

Abstract
Aims
The EMPA‐REG OUTCOME study showed reduced mortality and hospitalization due to heart failure (HF) in diabetic patients treated with empagliflozin. Overexpression and Ca2+‐dependent activation of Ca2+/calmodulin‐dependent kinase II (CaMKII) are hallmarks of HF, leading to contractile dysfunction and arrhythmias. We tested whether empagliflozin reduces CaMKII‐ activity and improves Ca2+‐handling in human and murine ventricular myocytes.
Methods and results
Myocytes from wild‐type mice, mice with transverse aortic constriction (TAC) as a model of HF, and human failing ventricular myocytes were exposed to empagliflozin (1 μmol/L) or vehicle. CaMKII activity was assessed by CaMKII–histone deacetylase pulldown assay. Ca2+ spark frequency (CaSpF) as a measure of sarcoplasmic reticulum (SR) Ca2+ leak was investigated by confocal microscopy. [Na+]i was measured using Na+/Ca2+‐exchanger (NCX) currents (whole‐cell patch clamp). Compared with vehicle, 24 h empagliflozin exposure of murine myocytes reduced CaMKII activity (1.6 ± 0.7 vs. 4.2 ± 0.9, P < 0.05, n = 10 mice), and also CaMKII‐dependent ryanodine receptor phosphorylation (0.8 ± 0.1 vs. 1.0 ± 0.1, P < 0.05, n = 11 mice), with similar results upon TAC. In murine myocytes, empagliflozin reduced CaSpF (TAC: 1.7 ± 0.3 vs. 2.5 ± 0.4 1/100 μm−1 s−1, P < 0.05, n = 4 mice) but increased SR Ca2+ load and Ca2+ transient amplitude. Importantly, empagliflozin also significantly reduced CaSpF in human failing ventricular myocytes (1 ± 0.2 vs. 3.3 ± 0.9, P < 0.05, n = 4 patients), while Ca2+ transient amplitude was increased (F/F0: 0.53 ± 0.05 vs. 0.36 ± 0.02, P < 0.05, n = 3 patients). In contrast, 30 min exposure with empagliflozin did not affect CaMKII activity nor Ca2+‐handling but significantly reduced [Na+]i.
Conclusions
We show for the first time that empagliflozin reduces CaMKII activity and CaMKII‐dependent SR Ca2+ leak. Reduced Ca2+ leak and improved Ca2+ transients may contribute to the beneficial effects of empagliflozin in HF.
Keywords: Empagliflozin, Heart failure, CaMKII, Calcium, Ca leak
Background
Treatment of diabetic patients with cardiovascular disease with the novel anti‐diabetic drug empagliflozin, a selective inhibitor of sodium‐dependent glucose transporter 2 (SGLT2) in the renal proximal tubules, reduced the incidence of cardiovascular death by 38% (EMPA‐REG OUTCOME trial, n = 7020 patients).1 The underlying mechanisms are hotly discussed. Surprisingly, the beneficial effects were not driven by a reduction in ischaemic endpoints (stroke or myocardial infarction) but by a reduction in the risk of hospitalization for heart failure (HF).1 In addition, empagliflozin exerts the same beneficial cardiovascular effects after adjustment for ischaemic risk factors (blood pressure, low‐density lipoprotein cholesterol, HbA1c).2 In a mediation analysis of this trial, changes in haematocrit (and haemoglobin) but not HbA1c appeared to be the variables with the largest impact on the risk of cardiovascular death.3 This argues strongly against glycaemic control as the underlying cardio‐protective mechanism.4 As a consequence, large Phase III trials are currently initiated that investigate empagliflozin in non‐diabetic patients with HF (EMPEROR and EMPERIAL trial programs). Interestingly, in a non‐diabetic mouse model of afterload‐induced HF, empagliflozin prevented worsening of left ventricular function.5 Overexpression and activation of Ca2+/calmodulin‐dependent kinase II (CaMKII) are hallmarks of HF3 and also found in diabetic patients,4 leading to increased diastolic Ca2+ leak from the sarcoplasmic reticulum (SR) and reduced SR Ca2+ load, causing contractile dysfunction and arrhythmias.3 Interestingly, CaMKII is activated by increased cytosolic Ca2+ concentrations.
Aims
We tested whether empagliflozin inhibits CaMKII activity and diastolic SR Ca2+ leak in cultured human and murine ventricular cardiomyocytes.
Methods
Experiments conform to the Declaration of Helsinki and were approved by local authorities. Left ventricular myocardium was acquired from explanted hearts of heart transplant recipients with end‐stage HF (patient characteristics in Table S1 ). Atrial tissue was acquired from patients undergoing coronary artery bypass grafting. Written consent had been given prior to tissue donation. For some experiments, C57BL/6 mice (8–12 weeks) were used, and HF was induced by transverse aortic constriction (TAC; 5 weeks, as previously described6). Echocardiographic data on heart structure and function of TAC mice are shown in Table S2. Isolated cardiomyocytes were exposed to either vehicle or 1 μmol/L empagliflozin. This concentration is in the range of the typical plasma concentration upon 25 mg q.d. oral treatment of patients.7 CaMKII activity was measured by CaMKII–histone deacetylase (HDAC4) pulldown normalized to CaMKII expression.8 Cell isolation, culture, Western blots, and measurement of Na+ and Ca2+ handling were employed as previously described.9, 10 Cardiomyocyte survival after 30 min or 24 h exposure to empagliflozin or vehicle control was not significantly different. Whole‐cell voltage‐clamp patch‐clamp experiments were performed as described previously, and subsarcolemmal [Na+]i was calculated using the Nernst equation.11 Liquid chromatography–mass spectrometry (LC–MS) from SGLT2 gel bands from non‐failing and failing human myocardium and high‐resolution selected reaction monitoring (HR‐SRM) for SGLT2 from human failing myocardium, and murine kidney and myocardium were performed as previously described. Data are shown as mean ± SEM; normality was tested using the Shapiro–Wilk normality test and parametric [ANOVA or (paired) Student's t‐test] or non‐parametric (Mann–Whitney test or Wilcoxon signed‐rank test) tests were used as appropriate.
An extended methods section can be found in the Supporting Information.
Results
CaMKII activity was measured using CaMKII pulldown by HDAC4 binding. Interestingly, 24 h exposure of murine wild‐type (WT) ventricular cardiomyocytes to empagliflozin (1 μmol/L) significantly reduced CaMKII activity from 4.2 ± 0.9 to 1.6 ± 0.7 (P < 0.05, n = 10 mice, Figure 1 A). As CaMKII is a critical regulator of Ca2+ handling, we assessed SR Ca2+ load (caffeine transients, 10 mmol/L) and diastolic Ca2+ leak (Ca2+ sparks) in isolated murine WT ventricular cardiomyocytes. Interestingly, compared with vehicle, 24 h empagliflozin exposure significantly increased SR Ca2+ load (Figure 1 B). Without post‐translational modifications of cardiac ryanodine receptor (RyR2) that directly reduce their open probability, increased luminal (SR) Ca2+ would lead to increased Ca2+ leak.12 However, compared with vehicle, 24 h empagliflozin exposure did not increase Ca2+ spark frequency (CaSpF) as a measure of Ca2+ leak but significantly reduced the SR Ca2+ leak/SR Ca2+ load ratio (Figure 1 C). Accordingly, CaMKII‐dependent phosphorylation of RyR2 (at serine 2814) was significantly decreased, while the protein kinase A (PKA)‐dependent phosphorylation (at S2809) was unaltered upon 24 h exposure to empagliflozin (Figure 1 D). Interestingly, phosphorylation of phospholamban (PLN) at threonine 17, perhaps the most frequently studied CaMKII phospho‐site, was significantly reduced by 24 h empagliflozin exposure (Figure 1 E).
Figure 1.

(A) Original Western blots of CaMKII–HDAC pulldown, total HDAC, CaMKII expression, and GADPH from lysates of murine ventricular cardiomyocytes exposed to empagliflozin (Empa, 1 μmol/L) for 24 h. (B) Mean data for caffeine‐induced Ca2+ transient amplitude (CaffTransAmpl) as a measure of SR Ca2+ load in murine ventricular cardiomyocytes after 24 h empagliflozin exposure (Fura‐2). (C) Original confocal linescans (left panel) and mean data for CaSpF (Fluo‐4, middle panel) and SR Ca2+ leak normalized to SR Ca2+ load (right panel) in murine ventricular myocytes upon 24 h empagliflozin exposure. Ca2+ leak was calculated as CaSpF × CaSpAmpl × CaSpWidth (full width at half maximum) × CaSpDuration (full duration at half maximum) and divided by CaffTransAmpl. (D) Original Western blots and mean data for phospho‐S2814/RyR2 ratio, phospho‐S2809/RyR2 ratio, and RyR2 expression from murine ventricular cardiomyocytes after 24 h empagliflozin (E) or vehicle (V) exposure. (E) Original Western blots and mean data for phospho‐T17/PLN ratio, as well as PLN and SERCA expression from murine ventricular cardiomyocytes after 24 h empagliflozin (E) or vehicle (V) exposure. (A–C) *P < 0.05 Mann–Whitney test. (D and E) *P < 0.05 Wilcoxon signed‐rank test or paired t‐test as appropriate. CaMKII, Ca2+/calmodulin‐dependent kinase II; CaSpF, Ca2+ spark frequency; HDAC, histone deacetylase; PLN, phospholamban; SR, sarcoplasmic reticulum.
In order to test if empagliflozin also affects CaMKII‐dependent SR Ca2+ leak in HF, ventricular myocytes were isolated from mice with HF upon TAC and from explanted hearts of patients with end‐stage HF. Intriguingly, compared with vehicle, 24 h empagliflozin exposure significantly reduced CaSpF in TAC myocytes and human failing ventricular cardiomyocytes (Figure 2 A). This empagliflozin effect was not restricted to HF. Treatment with empagliflozin also reduced CaSpF in human atrial cardiomyocytes (Figure 2 A). Accordingly, 24 h empagliflozin exposure significantly reduced CaMKII activity (CaMKII pulldown by HDAC binding) and CaMKII‐dependent RyR2 and PLN phosphorylation in failing TAC cardiomyocytes (Figure 2 B) and CaMKII‐dependent RyR2 phosphorylation in human failing ventricular myocytes (Figure 2 C). PKA‐dependent RyR2 phosphorylation (at 2809), on the other hand, was not affected by empagliflozin in both in TAC and human failing cardiomyocytes (Figure 2 B and C). Expression of neither RyR2 nor PLN was significantly altered.
Figure 2.

(A) Mean data for CaSpF in failing murine ventricular myocytes (TAC, left panel), human atrial myocytes (middle panel), or human failing ventricular cardiomyocytes (right panel, original line scans also shown). *P < 0.05 Mann–Whitney test. (B) Left panel: original Western blots of CaMKII–HDAC pulldown, total HDAC, CaMKII expression, and GADPH, as well as mean data of CaMKII activity normalized to CaMKII expression from lysates of murine TAC ventricular cardiomyocytes exposed to empagliflozin (Empa, 1 μmol/L) for 24 h. Middle panel: original Western blots and mean data for phospho‐S2814/RyR2 ratio and phospho‐S2809/RyR2 ratio from murine TAC ventricular cardiomyocytes after 24 h empagliflozin (E) or vehicle (V) exposure. Right panel: original Western blots and mean data for phospho‐T17/PLN ratio, as well as PLN and SERCA expression from murine ventricular cardiomyocytes after 24 h empagliflozin (E) or vehicle (V) exposure. (C) Original Western blots and mean data for phospho‐S2814/RyR2 ratio, phospho‐S2809/RyR2 ratio, and RyR2 expression from human failing ventricular cardiomyocytes. (B and C) *P < 0.05 Wilcoxon signed‐rank test or paired t‐test as appropriate. CaMKII, Ca2+/calmodulin‐dependent kinase II; CaSpF, Ca2+ spark frequency; HDAC, histone deacetylase; PLN, phospholamban; TAC, transverse aortic constriction.
Increased SR Ca2+ load can lead to increased systolic SR Ca2+ release and improved contractile function. We show here that, compared with vehicle, 24 h empagliflozin exposure significantly increased Ca2+ transient amplitude (CaTransAmpl) in both healthy murine ventricular myocytes and failing ventricular myocytes from TAC mice and human failing hearts (Figure 3 A). This increase was less pronounced in WT compared with β‐adrenergic stimulation (isoproterenol (ISO) 10−7 M). At 1 Hz, ISO increased CaTransAmpl 2.1 ± 0.65‐fold relative to vehicle (n = 9) but empagliflozin increased 1.6 ± 0.35‐fold only (n = 9 vs. 13 mice).
Figure 3.
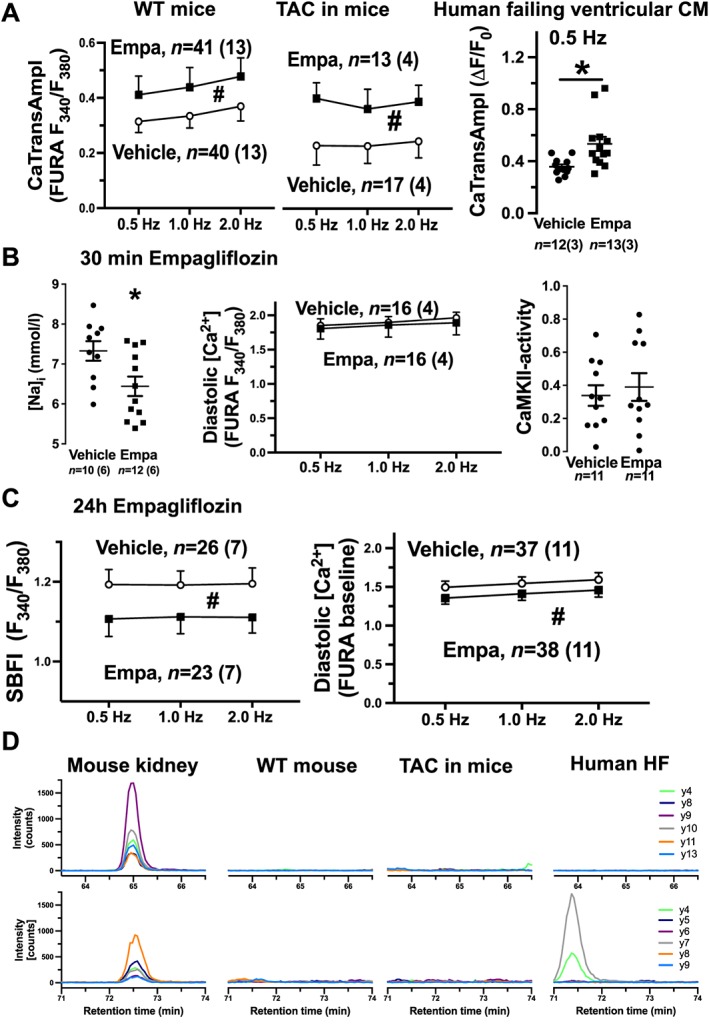
(A) Mean data for Ca2+ transient amplitude (CaTransAmpl) in healthy murine ventricular myocytes (left panel), failing murine ventricular myocytes (TAC, middle panel, both Fura‐2), or human failing ventricular cardiomyocytes (right panel, Fluo‐4) elicited by electrical field stimulation at indicated frequencies. (B) Mean data of subsarcolemmal [Na+]i in murine WT CM (left panel, calculated from Na+/Ca2+‐exchanger (NCX) current measured by whole‐cell patch clamp), diastolic [Ca2+]i (Fura‐2, middle panel) in murine WT CM (electrical field stimulation at indicated frequencies), and CaMKII activity (CaMKII–HDAC pulldown) in human failing ventricular tissue after 30 min incubation with empagliflozin. (C) Mean data of [Na+]i [SBFI (sodium‐binding benzofuran isophthalate = sodium indicator) fluorescence, left panel] and diastolic [Ca2+]i (Fura‐2, right panel) in murine WT CM after 24 h empagliflozin exposure. (D) Representative chromatographic images of HR‐SRM analyses of human and murine SGLT2 protein. Two proteotypic SGLT2 peptides that are identical between mouse and human were used for highly sensitive detection of SGLT2 (top panel, VCGTEVGCSNIAYPR; bottom panel, GTVGGYFLAGR). For each peptide, six transitions (y4, 8, 9, 10, 11, 13 or y4, 5, 6, 7, 8, 9, respectively) were detected. In contrast to clear SGLT2 expression in mouse kidney (at retention times of 65 or 72.5 min), no signal was detected neither in WT or TAC mouse hearts nor in human failing myocardium. Of note, the signal at 71.5 min retention time in human HF is unspecific. *P < 0.05 (Mann–Whitney test), #P < 0.05 two‐way ANOVA. CaMKII, Ca2+/calmodulin‐dependent kinase II; HDAC, histone deacetylase; HR‐SRM, high‐resolution selected reaction monitoring; TAC, transverse aortic constriction.
In contrast to 24 h exposure, acute empagliflozin exposure for 30 min did not reduce CaSpF (in healthy murine ventricular myocytes, TAC myocytes, or human failing ventricular myocytes) or SR Ca2+ load (murine ventricular myocytes; data not shown). Furthermore, acute empagliflozin exposure for 30 min also did not alter diastolic Ca2+ concentration (WT myocytes) or CaMKII activity (human failing ventricular tissue; Figure 3 B, n = 11 patients). This suggests that direct inhibitory effects of empagliflozin on CaMKII or RyR2 are unlikely. Interestingly, consistent with previous data,5, 6 acute (30 min) empagliflozin exposure significantly reduced subsarcolemmal [Na+]i (measured by Na+/Ca2+‐exchanger (NCX) currents; Figure 3 B). Therefore, empagliflozin may rather directly impair Na+ entry or elimination. To test if empagliflozin may inhibit SGLT2 in the heart, which is a Na+ transporter, we analysed SGLT2 expression in ventricular myocardium from three patients with end‐stage HF and three non‐failing donor hearts by LC–MS. No SGLT2 expression was detected using this methodology (data not shown). We further analysed SGLT2 expression by HR‐SRM in murine WT myocardium and kidney, TAC myocardium, and human failing myocardium. In contrast to a robust SGLT2 expression in murine kidney, no SGLT2 signal was detected in myocardium (Figure 3 D), full data set linked in the Supporting Information), which is in accordance with previous data.13 Thus, cardiac SGLT2 inhibition can be excluded as potential mechanism of action of empagliflozin on the heart. Baartscheer et al. recently proposed that empagliflozin may reduce cardiomyocyte [Na+]i and [Ca2+]i by inhibition of Na+/H+ exchange.14 As CaMKII is activated by increased diastolic [Ca2+], which may be a consequence of Na+ entry, we also measured Na+ and diastolic Ca2+. Interestingly, 24 h empagliflozin exposure significantly reduced cytosolic [Na+]i and diastolic [Ca2+]i (Figure 3 C).
Conclusions
We show for the first time that empagliflozin potently reduces CaMKII activity in isolated failing and non‐failing murine ventricular myocytes. More importantly, empagliflozin also reduced CaMKII‐dependent phosphorylation of RyR2 in not only murine but also failing human ventricular myocytes. This results in a significantly reduced SR Ca2+ leak and improved contractility as measured by significantly increased Ca2+ transient amplitude in murine (TAC) and human failing ventricular myocytes. The mechanism by which empagliflozin reduces CaMKII activity has yet to be discerned, but our data demonstrate that empagliflozin may be useful in the treatment of pathologies with increased CaMKII activity, such as HF.
Conflict of interest
Julian Mustroph, Olivia Wagemann, Charlotte M. Lücht, Maximilian Trum, Karin P. Hammer, Can Martin Sag, Simon Lebek, Daniel Tarnowski, Jörg Reinders, Filippo Perbellini, Cesare Terracciano, Christof Schmid, Simon Schoppka, Michael Hilker, York Zausig, Steffen Pabel, Samuel T. Sossalla, Frank Schweda, and Stefan Wagner declare that they have no conflict of interest. Lars S. Maier gives talks for Boehringer Ingelheim, the company that sells empagliflozin.
Funding
This work was supported by grants from Deutsche Forschungsgemeinschaft (WA 2539/4‐1, WA 2539/5‐1, and WA 2539/7‐1 to S.W., and MA 1982/5‐1 and MA 1982/7‐1 to L.S.M.), faculty grants from the University of Regensburg (ReForM C program, L.S.M., S.T.S., and S.W.), and grant from Marga und Walter Boll‐Stiftung (S.T.S.).
Supporting information
Table S1. Clinical characteristics of patient, from which atrial biopsies were obtained (upper table), or from which left ventricular tissue of explanted hearts (after heart transplantation, lower table) was used. y = yes, n = no, Nr = number, CAD = coronary artery disease, ICM = ischemic cardiomyopathy, DCM = dilated cardiomyopathy, EF = ejection fraction in %, LVEDD = left ventricular enddiastolic diameter in mm, m = male, SR = sinus rhythm, CABG = coronary artery bypass graft, valves = valvular dysfunction, MR = mitral regurgitation, TR = tricuspid regurgitation x° = indicated severity degree according to international standards, Art Hyp = arterial hypertension, KD = kidney disease, GFR = glomerular filtration rate in ml/min, NTproBNP in pg/ml (internal cutoff <150 pg/ml).
Table S2. Echocardiographic data obtained from 16 mice. OP = TAC surgery, bpm = beats per minute, LV = left ventricle, AWThd = anterior wall thickness in diastole, PWThd = posterior wall thickness in diastole, LVIDs = left ventricular internal diameter in systole, LVIDd = left ventricular internal diameter in diastole, *P < 0.05 versus respective pre‐OP‐measurement (Student's t‐test).
Acknowledgements
We thank Thomas Sowa and Felicia Radtke for their technical expertise.
Mustroph, J. , Wagemann, O. , Lücht, C. M. , Trum, M. , Hammer, K. P. , Sag, C. M. , Lebek, S. , Tarnowski, D. , Reinders, J. , Perbellini, F. , Terracciano, C. , Schmid, C. , Schopka, S. , Hilker, M. , Zausig, Y. , Pabel, S. , Sossalla, S. T. , Schweda, F. , Maier, L. S. , and Wagner, S. (2018) Empagliflozin reduces Ca/calmodulin‐dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Failure, 5: 642–648. 10.1002/ehf2.12336.
References
- 1. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373: 2117–2128. [DOI] [PubMed] [Google Scholar]
- 2. Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA‐REG OUTCOME trial. Eur Heart J 2016; 37: 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Inzucchi SE, Zinman B, Fitchett D, Wanner C, Ferrannini E, Schumacher M, Schmoor C, Ohneberg K, Johansen OE, George JT, Hantel S, Bluhmki E, Lachin JM. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA‐REG OUTCOME trial. Diabetes Care 2018; 41: 356–363. [DOI] [PubMed] [Google Scholar]
- 4. Wanner C, Lachin JM, Inzucchi SE, Fitchett D, Mattheus M, George J, Woerle HJ, Broedl UC, von EM, Zinman B, EMPA‐REG OUTCOME Investigators . Empagliflozin and clinical outcomes in patients with type 2 diabetes mellitus, established cardiovascular disease, and chronic kidney disease. Circulation 2018; 137: 119–129. [DOI] [PubMed] [Google Scholar]
- 5. Byrne NJ, Parajuli N, Levasseur JL, Boisvenue J, Beker DL, Masson G, Fedak PWM, Verma S, Dyck JRB. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload‐induced heart failure. JACC Basic Transl Sci 2017; 2: 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, Sag CM, Hund TJ, Mohler PJ, Belardinelli L, Hasenfuss G, Maier LS, Sossalla S. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure‐induced heart disease. J Mol Cell Cardiol 2013; 61: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co‐transporter 2 inhibitor. Clin Pharmacokinet 2014; 53: 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kreusser MM, Lehmann LH, Keranov S, Hoting M‐O, Oehl U, Kohlhaas M, Reil J‐C, Neumann K, Schneider MD, Hill JA, Dobrev D, Maack C, Maier LS, Gröne H‐J, Katus HA, Olson EN, Backs J. Cardiac CaM kinase II genes δ and γ contribute to adverse remodeling but redundantly inhibit calcineurin‐induced myocardial hypertrophy. Circulation 2014; 130: 1262–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species‐activated Ca/calmodulin kinase IIδ is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res 2011; 108: 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hofhuis J, Bersch K, Büssenschütt R, Drzymalski M, Liebetanz D, Nikolaev VO, Wagner S, Maier LS, Gärtner J, Klinge L, Thoms S. Dysferlin mediates membrane tubulation and links T‐tubule biogenesis to muscular dystrophy. J Cell Sci 2017; 130: 841–852. [DOI] [PubMed] [Google Scholar]
- 11. Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O'Rourke B. Role of sodium–calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res 2003; 93: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shannon TR. Quantitative assessment of the SR Ca2+ leak–load relationship. Circ Res 2002; 91: 594–600. [DOI] [PubMed] [Google Scholar]
- 13. von Lewinski D, Rainer PP, Gasser R, Huber M‐S, Khafaga M, Wilhelm B, Haas T, Mächler H, Rössl U, Pieske B. Glucose‐transporter‐mediated positive inotropic effects in human myocardium of diabetic and nondiabetic patients. Metabolism 2010; 59: 1020–1028. [DOI] [PubMed] [Google Scholar]
- 14. Baartscheer A, Schumacher CA, Wüst RCI, Fiolet JWT, Stienen GJM, Coronel R, Zuurbier CJ. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017; 60: 568–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical characteristics of patient, from which atrial biopsies were obtained (upper table), or from which left ventricular tissue of explanted hearts (after heart transplantation, lower table) was used. y = yes, n = no, Nr = number, CAD = coronary artery disease, ICM = ischemic cardiomyopathy, DCM = dilated cardiomyopathy, EF = ejection fraction in %, LVEDD = left ventricular enddiastolic diameter in mm, m = male, SR = sinus rhythm, CABG = coronary artery bypass graft, valves = valvular dysfunction, MR = mitral regurgitation, TR = tricuspid regurgitation x° = indicated severity degree according to international standards, Art Hyp = arterial hypertension, KD = kidney disease, GFR = glomerular filtration rate in ml/min, NTproBNP in pg/ml (internal cutoff <150 pg/ml).
Table S2. Echocardiographic data obtained from 16 mice. OP = TAC surgery, bpm = beats per minute, LV = left ventricle, AWThd = anterior wall thickness in diastole, PWThd = posterior wall thickness in diastole, LVIDs = left ventricular internal diameter in systole, LVIDd = left ventricular internal diameter in diastole, *P < 0.05 versus respective pre‐OP‐measurement (Student's t‐test).