

The title compounds were synthesized using the Claisen–Schmidt condensation method and characterized by UV–Vis spectroscopy. The geometrical parameters optimized using density functional theory (DFT) calculations show a good correlation with the experimental results. The small HOMO–LUMO energy gaps of 3.11 and 3.07 eV enhances the non-linear responses of these molecular systems.
Keywords: chalcone, anthracene, crystal structure, DFT, UV–Vis
Abstract
The title compounds, C24H18O2 and C24H17FO2, were synthesized using the Claisen–Schmidt condensation method and characterized by UV–Vis spectroscopy. Weak intermolecular C—H⋯O, C—H⋯π and π–π hydrogen-bonding interactions help to stabilize the crystal structures of both compounds. The geometrical parameters obtained from the molecular structure were optimized using density functional theory (DFT) calculations at the B3LYP/6–311++G(d,p) level, showing a good correlation with the experimental results. The small HOMO–LUMO energy gaps of 3.11 and 3.07 eV enhances the non-linear responses of these molecular systems.
Chemical context
Conjugated organic systems contain delocalized π electrons, which often show excellent NLO properties as they can easily be polarized. There are three features essential for high non-linear activity in an organic compound which are: a strong electron donor, a highly polarizable π-conjugated bridged moiety and a strong π-electron acceptor. Chalcones generally satisfy these criteria given their π-conjugated bridged structures that can be functionalized with a wide range of substitutions. Recently, we found that the presence of an anthracene fused-ring system positioned at the terminal ring of these derivative compounds is useful in getting good quality single crystals with an easily synthesizable method. The structure of anthracene is benzene-like, having three six-membered rings fused together in a planar-like arrangement. These polyaromatic hydrocarbons containing π-conjugated materials show unique properties in terms of conductivity that have led to significant advancements in the field of organic electronics (Li et al., 2016 ▸). In this work, we report the synthesis and combined experimental and theoretical studies of two new anthracene chalcones C24H18O2 (I) and C24H17FO2 (II), containing methoxyphenyl (I) and fluoromethoxyphenyl (II) groups as substituents. Additionally, the UV–Vis absorption and HOMO–LUMO analysis are also reported herein.
Structural commentary
The new chalcones C24H18O2 (I) and C24H17FO2 (II) consist of an anthracene fused-ring system and the substituent units 1-methoxy-2-methylbenzene (A) and 2-fluoro-1-methoxy-4-methylbenzene (B), respectively. These compounds represent D–A π intermolecular charge-transfer systems. Displacement ellipsoid plots and DFT optimized structures of the title compounds with their atom-labeling schemes are shown in Fig. 1 ▸. Compounds (I) and (II) crystallize in the monoclinic P21
/c and triclinic P
 space groups, respectively. Selected B3LYP/6-311++G(d,p) geometry-optimized calculated values (Frisch et al., 2009 ▸) for the bond lengths and angles of both compounds based on geometries in the gaseous state are compared to those of the crystalline structures in the solid state in Table S1 in the supporting information. The theoretical bond lengths and bond angles correlate well with the experimental data and are in normal ranges.
space groups, respectively. Selected B3LYP/6-311++G(d,p) geometry-optimized calculated values (Frisch et al., 2009 ▸) for the bond lengths and angles of both compounds based on geometries in the gaseous state are compared to those of the crystalline structures in the solid state in Table S1 in the supporting information. The theoretical bond lengths and bond angles correlate well with the experimental data and are in normal ranges.
Figure 1.

(a) The molecular structure for compounds (I) and (II) showing the atom-numbering schemes and 50% probability ellipsoids; (b) The DFT-optimized structures at the B3LYP 6–311++G(d,p) level for compounds (I) and (II).
Both molecular structures adopt an s-trans configuration with respect to the C16=C17 double bond across the ethylenic bridge (O1/C15–C17). The anthracene unit in both (I) and (II) is found to be twisted at the C14—C15 bond with the C1—C14—C15—C16 torsion angles being −95.91 (18)° in (I) and −106.3 (2)° in (II). This is probably due to the bulkiness of the strong electron donor. The corresponding DFT-calculated results give values of −95.94° (I) and −91.27°(II), respectively. The experimental and theoretical torsion-angle difference of 15.0° observed in (II) is most likely due to the formation of a weak intermolecular C12—H12 O2 interaction involving the anthracene fused-ring system with the terminal methoxy substituent unit.
The mean plane of the enone moiety in (I) [O1/C15–C17, maximum deviation of 0.0085 (18) Å at C16] forms dihedral angles of 88.15 (18) and 1.44 (19)° with the mean plane of the anthracene ring system (C1–C14) and the 1-methoxy-2-methylbenzene (A) ring, respectively. The DFT geometry-optimization calculations give the same values as the experimental values. In (II) the mean plane of the enone moiety [O1/C15–C17, maximum deviation of 0.0092 (18) Å at C16] forms dihedral angles of 73.65 (18) and 2.40 (19)° with the mean planes of the anthracene ring system (C1–C14) and the 2-fluoro-1-methoxy-4-methylbenzene ring (B). The corresponding DFT geometry-optimization calculation gives values of 89.99 and 0.01°, respectively. Additionally, the mean plane of the anthracene ring system (C1–C14) in the two compounds form dihedral angles of 87.52 (8)° (experimental and DFT) and 71.31 (7)° (experimental) and 90.00° (DFT) with the mean planes of A and B, respectively.
Supramolecular features
The crystal packing of both compounds is shown in Fig. 2 ▸ and details of the weak intermolecular hydrogen-bonding interactions are given in Table 1 ▸. No classical hydrogen bonds are observed in either structure. The crystal packing of (I) shows only weak π–π interactions (Table 2 ▸) with centroid–centroid distances of 3.8804 (12) and 3.6725 (13) Å. The molecules are further linked into infinite zigzag chains along the c-axis direction.
Figure 2.

(a) Crystal packing for compound (I) viewed along the a axis showing weak π–π interactions (dashed lines), where Cg1 and Cg2 are the centroids of the C1–C6 and C18–C22 rings, respectively, and (b) Weak C—H⋯ O, C—H⋯π and π–π interactions (dashed lines) for compound (II),, forming 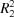 (14) ring graph-set motifs, where Cg3 and Cg4 are the centroids of the C8–C13 and C1–C6 rings, respectively.
(14) ring graph-set motifs, where Cg3 and Cg4 are the centroids of the C8–C13 and C1–C6 rings, respectively.
Table 1. Hydrogen-bond geometry (Å, °) for (II) .
Cg4 is the centroid of the C1–C6 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C12—H12A⋯O2i | 0.93 | 2.48 | 3.345 (2) | 154 |
| C19—H19A⋯O1ii | 0.93 | 2.48 | 3.393 (3) | 166 |
| C24—H24D⋯Cg4iii | 0.96 | 2.77 | 3.391 (3) | 123 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Table 2. Weak π–π interactions in compounds (I) and (II).
Cg1 and Cg2 are the centroids of the C1–C6 and C18–C23 rings, respectively, in compound (I). Cg3 and Cg4 are the centroids of the C8–C13 and C1–C6 rings, respectively, in compound (II).
| I | J | I⋯J | Symmetry |
|---|---|---|---|
| Cg1 | Cg1 | 3.8804 (12) | 1 − x, 2 − y, 2 − z |
| Cg2 | Cg2 | 3.6725 (13) | 1 − x, 2 − y, 1 − z |
| Cg3 | Cg3 | 3.7891 (12) | 1 − x, 1 − y, 2 − z |
| Cg3 | Cg4 | 3.8126 (11) | 1 − x, −y, 2 − z |
In (II), weak C19—H19A⋯O1ii and C12—H12A⋯O2i hydrogen bonds (Table 1 ▸) connect the molecules into centrosymmetric dimers with (14) ring motifs. These dimers are further linked into infinite sheets stacked along the a-axis direction. Weak C24—H24⋯Cg4iii (Table 1 ▸) and π–π interactions [centroid–centroid distances = 3.8126 (11) and 3.789 (12) Å; Table 2 ▸] are also observed in the crystal packing and further stabilize the crystal structure. These weak intermolecular C—H⋯O, C—H⋯π and π–π interactions are significant in bridging the molecules into a three-dimensional supramolecular network.
UV–Vis absorption analysis
Experimental electronic absorption spectra of (I) and (II) have been measured and compared to the ground state (HOMO) and excited state (LUMO) molecular orbital energies, calculated using time-dependent DFT B3LYP/6-311++G(d,p) theoretical calculations in the gas phase. The experimental absorption peaks (Fig. 3 ▸) of (I) and (II) are found at the same maximum wavelength of 387 nm, whereas the simulated values are observed at 386 nm and 394 nm, respectively. The shift of the theoretical values to higher wavelengths are due to the fact that the calculations are confined to a gaseous environment, whereas the observations are obtained from the solution state (Zainuri et al., 2017 ▸).
Figure 3.

UV–Vis absorption spectra of compounds (I) and (II).
The HOMO and LUMO energies characterize the ability of donating and accepting electrons, whereas the value of the energy gap between the HOMO and LUMO molecular orbitals characterizes the molecular chemical stability. The energy gaps are largely responsible for the chemical and spectroscopic properties of the compounds. In Fig. 4 ▸, the charge densities in the ground state (HOMO) are mainly delocalized over the entire anthrancenyl donor ring, while in the excited state (LUMO), the charge densities are accumulated on the π-conjugated enone bridge and the terminal electron-acceptor group. The HOMO and LUMO energy gaps were computed to be 3.24 eV for (I) and 3.25 eV for (II). Through an extrapolation of the linear trend observed in the optical spectra, the experimental energy band gaps for (I) and (II) become 3.11 eV and 3.07 eV, respectively. These optical band-gap values indicate the suitability of these compounds for opto-electronic applications as for structures of chalcones previously reported by Prabhu et al. (2016 ▸).
Figure 4.

Molecular orbital electron distributions of the HOMO and LUMO energy levels for (I) and (II).
Database survey
A survey of the Cambridge Structural Database (CSD, Version 5.39, last update November 2017; Groom et al., 2016 ▸) revealed several fused-ring substituted chalcones similar to (I) and (II). There are four compounds that have an anthrancene-ketone subtituent on the chalcone, including 9-anthryl styryl ketone and 9,10-anthryl bis(styryl ketone) reported by Harlow et al. (1975 ▸). (2E)-1-(Anthracen-9-yl)-3-[4-(propan-2-yl)phenyl]prop-2-en-1-one was reported by Girisha et al. (2016 ▸), while (E)-1-(anthracen-9-yl)-3-(2-chloro-6-fluorophenyl)prop-2-en-1-one was reported by Abdullah et al. (2016 ▸). Zainuri et al. (2018a ▸) reported a chalcone with two anthrancene substituents, viz. (E)-1,3-bis(anthracen-9-yl)prop-2-en-1-one. Other related compounds include 1-(anthracen-9-yl)-2-methylprop-2-en-1-one (Agrahari et al., 2015 ▸), 9-anthroylacetone (Cicogna et al., 2004 ▸), (E)-1-(anthracen-9-yl)-3-(naphthalen-2-yl)prop-2-en-1-one and (E)-1-(anthracen-9-yl)-3-(pyren-1-yl)prop-2-en-1-one (Zainuri et al., 2018b ▸,c ▸).
Synthesis and crystallization
A mixture of 9-acetylanthracene (0.5 mmol) and 2-methoxybenzaldehyde (0.5 mmol) and 3-fluoro-4-methoxybenzaldehyde (0.5 mmol) for compounds (I) and (II), respectively, was dissolved in methanol (20 ml). A catalytic amount of NaOH (5 ml, 20%) was added to the solutions, dropwise under vigorous stirring. The reaction mixtures were stirred for about 5-6 h at room temperature. After stirring, the contents of the flask were poured into ice-cold water (50 ml). The resultant crude products were filtered, washed successively with distilled water and recrystallized to get the corresponding chalcones (see scheme). Single crystals of (I) and (II) suitable for X-ray diffraction were obtained by the slow evaporation technique using acetone.
Refinement
Crystal data collection and structure refinement details are summarized in Table 3 ▸. All H atoms were positioned geometrically [C—H = 0.93 and 0.96 Å in (I) and (II)] and refined using a riding model with U
iso(H) = 1.2 or 1.5U
eq(C). A rotating group model was applied to the methyl group. In the final refinement of (I), one outlier ( 2 15) was omitted.
2 15) was omitted.
Table 3. Experimental details.
| (I) | (II) | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C24H18O2 | C24H17FO2 |
| M r | 338.38 | 356.37 |
| Crystal system, space group | Monoclinic, P21/c | Triclinic, P
|
| Temperature (K) | 294 | 296 |
| a, b, c (Å) | 9.0554 (8), 17.4260 (15), 12.9217 (9) | 8.6646 (5), 9.5752 (5), 11.5636 (6) |
| α, β, γ (°) | 90, 119.916 (5), 90 | 100.593 (2), 105.443 (2), 92.422 (2) |
| V (Å3) | 1767.3 (3) | 904.76 (9) |
| Z | 4 | 2 |
| Radiation type | Mo Kα | Mo Kα |
| μ (mm−1) | 0.08 | 0.09 |
| Crystal size (mm) | 0.60 × 0.23 × 0.15 | 0.99 × 0.31 × 0.25 |
| Data collection | ||
| Diffractometer | Bruker SMART APEXII DUO CCD area-detector | Bruker SMART APEXII DUO CCD area-detector |
| Absorption correction | Multi-scan (SADABS; Bruker, 2009 ▸) | Multi-scan (SADABS; Bruker, 2009 ▸) |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 30614, 4249, 2916 | 34988, 5394, 3307 |
| R int | 0.045 | 0.045 |
| (sin θ/λ)max (Å−1) | 0.661 | 0.711 |
| Refinement | ||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.054, 0.146, 1.08 | 0.059, 0.183, 1.02 |
| No. of reflections | 4249 | 5394 |
| No. of parameters | 236 | 245 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.17, −0.19 | 0.27, −0.20 |
Supplementary Material
Crystal structure: contains datablock(s) I, II. DOI: 10.1107/S205698901800974X/jj2199sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S205698901800974X/jj2199Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S205698901800974X/jj2199IIsup3.hkl
Supporting information file. DOI: 10.1107/S205698901800974X/jj2199Isup4.cml
Supporting information file. DOI: 10.1107/S205698901800974X/jj2199IIsup5.cml
Comparison of selected experimental and DFT-optimized data for compounds (I) and (II). DOI: 10.1107/S205698901800974X/jj2199sup6.pdf
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
(I). Crystal data
| C24H18O2 | F(000) = 712 |
| Mr = 338.38 | Dx = 1.272 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 9.0554 (8) Å | Cell parameters from 6535 reflections |
| b = 17.4260 (15) Å | θ = 2.6–28.0° |
| c = 12.9217 (9) Å | µ = 0.08 mm−1 |
| β = 119.916 (5)° | T = 294 K |
| V = 1767.3 (3) Å3 | Needle, yellow |
| Z = 4 | 0.60 × 0.23 × 0.15 mm |
(I). Data collection
| Bruker SMART APEXII DUO CCD area-detector diffractometer | 2916 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.045 |
| φ and ω scans | θmax = 28.0°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −11→11 |
| k = −22→23 | |
| 30614 measured reflections | l = −17→17 |
| 4249 independent reflections |
(I). Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.054 | H-atom parameters constrained |
| wR(F2) = 0.146 | w = 1/[σ2(Fo2) + (0.063P)2 + 0.2604P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.08 | (Δ/σ)max = 0.001 |
| 4249 reflections | Δρmax = 0.17 e Å−3 |
| 236 parameters | Δρmin = −0.19 e Å−3 |
(I). Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
(I). Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.53861 (18) | 0.72502 (7) | 0.79211 (12) | 0.0752 (4) | |
| O2 | 0.20209 (16) | 1.03186 (6) | 0.56513 (10) | 0.0642 (3) | |
| C1 | 0.53056 (19) | 0.86680 (8) | 0.94301 (13) | 0.0478 (3) | |
| C2 | 0.6917 (2) | 0.88894 (10) | 0.95792 (15) | 0.0592 (4) | |
| H2A | 0.7215 | 0.8740 | 0.9017 | 0.071* | |
| C3 | 0.8018 (3) | 0.93154 (11) | 1.05315 (18) | 0.0738 (5) | |
| H3A | 0.9052 | 0.9465 | 1.0604 | 0.089* | |
| C4 | 0.7618 (3) | 0.95337 (12) | 1.14117 (18) | 0.0802 (6) | |
| H4A | 0.8400 | 0.9815 | 1.2071 | 0.096* | |
| C5 | 0.6114 (3) | 0.93389 (11) | 1.13057 (16) | 0.0731 (5) | |
| H5A | 0.5870 | 0.9487 | 1.1896 | 0.088* | |
| C6 | 0.4886 (2) | 0.89110 (9) | 1.03106 (13) | 0.0552 (4) | |
| C7 | 0.3300 (2) | 0.87229 (10) | 1.01595 (15) | 0.0615 (4) | |
| H7A | 0.3036 | 0.8876 | 1.0738 | 0.074* | |
| C8 | 0.2100 (2) | 0.83166 (9) | 0.91831 (15) | 0.0538 (4) | |
| C9 | 0.0457 (3) | 0.81257 (11) | 0.90222 (19) | 0.0713 (5) | |
| H9A | 0.0176 | 0.8282 | 0.9591 | 0.086* | |
| C10 | −0.0685 (3) | 0.77249 (12) | 0.8065 (2) | 0.0787 (6) | |
| H10A | −0.1744 | 0.7609 | 0.7977 | 0.094* | |
| C11 | −0.0284 (2) | 0.74811 (11) | 0.7196 (2) | 0.0757 (6) | |
| H11A | −0.1087 | 0.7209 | 0.6535 | 0.091* | |
| C12 | 0.1255 (2) | 0.76379 (9) | 0.73108 (16) | 0.0605 (4) | |
| H12A | 0.1497 | 0.7466 | 0.6730 | 0.073* | |
| C13 | 0.25104 (19) | 0.80611 (8) | 0.83027 (14) | 0.0478 (3) | |
| C14 | 0.41240 (18) | 0.82319 (8) | 0.84557 (13) | 0.0445 (3) | |
| C15 | 0.4659 (2) | 0.78700 (8) | 0.76321 (14) | 0.0500 (4) | |
| C16 | 0.4322 (2) | 0.82415 (9) | 0.65306 (14) | 0.0526 (4) | |
| H16A | 0.4648 | 0.7987 | 0.6044 | 0.063* | |
| C17 | 0.35781 (19) | 0.89218 (8) | 0.61731 (12) | 0.0459 (3) | |
| H17A | 0.3278 | 0.9171 | 0.6678 | 0.055* | |
| C18 | 0.3182 (2) | 0.93191 (9) | 0.50726 (13) | 0.0489 (4) | |
| C19 | 0.3583 (2) | 0.90106 (11) | 0.42487 (15) | 0.0640 (5) | |
| H19A | 0.4128 | 0.8538 | 0.4405 | 0.077* | |
| C20 | 0.3189 (3) | 0.93900 (14) | 0.32089 (17) | 0.0804 (6) | |
| H20A | 0.3470 | 0.9176 | 0.2670 | 0.096* | |
| C21 | 0.2377 (3) | 1.00888 (14) | 0.29715 (17) | 0.0813 (6) | |
| H21A | 0.2101 | 1.0343 | 0.2265 | 0.098* | |
| C22 | 0.1965 (2) | 1.04188 (11) | 0.37628 (16) | 0.0686 (5) | |
| H22A | 0.1424 | 1.0893 | 0.3594 | 0.082* | |
| C23 | 0.2366 (2) | 1.00373 (9) | 0.48149 (13) | 0.0527 (4) | |
| C24 | 0.1245 (3) | 1.10560 (10) | 0.54615 (19) | 0.0769 (6) | |
| H24A | 0.1028 | 1.1170 | 0.6100 | 0.115* | |
| H24B | 0.0190 | 1.1057 | 0.4717 | 0.115* | |
| H24C | 0.1994 | 1.1437 | 0.5440 | 0.115* |
(I). Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0919 (9) | 0.0621 (7) | 0.0778 (9) | 0.0293 (7) | 0.0470 (8) | 0.0145 (6) |
| O2 | 0.0823 (8) | 0.0544 (6) | 0.0513 (7) | 0.0117 (6) | 0.0300 (6) | 0.0043 (5) |
| C1 | 0.0516 (8) | 0.0490 (8) | 0.0389 (8) | 0.0037 (6) | 0.0197 (7) | 0.0081 (6) |
| C2 | 0.0552 (10) | 0.0622 (10) | 0.0545 (10) | −0.0019 (8) | 0.0231 (8) | 0.0058 (8) |
| C3 | 0.0619 (11) | 0.0723 (12) | 0.0692 (13) | −0.0113 (9) | 0.0191 (10) | 0.0034 (10) |
| C4 | 0.0840 (15) | 0.0739 (12) | 0.0522 (12) | −0.0120 (11) | 0.0110 (10) | −0.0062 (9) |
| C5 | 0.0892 (15) | 0.0761 (12) | 0.0414 (10) | −0.0018 (10) | 0.0231 (10) | −0.0041 (9) |
| C6 | 0.0686 (11) | 0.0583 (9) | 0.0341 (8) | 0.0049 (8) | 0.0221 (8) | 0.0073 (7) |
| C7 | 0.0750 (12) | 0.0749 (11) | 0.0449 (9) | 0.0106 (9) | 0.0377 (9) | 0.0105 (8) |
| C8 | 0.0590 (10) | 0.0583 (9) | 0.0513 (10) | 0.0092 (7) | 0.0328 (8) | 0.0177 (7) |
| C9 | 0.0678 (12) | 0.0830 (12) | 0.0783 (13) | 0.0143 (10) | 0.0479 (11) | 0.0273 (11) |
| C10 | 0.0582 (11) | 0.0746 (12) | 0.1062 (17) | 0.0024 (10) | 0.0433 (12) | 0.0263 (12) |
| C11 | 0.0574 (11) | 0.0589 (10) | 0.0954 (16) | −0.0060 (8) | 0.0265 (11) | 0.0023 (10) |
| C12 | 0.0555 (10) | 0.0526 (9) | 0.0668 (11) | 0.0000 (7) | 0.0254 (9) | −0.0020 (8) |
| C13 | 0.0499 (8) | 0.0445 (7) | 0.0482 (9) | 0.0056 (6) | 0.0241 (7) | 0.0103 (6) |
| C14 | 0.0485 (8) | 0.0440 (7) | 0.0398 (8) | 0.0056 (6) | 0.0211 (7) | 0.0077 (6) |
| C15 | 0.0499 (8) | 0.0483 (8) | 0.0502 (9) | 0.0035 (7) | 0.0237 (7) | 0.0006 (7) |
| C16 | 0.0621 (10) | 0.0557 (9) | 0.0459 (9) | 0.0033 (7) | 0.0315 (8) | −0.0045 (7) |
| C17 | 0.0504 (8) | 0.0504 (8) | 0.0375 (8) | −0.0048 (6) | 0.0223 (7) | −0.0066 (6) |
| C18 | 0.0541 (9) | 0.0543 (8) | 0.0354 (8) | −0.0121 (7) | 0.0202 (7) | −0.0058 (6) |
| C19 | 0.0771 (12) | 0.0733 (11) | 0.0459 (9) | −0.0143 (9) | 0.0340 (9) | −0.0113 (8) |
| C20 | 0.0965 (15) | 0.1083 (17) | 0.0450 (11) | −0.0302 (13) | 0.0418 (11) | −0.0149 (11) |
| C21 | 0.0902 (15) | 0.1026 (16) | 0.0408 (10) | −0.0332 (13) | 0.0249 (10) | 0.0068 (10) |
| C22 | 0.0707 (12) | 0.0707 (11) | 0.0470 (10) | −0.0156 (9) | 0.0162 (9) | 0.0103 (8) |
| C23 | 0.0546 (9) | 0.0565 (9) | 0.0367 (8) | −0.0135 (7) | 0.0149 (7) | −0.0019 (7) |
| C24 | 0.0823 (13) | 0.0525 (10) | 0.0829 (14) | 0.0105 (9) | 0.0313 (11) | 0.0054 (9) |
(I). Geometric parameters (Å, º)
| O1—C15 | 1.2226 (18) | C11—H11A | 0.9300 |
| O2—C23 | 1.3574 (19) | C12—C13 | 1.423 (2) |
| O2—C24 | 1.426 (2) | C12—H12A | 0.9300 |
| C1—C14 | 1.401 (2) | C13—C14 | 1.405 (2) |
| C1—C2 | 1.426 (2) | C14—C15 | 1.509 (2) |
| C1—C6 | 1.431 (2) | C15—C16 | 1.451 (2) |
| C2—C3 | 1.355 (3) | C16—C17 | 1.327 (2) |
| C2—H2A | 0.9300 | C16—H16A | 0.9300 |
| C3—C4 | 1.408 (3) | C17—C18 | 1.457 (2) |
| C3—H3A | 0.9300 | C17—H17A | 0.9300 |
| C4—C5 | 1.343 (3) | C18—C19 | 1.393 (2) |
| C4—H4A | 0.9300 | C18—C23 | 1.406 (2) |
| C5—C6 | 1.421 (3) | C19—C20 | 1.375 (3) |
| C5—H5A | 0.9300 | C19—H19A | 0.9300 |
| C6—C7 | 1.389 (2) | C20—C21 | 1.376 (3) |
| C7—C8 | 1.381 (2) | C20—H20A | 0.9300 |
| C7—H7A | 0.9300 | C21—C22 | 1.377 (3) |
| C8—C13 | 1.433 (2) | C21—H21A | 0.9300 |
| C8—C9 | 1.435 (2) | C22—C23 | 1.389 (2) |
| C9—C10 | 1.346 (3) | C22—H22A | 0.9300 |
| C9—H9A | 0.9300 | C24—H24A | 0.9600 |
| C10—C11 | 1.406 (3) | C24—H24B | 0.9600 |
| C10—H10A | 0.9300 | C24—H24C | 0.9600 |
| C11—C12 | 1.354 (2) | ||
| C23—O2—C24 | 118.55 (13) | C14—C13—C8 | 119.06 (14) |
| C14—C1—C2 | 122.63 (14) | C12—C13—C8 | 117.99 (14) |
| C14—C1—C6 | 119.23 (14) | C1—C14—C13 | 121.04 (13) |
| C2—C1—C6 | 118.13 (15) | C1—C14—C15 | 119.37 (13) |
| C3—C2—C1 | 120.73 (17) | C13—C14—C15 | 119.27 (13) |
| C3—C2—H2A | 119.6 | O1—C15—C16 | 120.75 (14) |
| C1—C2—H2A | 119.6 | O1—C15—C14 | 117.85 (14) |
| C2—C3—C4 | 120.82 (19) | C16—C15—C14 | 121.40 (13) |
| C2—C3—H3A | 119.6 | C17—C16—C15 | 124.17 (13) |
| C4—C3—H3A | 119.6 | C17—C16—H16A | 117.9 |
| C5—C4—C3 | 120.41 (19) | C15—C16—H16A | 117.9 |
| C5—C4—H4A | 119.8 | C16—C17—C18 | 126.82 (14) |
| C3—C4—H4A | 119.8 | C16—C17—H17A | 116.6 |
| C4—C5—C6 | 121.37 (19) | C18—C17—H17A | 116.6 |
| C4—C5—H5A | 119.3 | C19—C18—C23 | 117.97 (14) |
| C6—C5—H5A | 119.3 | C19—C18—C17 | 122.13 (15) |
| C7—C6—C5 | 122.56 (16) | C23—C18—C17 | 119.90 (13) |
| C7—C6—C1 | 118.96 (15) | C20—C19—C18 | 121.46 (19) |
| C5—C6—C1 | 118.48 (16) | C20—C19—H19A | 119.3 |
| C8—C7—C6 | 122.45 (15) | C18—C19—H19A | 119.3 |
| C8—C7—H7A | 118.8 | C19—C20—C21 | 119.51 (18) |
| C6—C7—H7A | 118.8 | C19—C20—H20A | 120.2 |
| C7—C8—C13 | 119.20 (14) | C21—C20—H20A | 120.2 |
| C7—C8—C9 | 122.52 (16) | C20—C21—C22 | 121.12 (17) |
| C13—C8—C9 | 118.27 (17) | C20—C21—H21A | 119.4 |
| C10—C9—C8 | 121.30 (18) | C22—C21—H21A | 119.4 |
| C10—C9—H9A | 119.4 | C21—C22—C23 | 119.45 (19) |
| C8—C9—H9A | 119.4 | C21—C22—H22A | 120.3 |
| C9—C10—C11 | 120.29 (17) | C23—C22—H22A | 120.3 |
| C9—C10—H10A | 119.9 | O2—C23—C22 | 123.70 (16) |
| C11—C10—H10A | 119.9 | O2—C23—C18 | 115.80 (13) |
| C12—C11—C10 | 120.88 (19) | C22—C23—C18 | 120.49 (16) |
| C12—C11—H11A | 119.6 | O2—C24—H24A | 109.5 |
| C10—C11—H11A | 119.6 | O2—C24—H24B | 109.5 |
| C11—C12—C13 | 121.27 (18) | H24A—C24—H24B | 109.5 |
| C11—C12—H12A | 119.4 | O2—C24—H24C | 109.5 |
| C13—C12—H12A | 119.4 | H24A—C24—H24C | 109.5 |
| C14—C13—C12 | 122.95 (14) | H24B—C24—H24C | 109.5 |
| C14—C1—C2—C3 | 179.60 (15) | C2—C1—C14—C15 | 10.2 (2) |
| C6—C1—C2—C3 | 0.2 (2) | C6—C1—C14—C15 | −170.46 (13) |
| C1—C2—C3—C4 | 1.6 (3) | C12—C13—C14—C1 | 178.29 (13) |
| C2—C3—C4—C5 | −1.7 (3) | C8—C13—C14—C1 | −2.2 (2) |
| C3—C4—C5—C6 | −0.1 (3) | C12—C13—C14—C15 | −8.3 (2) |
| C4—C5—C6—C7 | −177.89 (18) | C8—C13—C14—C15 | 171.27 (13) |
| C4—C5—C6—C1 | 1.9 (3) | C1—C14—C15—O1 | 84.19 (18) |
| C14—C1—C6—C7 | −1.5 (2) | C13—C14—C15—O1 | −89.38 (19) |
| C2—C1—C6—C7 | 177.85 (14) | C1—C14—C15—C16 | −95.91 (18) |
| C14—C1—C6—C5 | 178.68 (14) | C13—C14—C15—C16 | 90.53 (18) |
| C2—C1—C6—C5 | −1.9 (2) | O1—C15—C16—C17 | −178.25 (16) |
| C5—C6—C7—C8 | 179.05 (16) | C14—C15—C16—C17 | 1.8 (2) |
| C1—C6—C7—C8 | −0.7 (2) | C15—C16—C17—C18 | −178.99 (14) |
| C6—C7—C8—C13 | 1.5 (2) | C16—C17—C18—C19 | −0.7 (2) |
| C6—C7—C8—C9 | −179.64 (16) | C16—C17—C18—C23 | 179.15 (15) |
| C7—C8—C9—C10 | −179.61 (17) | C23—C18—C19—C20 | −0.3 (3) |
| C13—C8—C9—C10 | −0.8 (3) | C17—C18—C19—C20 | 179.50 (16) |
| C8—C9—C10—C11 | 0.2 (3) | C18—C19—C20—C21 | −0.2 (3) |
| C9—C10—C11—C12 | 0.6 (3) | C19—C20—C21—C22 | 0.6 (3) |
| C10—C11—C12—C13 | −0.8 (3) | C20—C21—C22—C23 | −0.5 (3) |
| C11—C12—C13—C14 | 179.68 (15) | C24—O2—C23—C22 | −1.7 (2) |
| C11—C12—C13—C8 | 0.1 (2) | C24—O2—C23—C18 | 177.97 (15) |
| C7—C8—C13—C14 | −0.1 (2) | C21—C22—C23—O2 | 179.57 (16) |
| C9—C8—C13—C14 | −178.96 (13) | C21—C22—C23—C18 | −0.1 (2) |
| C7—C8—C13—C12 | 179.47 (14) | C19—C18—C23—O2 | −179.20 (14) |
| C9—C8—C13—C12 | 0.6 (2) | C17—C18—C23—O2 | 0.9 (2) |
| C2—C1—C14—C13 | −176.37 (13) | C19—C18—C23—C22 | 0.5 (2) |
| C6—C1—C14—C13 | 3.0 (2) | C17—C18—C23—C22 | −179.34 (14) |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Crystal data
| C24H17FO2 | Z = 2 |
| Mr = 356.37 | F(000) = 372 |
| Triclinic, P1 | Dx = 1.308 Mg m−3 |
| a = 8.6646 (5) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 9.5752 (5) Å | Cell parameters from 8625 reflections |
| c = 11.5636 (6) Å | θ = 2.2–30.2° |
| α = 100.593 (2)° | µ = 0.09 mm−1 |
| β = 105.443 (2)° | T = 296 K |
| γ = 92.422 (2)° | Block, yellow |
| V = 904.76 (9) Å3 | 0.99 × 0.31 × 0.25 mm |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Data collection
| Bruker SMART APEXII DUO CCD area-detector diffractometer | 3307 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.045 |
| φ and ω scans | θmax = 30.4°, θmin = 1.9° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −12→12 |
| k = −13→13 | |
| 34988 measured reflections | l = −16→16 |
| 5394 independent reflections |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.059 | H-atom parameters constrained |
| wR(F2) = 0.183 | w = 1/[σ2(Fo2) + (0.0672P)2 + 0.4075P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 5394 reflections | Δρmax = 0.27 e Å−3 |
| 245 parameters | Δρmin = −0.20 e Å−3 |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| F1 | −0.05579 (15) | 0.41714 (15) | 0.24083 (12) | 0.0805 (4) | |
| O1 | 0.1312 (2) | −0.03230 (17) | 0.68989 (16) | 0.0842 (6) | |
| O2 | 0.12761 (17) | 0.65677 (16) | 0.28147 (14) | 0.0642 (4) | |
| C1 | 0.49638 (19) | 0.04101 (17) | 0.81471 (15) | 0.0403 (4) | |
| C2 | 0.5049 (3) | −0.0702 (2) | 0.71696 (18) | 0.0572 (5) | |
| H2A | 0.4185 | −0.0931 | 0.6467 | 0.069* | |
| C3 | 0.6364 (3) | −0.1434 (2) | 0.7244 (2) | 0.0700 (6) | |
| H3A | 0.6391 | −0.2152 | 0.6589 | 0.084* | |
| C4 | 0.7689 (3) | −0.1126 (2) | 0.8295 (2) | 0.0661 (6) | |
| H4A | 0.8579 | −0.1644 | 0.8333 | 0.079* | |
| C5 | 0.7672 (2) | −0.0079 (2) | 0.92466 (19) | 0.0548 (5) | |
| H5A | 0.8551 | 0.0111 | 0.9940 | 0.066* | |
| C6 | 0.63319 (19) | 0.07396 (18) | 0.92079 (15) | 0.0409 (4) | |
| C7 | 0.63115 (19) | 0.18423 (19) | 1.01674 (15) | 0.0436 (4) | |
| H7A | 0.7199 | 0.2054 | 1.0854 | 0.052* | |
| C8 | 0.5005 (2) | 0.26367 (18) | 1.01297 (14) | 0.0413 (4) | |
| C9 | 0.5007 (2) | 0.3788 (2) | 1.11086 (17) | 0.0556 (5) | |
| H9A | 0.5913 | 0.4024 | 1.1779 | 0.067* | |
| C10 | 0.3724 (3) | 0.4537 (2) | 1.1079 (2) | 0.0659 (6) | |
| H10A | 0.3746 | 0.5280 | 1.1728 | 0.079* | |
| C11 | 0.2345 (3) | 0.4199 (2) | 1.0066 (2) | 0.0655 (6) | |
| H11A | 0.1461 | 0.4719 | 1.0059 | 0.079* | |
| C12 | 0.2286 (2) | 0.3125 (2) | 0.90999 (18) | 0.0533 (4) | |
| H12A | 0.1364 | 0.2924 | 0.8440 | 0.064* | |
| C13 | 0.36166 (19) | 0.23022 (17) | 0.90848 (15) | 0.0399 (3) | |
| C14 | 0.36231 (19) | 0.11926 (17) | 0.81074 (14) | 0.0390 (3) | |
| C15 | 0.2162 (2) | 0.07711 (19) | 0.70129 (17) | 0.0494 (4) | |
| C16 | 0.1775 (2) | 0.1640 (2) | 0.60830 (16) | 0.0516 (4) | |
| H16A | 0.0893 | 0.1315 | 0.5407 | 0.062* | |
| C17 | 0.2592 (2) | 0.28637 (19) | 0.61334 (15) | 0.0442 (4) | |
| H17A | 0.3492 | 0.3150 | 0.6803 | 0.053* | |
| C18 | 0.22475 (19) | 0.38103 (18) | 0.52646 (14) | 0.0412 (4) | |
| C19 | 0.0943 (2) | 0.35137 (19) | 0.42003 (16) | 0.0453 (4) | |
| H19A | 0.0259 | 0.2681 | 0.4022 | 0.054* | |
| C20 | 0.0689 (2) | 0.4455 (2) | 0.34319 (16) | 0.0477 (4) | |
| C21 | 0.1660 (2) | 0.57237 (19) | 0.36488 (16) | 0.0453 (4) | |
| C22 | 0.2941 (2) | 0.6011 (2) | 0.46933 (17) | 0.0498 (4) | |
| H22A | 0.3621 | 0.6847 | 0.4869 | 0.060* | |
| C23 | 0.3221 (2) | 0.50661 (19) | 0.54785 (15) | 0.0481 (4) | |
| H23A | 0.4094 | 0.5281 | 0.6175 | 0.058* | |
| C24 | 0.2160 (3) | 0.7934 (3) | 0.3095 (3) | 0.0818 (7) | |
| H24D | 0.1755 | 0.8436 | 0.2447 | 0.123* | |
| H24A | 0.3276 | 0.7817 | 0.3177 | 0.123* | |
| H24B | 0.2043 | 0.8470 | 0.3850 | 0.123* |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| F1 | 0.0667 (8) | 0.0865 (9) | 0.0653 (8) | −0.0140 (7) | −0.0250 (6) | 0.0267 (7) |
| O1 | 0.0745 (10) | 0.0659 (10) | 0.0862 (11) | −0.0277 (8) | −0.0240 (8) | 0.0265 (8) |
| O2 | 0.0579 (8) | 0.0682 (9) | 0.0672 (9) | 0.0042 (7) | 0.0063 (7) | 0.0323 (7) |
| C1 | 0.0414 (8) | 0.0376 (8) | 0.0392 (8) | −0.0023 (6) | 0.0078 (6) | 0.0073 (6) |
| C2 | 0.0593 (11) | 0.0508 (10) | 0.0517 (10) | 0.0007 (9) | 0.0088 (9) | −0.0033 (8) |
| C3 | 0.0745 (15) | 0.0583 (12) | 0.0732 (14) | 0.0090 (11) | 0.0272 (12) | −0.0069 (11) |
| C4 | 0.0548 (12) | 0.0603 (12) | 0.0862 (16) | 0.0168 (10) | 0.0260 (11) | 0.0099 (11) |
| C5 | 0.0412 (9) | 0.0592 (11) | 0.0631 (12) | 0.0090 (8) | 0.0107 (8) | 0.0148 (9) |
| C6 | 0.0360 (8) | 0.0430 (8) | 0.0430 (8) | 0.0015 (6) | 0.0078 (6) | 0.0118 (7) |
| C7 | 0.0357 (8) | 0.0511 (9) | 0.0380 (8) | 0.0005 (7) | 0.0010 (6) | 0.0086 (7) |
| C8 | 0.0413 (8) | 0.0440 (9) | 0.0353 (8) | 0.0010 (7) | 0.0054 (6) | 0.0080 (6) |
| C9 | 0.0580 (11) | 0.0593 (11) | 0.0410 (9) | 0.0038 (9) | 0.0063 (8) | 0.0002 (8) |
| C10 | 0.0721 (14) | 0.0636 (13) | 0.0555 (12) | 0.0125 (11) | 0.0162 (10) | −0.0036 (10) |
| C11 | 0.0611 (12) | 0.0653 (13) | 0.0708 (14) | 0.0223 (10) | 0.0195 (10) | 0.0102 (11) |
| C12 | 0.0448 (9) | 0.0571 (11) | 0.0535 (10) | 0.0096 (8) | 0.0043 (8) | 0.0127 (8) |
| C13 | 0.0373 (8) | 0.0400 (8) | 0.0399 (8) | 0.0028 (6) | 0.0050 (6) | 0.0104 (6) |
| C14 | 0.0379 (8) | 0.0378 (8) | 0.0367 (8) | −0.0014 (6) | 0.0015 (6) | 0.0102 (6) |
| C15 | 0.0455 (9) | 0.0454 (9) | 0.0474 (9) | −0.0023 (7) | −0.0021 (7) | 0.0083 (7) |
| C16 | 0.0462 (9) | 0.0560 (11) | 0.0412 (9) | −0.0004 (8) | −0.0056 (7) | 0.0085 (8) |
| C17 | 0.0378 (8) | 0.0524 (10) | 0.0355 (8) | 0.0037 (7) | 0.0019 (6) | 0.0039 (7) |
| C18 | 0.0384 (8) | 0.0469 (9) | 0.0349 (8) | 0.0055 (7) | 0.0076 (6) | 0.0036 (7) |
| C19 | 0.0417 (8) | 0.0443 (9) | 0.0440 (9) | −0.0001 (7) | 0.0048 (7) | 0.0057 (7) |
| C20 | 0.0379 (8) | 0.0561 (10) | 0.0411 (9) | 0.0029 (7) | −0.0011 (7) | 0.0077 (7) |
| C21 | 0.0419 (9) | 0.0502 (10) | 0.0451 (9) | 0.0073 (7) | 0.0115 (7) | 0.0128 (7) |
| C22 | 0.0485 (10) | 0.0479 (10) | 0.0490 (10) | −0.0036 (8) | 0.0097 (8) | 0.0072 (8) |
| C23 | 0.0432 (9) | 0.0553 (10) | 0.0372 (8) | −0.0027 (8) | 0.0017 (7) | 0.0037 (7) |
| C24 | 0.0764 (16) | 0.0712 (15) | 0.104 (2) | −0.0004 (12) | 0.0183 (14) | 0.0456 (14) |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Geometric parameters (Å, º)
| F1—C20 | 1.3485 (19) | C11—C12 | 1.361 (3) |
| O1—C15 | 1.220 (2) | C11—H11A | 0.9300 |
| O2—C21 | 1.354 (2) | C12—C13 | 1.426 (2) |
| O2—C24 | 1.425 (3) | C12—H12A | 0.9300 |
| C1—C14 | 1.404 (2) | C13—C14 | 1.403 (2) |
| C1—C2 | 1.424 (2) | C14—C15 | 1.511 (2) |
| C1—C6 | 1.436 (2) | C15—C16 | 1.457 (3) |
| C2—C3 | 1.354 (3) | C16—C17 | 1.327 (3) |
| C2—H2A | 0.9300 | C16—H16A | 0.9300 |
| C3—C4 | 1.407 (3) | C17—C18 | 1.455 (2) |
| C3—H3A | 0.9300 | C17—H17A | 0.9300 |
| C4—C5 | 1.349 (3) | C18—C23 | 1.383 (2) |
| C4—H4A | 0.9300 | C18—C19 | 1.406 (2) |
| C5—C6 | 1.424 (2) | C19—C20 | 1.363 (3) |
| C5—H5A | 0.9300 | C19—H19A | 0.9300 |
| C6—C7 | 1.388 (2) | C20—C21 | 1.391 (3) |
| C7—C8 | 1.386 (2) | C21—C22 | 1.380 (2) |
| C7—H7A | 0.9300 | C22—C23 | 1.380 (3) |
| C8—C9 | 1.427 (2) | C22—H22A | 0.9300 |
| C8—C13 | 1.436 (2) | C23—H23A | 0.9300 |
| C9—C10 | 1.345 (3) | C24—H24D | 0.9600 |
| C9—H9A | 0.9300 | C24—H24A | 0.9600 |
| C10—C11 | 1.409 (3) | C24—H24B | 0.9600 |
| C10—H10A | 0.9300 | ||
| C21—O2—C24 | 117.53 (17) | C14—C13—C8 | 119.19 (14) |
| C14—C1—C2 | 123.01 (15) | C12—C13—C8 | 117.62 (15) |
| C14—C1—C6 | 119.47 (15) | C13—C14—C1 | 120.71 (14) |
| C2—C1—C6 | 117.52 (16) | C13—C14—C15 | 120.79 (15) |
| C3—C2—C1 | 121.23 (19) | C1—C14—C15 | 118.45 (15) |
| C3—C2—H2A | 119.4 | O1—C15—C16 | 119.73 (16) |
| C1—C2—H2A | 119.4 | O1—C15—C14 | 119.46 (16) |
| C2—C3—C4 | 121.1 (2) | C16—C15—C14 | 120.80 (15) |
| C2—C3—H3A | 119.5 | C17—C16—C15 | 124.58 (16) |
| C4—C3—H3A | 119.5 | C17—C16—H16A | 117.7 |
| C5—C4—C3 | 120.13 (19) | C15—C16—H16A | 117.7 |
| C5—C4—H4A | 119.9 | C16—C17—C18 | 127.67 (15) |
| C3—C4—H4A | 119.9 | C16—C17—H17A | 116.2 |
| C4—C5—C6 | 121.11 (19) | C18—C17—H17A | 116.2 |
| C4—C5—H5A | 119.4 | C23—C18—C19 | 117.44 (16) |
| C6—C5—H5A | 119.4 | C23—C18—C17 | 119.75 (14) |
| C7—C6—C5 | 121.83 (16) | C19—C18—C17 | 122.81 (15) |
| C7—C6—C1 | 119.24 (15) | C20—C19—C18 | 119.55 (16) |
| C5—C6—C1 | 118.93 (16) | C20—C19—H19A | 120.2 |
| C8—C7—C6 | 121.79 (15) | C18—C19—H19A | 120.2 |
| C8—C7—H7A | 119.1 | F1—C20—C19 | 119.69 (16) |
| C6—C7—H7A | 119.1 | F1—C20—C21 | 117.16 (16) |
| C7—C8—C9 | 121.43 (15) | C19—C20—C21 | 123.15 (15) |
| C7—C8—C13 | 119.59 (15) | O2—C21—C22 | 125.48 (17) |
| C9—C8—C13 | 118.98 (16) | O2—C21—C20 | 117.35 (15) |
| C10—C9—C8 | 121.19 (18) | C22—C21—C20 | 117.17 (16) |
| C10—C9—H9A | 119.4 | C23—C22—C21 | 120.44 (17) |
| C8—C9—H9A | 119.4 | C23—C22—H22A | 119.8 |
| C9—C10—C11 | 120.17 (19) | C21—C22—H22A | 119.8 |
| C9—C10—H10A | 119.9 | C22—C23—C18 | 122.25 (15) |
| C11—C10—H10A | 119.9 | C22—C23—H23A | 118.9 |
| C12—C11—C10 | 121.12 (19) | C18—C23—H23A | 118.9 |
| C12—C11—H11A | 119.4 | O2—C24—H24D | 109.5 |
| C10—C11—H11A | 119.4 | O2—C24—H24A | 109.5 |
| C11—C12—C13 | 120.89 (17) | H24D—C24—H24A | 109.5 |
| C11—C12—H12A | 119.6 | O2—C24—H24B | 109.5 |
| C13—C12—H12A | 119.6 | H24D—C24—H24B | 109.5 |
| C14—C13—C12 | 123.18 (15) | H24A—C24—H24B | 109.5 |
| C14—C1—C2—C3 | −179.92 (19) | C8—C13—C14—C15 | 177.67 (15) |
| C6—C1—C2—C3 | 0.8 (3) | C2—C1—C14—C13 | −178.46 (16) |
| C1—C2—C3—C4 | 0.4 (4) | C6—C1—C14—C13 | 0.8 (2) |
| C2—C3—C4—C5 | −0.6 (4) | C2—C1—C14—C15 | 4.1 (2) |
| C3—C4—C5—C6 | −0.5 (3) | C6—C1—C14—C15 | −176.59 (15) |
| C4—C5—C6—C7 | −178.44 (19) | C13—C14—C15—O1 | −104.9 (2) |
| C4—C5—C6—C1 | 1.7 (3) | C1—C14—C15—O1 | 72.5 (3) |
| C14—C1—C6—C7 | −1.0 (2) | C13—C14—C15—C16 | 76.3 (2) |
| C2—C1—C6—C7 | 178.36 (16) | C1—C14—C15—C16 | −106.3 (2) |
| C14—C1—C6—C5 | 178.85 (16) | O1—C15—C16—C17 | 178.2 (2) |
| C2—C1—C6—C5 | −1.8 (2) | C14—C15—C16—C17 | −3.0 (3) |
| C5—C6—C7—C8 | −179.87 (16) | C15—C16—C17—C18 | −177.76 (17) |
| C1—C6—C7—C8 | 0.0 (3) | C16—C17—C18—C23 | 178.73 (18) |
| C6—C7—C8—C9 | −178.66 (17) | C16—C17—C18—C19 | −0.9 (3) |
| C6—C7—C8—C13 | 1.2 (3) | C23—C18—C19—C20 | 0.0 (3) |
| C7—C8—C9—C10 | −178.64 (19) | C17—C18—C19—C20 | 179.61 (16) |
| C13—C8—C9—C10 | 1.5 (3) | C18—C19—C20—F1 | 179.80 (16) |
| C8—C9—C10—C11 | −0.4 (3) | C18—C19—C20—C21 | −0.5 (3) |
| C9—C10—C11—C12 | −0.5 (4) | C24—O2—C21—C22 | −6.9 (3) |
| C10—C11—C12—C13 | 0.3 (3) | C24—O2—C21—C20 | 173.58 (19) |
| C11—C12—C13—C14 | −179.42 (18) | F1—C20—C21—O2 | −0.1 (3) |
| C11—C12—C13—C8 | 0.8 (3) | C19—C20—C21—O2 | −179.88 (17) |
| C7—C8—C13—C14 | −1.3 (2) | F1—C20—C21—C22 | −179.69 (17) |
| C9—C8—C13—C14 | 178.55 (16) | C19—C20—C21—C22 | 0.6 (3) |
| C7—C8—C13—C12 | 178.46 (16) | O2—C21—C22—C23 | −179.74 (17) |
| C9—C8—C13—C12 | −1.7 (2) | C20—C21—C22—C23 | −0.2 (3) |
| C12—C13—C14—C1 | −179.46 (16) | C21—C22—C23—C18 | −0.2 (3) |
| C8—C13—C14—C1 | 0.3 (2) | C19—C18—C23—C22 | 0.3 (3) |
| C12—C13—C14—C15 | −2.1 (3) | C17—C18—C23—C22 | −179.30 (16) |
(E)-1-(Anthracen-9-yl)-3-(3-fluoro-4-methoxyphenyl)prop-2-en-1-one (II) . Hydrogen-bond geometry (Å, º)
Cg4 is the centroid of the C1–C6 ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12A···O2i | 0.93 | 2.48 | 3.345 (2) | 154 |
| C19—H19A···O1ii | 0.93 | 2.48 | 3.393 (3) | 166 |
| C24—H24D···Cg4iii | 0.96 | 2.77 | 3.391 (3) | 123 |
Symmetry codes: (i) −x, −y+1, −z+1; (ii) −x, −y, −z+1; (iii) −x+1, −y+1, −z+1.
Funding Statement
This work was funded by Ministry of Higher Education, Malaysia grants 203/PFIZIK/6711606, 203/PFIZIK/6711572, 304/PFIZIK/6313336, and My Brain15.
References
- Abdullah, A. A., Hassan, N. H. H., Arshad, S., Khalib, N. C. & Razak, I. A. (2016). Acta Cryst. E72, 648–651. [DOI] [PMC free article] [PubMed]
- Agrahari, A., Wagers, P. O., Schildcrout, S. M., Masnovi, J. & Youngs, W. J. (2015). Acta Cryst. E71, 357–359. [DOI] [PMC free article] [PubMed]
- Bruker (2009). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Cicogna, F., Ingrosso, G., Lodato, F., Marchetti, F. & Zandomeneghi, M. (2004). Tetrahedron, 60, 11959–11968.
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, V., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. C., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J. & Fox, D. J. Gaussian 09, Revision A1. Gaussian, Inc., Wallingford CT, USA.
- Girisha, M., Yathirajan, H. S., Jasinski, J. P. & Glidewell, C. (2016). Acta Cryst. E72, 1153–1158. [DOI] [PMC free article] [PubMed]
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Harlow, R. L., Loghry, R. A., Williams, H. J. & Simonsen, S. H. (1975). Acta Cryst. B31, 1344–1350.
- Li, X. C., Wang, C. Y., Lai, W. Y. & Huang, W. (2016). J. Mater. Chem. C, 4, 10574–10587.
- Prabhu, A. N., Upadhyaya, V., Jayarama, A. & Bhat, K. B. (2016). Mol. Cryst. Liq. Cryst. 637, 76–86.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Zainuri, D. A., Arshad, S., Khalib, N. C., Razak, A. I., Pillai, R. R., Sulaiman, F., Hashim, N. S., Ooi, K. L., Armaković, S., Armaković, S. J., Panicker, Y. & Van Alsenoy, C. (2017). J. Mol. Struct. 1128, 520–533.
- Zainuri, D. A., Razak, I. A. & Arshad, S. (2018a). Acta Cryst. E74, 492–496. [DOI] [PMC free article] [PubMed]
- Zainuri, D. A., Razak, I. A. & Arshad, S. (2018b). Acta Cryst. E74, 650–655. [DOI] [PMC free article] [PubMed]
- Zainuri, D. A., Razak, I. A. & Arshad, S. (2018c). Acta Cryst. E74, 780–785. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, II. DOI: 10.1107/S205698901800974X/jj2199sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S205698901800974X/jj2199Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S205698901800974X/jj2199IIsup3.hkl
Supporting information file. DOI: 10.1107/S205698901800974X/jj2199Isup4.cml
Supporting information file. DOI: 10.1107/S205698901800974X/jj2199IIsup5.cml
Comparison of selected experimental and DFT-optimized data for compounds (I) and (II). DOI: 10.1107/S205698901800974X/jj2199sup6.pdf
Additional supporting information: crystallographic information; 3D view; checkCIF report