK, Lam Y, Yip GWK, Yu M. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2013;15:622–9. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yap TA, Walton MI, Grimshaw KM, Te Poele RH, Eve PD, Valenti MR, et al. AT13148 is a novel, oral multi-AGC kinase inhibitor with potent pharmacodynamic and antitumor activity. Clin Cancer Res. 2012;18:3912–23. doi: 10.1158/1078-0432.CCR-11-3313. [DOI] [PubMed] [Google Scholar]
- 9.Cai A, Zhou Y, Li L. Rho-GTPase and Atherosclerosis: A Effects of Statins. J Am Heart Assoc. 2015;4:e002113. doi: 10.1161/JAHA.115.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther. 1999;84:413–28. doi: 10.1016/s0163-7258(99)00045-5. [DOI] [PubMed] [Google Scholar]
- 11.Liao JK, Seto M, Noma K. Rho Kinase (ROCK) Inhibitors. J Cardiovasc Pharmacol. 2007;50:17–24. doi: 10.1097/FJC.0b013e318070d1bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97:1232–5. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buzková H, Pechandová K, Danzig V, Vareka T, Perlik F, Zak A, et al. Lipid-lowering effect of fluvastatin in relation to cytochrome P450 2C9 variant alleles frequently distributed in the Czech population. Med Sci Monit. 2012;18:CR512–517. doi: 10.12659/MSM.883272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim WS, Kim MM, Choi HJ, Yoon SS, Lee MH, Park K, et al. Phase II study of high-dose lovastatin in patients with advanced gastric adenocarcinoma. Invest New Drugs. 2001;19:81–3. doi: 10.1023/a:1006481423298. [DOI] [PubMed] [Google Scholar]
- 15.Larner J, Jane J, Laws E, Packer R, Myers C, Shaffrey M. A phase I-II trial of lovastatin for anaplastic astrocytoma and glioblastoma multiforme. Am J Clin Oncol. 1998;21:579–83. doi: 10.1097/00000421-199812000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Sutphin PD, Chan Da, Li JM, Turcotte S, Krieg AJ, Giaccia AJ. Targeting the loss of the von Hippel-Lindau tumor suppressor gene in renal cell carcinoma cells. Cancer Res. 2007;67:5896–905. doi: 10.1158/0008-5472.CAN-07-0604. [DOI] [PubMed] [Google Scholar]
- 17.Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer Nature Publishing Group. 2014;15:55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- 18.Reilly JE, Neighbors JD, Tong H, Henry MD, Hohl RJ. Targeting geranylgeranylation reduces adrenal gland tumor burden in a murine model of prostate cancer metastasis. Clin Exp Metastasis. 2015;32:555–66. doi: 10.1007/s10585-015-9727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menter DG, Ramsauer VP, Harirforoosh S, Chakraborty K, Yang P, Hsi L, et al. Differential effects of pravastatin and simvastatin on the growth of tumor cells from different organ sites. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu P, Liu Y, Lin L, Chen J. Evidence for Statin Pleiotropy in Humans: Differential Effects of Statins and Ezetimibe on Rho-Associated Coiled-Coil Containing Protein Kinase Activity, Endothelial Function, and Inflammation. Inflammation. 2009;119:131–8. doi: 10.1161/CIRCULATIONAHA.108.813311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collisson Ea, Kleer C, Wu M, De A, Gambhir SS, Merajver SD, et al. Atorvastatin prevents RhoC isoprenylation, invasion, and metastasis in human melanoma cells. Mol Cancer Ther. 2003;2:941–8. [PMC free article] [PubMed] [Google Scholar]
- 22.Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, et al. Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem. 1999;274:3744–52. doi: 10.1074/jbc.274.6.3744. [DOI] [PubMed] [Google Scholar]
- 23.Garcia MC, Williams J, Johnson K, Olden K, Roberts JD. Arachidonic acid stimulates formation of a novel complex containing nucleolin and RhoA. FEBS Lett. 2011;585:618–22. doi: 10.1016/j.febslet.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 25.Chan DA, Sutphin PD, Denko NC, Giaccia AJ. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1α. J Biol Chem. 2002;277:40112–7. doi: 10.1074/jbc.M206922200. [DOI] [PubMed] [Google Scholar]
- 26.Chan Da, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3:94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masoud GN, Li W. HIF-1a pathway: Role, regulation and intervention for cancer therapy. Acta Pharm Sin B Elsevier. 2015;5:378–89. doi: 10.1016/j.apsb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khurana V, Caldito G, Ankem M. Statins Might Reduce Risk of Renal Cell Carcinoma in Humans: Case-Control Study of 500,000 Veterans. Urology. 2008;71:118–22. doi: 10.1016/j.urology.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 29.Kaffenberger SD, Morgan TM, Stratton KL, Boachie AM, Barocas Da, Chang SS, et al. Statin use is associated with improved survival in patients undergoing surgery for renal cell carcinoma. BJU Int Elsevier. 2012;110:21.e11–21.e17. doi: 10.1016/j.urolonc.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamilton RJ, Morilla D, Cabrera F, Leapman M, Chen LY, Bernstein M, et al. The association between statin medication and progression after surgery for localized renal cell carcinoma. J Urol. 2014;191:914–9. doi: 10.1016/j.juro.2013.10.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoque A, Chen H, Xu X. Statin Induces Apoptosis and Cell Growth Arrest in Prostate Cancer Cells. Cancer Epidemiol Biomarkers Prev. 2008;17:88–94. doi: 10.1158/1055-9965.EPI-07-0531. [DOI] [PubMed] [Google Scholar]
- 32.Turcotte S, Chan DDa, Sutphin PPD, Hay MMP, Denny Wa, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14:90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bommi-Reddy A, Almeciga I, Sawyer J, Geisen C, Li W, Harlow E, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci U S A. 2008;105:16484–9. doi: 10.1073/pnas.0806574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woldemichael GM, Turbyville TJ, Vasselli JR, Linehan WM, McMahon JB. Lack of a functional VHL gene product sensitizes renal cell carcinoma cells to the apoptotic effects of the protein synthesis inhibitor verrucarin A. Neoplasia. 2012;14:771–7. doi: 10.1593/neo.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolff NC, Pavía-Jiménez A, Tcheuyap VT, Alexander S, Vishwanath M, Christie A, et al. High-throughput simultaneous screen and counterscreen identifies homoharringtonine as synthetic lethal with von Hippel-Lindau loss in renal cell carcinoma. Oncotarget. 2015;6:16951–62. doi: 10.18632/oncotarget.4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araki M, Maeda M, Motojima K. Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur J Pharmacol Elsevier BV. 2012;674:95–103. doi: 10.1016/j.ejphar.2011.10.044. [DOI] [PubMed] [Google Scholar]
- 37.Malenda A, Skrobanska A, Issat T, Winiarska M, Bil J, Oleszczak B, et al. Statins impair glucose uptake in tumor cells. Neoplasia. 2012;14:311–23. doi: 10.1593/neo.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sala SG. HMG-CoA Reductase Inhibitor Simvastatin Inhibits Cell Cycle Progression at the G 1/S Checkpoint in Immortalized Lymphocytes from Alzheimer’s Disease Patients Independently of Cholesterol-Lowering Effects. Pharmacology. 2008;324:352–9. doi: 10.1124/jpet.107.128959. [DOI] [PubMed] [Google Scholar]
- 39.Tsubaki M, Takeda T, Sakamoto K, Shimaoka H, Fujita A, Itoh T, et al. Bisphosphonates and statins inhibit expression and secretion of MIP-1α via suppression of Ras/MEK/ERK/AML-1A and Ras/PI3K/Akt/AML-1A pathways. Am J Cancer Res. 2015;5:168–79. [PMC free article] [PubMed] [Google Scholar]
- 40.Siekmeier R, Lattke P, Mix C, Park JW, Jaross W. Dose dependency of fluvastatin pharmacokinetics in serum determined by reversed phase HPLC. J Cardiovasc Pharmacol Ther. 2001;6:137–45. doi: 10.1177/107424840100600205. [DOI] [PubMed] [Google Scholar]
- 41.Hartman Ha, Myers La, Evans M, Robison RL, Engstrom RG, Tse FL. The safety evaluation of fluvastatin, an HMG-CoA reductase inhibitor, in beagle dogs and rhesus monkeys. Fundam Appl Toxicol. 1996;29:48–62. doi: 10.1006/faat.1996.0005. [DOI] [PubMed] [Google Scholar]
- 42.López-Aguilar E, Sepúlveda-Vildósola AC, Rivera-Márquez H, Cerecedo-Diaz F, Valdez-Sánchez M, Villasis-Keever MA. Security and maximal tolerated doses of fluvastatin in pediatric cancer patients. Arch Med Res. 1999;30:128–31. doi: 10.1016/s0188-0128(98)00018-9. [DOI] [PubMed] [Google Scholar]
- 43.Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res. 1996;2:483–91. [PubMed] [Google Scholar]
- 44.Fang Z, Tang Y, Fang J, Zhou Z, Xing Z, Guo Z, et al. Simvastatin Inhibits Renal Cancer Cell Growth and Metastasis via AKT/mTOR, ERK and JAK2/STAT3 Pathway. PLoS One. 2013;8 doi: 10.1371/journal.pone.0062823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruth MC, Xu Y, Maxwell IH, Ahn NG, Norris Da, Shellman YG. J Invest Dermatol. Vol. 126. Elsevier; Masson SAS: 2006. RhoC promotes human melanoma invasion in a PI3K/Akt-dependent pathway; pp. 862–8. [DOI] [PubMed] [Google Scholar]
- 46.Gordan J, Lal P, Dondeti V, Letrero R. HIF-α Effects on c-Myc Distinguish Two Subtypes of Sporadic VHL-Deficient Clear Cell Renal Carcinoma. Cancer Cell. 2008;14:435–46. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y, et al. The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep. 2017 doi: 10.1016/j.celrep.2017.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harlander S, Schönenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, et al. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat Med. 2017;23:869–77. doi: 10.1038/nm.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi S-K, Min GE, Jeon SH, Lee H-L, Chang S-G, Yoo KH. Effects of statins on the prognosis of local and locally advanced renal cell carcinoma following nephrectomy. Mol Clin Oncol. 2013;1:365–8. doi: 10.3892/mco.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viers BR, Houston Thompson R, Psutka SP, Lohse CM, Cheville JC, Leibovich BC, et al. The association of statin therapy with clinicopathologic outcomes and survival among patients with localized renal cell carcinoma undergoing nephrectomy. Urol Oncol Semin Orig Investig Elsevier. 2015;33:388.e11–388.e18. doi: 10.1016/j.urolonc.2015.01.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.