

Abstract
Cooperativity between WNT and FGF signaling is well documented in embryonic development and cancer progression, but the molecular mechanisms underlying this crosstalk remain elusive. In this study, we interrogated the dynamics of RNA levels, ribosome occupancy, and protein expression as a function of inducible FGF signaling in mouse mammary glands with constitutive WNT hyperactivation. Multi-omics correlation analysis revealed a substantial discrepancy between RNA and ribosome occupancy levels versus protein levels. However, this discrepancy decreased as cells became pre-malignant and dynamically responded to FGF signaling, implicating the importance of stringent gene regulation in non-transformed cells. Analysis of individual genes demonstrated that acute FGF hyperactivation increased translation of many stem cell self-renewal regulators, including WNT signaling components, and decreased translation of genes regulating cellular senescence. WNT pathway components translationally upregulated by FGF signaling had long and structured 5′ UTRs with a high frequency of polypurine sequences, several of which harbored (CGG)4 motifs that can fold into either stable G-quadruplexes or other stable secondary structures. The FGF-mediated increase in translation of WNT pathway components was compromised by silvestrol, an inhibitor of EIF4A that clamps EIF4A to polypurine sequences to block 43S scanning and inhibits its RNA-unwinding activity important for translation initiation. Moreover, silvestrol treatment significantly delayed FGF-WNT-driven tumorigenesis. Taken together, these results suggest that FGF signaling selectively enhances translation of structured mRNAs, particularly WNT signaling components, and highlight their vulnerability to inhibitors that target the RNA helicase EIF4A.
Introduction
Both the WNT and FGF pathways play important roles in embryonic development and stem cell self-renewal and are frequently deregulated in breast cancer. WNT signaling is often activated in basal-like breast cancers and is associated with poor prognosis (1). Likewise, FGFR1 is frequently amplified in breast tumors and is associated with therapeutic resistance (2). Genetic studies using the mouse mammary tumor virus (MMTV) demonstrate co-activation of FGF and WNT pathway components as the most common occurrence in resulting tumors (3), providing genetic evidence for the cooperativity between these two pathways. In breast cancer, tumors with FGFR1 amplification and a high level of WNT signaling activity have the worst outcome compared with those that have deregulation of either pathway alone or normal levels of FGF and WNT signaling (4). Moreover, in vitro maintenance and expansion of stem cells and organoids typically require addition of ligands that activate both FGF and WNT signaling (5). Despite extensive evidence for the cooperation between these two pathways in normal development and cancer, the underlying molecular mechanisms remain poorly understood.
To elucidate the molecular crosstalk between FGF and WNT signaling, we generated a bigenic mouse model, MMTV-Wnt1/MMTV-iFgfr1 (WNT/iR1), in which iFGFR1 (inducible FGFR1) signaling can be activated by a chemical dimerizer in a ligand-independent manner (6,7), specifically in the mouse mammary gland with constitutive hyperactivation of WNT signaling (4,8). Dual FGF-WNT hyperactivation rapidly induced the formation of mammary tumors, which exhibited enhanced activity of the translation machinery (8,9) and were reversible by specific FGFR inhibitors (4). In the current study, we leveraged the WNT/iR1 model for real-time monitoring of the cellular response to acute iFGFR1 activation in mammary epithelial cells (MECs) in mice, allowing us to track the dynamics of RNA, translational regulation and protein levels as a function of iFGFR1 signaling. We found that when cells dynamically respond to a potent growth signal, such as iFGFR1 signaling, in mice, the correlation between RNA, ribosome occupancy and protein abundance unexpectedly increases, providing novel insights into gene regulation in pre-malignant cells.
We further explored the hypothesis that iFGFR1 activation regulates WNT signaling by selectively enhancing the translation of WNT pathway-regulated signaling components. We observed that iFGFR1 signaling increased the translation of WNT pathway components, several of which have structured 5′ untranslated regions (UTRs) with a high frequency of polypurine sequences and contain (CGG)4 motifs that can fold into either stable G-quadruplexes or other stable secondary structures. Silvestrol, which targets the RNA helicase EIF4A important for unwinding structured 5′ UTRs to initiate translation and also can clamp EIF4A to polypurine sequences to inhibit 43S scanning, delayed iFGFR1-induced tumorigenesis in WNT-hyperactive cells. This delay was accompanied by a concomitant reduction in the translation of WNT signaling components that were translationally upregulated by iFGFR1 signaling. These studies suggest that breast tumors with hyperactivation of WNT signaling together with FGF signaling may be vulnerable to EIF4A inhibition.
Materials and Methods
Further details of the study methods are provided in Supplementary Methods.
Animals and in vivo iFGFR1 induction
Bigenic MMTV-Wnt1+/−/MMTV-iFgfr1+/− mice were generated and maintained in FVB/N background as previously described (4). Four-week-old WNT/iR1 mice were injected i.p. with vehicle (= -iFGFR1) or 1mg/kg AP20187 (Clontech, 635069) for 6 hrs (= +iFGFR1). AP20187 was formulated in PBS with 2% Tween-20 and 5% polyethylene glycol (Sigma-Aldrich, 202371). Mammary glands were harvested and processed for downstream analyses. All animal experiments were performed in compliance with institutional guidelines as approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Ex vivo ribosome profiling (Ribo-Seq) on MECs
The 3rd, 4th and 5th mammary glands were isolated from mice with or without iFGFR1 activation and immediately transferred to DMEM/F-12 (Thermo Scientific, 11330032) media with 100 μg/ml Cycloheximide (CHX) (Sigma-Aldrich, C4859) at room temperature. The glands were then processed as previously described for isolation of primary MECs (10) in the presence of 100 ug/ml CHX. The resulting MEC pellet was split into three fractions and subsequently processed for purification of ribosome-protected mRNA fragments (RPFs), RNA and protein. MECs from multiple mice with the same treatment were pooled together to generate enough materials for multi-omics analyses, which were reproduced in triplicates. RPFs were isolated using a published protocol (11) with a few modifications. Instead of sucrose gradient centrifugation, recovery of RPFs following nuclease treatment was performed using Illustra MicroSpin S-400 HR columns (GE Healthcare, 27514001) and the RNA Clean & Concentrator™-25 kit (Zymo Research, R1017) as described in the protocol of the TrueSeq® Ribo Profile kit (Illumina, RPHMR12126). Deep sequencing libraries were generated from these fragments and sequenced on the HiSeq 2500 platform. Each sequencing run generated approximately 100 million reads per sample, of which about 27 million reads were used for subsequent analysis. Ribo-Seq and RNA-Seq read counts were normalized for translation efficiency (TE) quantification and differential analysis using Xtail algorithm v.1.1.5 (12). The raw Ribo-Seq and RNA-Seq data have been deposited to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE107926.
Library preparation and RNA-Seq analysis
RNA was isolated from MECs using TRIzol Reagent in accordance with manufacturer’s protocol (Thermo Scientific, 15596018). The Genomic and RNA Profiling core at Baylor College of Medicine performed quality control, selection of poly(A) mRNA and preparation of cDNA library using TruSeq® Stranded mRNA Library Prep Kit Set A (Illumina, RS1222101). Equimolar amounts of the library for each sample were loaded and sequenced on the HiSeq 2500 platform. Average reads derived per sample were approximately 200 million, and 180 million reads were used for downstream analysis. RNA-Seq read counts were normalized for differential analysis of RNA level changes using algorithm from limma v.3.30.13 (13).
Proteome profiling with LC-MS/MS
Purified MECs were processed and analyzed as previously described (14) for proteome profiling. The iBAQ algorithm (15) was used to calculate protein abundance to compare relative amount between different proteins in the sample using in house data processing algorithm (14). The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the MASSIVE repository (MSV000081730) with the dataset identifier PXD008268.
Multi-omics correlation analysis
RNA-Seq and Ribo-Seq reads were normalized by DESeq for RNA and ribosome occupancy expression correlations. Quantile normalization was used to normalize iBaq values for protein expression correlations. The dynamic ranges are comparable between the three omics data types. Inter- and intra-omic correlations were performed using the Spearman correlation method, which is non-parametric method and is less sensitive to data distribution and outliers as a way to control for spurious correlation.
Quality control of Ribo-Seq data
Quality control was performed by Ribo-TISH (16), using all of the uniquely mapped RPF reads in the annotated mouse protein-coding genes (PCGs). Each aligned RPF read was represented by its 5′ end before estimation of the P-site offset. The P-site offset estimation was performed as previously described (16). The RPF count between the 15 nucleotides (nt) upstream of the first base of the start codon and the 12 nt upstream of the first base of the stop codon were used to calculate the RPF count distributions across three reading frames for different RPF lengths. The ratio between the maximum RPF count among all three reading frames and the sum of the RPF counts from all reading frames were calculated for different RPF lengths to assess the 3-nt periodicity. The metagene RPF count profile near the start/stop codon was constructed by summing the RPF count between −40 and +20 nt of the first base of the start/stop codon across all annotated PCGs. The CDS metagene profile was constructed for all three reading frames, using the RPF counts in the region between 15 nt upstream of the first base of the start codon and 12 nt upstream of the first base of the stop codon. For each frame, the CDS region was divided into 20 bins, and the average RPF count across all annotated PCGs was calculated for each bin.
Polysome analysis
MECs were purified from mammary glands of mice with or without iFGFR1 activation in the presence of CHX. Cell pellets were processed at 4 °C for polysome analysis as previously described (17). Polysome profiles were obtained with an ISCO gradient fractionator with UV absorbance monitoring. RNA was extracted from all fractions with TRIzol Reagent (Thermo Scientific, 15596018) for qPCR. Changes in RNA proportion between pre-polysome and polysome fractions were compared using paired t-test.
5′ UTR analysis
5′ UTRs of TE UP, TE DOWN and background genes were obtained from UCSC mm10 database and compared for 5′ UTR length and minimum folding free energy by two-tailed Student’s t-tests. The length of the 5′ UTR sequences were computed using EMBOSS v.6.6.0.0. The minimum free energy of secondary structure formation was calculated by Vienna RNA Package RNAfold (v.2.4.3), which uses nearest-neighbor thermodynamic parameters to predict the minimum folding free energy of 5′ UTR sequences (18). MEME (v.4.11.4) with window lengths of 9 to 12 was used to find motifs in 5′ UTR sequences. FIMO (v.4.11.4) was used to match motifs to individual 5′ UTR sequences and to determine enrichment of the (CGG)4 motif in TE UP genes. Detailed RNA secondary structure predictions and analyses were performed using RNAstructure v.6.0 (19). For quantification of polypurine sequence frequency, 6-nt motifs with different combinations of purines A and G were input into FIMO (v.4.11.4) to scan for polypurine sequences on 5′ UTRs of TE UP, TE DOWN and background genes.
Silvestrol treatment
Three-week-old FVB/N mice (Jax® Mice) with cleared mammary fat pads were transplanted with mammary tissue chunks from WNT/iR1 mice. Three days following orthotopic transplantation, mice were randomly assigned into vehicle control or silvestrol treatment groups; n was determined empirically based on previous experiments. Silvestrol was formulated in 20% 2-hydroxypropyl-β-cyclodextrin (CTD, Inc., THPB-P) and administered i.p. at 1.5 mg/kg. After 24 hrs of pretreatment with vehicle or silvestrol, mice were injected i.p. with 1 mg/kg AP20187 every 3 days and 1.5 mg/kg vehicle or silvestrol every 2 days. Mouse weight and tumor size were measured every 2 to 3 days. Tumor volume was calculated using the following formula: Volume = (Length × Width × Width)/2. P values were calculated using two-tailed Student’s t-tests for paired data.
Correlation between receptor tyrosine kinase (RTK) activation and translation signatures
RTK activation signature was derived based on gene expression profiles of 4T1 cells upon TKI258 treatment to inhibit FGFR signaling (9). The signature was determined by Sum(genes downregulated by TKI258) – Sum(gene upregulated by TKI258). The “translation” signature was obtained from the MSigDB v.6.1 collection. The score for each signature was generated for each breast tumor in the METABRIC dataset and normalized to 0-1 region linearly with 0 meaning the minimum and 1 representing the maximum. The Spearman correlation was calculated by R statistical programing software and plotted using the R package “ggplot2”. P values were computed based on Student’s t-tests.
Statistical analysis
Statistical tests with suitable underlying assumptions on variance characteristics and data distribution were employed. Unless otherwise noted, two-tailed Student’s t-tests were used for comparisons between groups.
Results
An ex vivo Ribo-Seq strategy to interrogate translatome dynamics in mammary epithelial cells
To elucidate mechanisms underlying FGF-WNT cooperativity and to assess the dynamics of RNA, ribosome occupancy and protein levels in vivo, 4-week-old WNT/iR1 mice were treated i.p. for 0 (steady state) or 6 hrs (dynamic activation) with AP20187, a chemical dimerizer, to induce iFGFR1 tyrosine kinase activity in MECs in vivo (6) (Supplementary Fig. S1A and S1B). AP20187 selectively binds to the two F36V-FKBP motifs fused to the tyrosine kinase domains of FGFR1 with sub-nanomolar affinity (20) and has been used widely for characterization of FGF signaling (21,22) without any known off-target effects. Following 6 hrs of treatment, iFGFR1 signaling resulted in increased proliferation, enhanced phosphorylation of the translation regulator, ribosomal protein S6 (RPS6), and a decrease in lumen size (Supplementary Fig. S1C).
In addition to RNA and protein changes, we interrogated the dynamics of ribosome occupancy on mRNAs, which should reflect changes in translational regulation following iFGFR1 activation. To achieve this, we employed a Ribo-Seq technique modified for ex vivo analysis that captures RPFs for deep sequencing (details in Materials and Methods) in addition to RNA-Seq and LC-MS/MS (Fig. 1A). To ensure the quality of RPF reads, we examined whether the reads exhibit 3-nt periodicity, a unique property of actively translating ribosomes (16). Metagene analysis of RPF and RNA-Seq reads mapping to the coding sequence (CDS) demonstrated that while RNA-Seq reads did not show any bias in their distribution, 77% of RPF reads distributed in the same frame relative to their 5′ ends (i.e. position 1 in the codon of the P site with +12-nt offset) (Fig. 1B). We confirmed that the majority of the RPF reads mapped to the CDS with 3-nt periodicity, whereas RNA-Seq reads did not exhibit any periodic distribution and mapped to both the CDS and the UTRs (Fig. 1C; Supplementary Fig. S2). Additionally, most RPF reads within the annotated open reading frame (ORF) had a strong in-frame distribution as illustrated for β-actin (Fig. 1D). Together, these data indicate that our ex vivo Ribo-Seq strategy for MECs in mice captures high quality RPFs with 3-nt periodicity representative of the stepwise translocation of active ribosomes, and that these RPF reads can be reliably used for multi-omics correlation analysis and identification of translationally altered genes.
Figure 1.
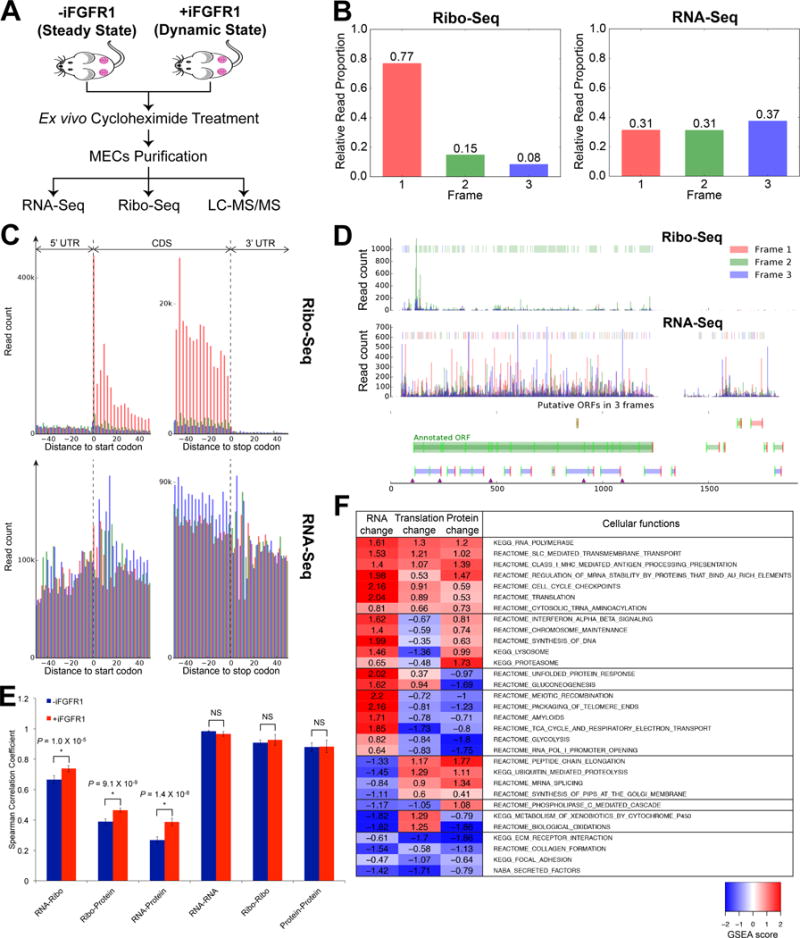
Ex vivo Ribo-Seq on MECs and multi-omics correlation analysis. A, Schematic overview of tissue processing for multi-omics profiling on MECs from the WNT/iR1 mouse model. B, Distribution of Ribo-Seq RPF reads (left) and RNA-Seq reads (right) across 3 possible reading frames relative to their 5′ ends in all annotated codons; showing proportion of reads from each reading frame. C, RPF and RNA-Seq reads aligned to a composite Refseq transcript, including the CDS, and 5′ and 3′ UTRs. Reads are colored as in (B) corresponding to respective reading frames. D, RPF and RNA-Seq read count profiles of β-actin. Color-coded boxes in the upper area of each plot highlight the dominant reading frame. Note that majority of RPF reads within the annotated ORF are in Frame 2, which is the same frame as the annotated ORF, whereas RNA-Seq reads are not predominantly distributed in any particular frames. E, Comparison of Spearman correlation coefficients for inter-omic and intra-omic (i.e. between biological replicates) correlations in MECs without and with 6-hrs iFGFR1 activation. Note that intra-omic correlations were not significantly different between cells at the steady and dynamic states. *P < 0.05; NS, not significant (P > 0.05); error bars indicate standard deviation. F, Cellular functions differentially or similarly controlled by distinct regulatory processes. For each regulatory step (columns) and cellular function (rows) shown is the magnitude of the normalized gene set enrichment analysis (GSEA) score of the cellular function’s signature in the differential fold changes of RNA, TE and protein levels upon acute iFGFR1 activation. Shown are the most enriched cellular functions; redundant functions were excluded.
FGFR1 signaling strengthens the correlation between RNA, ribosome occupancy and protein abundance
Before probing the translationally regulated genes following iFGFR1 activation, we first assessed the relationship between the dynamics of RNA, ribosome occupancy and protein in the steady (-iFGFR1) and dynamic states (+iFGFR1) in vivo. A total of 7318 genes reliably detected by RNA-Seq, Ribo-Seq and LC-MS/MS in three independent biological replicates were used for multi-omics correlation analysis (Supplementary Table S1A). Biological replicates were highly reproducible across all omic datasets with average Spearman correlation coefficients of 0.98 for RNA-Seq, 0.92 for Ribo-Seq and 0.88 for LC-MS/MS and were equivalent between cells at the steady and dynamic states (Fig. 1E). Ribosome occupancy levels correlated better with protein abundance than RNA expression correlated with protein abundance in both the steady (R = 0.35 vs. 0.25) and dynamic states (R = 0.47 vs. 0.42) (Supplementary Fig. S3A and S3B). However, the correlation between RNA and ribosome occupancy levels was the highest in both the steady (R = 0.66) and dynamic states (R = 0.77) (Supplementary Fig. S3A and S3B). Together, these results demonstrate that, in both the steady and dynamic states in vivo, although the translatome displayed a better correlation with the proteome (i.e. protein), it still largely resembles the transcriptome (i.e. RNA). It is important to note that many low-abundant proteins with interesting biological functions, such as cytokines and growth factors, were not reliably detected by LC-MS/MS, and hence the correlation values reported in this study are limited to relatively more abundant proteins.
Unexpectedly, we found that when cells switched from a steady state to a dynamic state in response to acute iFGFR1 activation, Spearman correlation coefficients significantly increased (Fig. 1E). The RNA vs. protein correlation exhibited the greatest increase (ΔR = 0.12), whereas the correlation increased equivalently between RNA vs. Ribo (ΔR = 0.072) and Ribo vs. Protein (ΔR = 0.076) (Supplementary Fig. S3C). These data suggest that both post-transcriptional and post-translational regulation were altered or deregulated under the influence of a potent growth signal like iFGFR1.
We further determined how cellular functions were differentially regulated with respect to changes in RNA levels, translational regulation and protein levels in response to acute iFGFR1 activation. Changes in translational regulation were evaluated based on variations in TE, which was quantified by normalizing the RPF frequency (i.e. ribosome occupancy) to transcript length and total transcript abundance. It is common to find cellular processes with RNA levels and translational regulation changing in the same direction (Fig. 1F). However, very few cellular functions have protein levels that change in the opposite direction to both RNA and translational regulation changes (Fig. 1F). Furthermore, we found that changes in levels of proteins involved in regulation of DNA synthesis, type-I interferon-dependent immune response, lysosomal functions and biological oxidations were particularly dominated by RNA abundance changes, which were antagonized by translational regulation (Fig. 1F). In contrast, changes in levels of proteins that regulate the citric acid cycle, the electron transport chain, peptide chain elongation and RNA splicing were predominantly regulated by translational changes, when RNA levels changed in the opposite direction (Fig. 1F), suggesting that translational regulation changes can dominate RNA variations to modulate changes in protein output in vivo following acute iFGFR1 activation.
FGFR1 signaling enhances translation of both positive and negative regulators of WNT signaling components and stem cell regulators
To study changes of mRNA and TE levels in MECs following iFGFR1 activation, we applied Voom (13) and Xtail (12) algorithms to all genes that were reliably detected by both RNA-Seq and Ribo-Seq for differential expression analysis. Genes with homodirectional changes between mRNA and TE were excluded from the analysis of translationally altered genes in response to iFGFR1 activation to eliminate potential confounding effects of mRNA variability on TE changes. Therefore, genes with increased TE and decreased or unchanged mRNA were categorized as TE UP genes (TE log2 fold change (FC) ≥ 0.5 and mRNA log2FC < 0.5; n = 1976), whereas those with decreased TE and increased or unchanged mRNA were defined as TE DOWN genes (TE log2FC ≤ −0.5 and mRNA log2FC > −0.5; n = 1427) (Fig. 2A; Supplementary Tables S1B-D). Functional pathway enrichment analysis revealed that many TE UP genes were involved in transport of amino acids, G2/M transition, WNT signaling and other cancer-related pathways such as TGF-β, SCF-KIT, and NOTCH (Fig. 2B). Moreover, both positive and negative regulators of WNT pathway were translationally upregulated by iFGFR1 activation suggesting that FGF signaling possibly regulates both positive and negative feedback loops to maintain a durable level of WNT activation (Supplementary Table S2A). In contrast, TE DOWN genes primarily regulated DNA methylation and cellular senescence (Fig. 2C). Interestingly, the TE UP gene set was enriched for embryonic stem cell signature (23) as well as G2/M-checkpoint-hallmark signature (Fig. 2D). These findings are consistent with the roles of FGF and WNT signaling in embryonic development, cancer stem cells, and stem cell self-renewal (24–26). Polysome analysis validated a decrease in the fraction of pre-polysome RNA and an increase in the fraction of polysomal RNA in TE UP genes including a cell cycle regulator, Cdk13, and Prkca, a component of WNT pathway with roles in breast cancer stem cells (27), whereas the opposite was observed for TE DOWN genes (Supplementary Fig. S4A-C).
Figure 2.
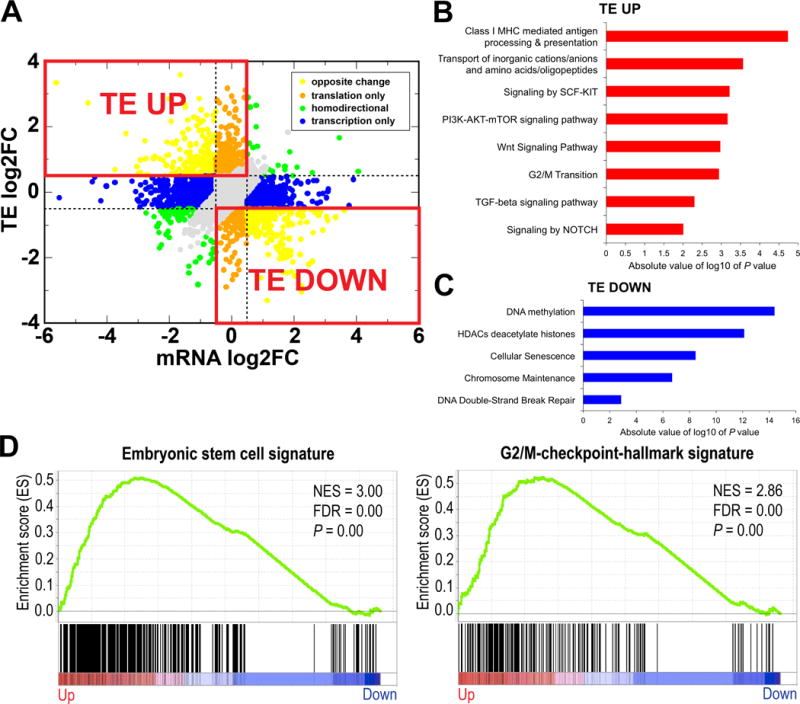
iFGFR1 signaling enhances translation of WNT signaling components and stem cell regulators. A, TE fold changes plotted against mRNA fold changes following 6-hrs iFGFR1 induction. Genes that changed with respect to log2FC threshold of 0.5 were color-coded. The TE UP quadrant is comprised of genes with increased TE and either a decrease or no change in mRNA (TE log2FC ≥ 0.5, mRNA log2FC < 0.5). The TE DOWN quadrant is comprised of genes with decreased TE and either an increase or no change in mRNA (TE log2FC ≤ −0.5, mRNA log2FC > −0.5). B and C, Non-redundant cellular functions and pathways overrepresented in TE UP and TE DOWN genes, respectively, using ConsensusPathDB’s databases. D, GSEA plots for embryonic stem cell (left) and G2/M-checkpoint-hallmark (right) signatures in TE UP genes. All P values are less than 0.01; NES, normalized enrichment score; FDR, false discovery rate.
WNT signaling components translationally upregulated by FGFR1 signaling have structured 5′ UTRs with a high frequency of polypurine sequences and harbor (CGG)4 motifs that can form either stable G-quadruplexes or other stable secondary structures
The 5′ UTRs of mRNAs are a major determinant of translational efficiency (28). Accordingly, we compared the 5′ UTRs of TE UP with TE DOWN genes and those genes with no change in TE, designated here as background (bknd) genes (Supplementary Table S1D). Unexpectedly, we found a significant enrichment of a 12-nt (CGG)4 motif at the 5′ UTR of TE UP genes, whereas the same motif was not enriched in TE DOWN and background gene sets (Fig. 3A). These 12-nt motifs can form either stable hairpin structures (29) or G-quadruplexes (30), which consist of G tetrads that form a stable structure through hydrogen bonding and cation coordination and are predicted to be present in many oncogenes (31). The 5′ UTR of Ep300, a component of WNT pathway, provides an example of a computationally predicted G-quadruplex structure that coincided with the motif (Fig. 3B). Interestingly, WNT signaling components were overrepresented in TE UP genes with the (CGG)4 motif at the 5′ UTR (P = 4.0 × 10−4), suggesting that they may form G-quadruplexes or stable secondary structures (Fig. 3C; Supplementary Table S2B and S2C). There was no (CGG)4 motif enrichment observed for genes involved in transport of amino acids, SCF-KIT pathway or PI3K-AKT-mTOR signaling, which were the top enriched functional pathways in the TE UP gene set (Fig. 2B). These data indicate that the overrepresentation for WNT signaling components among genes with the (CGG)4 motif was not simply due to the abundance of these genes in the TE UP gene set.
Figure 3.
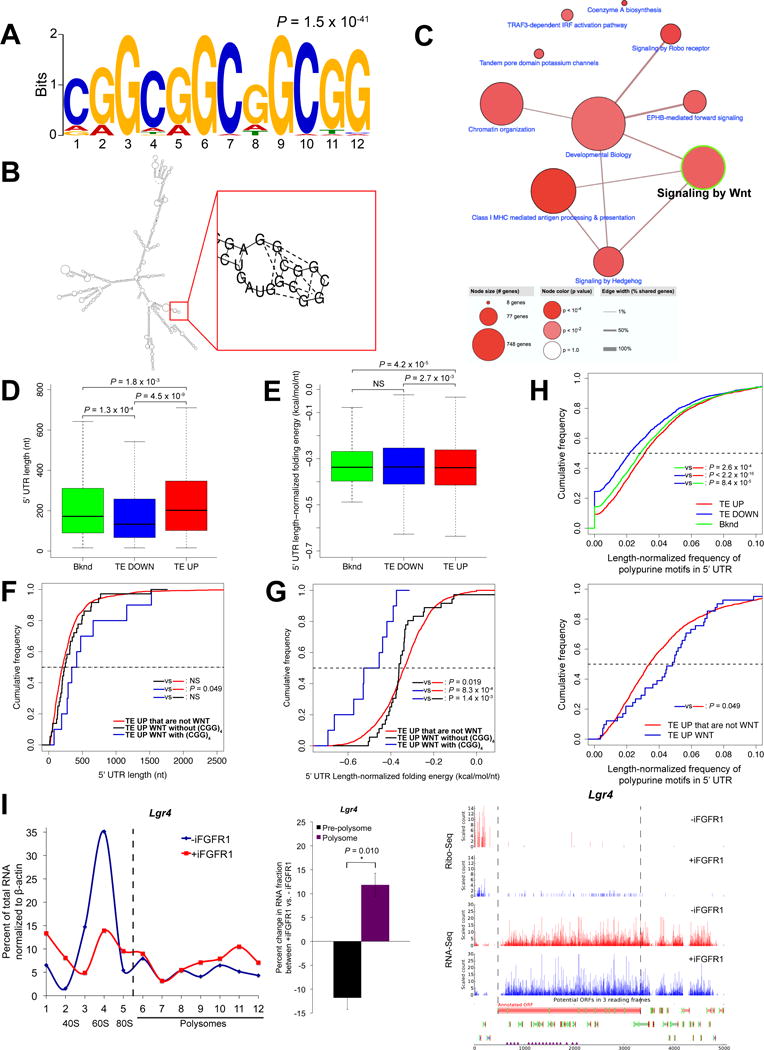
WNT pathway components translationally upregulated by iFGFR1 signaling have structured 5′ UTRs with a high frequency of polypurine sequences and harbor (CGG)4 motifs that can potentially form G-quadruplexes or stable secondary structures. A, The 12-nt (CGG)4 motif enriched in the 5′ UTRs of TE UP genes but not TE DOWN or background genes. B, The 5′ UTR of Ep300 illustrates the (CGG)4 motif and G-quadruplex structure formation using RNAstructure v.6.0 algorithm. C, Pathway annotation networks enriched in TE UP genes that can potentially form G-quadruplexes or stable hairpin structures through the (CGG)4 motif at the 5′ UTR. Note that the “Signaling by Wnt” node highlighted with green border is a major node. D and E, Comparison of 5′ UTR lengths and length-normalized minimum folding free energy, respectively, between TE UP, TE DOWN and background genes. The horizontal bar within each box indicates mean value. All P values shown are less than 0.05; NS, not significant (P > 0.05). F and G, Cumulative distribution of 5′ UTR lengths and length-normalized minimum folding free energy, respectively, in TE UP genes that are WNT signaling components either with (blue) or without (black) the (CGG)4 motif versus other TE UP genes that are not WNT signaling components (red). All P values (Kolmogorov-Smirnov test) shown are less than 0.05; NS, not significant (P > 0.05). H, Cumulative distribution of frequency of polypurine motifs in 5′ UTRs normalized by length in Upper: TE UP, TE DOWN and background genes; Lower: TE UP genes that are WNT signaling components (blue) versus TE UP genes that are not WNT signaling components (red). All P values (Kolmogorov-Smirnov test) shown are less than 0.05. I, Polysome profile (left) shows the proportion of Lgr4 mRNA in each pre-polysome and polysome fraction generated by sucrose gradient fractionation of purified mouse MECs without and with 6-hrs iFGFR1 activation. Relative mRNA levels were measured by qPCR. Shown is a representative of 3 biological replicates. Bar graph (middle) compares changes in proportion of mRNA between fractions. Fractions 1-5 are pre-polysome, and 6-12 are polysome fractions. *P < 0.05; n = 3 biological replicates; error bars indicate standard deviation. Ribo-Seq and RNA-Seq read count profiles (right) of Lgr4. Dashed lines indicate the boundary of the annotated ORF. Read counts were normalized using DESeq size factors.
We also found that the 5′ UTRs of TE UP genes were longer than those of TE DOWN and background genes, among which TE DOWN genes had the shortest 5′ UTRs (Mean UTR length, 286nt in TE UP, 221nt in TE DOWN, and 258nt in background) (Fig. 3D). Moreover, TE UP genes had the smallest length-normalized minimum folding free energy compared with TE DOWN and background genes, whereas TE DOWN and background genes exhibited no difference (Fig. 3E). Since the smaller the minimum folding free energy, the more structured the RNA sequence, these results suggest that TE UP genes are more structured than TE DOWN and background genes independent of length. Interestingly, TE UP genes that are WNT signaling components, particularly those with the (CGG)4 motif that can potentially fold into G-quadruplexes or stable hairpin structures, have longer and more structured 5′ UTRs compared with TE UP genes that are not WNT signaling components as indicated by the right and left shifts in cumulative distribution of 5′ UTR length and minimum folding free energy, respectively (Fig. 3F and 3G). Translationally upregulated WNT signaling components without the (CGG)4 motif also have more structured 5′ UTRs than TE UP genes that are not involved in WNT signaling regulation, suggesting that iFGFR1 activation in vivo preferentially drives the translation of WNT signaling mRNAs with structured 5′ UTRs (Fig. 3G).
Additionally, studies, using luciferase reporters, have demonstrated that 5′ UTRs with tandem repeats of the same (CGG)4 motif identified in TE UP genes are hypersensitive to fluctuation in EIF4A activity (29,30), despite current debates on whether G-quadruplexes or classical secondary structures derived from these motifs confer hypersensitivity to EIF4A activity. Rocaglates, a class of protein synthesis inhibitors that targets EIF4A, can clamp EIF4A onto polypurine sequences to interfere with 43S scanning and subsequent translation (32). We searched for these polypurine sequences and found that TE UP genes indeed have the highest frequency of 6-nt polypurine sequences at the 5′ UTR, previously experimentally validated to confer mRNA hypersensitivity to rocaglates using reporter assays (32) (Fig. 3H). This frequency was more than background genes, whereas TE DOWN genes have the lowest frequency (Fig. 3H). TE UP genes that are WNT signaling components also have a higher frequency of the sequences than the other TE UP genes (Fig. 3H, Supplementary Table S2D), suggesting potential therapeutic benefits of rocaglates in this tumor model of FGF-WNT hyperactivation. Polysome analysis validated the increased polysome association of Lgr4 mRNA, a FGF-upregulated WNT signaling component and modulator of breast cancer metastasis, harboring the (CGG)4 motif with a relatively high frequency of polypurine sequences (Fig. 3I).
Inhibition of the RNA helicase EIF4A compromises FGFR1-mediated increase in translation of WNT pathway components and delays FGFR1-WNT-driven tumorigenesis, potentially through perturbation of EIF4A1 activity
The primary function of EIF4A, an ATP-dependent DEAD-box RNA helicase, is to unwind structured 5′ UTRs for translation initiation (30). Moreover, mRNAs with more complex 5′ UTRs are more dependent on EIF4A’s RNA-helicase activity (33).Therefore, inhibition of EIF4A should selectively diminish translation of structured mRNAs translationally upregulated by iFGFR1 signaling. To target EIF4A, we used silvestrol, a natural compound with promising anticancer properties that crosslinks EIF4A to the mRNA and sequesters EIF4A from EIF4E and EIF4G (34). The high frequency of polypurine sequences in TE UP genes also makes silvestrol an attractive drug. It is a member of the rocaglate family that can clamp EIF4A to polypurine sequences for translation inhibition in addition to inhibition of EIF4A’s RNA-helicase activity (32). Therefore, we asked if silvestrol would impact iFGFR1-induced tumorigenesis in cells with constitutive WNT hyperactivation (Fig. 4A). Mice transplanted with WNT/iFGFR1 mammary epithelial cells into the mammary fat pad were simultaneously treated with AP20187 to induce iFGFR1 hyperactivation for rapid tumorigenesis and 1.5 mg/kg silvestrol. This dose of silvestrol was selected based on previous studies for its known safety in non-tumor-bearing animals (35), and robust anti-tumor activity while preserving innate and adaptive immunity against lymphomas (36). We found that silvestrol significantly delayed iFGFR1-WNT-driven tumorigenesis (Fig. 4B), with minimal toxicity as reflected by the analysis of the mouse’s body weight (Supplementary Fig. S5A). Silvestrol-treated tumors displayed an enlarged stromal compartment with smaller islands of cells (Fig. 4C) and a significantly increased level of apoptosis with a non-significant decrease in proliferation (Fig. 4D; Supplementary Fig. S5B). Silvestrol treatment also reduced translation and protein expression of FGF-upregulated WNT signaling components including Lgr4, Fzd6, Prkca and Ep300 as well as WNT-target oncogenes including Myc and Cyclin D1 (Fig. 4E and 4F; Supplementary Fig. S5C). The inhibition of Myc and Cyclin D1 is consistent with other studies, particularly Myc, whose translation and protein levels, are robustly inhibited by silvestrol and inhibition of EIF4A’s RNA-helicase activity across different cancer types (30,34,37), suggesting that EIF4A function and its association with the EIF4E and EIF4G were compromised by silvestrol. In contrast, house-keeping genes, Gapdh and β-actin, were largely unaffected (Fig. 4F; Supplementary Fig. S5C), suggesting selective inhibition of translation of WNT pathway components through inhibition of EIF4A function.
Figure 4.
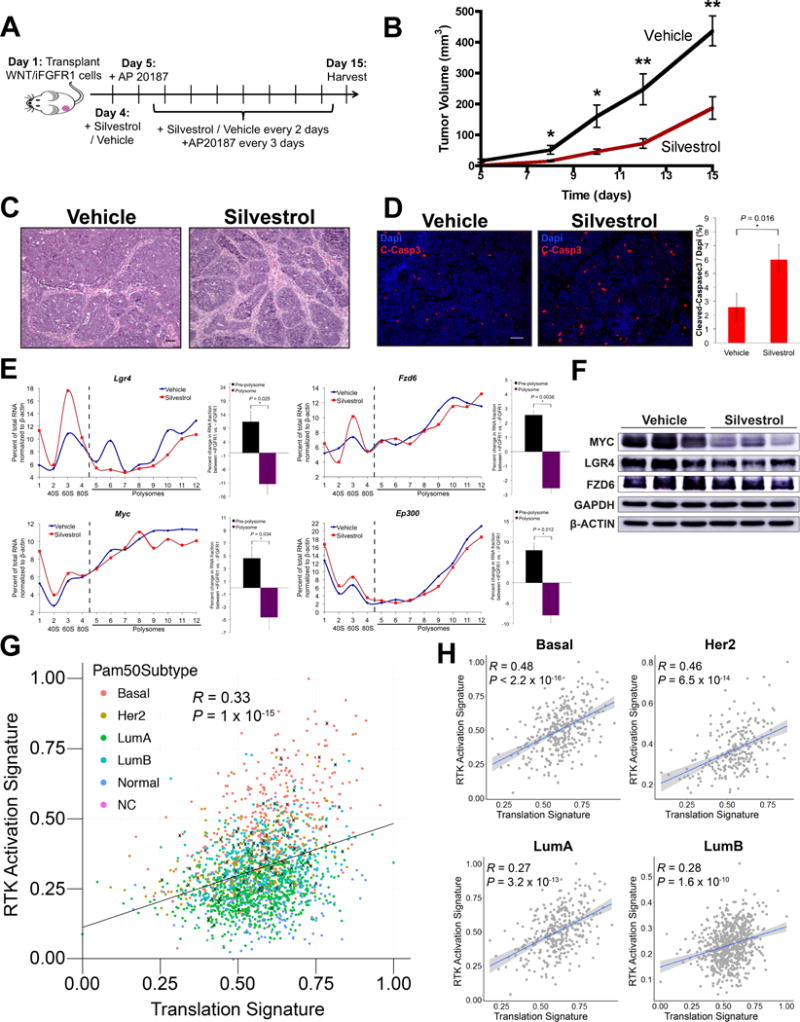
Inhibition of the RNA helicase EIF4A compromises iFGFR1-mediated increase in translation of WNT pathway components and delays iFGFR1-WNT-driven tumorigenesis. A, Treatment scheme with silvestrol against iFGFR1-WNT-driven tumorigenesis. B, Tumor growth curves of mice that were administered vehicle or 1.5 mg/kg silvestrol as described in (A). *P < 0.05; **P < 0.01; error bars indicate standard deviation; n = 9 biological replicates. C, Hematoxylin and eosin staining and D, cleaved-caspase 3 (C-Casp3) immunofluorescence staining of cross-sections from vehicle and silvestrol treated tumors collected at day 15 (scale bar = 50 μm). Bar graph in (D) shows percentage of C-Casp3 positive cells. *P < 0.05; error bars indicate standard deviation; n = 3 biological replicates. E, Polysome profiles show the proportion of mRNAs of WNT signaling components and target genes in each pre-polysome and polysome fraction generated by sucrose gradient fractionation of purified tumor MECs treated with silvestrol or vehicle. Relative mRNA levels were measured by qPCR. Data shown are representative of 3 biological replicates. Bar graph compares changes in proportion of mRNA between fractions. Fractions 1-4 are pre-polysome, and 5-12 are polysome fractions. *P < 0.05; n = 3 biological replicates; error bars indicate standard deviation. F, Western blots of vehicle- and silvestrol-treated tumors in biological triplicates. G, Correlation plot between RTK activation and translation signatures using the METABRIC dataset (n = 1992). Breast cancer subtypes including basal-like (basal), Her2, luminal A (LumA), luminal B (LumB), normal and non-classified (NC) are color-coded. “X” indicates tumors with FGFR1 amplification (copy number > 4). Note that common genes between RTK activation and translation signatures were removed in this analysis, and hence the correlations were not due to overlapping gene identity. H, Correlation plots generated as in (G) for individual breast cancer subtypes.
Although silvestrol targets both EIF4A1 and EIF4A2 isoforms of EIF4A, EIF4A1 protein expression is 8.1-fold higher than EIF4A2 levels in MECs without iFGFR1 activation and is 39-fold higher than EIF4A2 expression following acute iFGFR1 activation (Supplementary Fig. S5D). This difference is due to a 1.8-fold increase in EIF4A1 expression and a 2.5-fold decrease in EIF4A2 protein levels upon iFGFR1 induction (Supplementary Fig. S5E). These results suggest that EIF4A1 expression predominates over EIF4A2 levels and that EIF4A1 is potentially more important than EIF4A2 as a downstream regulator of translational effects induced by FGF signaling in this model. Taken together, iFGFR1 induction of tumorigenesis in cooperation with WNT signaling is vulnerable to inhibition of EIF4A, and WNT pathway components are selectively inhibited. Perturbation of EIF4A1 function is likely to be the main determinant of the observed effects.
FGFR1-WNT-driven tumors share molecular signatures with basal-like and Her2 breast cancers in human
Because the murine iFGFR1-WNT-driven tumors contain both activated FGFR1 receptor tyrosine kinase signaling and an increased activity of the translation machinery, we wondered if there was any correlation between these two pathways in human breast cancer. Analysis of the Metabric dataset of 1992 tumors demonstrated that the RTK activation signature (41) positively correlated with a translation signature, which is comprised of components of the translation machinery (Fig. 4G). Consistently, the majority of tumors with FGFR1 amplification fell above the trend line, suggesting that the RTK activation signature correlates with genomic amplification of FGFR1 (P = 0.0007) (Fig. 4G). Surprisingly, basal-like and Her2 breast cancer subtypes displayed the best correlation, approximately twice that of luminal-A and luminal-B breast cancer subtypes (Fig. 4H). Coincidentally, the iFGFR1-WNT-driven tumors in the Wnt/iR1 mouse model are enriched with molecular signatures of both basal-like and Her2 subtypes (Supplementary Fig. S5F).
Discussion
In the current study, we elucidated a novel mechanism underlying the cooperation between FGF and WNT signaling in breast cancer development and identified cellular functions likely to be dominated by changes in translational regulation or RNA expression when cells dynamically respond to a potent growth signal like iFGFR1 in their physiological microenvironment. This study for the first time correlated RNA expression and ribosome occupancy levels with protein abundance in a dynamic system in the mice, where the cells under prolonged exposure to iFGFR1 activation become pre-malignant. The results of our multi-omics correlation analysis are largely in agreement with other studies on cell lines (15,38). However, our multi-omics analysis unexpectedly found a significant increase and improved correlation between RNA, ribosome occupancy and protein levels in cells dynamically responding to iFGFR1 signaling as compared with those analyzed in a relatively steady state. Previous studies on dynamic responses of cells to various perturbations including oxidative stress and cytokine stimulation in vitro did not report such an improvement in correlation (39,40). These differences may reflect the type of perturbation (e.g. oxidative stress vs. growth signaling), cell types (e.g. yeast vs. MECs) and systems (i.e. in vitro on tissue-culture dishes vs. in vivo in mice). Our observations suggest that in the presence of a potent growth signal, the system of checks and balances, including those involved in the regulation of protein synthesis and degradation required for maintaining RNA and protein homeostasis, may be affected, potentially giving rise to pre-malignant cells as illustrated by our aggressive tumor model driven by FGF-WNT cooperativity.
Although the crosstalk between the FGF and WNT pathways has been reported previously (41,42), underlying molecular mechanisms remain poorly understood. In this study, we for the first time demonstrate that FGF and WNT signaling cooperates at the translational level where FGF signaling selectively enhances translation of both positive and negative regulators of WNT signaling with structured 5′ UTRs that may form either G-quadruplexes or stable secondary structures derived from (CGG)4 motifs, ultimately contributing to driving rapid tumorigenesis. This is not surprising since WNT signaling requires both positive and negative feedback loops to achieve robust control of gene expression amplitude and duration (43), especially in a physiological microenvironment, where the cells constantly interact with different hormonal factors and cell types (e.g. immune cells or fibroblasts) (9). In fact, many negative regulators are direct targets of WNT pathway (e.g. Axin2 and DKK1). Therefore, FGF signaling regulates the network of interlocking positive and negative feedback loops of the WNT pathway that drive subsequent oncogenic events to promote tumorigenesis.
Furthermore, we discovered a unique connection between FGF-WNT cooperativity and the RNA helicase EIF4A, important for translation initiation, particularly mRNAs with structured 5′ UTRs. We demonstrated that FGF-mediated increase in translation of WNT pathway components and the subsequent tumorigenesis process are vulnerable to inhibition of EIF4A function by silvestrol. In vivo iFGFR1 activation in this WNT/iR1 model has previously been shown to trigger hyperactivation of the PI3K-AKT-mTOR axis, characteristic of FGF signaling. This includes many of its downstream regulators of translation, particularly increased phosphorylation of S6K (8,9), which can in turn phosphorylate PDCD4 resulting in its degradation (44). PDCD4 is thought to inhibit EIF4A activity and subsequent translation by competing with EIF4G and with RNA for binding to EIF4A (45). Therefore, FGF signaling potentially enhances EIF4A activity through activation of the PI3K-AKT-mTOR axis, which subsequently diminishes the inhibitory effect of PDCD4 on EIF4A activity. Furthermore, our data suggest that FGF signaling relies more on EIF4A1 activity rather than EIF4A2 activity for enhancing translation initiation of transcripts with structured 5′ UTRs and that the observed effects following silvestrol treatment are more likely due to inhibition of EIF4A1 function. High EIF4A1 expression is associated with a poor outcome in ER-negative breast cancers, which is consistent with this aggressive tumor model that is enriched for signatures of basal-like and Her2 breast cancer subtypes but not ER-positive subtypes like luminal A and luminal B (46). EIF4A1 depletion also resulted in decreased translation of genes harboring (GGC)-repeat motifs (46), similar to the (CGG)4 motif identified in FGF-regulated genes, supporting the conclusion that FGF signaling potentially acts predominantly through EIF4A1 to selectively enhance translation of mRNAs with structured 5′ UTRs. Nevertheless, EIF4A2 may predominate in other cancer types, e.g. it is highly expressed in T-cell acute lymphoblastic leukemia as compared with EIF4A1 (30).
The (CGG)4 motif identified in FGF-regulate genes is unlikely to be the only determinant of hypersensitivity to changes in EIF4A activity as only 7.8% of TE UP genes harbor the motif. Other mRNAs without the (CGG)4 motif may have other motifs that are potentially capable of forming other stable tertiary or secondary structures that are dependent on eIF4A activity for translation initiation, although they may not be as prevalent as the (CGG)4 motif. In fact, a recent study argued that stable hairpin structures derived from the (CGG)4 motif rather than G-quadruplexes confer hypersensitivity to fluctuations in EIF4A activity (29). The fact that 5′ UTRs of TE UP genes are highly structured independent of length (Fig. 3E) supports this notion. Moreover, the data support the previously proposed model that polypurine sequences confer sensitivity to rocaglates (32), as shown by the higher frequency of these sequences at the 5′ UTR of FGF-upregulated genes, which are subsequently inhibited by silvestrol. It is important to emphasize that these short sequences are present in nearly every gene. However, the higher frequency of the sequences can distinguish FGF-upregulated genes from FGF-downregulated genes, suggesting that the frequency of these motifs may potentially be linked to EIF4A function, which is enhanced by FGF signaling.
The delay in iFGFR1-WNT-driven tumor progression induced by silvestrol is not likely due only to the inhibition of translation of WNT pathway components alone, but most likely involves other tumor-associated genes which contain structured 5′ UTRs. Interestingly, a genome-wide study on human medulloblastoma reported that recurrent mutations of DDX3, another member of the DEAD-box RNA helicase family essential for unwinding long and complex 5′ UTRs, synergize with mutated β-catenin to regulate WNT signaling (47). Acute activation of iFGFR1 may potentially function in a similar manner to the recurrent mutations detected in medulloblastoma to dysregulate WNT signaling and DDX3’s helicase activity. Cencic et al. (2009) previously demonstrated sensitivity of the human basal-like breast cancer cell line MDA-MB-231 to silvestrol (34). MDA-MB-231 cells also have active WNT signaling (48) and elevated expression of FGFR1 (49), consistent with the responsiveness of the iFGFR1-WNT-driven tumor model, a basal-like and HER2 mixed tumor type, to silvestrol. Hyperactivation of WNT signaling and FGF signaling may be useful molecular biomarkers to determine the sensitivity to a new generation of inhibitors that are under development to target the RNA helicase EIF4A in breast cancer.
In conclusion, the current study provides new insights regarding the translational cooperation exhibited between FGF and WNT signaling in vivo, which we demonstrated could be perturbed by inhibition of the RNA helicase EIF4A. Using a newly-developed assay, we were able to specifically track changes in ribosome occupancy ex vivo for MECs, allowing for the generation of high quality ribosome-protected RNA fragments with 3-nt periodicity. This approach can be readily applied for the interrogation of the translatome in mouse models, and potentially in human breast tumors, especially those maintained as patient derived xenografts. Lastly, the mechanism of translation cooperativity observed between FGF and WNT signaling in the WNT/iR1 bigenic mice likely plays an important role in the regulation of stem cell dynamics in normal tissue homeostasis, particularly in normal mammary gland development.
Supplementary Material
Significance.
The RNA helicase EIF4A may serve as a therapeutic target for breast cancers that require FGF and WNT signaling.
Acknowledgments
We thank Drs. David Lucas and A. Douglas Kinghorn at The Ohio State University (Columbus, OH) for generously providing silvestrol and Dr. Qingyun Liu at The University of Texas Health Science Center at Houston (Houston, TX) for providing the LGR4 antibody. This work was supported by grants from National Cancer Institute (NCI) of the National Institute of Health (NIH) under award number NIH NCI R01 CA16303-42 (to J.M. Rosen); Cancer Prevention and Research Institute of Texas (CPRIT) Research Award RP160283 (to T.M. Nguyen); NIH NCI U24 CA210954 and CPRIT Grant RR160027 (to B. Zhang); American Cancer Society-Athena Water Breast Cancer Research Scholar Grant RSG-15-088-01RMC and NIH NCI CA190467 (to J.R. Neilson); NIH AI50237 (to R.E. Lloyd); NIH NCI R00 CA175290 and CPRIT Grant RR140071 (to Y. Chen); and NIH NCI R01 CA148761 (to C.M. Perou and J.M. Rosen). This work was conducted with the help of the Baylor College of Medicine Lester and Sue Smith Breast Center Pathology Core, the Genomic and RNA Profiling Core and the Mass Spectrometry Proteomics Core with funding from the CPRIT Core Facility Award RP170005 (to D.P. Edwards) and the Dan L. Duncan Comprehensive Cancer Center NIH NCI P30 CA125123 (to C.K. Osborne).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare no conflict of interest.
References
- 1.Lin S-Y, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, et al. β-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A. 2000;97:4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J, et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet. 2007;39:759–769. doi: 10.1038/ng2034. [DOI] [PubMed] [Google Scholar]
- 4.Holdman XB, Welte T, Rajapakshe K, Pond A, Coarfa C, Mo Q, et al. Upregulation of EGFR signaling is correlated with tumor stroma remodeling and tumor recurrence in FGFR1-driven breast cancer. Breast Cancer Res. 2015:1–17. doi: 10.1186/s13058-015-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Christin JR, Wang C, Ge K, Oktay MH, Guo W. Mammary-Stem-Cell-Based Somatic Mouse Models Reveal Breast Cancer Drivers Causing Cell Fate Dysregulation. Cell Rep. 2016;16:3146–3156. doi: 10.1016/j.celrep.2016.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welm BE. Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J Cell Biol. 2002;157:703–714. doi: 10.1083/jcb.200107119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xian W, Schwertfeger KL, Vargo-Gogola T, Rosen JM. Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J Cell Biol. 2005;171:663–673. doi: 10.1083/jcb.200505098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pond AC, Herschkowitz JI, Schwertfeger KL, Welm B, Zhang Y, York B, et al. Fibroblast Growth Factor Receptor Signaling Dramatically Accelerates Tumorigenesis and Enhances Oncoprotein Translation in the Mouse Mammary Tumor Virus-Wnt-1 Mouse Model of Breast Cancer. Cancer Res. 2010;70:4868–4879. doi: 10.1158/0008-5472.CAN-09-4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632–644. doi: 10.1038/ncb3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roarty K, Shore AN, Creighton CJ, Rosen JM. Ror2 regulates branching, differentiation, and actin-cytoskeletal dynamics within the mammary epithelium. J Cell Biol. 2015;208:351–66. doi: 10.1083/jcb.201408058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao Z, Zou Q, Liu Y, Yang X. Genome-wide assessment of differential translations with ribosome profiling data. Nat Commun. 2016;7:11194. doi: 10.1038/ncomms11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung SY, Choi JM, Rousseaux MWC, Malovannaya A, Kim JJ, Kutzera J, et al. An Anatomically Resolved Mouse Brain Proteome Reveals Parkinson Disease-relevant Pathways. Mol Cell Proteomics. 2017;16:581–93. doi: 10.1074/mcp.M116.061440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. 2012;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 16.Zhang P, He D, Xu Y, Hou J, Pan B-F, Wang Y, et al. Genome-wide identification and differential analysis of translational initiation. Nat Commun. 2017;8:1749. doi: 10.1038/s41467-017-01981-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereboom TC, Bondt A, Pallaki P, Klasson TD, Goos YJ, Essers PB, et al. Translation of branched-chain aminotransferase-1 transcripts is impaired in cells haploinsufficient for ribosomal protein genes. Exp Hematol. 2014;42:394–403.e4. doi: 10.1016/j.exphem.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 18.Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci U S A. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohrer LR, Chuntova P, Bade LK, Beadnell TC, Leon RP, Brady NJ, et al. Activation of the FGFR-STAT3 pathway in breast cancer cells induces a hyaluronan-rich microenvironment that licenses tumor formation. Cancer Res. 2014;74:374–86. doi: 10.1158/0008-5472.CAN-13-2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed JR, Stone MD, Beadnell TC, Ryu Y, Griffin TJ, Schwertfeger KL. Fibroblast Growth Factor Receptor 1 Activation in Mammary Tumor Cells Promotes Macrophage Recruitment in a CX3CL1-Dependent Manner. PLoS ONE. 2012;7:e45877. doi: 10.1371/journal.pone.0045877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module Map of Stem Cell Genes Guides Creation of Epithelial Cancer Stem Cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawakami Y, Capdevila J, Büscher D, Itoh T, Rodríguez Esteban C, Izpisúa Belmonte JC. WNT signals control FGF-dependent limb initiation and AER induction in the chick embryo. Cell. 2001;104:891–900. doi: 10.1016/s0092-8674(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M, Atkinson RL, Rosen JM. Selective targeting of radiation-resistant tumor-initiating cells. Proc Natl Acad Sci U S A. 2010;107:3522–3527. doi: 10.1073/pnas.0910179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell. 2013;24:347–64. doi: 10.1016/j.ccr.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waldron JA, Raza F, Le Quesne J. eIF4A alleviates the translational repression mediated by classical secondary structures more than by G-quadruplexes. Nucleic Acids Res. 2018;46:3075–87. doi: 10.1093/nar/gky108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2015;513:65–70. doi: 10.1038/nature13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwasaki S, Floor SN, Ingolia NT. Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature. 2016;534:558–61. doi: 10.1038/nature17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, et al. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA. 2001;7:382–94. doi: 10.1017/s135583820100108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cencic R, Carrier M, Galicia-Vázquez G, Bordeleau M-E, Sukarieh R, Bourdeau A, et al. Antitumor Activity and Mechanism of Action of the Cyclopenta[b]benzofuran, Silvestrol. PLoS ONE. 2009;4:e5223. doi: 10.1371/journal.pone.0005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alinari L, Prince CJ, Edwards RB, Towns WH, Mani R, Lehman A, et al. Dual targeting of the cyclin/Rb/E2F and mitochondrial pathways in mantle cell lymphoma with the translation inhibitor silvestrol. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:4600–11. doi: 10.1158/1078-0432.CCR-12-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patton JT, Lustberg ME, Lozanski G, Garman SL, Towns WH, Drohan CM, et al. The translation inhibitor silvestrol exhibits direct anti-tumor activity while preserving innate and adaptive immunity against EBV-driven lymphoproliferative disease. Oncotarget. 2015;6:2693–708. doi: 10.18632/oncotarget.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiegering A, Uthe FW, Jamieson T, Ruoss Y, Huttenrauch M, Kuspert M, et al. Targeting Translation Initiation Bypasses Signaling Crosstalk Mechanisms That Maintain High MYC Levels in Colorectal Cancer. Cancer Discov. 2015;5:768–781. doi: 10.1158/2159-8290.CD-14-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK, et al. Genomic variation. Impact of regulatory variation from RNA to protein. Science. 2015;347:664–667. doi: 10.1126/science.1260793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R, et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015;347:1259038. doi: 10.1126/science.1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogel C, Silva GM, Marcotte EM. Protein expression regulation under oxidative stress. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.009217. M111.009217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mansukhani A. Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J Cell Biol. 2005;168:1065–1076. doi: 10.1083/jcb.200409182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aman A, Piotrowski T. Wnt/β-Catenin and Fgf Signaling Control Collective Cell Migration by Restricting Chemokine Receptor Expression. Dev Cell. 2008;15:749–761. doi: 10.1016/j.devcel.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Ji N, Middelkoop TC, Mentink RA, Betist MC, Tonegawa S, Mooijman D, et al. Feedback control of gene expression variability in the Caenorhabditis elegans Wnt pathway. Cell. 2013;155:869–80. doi: 10.1016/j.cell.2013.09.060. [DOI] [PubMed] [Google Scholar]
- 44.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–71. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki C, Garces RG, Edmonds KA, Hiller S, Hyberts SG, Marintchev A, et al. PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc Natl Acad Sci U S A. 2008;105:3274–9. doi: 10.1073/pnas.0712235105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Modelska A, Turro E, Russell R, Beaton J, Sbarrato T, Spriggs K, et al. The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape. Cell Death Dis. 2015;6:e1603. doi: 10.1038/cddis.2014.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pugh TJ, Weeraratne SD, Archer TC, Krummel DAP, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2013;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamb R, Ablett MP, Spence K, Landberg G, Sims AH, Clarke RB. Wnt Pathway Activity in Breast Cancer Sub-Types and Stem-Like Cells. PLoS ONE. 2013;8:e67811. doi: 10.1371/journal.pone.0067811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng CL, Thike AA, Tan SYJ, Chua PJ, Bay BH, Tan PH. Expression of FGFR1 is an independent prognostic factor in triple-negative breast cancer. Breast Cancer Res Treat. 2015;151:99–111. doi: 10.1007/s10549-015-3371-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.