

To the Editor
Autoimmune cytopenia (AIC) can occur after viral infection as a co-morbid condition with primary immunodeficiencies (PID). Patients with PID may develop severe complications with acute viral infections and AIC.(1,2) Early detection of an underlying PID may promote definitive treatment, such as hematopoietic stem cell transplantation (HSCT).
Patients with partial defects in severe combined immunodeficiency (SCID) associated genes, such as recombination activating gene (RAG), are increasingly reported with autoimmunity, such as (AIC) beyond infections.(3,4) The RAG1/2 proteins are essential components of the V(D)J recombination process that diversifies the repertoires of the T and B cell receptors. RAG1/2 deficiency has a broad phenotypic spectrum including combined immunodeficiency with granulomatous disease and/or autoimmunity (CID-G/AI) where milder impairment of RAG1/2 recombinase activity results in relatively preserved T and B lymphocyte counts and immunoglobulin levels however naïve T cell counts progressively decrease.(5) Therefore, measurement of T cell receptor excision circles (TREC), reflecting naïve T cell count, may identify these patients at birth in asymptomatic stage (via newborn screening) or later in life in the midst of infectious or autoimmune complications. However, in countries, where NBS for SCID is not available, serological testing with a panel of anti-cytokine autoantibodies targeting IFNα, IFNω and IL-12 may serve as complementary tool to raise suspicion for an underlying RAG deficiency with CID-G/AI phenotype in a child with history of refractory AIC with severe viral infections.(3)
Hereby we report a 26-month-old female with partial RAG deficiency, non-vaccine strain varicella infection and severe refractory AIC and discuss our diagnostic approach in Hungary where newborn screening for SCID is not available.
A previously healthy 26-month-old Caucasian female was referred to our Immunology group for immune evaluation of prolonged varicella infection. The patient had extensive vesicular rash on the entire body and continued to develop new vesicular eruptions up to two weeks after onset of symptoms. No other organ involvements were noted. Two months after the onset of varicella, she developed immune thrombocytopenia (platelet count 2×109/L), recalcitrant to high-dose intravenous immunoglobulin and pulse glucocorticosteroid treatment. The family history was negative for consanguinity, PID and autoimmunity.
Immune phenotyping was notable for decreased total and naive T cell count, but preserved B and NK cell compartments. Polyclonal gammopathy, low IgA and normal IgM were also noted. (Table 1) Lymphocyte proliferation assay revealed low response to phytohemagglutinin (SI 3), concanavalin A (SI 8), and a low normal response to pokeweed mitogen (SI 11) (SI normal value >10.) Infection with Human Immunodeficiency Virus, Epstein-Barr Virus and Cytomegalovirus were ruled out by serologic assay. The patient generated appropriate varicella zoster antibody response with seroconversion on day 8. (Testing with Virotech ELISA anti-varicella zoster virus IgM positive OD/CO 3.396, IgG positive: OD/CO 1.384, IgA negative). Retrospective testing of T cell receptor excision circles (TREC) from newborn screening blood spot specimen was undetectable (<252 copies/μL) in collaboration with the New England Newborn Screening Program.
Table 1.
Lymphocyte subsets and immunoglobulin levels at first assessment of the child
| Results | Normal value | |
|---|---|---|
| CD3 | 152/μL | 900–4500/μ |
| CD4+ | 92.8/μL | 500–2400/μl |
| CD8+ | 32.5/μL | 300–1600/μl |
| CD4+CD45RA+ | 19% | >60% |
| CD19+ | 213.8/μL | 200–2100/μl |
| CD16+ CD 56+ | 444.7/μL | 100–1000/μl |
| IgG | 1997 mg/dl | 453–916 mg/dl |
| IgA | 0.01 mg/dl | 20–100 mg/dl |
| IgM | 128 mg/dl | 19–146 mg/dl |
Polyclonal gammopathy prompted screening for autoantibodies. There was a borderline positive anti-nuclear antibody titer at 1:40. Anti-platelet antibodies were not tested by clinical laboratory testing as this laboratory assay is not available for routine testing for ITP in Hungary. Antibodies against double stranded deoxyribonucleic acid, anti-smooth-muscle antibody, anti-mitochondrial antibody, anti-neutrophil cytoplasmic antibody, and antibodies against cytoplasmic antigens, centromere antigens, and parietal cells were all negative by clinical laboratory testing. Autoantibodies targeting cytokines IFN-α, IFN-ω and IL-12 were detected by enzyme linked immunosorbent assay (Figure 1). The patient’s history of prolonged varicella infection with AIC, reduced naïve T cell compartment and impaired T cell proliferation was concerning for a severe form of PID and prompted preparation for emergent allogeneic HSCT. The combination of clinical and laboratory features with naïve T cell lymphopenia and the presence of autoantibodies targeting IFN-α, IFN-ω and IL-12 raised the possibility of RAG deficiency. (3)
Figure 1.
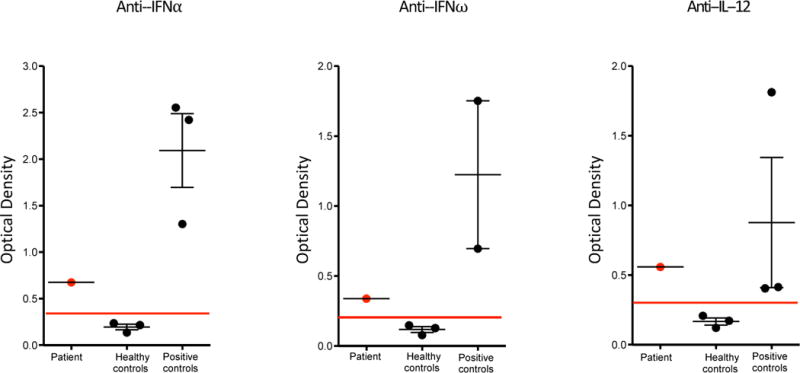
Anti-cytokine autoantibodies targeting IFN-α, IFN-ω and IL-12 were detected by enzyme linked immunosorbent assay. Our patient had higher levels of anti-cytokine autoantibodies than the normal healthy controls. Positive controls are patients (ages of 30, 18 and 6 years) with hypomorphic RAG deficiency and persistence of anticytokine antibodies, as previously described (3)
A search for an underlying genetic mutation was initiated concomitantly with preparation for hematopoietic stem cell transplantation (HSCT). Genomic DNA separated from peripheral blood samples from the patient and parents was used for library preparation and sequenced with Trusight One clinical exome kit (Illumina) on Illumina MiSeq platform for next generation sequencing (NGS). This clinical exome kit covers the coding region of 4813 clinically relevant, disease-associated genes.
Three missense variants in the RAG1 gene were identified. We excluded NM_000448.2:c.746A>G (rs3740955) as a possible causative mutation as it has high minor allele frequency (MAF=44.70) in the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org). Two rare missense variants in exon 2 (NM_000448.2:c.1331C>T, rs199474685 with MAF of =0.0008 and NM_000448.2:c.2974A>G, rs539590514 with MAF of =0.000007) were identified by NGS and confirmed by targeted Sanger sequencing. Patient was confirmed with a compound heterozygous RAG1 mutation by inheriting RAG1 c.2974A>G p.Lys992Glu from the mother and RAG1 c.1331C>T p.Ala444Val from the father. Residual Rag1 recombination activity was 9.2% (p.Lys992Glu) and 1.4% (p.Ala444Val), as measured by an in vitro recombinase assay.(9) No mutations were identified in the RAG2 gene. The mutation p.Lys992Glu has previously been reported in a homozygous and compound heterozygous form in Omenn syndrome (OS) of 5–6 months of age.(6) The mutation p.Ala444Val has been reported in both a homozygous and compound heterozygous form in young infants with the leaky SCID, OS and other SCID phenotypes of 0–4 months of age.(6, 7). All patients had B cell lymphopenia and no history of autoimmunity. This is in contrast with our previously healthy patient presenting at 26 months of age with varicella infection followed by severe AIC and laboratory phenotype of preserved B cell compartment and polyclonal gammopathy. We postulate that our patient had polyclonal B cells activation and AIC secondary to viral infection. The low naïve T cell compartment, undetectable TRECs and poor T cell proliferation are consistent with definition of LS, however preserved B cell lymphocyte count, polyclonal gammopathy and autoimmune complications suggest a shift in phenotype to CID-G/AI. Our patient underwent HSCT with a 10/10 matched unrelated donor. Thrombocytes engrafted day 43 and CD3+/CD4+ reached the normal range on day 129 post-transplant. Patient is currently 13 months after transplantation with no evidence of cytopenia.
Regarding the clinical history, our case had some similarities with three additional cases that we previously published. (3) The comparison of cases is listed in Supplementary Table 1 for Online Repository. There is close timing of 2–4 months between the onset of viral infection and AIC with emergence of anti-cytokine antibodies. These antibodies were absent in young infants with hypomorphic RAG and absence of infection. (3, 8)
Evaluation of a child with autoimmune cytopenia for CID, such as our case, can be difficult if lymphocyte immunophenotyping is atypical and immunoglobulin levels normal or elevated. The low number of naïve T cells is key finding and can be expedited by using the TREC assay. However, in countries where TREC screening is not available, such as Hungary, utilizing autoantibody profiling with anti-cytokine antibody testing may help to identify those with an underlying combined immunodeficiency, such as partial RAG deficiency; and expedite early recognition and confirmation by genetic testing that is essential for timely preparation for stem cell transplantation and favorable outcomes.
Supplementary Material
Clinical Implications.
Combined immunodeficiencies may initially present with herpes virus infections, autoimmune cytopenia and antibodies to cytokines. Beyond the assessment of naïve T cells, screen for anti-cytokine antibodies is useful to identify an underlying combined immunodeficiency ultimately confirmed by genetic testing.
Acknowledgments
This work was partly supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant no. 5K08AI103035 to J.E.W.), the Jeffrey Modell Foundation (to J.E.W.). “Research and publication grant” support from CSL Behring Canada, Inc. #54054.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors have no conflict of interest to disclose.
Conflict of Interest: Jolan Walter serves on speaker Bureau for CSL-Behring and Shire.
References
- 1.Ramos-Casals M, Garcia-Carrasco M, Lopez-Medrano F, Trejo O, Forns X, Lopez-Guillermo A, et al. Severe autoimmune cytopenias in treatment-naive hepatitis C virus infection: clinical description of 35 cases. Medicine. 2003;82(2):87–96. doi: 10.1097/00005792-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Seidel MG. Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood. 2014;124(15):2337–44. doi: 10.1182/blood-2014-06-583260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. 2015;125(11):4135–48. doi: 10.1172/JCI80477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016;16(4):234–46. doi: 10.1038/nri.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee YN, Frugoni F, Dobbs K, Tirosh I, Du L, Ververs FA, et al. Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Science Immunology. 2016;1(6):1–12. doi: 10.1126/sciimmunol.aah6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133(4):1099–108. doi: 10.1016/j.jaci.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93(5):885–96. doi: 10.1016/s0092-8674(00)81448-8. [DOI] [PubMed] [Google Scholar]
- 8.Henderson LA, Frugoni F, Hopkins G, de Boer H, Pai SY, Lee YN, et al. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity. J Allergy Clin Immunol. 2013;132(4):969–71 e1. 2. doi: 10.1016/j.jaci.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen K, Wu W, Mathew D, Zhang Y, Browne SK, Rosen LB, et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J Allergy Clin Immunol. 2014;133(3):880–882.e10. doi: 10.1016/j.jaci.2013.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.