

Abstract
Aims
Chronic kidney disease (CKD) in heart failure (HF) increases the risk of hyperkalaemia (HK), limiting angiotensin‐converting enzyme inhibitor (ACE‐I) or angiotensin receptor blocker (ARB) use. Patiromer is a sodium‐free, non‐absorbed potassium binder approved for HK treatment. We retrospectively evaluated patiromer's long‐term safety and efficacy in HF patients from AMETHYST‐DN.
Methods and results
Patients with Type 2 diabetes, CKD, and HK [baseline serum potassium >5.0–5.5 mmol/L (mild) or >5.5–<6.0 mmol/L (moderate)], with or without HF (New York Heart Association Class I and II, by investigator judgement), on ACE‐I/ARB, were randomized to patiromer 8.4–33.6 g to start, divided twice daily. Overall, 105/304 (35%) patients had HF (75%, Class II). Mean (standard deviation) ejection fraction (EF) was 44.9% (8.2) (n = 81) in patients with HF; 26 had EF ≤40%. In HF patients, mean serum potassium decreased by Day 3 through Week 52. At Week 4, estimated mean (95% confidence interval) change in serum potassium was −0.64 mmol/L (−0.72, −0.55) in mild and −0.97 mmol/L (−1.14, −0.80) in moderate HK (both P < 0.0001). Most HF patients with mild (>88%) and moderate (≥73%) HK had normokalaemia at each visit from Weeks 12 to 52. Three HF patients were withdrawn because of high (n = 1) or low (n = 2) serum potassium. The most common patiromer‐related adverse event was hypomagnesaemia (8.6%).
Conclusions
In patients with a clinical diagnosis of HF, diabetes, CKD, and HK on ACE‐I/ARB, patiromer was well tolerated and effective for HK treatment over 52 weeks.
Keywords: Heart failure, Hyperkalaemia, RAASi, Patiromer
Introduction
Renin–angiotensin–aldosterone system inhibitors (RAASi), such as an angiotensin‐converting enzyme inhibitor (ACE‐I) or an angiotensin receptor blocker (ARB) and a mineralocorticoid receptor antagonist (MRA), are indicated in patients with heart failure (HF) to reduce cardiovascular morbidity and mortality.1, 2, 3, 4, 5 However, these patients often have co‐morbid conditions such as chronic kidney disease (CKD) and/or Type 2 diabetes mellitus (DM) that increase their risk of developing hyperkalaemia.6, 7, 8, 9 The use of RAASi further amplifies the risk of developing hyperkalaemia in these patients, especially those with CKD or who receive ≥1 RAASi.10, 11, 12, 13 The fear of inducing hyperkalaemia has led to underuse of RAASi in these patients.14
Patiromer is a sodium‐free, non‐absorbed potassium binder that reduces serum potassium by exchanging calcium for potassium throughout the gut lumen but predominantly in the colon, where potassium concentration is high.14, 15 The use of calcium rather than sodium as the counter‐exchange ion in patiromer prevents the risk of adding a sodium load to patients who are already at greater risk for fluid overload because of their underlying heart or kidney disease.14, 15 Patiromer has been approved for the treatment of hyperkalaemia in the USA16 and in the European Union.17 Patiromer has been demonstrated to be well tolerated and effective over the short term in reducing serum potassium in patients with hyperkalaemia (serum potassium >5.0 to <6.5 mmol/L) or preventing hyperkalaemia in patients with CKD and HF.18, 19, 20 In one of these short‐term studies, a significantly higher proportion of patients with HF on patiromer were able to have their spironolactone dose increased vs. those on placebo.20
More recently, the reduction in serum potassium that has been observed after 4 weeks of patiromer treatment has been shown to be maintained for up to 52 weeks in the AMETHYST‐DN study in hyperkalaemic patients with DM and CKD on RAASi therapy.21 Mean serum potassium levels increased in patients discontinuing patiromer, either prematurely or at Week 52, suggesting that chronic therapy may be needed to control serum potassium in these patients.21 However, the long‐term safety and effectiveness of patiromer in patients with HF has not previously been examined. We therefore conducted a retrospective subanalysis of the AMETHYST‐DN study21 in patients with or without a clinical diagnosis of HF at baseline.
Methods
Study populations
The AMETHYST‐DN study has been described previously.21 Briefly, eligible patients were 30 to 80 years of age, had DM and CKD (estimated glomerular filtration rate 15 to <60 mL/min/1.73 m2), and were hyperkalaemic (mild: >5.0 to 5.5 mmol/L; moderate: >5.5 to <6.0 mmol/L; at screening or after an up‐to‐4‐week run‐in period). Patients with or without a clinical diagnosis of HF on a stable dose of RAASi for ≥28 days at screening were enrolled. The presence of HF was determined by the investigator using the patients' reported history and medical records (when available). The date of HF diagnosis and left ventricular ejection fraction (EF) (when available) and current New York Heart Association (NYHA) class were recorded in the case report form. No specific diagnostic criteria were provided to investigators by the sponsor.
Key exclusion criteria included current diagnosis of NYHA Class III or IV HF; occurrence of unstable angina (as assessed by the investigator), unresolved acute coronary syndrome, cardiac arrest, clinically significant ventricular arrhythmia, transient ischaemic attack, or stroke during the 2 months prior to screening; type 1 diabetes; prior kidney transplant; haemoglobin A1c >12% at screening (except for patients with hyperkalaemia at screening); urine albumin‐to‐creatinine ratio ≥1130 mg/mmol (except for patients with hyperkalaemia at screening); and serum magnesium <0.58 mmol/L at screening.21
Study design
AMETHYST‐DN was a multicentre, randomized, open‐label Phase 2B study performed at 48 sites in five European countries.20 The protocol was approved by local or national independent ethics committees at each study site and performed in accordance with the International Conference on Harmonisation E6 Guideline for Good Clinical Practice, the Declaration of Helsinki principles, and the European Union Clinical Trials Directive 2001/20/EC for sites in the European Union.22, 23, 24 All patients provided written informed consent before any study‐specific procedures were performed.
The 52‐week study included an 8‐week treatment period and a 44‐week long‐term maintenance period (LTMP) (Figure 1). The treatment period was preceded by an up‐to‐4‐week run‐in period, and LTMP was followed by an up‐to‐4‐week follow‐up period. At screening, patients on RAASi who were normokalaemic (serum potassium of 4.3–5.0 mmol/L) and had uncontrolled hypertension [sitting systolic blood pressure (BP) of >130 to ≤180 mmHg and diastolic BP of >80 to ≤110 mmHg] entered the run‐in period.21 The patients were randomly assigned in a 3:1 ratio to either switch from their current ACE‐I or ARB medications to losartan 100 mg/day (Cohort 1) or receive spironolactone 25 mg/day in addition to their current ACE‐I or ARB medications (Cohort 2). Additionally, patients in Cohorts 1 and 2 could receive up to 50 mg/day spironolactone if necessary for managing hypertension. Cohort 3 patients [with pre‐existing hyperkalaemia (baseline serum potassium >5.0 to <6.0 mmol/L)] did not enter the run‐in period and continued to take their ACE‐I or ARB medications.
Figure 1.
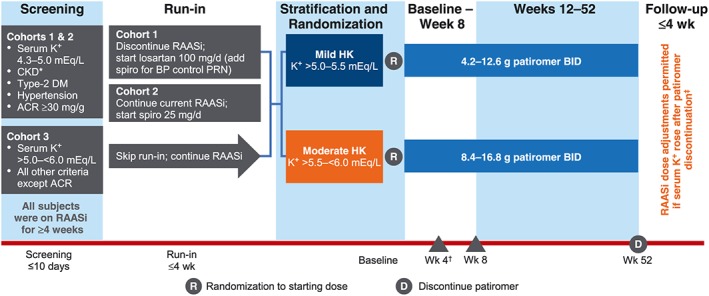
AMETHYST‐DN study design. *eGFR 15 to <60 mL/min/1.73 m2. †Primary endpoint. ‡RAASi therapy was continued after patiromer discontinuation only in patients who were normokalaemic (serum K+ ≤ 5.0 mEq/L) at the last on‐treatment visit. ACR, albumin‐to‐creatinine ratio; BID, twice daily; BP, blood pressure; CKD, chronic kidney disease; DM, diabetes mellitus; eGFR, estimated glomerular filtration rate; K+, potassium; PRN, as needed; RAASi, renin–angiotensin–aldosterone system inhibitors; spiro, spironolactone; Wk, week.
Patients who developed hyperkalaemia (serum potassium >5.0 to <6.0 mmol/L) during the run‐in period (Cohorts 1 and 2) and patients with pre‐existing hyperkalaemia at screening (Cohort 3) entered the treatment period and were assigned to one of two strata: mild hyperkalaemia (serum potassium >5.0 to 5.5 mmol/L) and moderate hyperkalaemia (>5.5 to <6.0 mmol/L). The patients were randomized to receive one of three starting doses of patiromer in each stratum: mild hyperkalaemia patients received 4.2, 8.4, or 12.6 g twice daily; and moderate hyperkalaemia patients received 8.4, 12.6, or 16.8 g twice daily. Following the baseline visit (Day 1), patients were assessed at Day 3 (approximately 48 h after the first patiromer dose) and weekly thereafter during the 8‐week treatment period and monthly during the 44‐week LTMP. The starting dose could be titrated up or down in a stepwise manner, starting at Day 3 and up to the Week 52 visit to achieve and maintain serum potassium ≤5.0 mmol/L (4.0–5.0 mmol/L during the treatment period and 3.8–5.0 mmol/L during the LTMP). By protocol, RAASi could not be down‐titrated or discontinued secondary to hyperkalaemia, although patiromer could be up‐titrated using the study‐defined dosing algorithm. Irrespective of whether they completed the entire 52‐week study period, all patients continued into the follow‐up period. As reported previously, patiromer decreased the mean serum potassium significantly from baseline to Week 4 in all starting‐dose groups in both strata (P < 0.001 for each comparison); the reductions were maintained through Week 52.21 In the present analyses, the data for the three starting‐dose groups with each baseline hyperkalaemia strata were combined for the purpose of comparing the patiromer effect on serum potassium in patients with and without a clinical diagnosis of HF.
Patients with serum potassium >5.0 mmol/L at the end‐of‐treatment visit discontinued patiromer and all RAASi medications and returned for two follow‐up visits within 7 days. Patients who were normokalaemic (serum potassium ≤5.0 mmol/L) at the end‐of‐treatment visit discontinued patiromer but remained on RAASi for 28 days and returned for five follow‐up visits (Day 3 and weekly visits through Week 4 post‐treatment) during this period. Patients who developed significant hyperkalaemia during the follow‐up period were treated as per standard of care (as determined by the investigator). This follow‐up design helped assess the effect of patiromer withdrawal on serum potassium in patients receiving RAASi.
Efficacy and safety assessments
In the HF subgroup, the same efficacy endpoints as in the AMETHYST‐DN study were used.21 We studied change in serum potassium from baseline to Week 4 (or prior to the first dose titration if it occurred before 4 weeks) and change in serum potassium from baseline to other post‐baseline visits. All serum potassium assessments (including baseline values) were based on central laboratory serum potassium.
Long‐term safety and tolerability were assessed over 52 weeks by frequency and severity of adverse events (AEs), clinical laboratory measurements, vital signs, and electrocardiogram parameters. Safety measures also included the proportion of patients who met the withdrawal criterion of serum potassium <3.5 mmol/L. Results are presented for the combined baseline hyperkalaemia strata in each subgroup (HF vs. no HF), except for serum potassium over time.
Statistical analyses
The detailed statistical analyses for the AMETHYST‐DN study have been described previously.21 For this analysis, serum potassium levels at post‐baseline time points through Week 52 were estimated for each stratum using a mixed‐effects repeated‐measures model with central laboratory serum potassium value as the dependent variable, time point and starting dose as fixed‐effect predictors, baseline central laboratory serum potassium value as a continuous covariate, and patient as a random effect. A compound symmetry correlation matrix was fit. All analyses were conducted using SAS Version 9.3 (SAS Institute Inc.).
Results
Patient disposition
Of the 306 patients who were randomized in the AMETHYST‐DN study, 105 were assessed by the investigators as having a clinical diagnosis of NYHA Class I or II HF. In total, 78 (74.3%) patients with HF and 133 (66.2%) without HF completed the entire study. The most common reasons for discontinuation in patients with and without HF were consent withdrawal [7 (6.7%) and 24 (11.9%), respectively] and AEs [7 (6.7%) and 12 (6.0%), respectively] (Supporting Information, Table S1 ). Discontinuations due to low serum potassium occurred in two (1.9%) patients with HF and five (2.5%) patients without HF. Discontinuations due to high serum potassium occurred in one (1.0%) patient with HF and six (3.0%) patients without HF.
Demographic and baseline characteristics
All 105 patients with HF and 199 of 201 patients without HF were treated with at least one dose of patiromer and are included in the safety and efficacy analyses. All patients were White, and the majority (68.6% with HF) were men (Table 1). In the HF subgroup, the mean [standard deviation (SD)] age was 67.7 (7.53) years (range: 49–80 years), and the mean (SD) serum potassium was 5.08 (0.42) mmol/L. Seventy‐seven (73.3%) HF patients had serum potassium >5.0 to 5.5 mmol/L (mild hyperkalaemia) at baseline, and 28 (26.7%) had serum potassium >5.5 to 6.0 mmol/L (moderate hyperkalaemia) at baseline. Of the patients with HF, 79 (75%) had NYHA Class II. Eighty‐one patients with HF had EF recorded at baseline (Table 2); mean (SD) EF was 44.9% (8.2%) in these patients. Of the 105 patients with HF, 16 (15.2%) had electrocardiographic evidence of atrial fibrillation at baseline. All patients with HF were on RAASi medications, mostly monotherapy with an ACE‐I or an ARB (Table 1).
Table 1.
Demographic and clinical characteristics at baseline or screening
| Parameter | Patients with HF (n = 105) | Patients without HF (n = 199) |
|---|---|---|
| Age at screening (years), mean ± SD | 67.7 ± 7.3 | 65.6 ± 9.1 |
| Male, n (%) | 72 (68.6) | 120 (60.3) |
| White, n (%) | 105 (100) | 199 (100) |
| Body mass index (kg/m2), mean ± SD | 29.0 (3.2) | 30.0 (4.6) |
| Serum potassium (mmol/L), mean ± SD (n) | 5.08 ± 0.42 (103) | 5.12 ± 0.46 (198) |
| 5.0 to <5.5, n (%) | 77 (73.3) | 143 (71.9) |
| 5.5 to <6.0, n (%) | 28 (26.7) | 56 (28.1) |
| eGFR at screening (mL/min/1.73 m2), mean ± SD (n) | 40.0 ± 14.89 (102) | 40.9 ± 16.1 (196) |
| CKD stage (eGFR, mL/min/1.73 m2) at screening, n (%) | ||
| 1 (≥90) | 0 | 1 (0.5) |
| 2 (60–89) | 8 (7.6) | 21 (10.6) |
| 3a (45–59) | 32 (30.5) | 53 (26.6) |
| 3b (30–44) | 37 (25.2) | 74 (37.2) |
| 4 (15–29) | 21 (20.0) | 45 (22.6) |
| 5 (<15) | 4 (3.8) | 2 (1.0) |
| NYHA class at screening, n (%) | ||
| I | 26 (24.8) | 0 |
| II | 79 (75.2) | 0 |
| No | 0 | 199 (100) |
| Hypertension, n (%) | 105 (100) | 199 (100) |
| Sitting BP at screening (mmHg), mean ± SD | ||
| Systolic | 155.4 ± 12.4 | 155.6 ± 11.8 |
| Diastolic | 86.4 ± 11.3 | 82.7 ± 11.2 |
| Ejection fraction (%), mean ± SD (n) | 44.9 ± 8.2 (81) | 54.7 ± 6.6 (84) |
| Urine ACR (mg/mmol), mean ± SD (n) | 118.0 ± 200.9 (100) | 136.1 ± 211.1 (193) |
| RAASi medication, n (%) | ||
| ACE‐I | 71 (67.6) | 128 (64.3) |
| ARB | 47 (44.8) | 95 (47.7) |
| Aldosterone antagonist | 5 (4.8) | 20 (10.1) |
| On any single RAASi | 88 (87.3) | 160 (80.4) |
| On ≥2 RAASia | 17 (16.3) | 39 (19.5) |
| On losartan 100 mg ± any dose of spironolactoneb | 15 (14.3) | 43 (21.6) |
| Digitalis/digoxin, n (%) | 22 (21.0) | 1 (0.5) |
| Beta‐blocker, n (%) | 62 (59.0) | 101 (50.8) |
| Non‐RAASi diuretic, n (%)c | 67 (63.8) | 63 (31.7) |
ACE‐I, angiotensin‐converting enzyme inhibitor; ACR, albumin‐to‐creatinine ratio; ARB, angiotensin receptor blocker; BP, blood pressure; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; HF, heart failure; NYHA, New York Heart Association; RAASi, renin–angiotensin–aldosterone system inhibitor; SD, standard deviation.
Of these patients, one patient with HF and five patients without HF were on triple‐RAASi therapy (i.e. ACE‐I, ARB, and mineralocorticoid receptor antagonist).
This category includes only patients in Cohort 1 (n = 58), of whom 24 (five with HF) were on at least 25 mg of spironolactone daily.
Includes loop and thiazide diuretics.
Table 2.
Heart failure classification and ejection fraction data in the 81 heart failure patients with ejection fraction available at baseline
| Ejection fraction | |||
|---|---|---|---|
| rEF EF ≤ 40% (n = 26) | Borderline pEF 40% < EF < 50% (n = 40) | pEF 50% ≥ EF (n = 15) | |
| Heart failure | |||
| Yes, NYHA Class I, n (%) | 1 (3.8) | 3 (7.5) | 8 (53.3) |
| Yes, NYHA Class II, n (%) | 25 (96.2) | 37 (92.5) | 7 (46.7) |
| No | 0 | 0 | 0 |
| Ejection fraction (%) | |||
| N | 26 | 40 | 15 |
| Mean (SD) | 37.4 (2.7) | 44.8 (2.6) | 58.1 (7. 8) |
| Median | 38.0 | 45.0 | 56.0 |
| Quartiles (25th, 75th) | 36.0, 39.0 | 42.0, 47.0 | 51.0, 60.0 |
EF, ejection fraction; HF, heart failure; NYHA, New York Heart Association; pEF, preserved ejection fraction; rEF, reduced ejection fraction; SD, standard deviation.
Efficacy
Mean serum potassium was reduced at the first post‐baseline visit (Day 3, when patients remained on their starting dose) through Week 52 in patients with and without HF, regardless of baseline hyperkalaemia severity (Figure 2). At Week 4 (or at the time of first titration if it occurred before Week 4), the least squares mean (LSM) (95% confidence interval [CI]) change in serum potassium from baseline in HF patients was −0.64 (−0.72, −0.55) mmol/L for those with mild hyperkalaemia and −0.97 (−1.14, −0.80) mmol/L for those with moderate hyperkalaemia (P < 0.0001 for both). Results in patients without HF were −0.60 (−0.67, −0.53) mmol/L for mild hyperkalaemia and −1.01 (−1.15, −0.88) mmol/L for moderate hyperkalaemia (P < 0.0001 for both). From Week 4 through Week 52, significant (P < 0.0001) mean decreases from baseline in mean serum potassium levels were observed at each monthly time point in patients with or without HF, irrespective of hyperkalaemia stratum (Figure 2). In the HF subgroup overall, ≥88% of patients with mild hyperkalaemia and ≥73% with moderate hyperkalaemia had serum potassium levels within the target range of 3.8 to 5.0 mmol/L at each scheduled visit during the LTMP (Weeks 12 to 52). In the subgroup without HF, the proportions were ≥84% and ≥79%, respectively.
Figure 2.
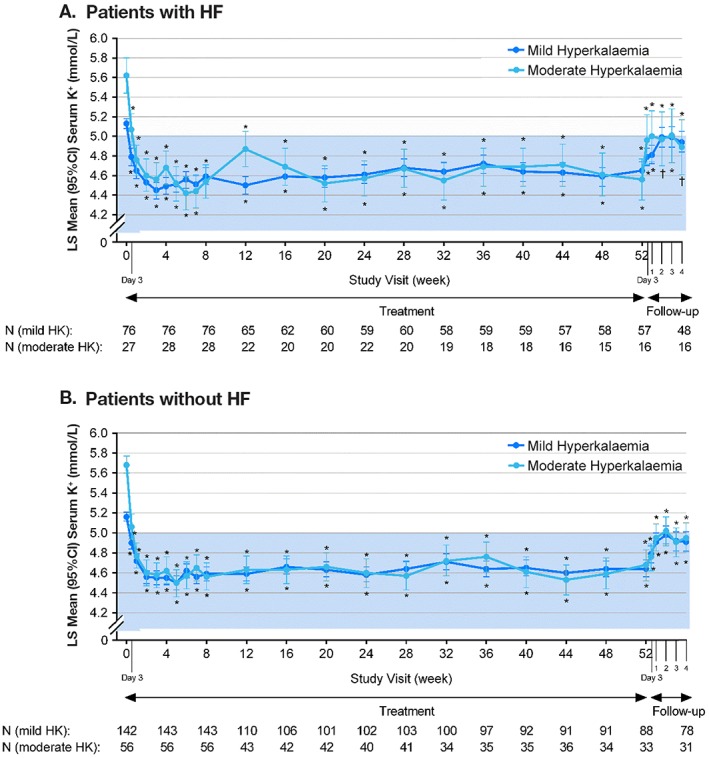
Least squares mean (95% CI) serum potassium levels over 52 weeks and during the follow‐up period in patients with (A) or without (B) HF. Serum potassium values [mmol/L (95% CI)] at baseline were as follows: in the HF subgroup, 5.11 (5.03, 5.20) for mild and 5.61 (5.28, 5.94) for moderate hyperkalaemia; and in the non‐HF subgroup, 5.16 (5.08, 5.23) for mild and 5.74 (5.59, 5.89) for moderate hyperkalaemia. All serum potassium analyses are based on central laboratory values. P‐values for least squares mean changes from baseline (for 52 weeks of treatment period) and from Week 52 (or time of last dose of patiromer; for follow‐up period). Least squares mean values are based on a mixed‐effects repeated‐measures model with serum potassium value as the dependent variable, time point and starting dose as fixed‐effect predictors, baseline central laboratory serum potassium value as a continuous covariate, and patient as a random effect. A compound symmetry matrix was fit. Least squares mean values for the follow‐up period are based on the same model described, while the baseline covariate used was the last potassium value before study termination. The blue shaded area denotes the target range for serum potassium (4.0–5.0 mmol/L during the treatment period and 3.8–5.0 mmol/L during the long‐term maintenance period). CI, confidence interval; HF, heart failure; HK, hyperkalaemia; K+, potassium; LS, least squares; N, number of patients at baseline and each successive 4‐week interval. *P ≤ 0.001; † P < 0.01.
Efficacy results in the 26 patients with EF ≤40% (both strata combined) were consistent with the overall HF subgroup, with an LSM change in serum potassium of −0.37 mmol/L (95% CI: −0.74, −0.00) at Day 3 and −0.66 mmol/L (95% CI: −1.12, −0.20) at Week 4. Between Weeks 1 and 52, mean serum potassium remained between 4.5 and <5.0 mmol/L in all 26 with EF ≤40%.
Discontinuation of patiromer led to a significant rise in serum potassium in patients with or without HF (Figure 2). Three days after patiromer was discontinued, the LSM (95% CI) increase in serum potassium from the Week 52 time point in HF patients was 0.27 (0.18, 0.36) mmol/L for those with mild hyperkalaemia (P < 0.0001) and 0.46 (0.20, 0.71) mmol/L for those with moderate hyperkalaemia (P = 0.0011). The corresponding LSM increase in serum potassium in patients without HF was 0.24 (95% CI: 0.15, 0.32) mmol/L for those with mild hyperkalaemia (P < 0.0001) and 0.27 (95% CI: 0.14, 0.40) mmol/L for those with moderate hyperkalaemia (P = 0.0001). Similar increases in serum potassium after patiromer discontinuation were observed in the subgroup with EF ≤40%.
The majority of patients with HF had either no titrations or one titration during the first 8 weeks of the study, with a dose increase being more common than a dose decrease. During the first 4 weeks, 65 (61.9%) HF patients had no dose adjustments, while 30 (28.6%) had one dose adjustment. During Weeks 4 to 8, the proportions with no titration or one titration were 62.6% and 24.2%, respectively ( Table S2 ).
Safety
Patiromer was generally well tolerated (Table 3). Among the patients with HF, 74 (71%) reported at least one AE during the study; 13% reported AEs that were considered by the investigators to be related to patiromer. Hypomagnesaemia (9.5%) and worsening of CKD (6.6%) were the two most common AEs (as AEs are defined by the investigator, these events are not based on pre‐specified laboratory criteria; see below for details on laboratory‐defined hypomagnesaemia, hypokalaemia, and changes in renal function). AEs of hypomagnesaemia did not lead to any discontinuations, while worsening of CKD did in three (2.9%) patients (with onset during Week 3 in one patient, Week 4 in the second patient, and Week 41 in the third patient). Diarrhoea (none severe) occurred in five (4.8%) and constipation (none severe) occurred in four (3.8%) HF patients. The three most common treatment‐related AEs in HF patients were hypomagnesaemia (8.6%), abdominal discomfort (1.9%), and constipation (1.9%). Non‐dose‐dependent peripheral oedema was reported in eight patients overall (8/304, 2.6%, one patient with HF and seven patients without HF); none were related to patiromer based on investigator assessment. In no patient was peripheral oedema associated with events of cardiac failure or worsening of CKD. Two (1.9%) patients with HF had AEs of atrial fibrillation (new onset).
Table 3.
Safety summary over 52 weeks
| Parameter, n (%) | Patients with HF (n = 105) | Patients without HF (n = 199) |
|---|---|---|
| At least one AE | 74 (70.5) | 137 (68.8) |
| Most common AEs (≥5% of patients in any subgroup with or without HF) | ||
| Hypomagnesaemia | 10 (9.5) | 16 (8.0) |
| Worsening of CKD | 7 (6.7) | 21 (20.0) |
| Ventricular extrasystoles | 7 (6.7) | 4 (2.0) |
| Influenza | 6 (5.7) | 3 (1.5) |
| Hypoglycaemia | 6 (5.7) | 4 (2.0) |
| Diarrhoea | 5 (4.8) | 12 (6.0) |
| Worsening of HTN | 4 (3.8) | 20 (10.1) |
| Constipation | 4 (3.8) | 15 (7.5) |
| Most common patiromer‐related AEs (≥5% of patients in any subgroup with or without HF) | ||
| Hypomagnesaemia | 9 (8.6) | 13 (6.5) |
| Constipation | 2 (1.9) | 12 (6.0) |
| Discontinuations due to any AE | 8 (7.6) | 20 (10.1) |
| Discontinuations due to gastrointestinal AEs | 1 (1.0) | 4 (2.0) |
| Abdominal discomfort | 1 (1.0) | 0 (0.0) |
| Constipation | 0 (0.0) | 2 (1.0) |
| Diarrhoea | 0 (0.0) | 1 (0.5) |
| Vomiting | 0 (0.0) | 1 (0.5) |
| Electrolytes of interest | ||
| Hypomagnesaemiaa | ||
| <0.58 mmol/L | 15 (14.3) | 31 (15.6) |
| <0.49 mmol/L | 2 (1.9) | 10 (5.0) |
| Hypokalaemiaa | ||
| <3.5 mmol/L | 4 (3.8) | 13 (6.5) |
| Other AEs of interest | ||
| Worsening of HF | 3 (2.9) | 1 (0.5) |
| Atrial fibrillation | 2 (1.9) | 5 (2.5) |
| Oedema | 1 (1.0) | 7 (3.5) |
| At least one serious AE | 18 (17.1) | 26 (13.1) |
| Deaths | 9 (8.6) | 6 (3.0) |
AE, adverse event; CKD, chronic kidney disease; HF, heart failure; HTN, hypertension. In each subsection, AEs are presented in descending incidence in the total HF group.
Based on predefined laboratory criteria.
Of the 26 patients with EF ≤40%, 65% reported at least one AE during the study. The most common AE in these 26 patients was ventricular extrasystoles, occurring in three patients; two patients each had AEs of hypoglycaemia, hypomagnesaemia, thrombocytopaenia, and worsening of CKD. Mild or moderate gastrointestinal AEs were the most common class (occurring in two patients) of treatment‐related AEs, with abdominal discomfort, nausea, and vomiting each occurring in one patient with EF ≤40%.
Eight (7.6%) patients with HF (of whom five had EF ≤40%) discontinued patiromer because of AEs. In seven of eight patients, the AE leading to discontinuation was not related to patiromer in the judgement of the investigator. The most common AE leading to discontinuation in patients with HF was worsening of CKD [three (2.8%)]. Discontinuations due to gastrointestinal AEs were infrequent, occurring in one patient with HF (abdominal discomfort).
Serious AEs (SAEs) were observed in 18 (17.1%) HF patients (four with EF ≤40%) and in 26 (13.1%) patients without HF; none of these were attributed to patiromer, according to investigators. The SAEs occurring in at least one patient with HF are shown in Table S3 . The most common SAEs in patients with HF were sudden cardiac death [four (3.8%) patients] and worsening of CKD [three (2.9%) patients]. In addition, events of cardiac failure, regardless of seriousness, were summarized as they are of special interest in an HF population. These included AEs of cardiac failure or chronic HF (or worsening of chronic HF), left ventricular failure, or acute left ventricular failure and occurred in three (2.9%) patients with HF (two with EF ≤40%) and in nine (4.5%) patients without HF. In one of the three patients with HF (a patient with EF ≤40%) and five of the nine patients without HF, the events were serious (i.e. required hospitalization). No events of cardiac failure were considered by the investigators to be related to patiromer treatment. There were no reported events of sustained ventricular arrhythmias. The safety profile in patients without HF was generally consistent with that observed in HF patients (Tables 3 and S3 ).
Over 52 weeks through follow‐up, nine patients with HF (three with EF ≤40%) and six without HF died. For seven of nine patients with HF and five of six without HF, the cause—as adjudicated by an independent safety review board (SRB)—was cardiovascular (sudden cardiac death: four with HF and three without HF; acute myocardial infarction: two with HF and two without HF; stroke: one with HF). None of the deaths were considered by the SRB to be related to hyperkalaemia or hypokalaemia; none were considered by the investigators to be related to patiromer.
Mean serum calcium, magnesium, and phosphate levels remained within the normal ranges during the study ( Table S4 ). Mean serum calcium levels decreased slightly during the first 4 weeks of patiromer treatment (by 0.2 mmol/L) in patients with HF and remained either slightly decreased (in those with mild hyperkalaemia at baseline) or increased slightly by 0.2 mmol/L (in those with moderate hyperkalaemia at baseline) at Week 52. In patients with HF, the mean [standard error (SE)] change in serum magnesium from baseline to Week 52 was −0.05 (0.02) mmol/L in patients with mild hyperkalaemia and −0.04 (0.03) mmol/L in those with moderate hyperkalaemia; the mean reductions in serum magnesium in patients without HF were similarly small ( Table S4 ). In patients with EF ≤40% (n = 26), the mean (SE) change in serum magnesium from baseline to Week 52 was −0.05 (0.03) mmol/L. By central laboratory measures, serum magnesium <0.58 mmol/L was observed in 15 (14.3%) with HF (one with EF ≤40%) and in 31 (15.6%) patients without HF during the entire 52‐week treatment period (Table 3). Serum magnesium levels <0.49 mmol/L were seen in two (1.9%) HF (none with EF ≤40%) and 10 (5.0%) non‐HF patients, and none had serum magnesium <0.41 mmol/L. No patients with serum magnesium levels of <0.58 or <0.49 mmol/L had cardiac arrhythmias temporally associated with hypomagnesaemia or neuromuscular abnormalities other than myalgias. Hypokalaemia (pre‐defined as serum potassium <3.5 mmol/L by central laboratory measures) was seen in four (3.8%) patients with HF (none with EF ≤40%) and 13 (6.5%) patients without HF (Table 3). There were no clinically meaningful alterations in the mean estimated glomerular filtration rate and mean urine albumin‐to‐creatinine ratio during the study ( Table S5 ).
In patients with HF, the mean systolic/diastolic BP at baseline was 154/87 mmHg in patients with mild hyperkalaemia and 155/83 mmHg in those with moderate hyperkalaemia. A mean reduction from baseline to Week 52 in the systolic/diastolic BP of 18/12 and 25/8 mmHg, respectively, was observed ( Table S6 ). One HF patient reported an AE of mild dizziness. The mean baseline systolic/diastolic BP and change over 52 weeks in patients without HF were generally similar ( Table S6 ). In the small group of patients with EF ≤40%, mean BP was 151/91 mmHg at baseline; 17/26 patients had baseline systolic BP ≥140 mmHg. Mean BP decreased from baseline to Week 52 by 17/16 in the patients with EF ≤40%, with no reported AEs of hypotension or dizziness and no recorded systolic BP <107 mmHg.
Discussion
The results of this study confirm that patiromer is effective in normalizing serum potassium in hyperkalaemic patients with a clinical diagnosis of HF and coexisting CKD and diabetes, with reductions in serum potassium at Week 4 (the primary endpoint in the study21) consistent with those observed in patients without HF based on overlapping CIs. The results also show that serum potassium can be maintained within the normal range in these patients for a period of up to 1 year despite maintenance of ACE‐I and ARB medications. The HF patients enrolled in AMETHYST‐DN had predominantly mild symptoms and EF >40%. However, long‐term efficacy results were consistent in the small group of HF patients with EF ≤40%. The efficacy profile of patiromer was achieved with no or only one titration in the majority of patients, which is consistent with the titration data in the Phase 3 study (OPAL‐HK).25 This titration frequency may be overestimated given that dose adjustments were driven by a treatment algorithm that required a dose change for even minor excursions above a threshold, an action not likely to be commonly taken in the clinic. Approximately 88% or more of HF patients with CKD, diabetes, and mild hyperkalaemia and 73% or more of those with moderate hyperkalaemia maintained normokalaemia at each visit between Weeks 12 and 52.
Of note, this is the first analysis that reports the long‐term safety of patiromer in patients with HF. Patiromer was generally well tolerated, and its long‐term safety and tolerability profile was similar in patients with and without HF at baseline. Eight (7.6%) patients with HF, of whom five had EF ≤40%, discontinued patiromer because of AEs, with the most common reason being worsening of CKD (three patients; 2.9%); however, none of the three cases in which worsening of CKD occurred were deemed by the investigator to be related to patiromer. Overall, the majority of discontinuations due to AEs were not patiromer related. The most common gastrointestinal AE in patients with HF was diarrhoea (4.8%), which was mild in all cases except for one patient with moderate symptoms, and did not result in discontinuation of patiromer. There was a low incidence of hypokalaemia [serum potassium <3.5 mmol/L (3.8%)] and hypomagnesaemia [serum Mg <0.49 mmol/L (1.9%)] over the 1‐year study duration; importantly, there were no episodes of sustained ventricular arrhythmias or sudden cardiac death associated with either hypomagnesaemia or hypokalaemia, including in the patients with EF ≤40%. These results suggest that hyperkalaemic patients with predominantly mild HF can take patiromer chronically while maintaining their ACE‐I and ARB medication(s) without significantly increasing their risk of cardiac arrhythmias due to shifts in serum electrolytes. Serum calcium levels were measured during the study, and mean levels remained within the normal range, with slight (0.2 mmol/L) increases observed at Week 52 in patients with HF. An 8.4 g dose of patiromer contains approximately 1.6 g of calcium. As previously reported, at the maximal recommended daily dose (25.2 g/day), patiromer administration in healthy adults increased urine calcium by 1.82 mmol/day (73 mg/day) and decreased urine phosphate by 0.67 mmol/day (64 mg/day), suggesting that only a small fraction of the calcium released from patiromer is available for absorption and that some of the released calcium may bind phosphate.26
Of interest, mean systolic and diastolic BP were reduced over 52 weeks in patients with and without HF. However, this reduction is difficult to assess owing to a lack of a comparator group (placebo) but may be related to decreases in aldosterone associated with a reduction in serum potassium, as seen in OPAL‐HK,27 and/or to an increase in binding of sodium to patiromer and a consequent increase in the sodium excretion in the faeces.26, 28
Currently, patients with HF who develop hyperkalaemia are advised to reduce their RAASi dose if their serum potassium is >5.5 mmol/L and to discontinue RAASi therapy if their serum potassium is ≥6.0 mmol/L.2 While this strategy may avoid the consequences of hyperkalaemia such as an increase in ventricular arrhythmias and sudden cardiac death, it exposes patients to the long‐term risks of progressive HF and death, which prior studies have indicated are reduced by chronic, continuous use of RAASi therapies.1, 2, 3, 4, 5, 29 An analysis of a large health records database for patients with ≥2 serum potassium readings and ≥1 RAASi prescription showed that nearly two‐thirds of patients who had HF with or without CKD were on lower than their target doses of RAASi and that 15% to 16% had discontinued RAASi therapy altogether.29 RAASi doses were reduced following 16% to 21% of all hyperkalaemia events and discontinued following 22% to 27% of hyperkalaemia events in patients with cardiorenal co‐morbidities, suggesting that hyperkalaemia was an important reason for RAASi dose reduction or discontinuation.30 A recent follow‐up analysis further showed that patients with HF on maximum‐recommended‐dose RAASi had significantly lower rates of adverse outcomes or death compared with those on submaximum‐dose RAASi.31 In HF patients ≥65 years, annualized total all‐cause medical and pharmacy costs were also significantly lower in the group on maximum‐recommended‐dose RAASi compared with those on submaximum‐dose RAASi. Moreover, hyperkalaemia was a strong predictor of annualized total all‐cause medical and pharmacy costs; and in patients with HF, it was associated with a 50% increase in costs for patients <65 years and a 29% increase for those ≥65 years.31 Thus, a reduction to less than the RAASi target dose or its discontinuation in a patient with HF appears to increase the risk of serious adverse outcomes, including death, as well as total costs.30, 31
An alternative to reducing RAASi medications in patients with HF complicated by hyperkalaemia would be to add a potassium‐binding polymer such as sodium polystyrene sulfonate (SPS).32 However, this drug is poorly tolerated over the long term and has been associated with the occurrence of bowel necrosis.33, 34 Furthermore, SPS exchanges sodium for potassium and may increase total body sodium content;35 therefore, it must be used with caution in patients with chronic HF, especially those with right HF who are at risk for volume overload. An increase in sodium absorption in a patient with volume overload (such as those with chronic HF and/or CKD) could further exacerbate the symptoms and consequences of volume overload with both short‐ and long‐term adverse consequences, including hospitalization for HF and death. Patiromer, in contrast, exchanges calcium for potassium, and its use in the prevention or treatment of hyperkalaemia in patients with HF and/or CKD has not been associated with any evidence of increased oedema or other clinical evidence of volume overload.18, 20, 21, 25
The availability of a well‐tolerated and effective serum‐potassium‐lowering agent, such as patiromer, that exchanges calcium rather than sodium for potassium may have important implications for the management of patients with HF. Such an agent may enable more patients with HF and a clinical indication for RAASi therapy to initiate and maintain clinical‐trial‐proven doses of RAASi over the long term. However, it will be important to educate patients with HF who are on RAASi and initiate a potassium binder that they should not discontinue the latter without consulting their physician. As shown in the current analysis, discontinuing patiromer without concomitant modification (dose reduction or discontinuation) of RAASi may lead to a recurrence of hyperkalaemia and its clinical consequences.
Limitations
The study, although randomized, was not blinded. However, it would be difficult to adequately blind a long‐term study such as this one because patients are likely to have their serum potassium measured multiple times over the course of a year, which may reveal their assignment to the study drug. Second, the patients were identified as having HF based on physician diagnosis and quantitative assessment of HF severity (e.g. pro‐BNP) was not available. Moreover the majority of HF patients had EF >40% and approximately one‐quarter had NYHA Class I symptoms. The mild nature of the HF in the present analysis is consistent with the low use of MRA in this subgroup (~5%). Therefore, further data will be required to evaluate patiromer for the treatment of hyperkalaemia in larger numbers of HF patients, including those with more severe signs and symptoms of HF and an indication to receive an MRA. Finally, by protocol, changes in RAASi medications were allowed only for the management of BP, and so inferences related to changes in RAASi use or doses in the context of the treatment of hyperkalaemia in routine clinical practice cannot be made. It should, however, be pointed out that 89 (85%) patients with HF enrolled in the present study were able to maintain their RAASi for a period of up to 1 year without the need to discontinue them owing to safety concerns ( Table S1 ).
Conclusions
The long‐term administration of patiromer to hyperkalaemic patients with a clinical diagnosis of HF appears to be effective and well tolerated, consistent with the findings in patients without a history of HF at baseline. The availability of an effective potassium‐lowering agent such as patiromer, which exchanges potassium for calcium rather than sodium, may increase the number of HF patients who can maintain their ACE‐I or ARB at target doses over the long term. Further studies will be required to determine if patiromer, when used to treat hyperkalaemia, allows maintenance of optimal doses of MRAs in those indicated to receive them, especially patients with EF ≤40%, as well as to determine if this strategy improves clinical outcomes in HF patients in comparison with the current approach of dose reduction or discontinuation of RAASi in patients who develop hyperkalaemia.
Conflict of interest
Dr Pitt reports personal fees for consulting with Bayer, Astra Zeneca, Sanofi, Relypsa/Vifor, scPharmaceuticals, Tricida, KBP Biosciences, Sarfez, and Stealth Peptides. He has stock options with scPharmaceuticals, KBP Biosciences, Tricida, and Sarfez. He serves on a data safety monitoring committee for Johnson & Johnson. In addition, he has a pending patent EFS ID 14916043, application number 61762661/UM‐33001/US‐1PRO, for the site‐specific delivery of eplerenone to the myocardium.
Dr Bakris reports personal fees from Relypsa and other from Relypsa during the conduct of the study; personal fees from AbbVie, Takeda, Relypsa, Janssen, and Bayer; and grants from Takeda outside the submitted work.
Dr Weir reports personal fees from scientific advisory boards from Relypsa, Inc., and Vifor Pharma Management Ltd, both Vifor Pharma Group Companies, and from Akebia, Janssen, AstraZeneca, Sanofi, MSD, AbbVie, and Boston Scientific outside the submitted work.
Dr Freeman reports personal fees from Relypsa during the conduct of the study; personal fees from Relypsa outside the submitted work; and stock option grants for involvement in helping design the overall clinical trial programme for patiromer.
Dr Lainscak reports personal fees from Relypsa and other from Relypsa during the conduct of the study and personal fees from Novartis, Relypsa, and Pfizer outside the submitted work.
Dr Mayo reports employment by Relypsa during the conduct of the study.
Dr Garza reports employment by Relypsa during the conduct of the study.
Dr Zawadzki reports personal fees from Relypsa during the conduct of the study and personal fees from Relypsa outside the submitted work.
Dr Berman reports employment by Relypsa and other from Relypsa during the conduct of the study. In addition, Dr Berman has a patent WO 2014/058905 pending.
Dr Bushinsky reports personal fees from Relypsa during the conduct of the study; stock and stock options from Amgen and Tricida; personal fees for consulting outside the submitted work from Amgen, Sanofi/Genzyme, OPKO, and Tricida; and stock in Amgen outside of the submitted work. Dr Bushinsky reports research support from the National Institutes of Health and from the Renal Research Institute outside of the submitted work.
Funding
Relypsa, Inc., a Vifor Pharma Group Company
Supporting information
Table S1. Reasons for discontinuation over 52 weeks*.
Table S2. Patiromer titration over 8 weeks.
Table S3. Serious adverse events occurring in at least one patient with HF over 52 weeks*.
Table S4. Mean (SE) change from baseline to week 4 and to week 52 in serum calcium, magnesium, and phosphate in patients with HF.
Table S5. Mean (SE) change from baseline to week 52 in eGFR and ACR.
Table S6. Mean (SE) change in systolic and diastolic BP in patients with HF from baseline to week 4 and to week 52.
Acknowledgements
Editorial assistance was provided by Narender Dhingra, MBBS, PhD, of AlphaBioCom, LLC; and Julie Obeid, of Relypsa, Inc.; and was funded by Relypsa, Inc., a Vifor Pharma Group Company.
Pitt, B. , Bakris, G. L. , Weir, M. R. , Freeman, M. W. , Lainscak, M. , Mayo, M. R. , Garza, D. , Zawadzki, R. , Berman, L. , and Bushinsky, D. A. (2018) Long‐term effects of patiromer for hyperkalaemia treatment in patients with mild heart failure and diabetic nephropathy on angiotensin‐converting enzymes/angiotensin receptor blockers: results from AMETHYST‐DN. ESC Heart Failure, 5: 592–602. 10.1002/ehf2.12292.
Clinical trial registration: ClinicalTrials.gov registry identifier NCT01371747
References
- 1. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: An update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 2016; 22: 659–669. [Google Scholar]
- 2. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Rouilope LM, Ruschitzka F, Rutten FH, van der Meer P, Authors/Task Force Members, Document Reviewers . ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18: 891–975. [DOI] [PubMed] [Google Scholar]
- 3. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M, Eplerenone Post‐Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators . Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348: 1309–1321. [DOI] [PubMed] [Google Scholar]
- 4. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341: 709–717. [DOI] [PubMed] [Google Scholar]
- 5. Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B, EMPHASIS‐HF Study Group . Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364: 11–21.21073363 [Google Scholar]
- 6. Kuijvenhoven MA, Haak EA, Gombert‐Handoko KB, Crul M. Evaluation of the concurrent use of potassium‐influencing drugs as risk factors for the development of hyperkalemia. Int J Clin Pharm 2013; 35: 1099–1104. [DOI] [PubMed] [Google Scholar]
- 7. Jain N, Kotla S, Little BB, Weideman RA, Brilakis ES, Reilly RF, Banerjee S. Predictors of hyperkalemia and death in patients with cardiac and renal disease. Am J Cardiol 2012; 109: 1510–1513. [DOI] [PubMed] [Google Scholar]
- 8. Einhorn LM, Zhan M, Hsu VD, Walker LD, Moen MF, Seliger SL, Weir MR, Fink JC. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med 2009; 169: 1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pitt B, Bakris GL. New potassium binders for the treatment of hyperkalemia: current data and opportunities for the future. Hypertension 2015; 66: 731–738. [DOI] [PubMed] [Google Scholar]
- 10. Palmer BF. Managing hyperkalemia caused by inhibitors of the renin–angiotensin–aldosterone system. N Engl J Med 2004; 351: 585–592. [DOI] [PubMed] [Google Scholar]
- 11. Albert NM, Yancy CW, Liang L, Zhao X, Hernandez AF, Peterson ED, Cannon CP, Fonarow GC. Use of aldosterone antagonists in heart failure. JAMA 2009; 302: 1658–1665. [DOI] [PubMed] [Google Scholar]
- 12. Perazella MA, Mahnensmith RL. Hyperkalemia in the elderly: drugs exacerbate impaired potassium homeostasis. J Gen Intern Med 1997; 12: 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin–angiotensin system: meta‐analysis of randomised trials. BMJ 2013; 346: f360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Epstein M, Pitt B. Recent advances in pharmacological treatments of hyperkalemia: focus on patiromer. Expert Opin Pharmacother 2016; 17: 1435–1448. [DOI] [PubMed] [Google Scholar]
- 15. Li L, Harrison SD, Cope MJ, Park C, Lee L, Salaymeh F, Madsen D, Benton WW, Berman L, Buysse J. Mechanism of action and pharmacology of patiromer, a nonabsorbed cross‐linked polymer that lowers serum potassium concentration in patients with hyperkalemia. J Cardiovasc Pharmacol Ther 2016; 21: 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Veltassa (patiromer) for oral suspension [package insert]. Redwood City, CA: Relypsa, Inc.; 2016. [Google Scholar]
- 17. European Medicines Agency . Veltassa. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004180/human_med_002141.jsp&mid=WC0b01ac058001d124. (24 August 2017).
- 18. Pitt B, Bakris GL, Bushinsky DA, Garza D, Mayo MR, Stasiv Y, Christ‐Schmidt H, Berman L, Weir MR. Effect of patiromer on reducing serum potassium and preventing recurrent hyperkalaemia in patients with heart failure and chronic kidney disease on RAAS inhibitors. Eur J Heart Fail 2015; 17: 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bushinsky DA, Williams GH, Pitt B, Weir MR, Freeman MW, Garza D, Stasiv Y, Li E, Berman L, Bakris GL. Patiromer induces rapid and sustained potassium lowering in patients with chronic kidney disease and hyperkalemia. Kidney Int 2015; 88: 1427–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ, PEARL‐HF Investigators . Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double‐blind, placebo‐controlled study in patients with chronic heart failure (the PEARL‐HF) trial. Eur Heart J 2011; 32: 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bakris GL, Pitt B, Weir MR, Freeman MW, Mayo MR, Garza D, Stasiv Y, Zawadzki R, Berman L, Bushinsky DA, AMETHYST‐DN Investigators . Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST‐DN randomized clinical trial. JAMA 2015; 314: 151–161. [DOI] [PubMed] [Google Scholar]
- 22. ICH Harmonised Tripartite Guideline . Guideline for good clinical practice E6. International Conference on Harmonisation; 1996.
- 23. World Medical Association Declaration of Helsinki . Ethical principles for medical research involving human subjects. JAMA 2013; 310: 2191–2194. [DOI] [PubMed] [Google Scholar]
- 24. Directive 2001/20/EC of the European Parliament and of the Council. 2001.
- 25. Weir MR, Bakris GL, Bushinsky DA, Mayo MR, Garza D, Stasiv Y, Wittes J, Christ‐Schmidt H, Berman L, Pitt B, Investigators OPAL‐HK. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372: 211–221. [DOI] [PubMed] [Google Scholar]
- 26. Bushinsky DA, Spiegel DM, Gross C, Benton WW, Fogli J, Hill Gallant KM, Du Mond C, Block GA, Weir MR, Pitt B. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11: 1769–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weir MR, Bakris GL, Gross C, Mayo MR, Garza D, Stasiv Y, Yuan J, Berman L, Williams GH. Treatment with patiromer decreases aldosterone in patients with chronic kidney disease and hyperkalemia on renin–angiotensin system inhibitors. Kidney Int 2016; 90: 696–704. [DOI] [PubMed] [Google Scholar]
- 28. Weir MR, Spiegel DM, Wilson D, Benton WW, Yuan J, Gross C. Aldosterone, renin and blood pressure during patiromer treatment of hyperkalemia in CKD. J Am Soc Nephrol 2016; 17: 201A. [Google Scholar]
- 29. The SOLVD Investigators , Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325: 293–302. [DOI] [PubMed] [Google Scholar]
- 30. Epstein M, Reaven NL, Funk SE, McGaughey KJ, Oestreicher N, Knispel J. Evaluation of the treatment gap between clinical guidelines and the utilization of renin–angiotensin–aldosterone system inhibitors. Am J Manag Care 2015; 21: S212–S220. [PubMed] [Google Scholar]
- 31. Epstein M, Alvarez PJ, Reaven NL, Funk SE, McGaughey KJ, Brenner MS, Benton W, Golestaneh L. Evaluation of clinical outcomes and costs based on prescribed dose level of renin–angiotensin–aldosterone system inhibitors. Am J Manag Care 2016; 22: S313–S326. [PubMed] [Google Scholar]
- 32. Chernin G, Gal‐Oz A, Ben‐Assa E, Schwartz IF, Weinstein T, Schwartz D, Silverberg DS. Secondary prevention of hyperkalemia with sodium polystyrene sulfonate in cardiac and kidney patients on renin–angiotensin–aldosterone system inhibition therapy. Clin Cardiol 2012; 35: 32–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hagan AE, Farrington CA, Wall GC, Belz MM. Sodium polystyrene sulfonate for the treatment of acute hyperkalemia: a retrospective study. Clin Nephrol 2016; 85: 38–43. [DOI] [PubMed] [Google Scholar]
- 34. Harel Z, Harel S, Shah PS, Wald R, Perl J, Bell CM. Gastrointestinal adverse events with sodium polystyrene sulfonate (Kayexalate) use: a systematic review. Am J Med 2013; 126: 264.e9–e24. [DOI] [PubMed] [Google Scholar]
- 35. Kayexalate (Sodium Polystyrene Sulfonate) Cation‐Exchange Resin [Prescribing Information]. Bridgewater, NJ: sanofi‐aventis; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Reasons for discontinuation over 52 weeks*.
Table S2. Patiromer titration over 8 weeks.
Table S3. Serious adverse events occurring in at least one patient with HF over 52 weeks*.
Table S4. Mean (SE) change from baseline to week 4 and to week 52 in serum calcium, magnesium, and phosphate in patients with HF.
Table S5. Mean (SE) change from baseline to week 52 in eGFR and ACR.
Table S6. Mean (SE) change in systolic and diastolic BP in patients with HF from baseline to week 4 and to week 52.