

Abstract
Hypertrophic cardiomyopathy (HCM) is a primary autosomal‐dominant disorder of the myocardium with variable expressivity and penetrance. Occasionally, homozygous sarcomere genetic variants emerge while genotyping HCM patients. In these cases, a more severe HCM phenotype is generally seen. Here, we report a case of HCM that was diagnosed clinically at 39 years of age. Initial symptoms were shortness of breath during exertion. Successively, he developed a wide array of severe clinical manifestations, which progressed to an ominous end‐stage heart failure that resulted in heart transplantation. Genotype analysis revealed a missense MYBPC3 variant NM_000256.3:c.2618C>A,p.(Pro873His) that presented in the homozygous form. Conflicting interpretations of pathogenicity have been reported for the Pro873His MYBPC3 variant described here. Our patient, presenting with two copies of the variant and devoid of a normal allele, progressed to end‐stage heart failure, which supports the notion of a deleterious effect of this variant in the homozygous form.
Keywords: HCM, MYBPC3, Homozygous, Heart failure
Introduction
Although most patients with hypertrophic cardiomyopathy (HCM) have a good prognosis, a significant number suffer from life‐threatening complications, primarily sudden cardiac death and end‐stage heart failure.1, 2, 3 The heterogeneous phenotypic expression of HCM has been related to its diverse genetic profile. In larger series, ~3–5% of adult HCM patients prove to be compound or double heterozygotes for two disease‐causing variants in the same or different sarcomeric protein genes.4, 5, 6, 7 Although data are still limited, indications are that patients with more than one sarcomere genetic variant, either in a compound heterozygote state or as a homozygous variant, have a more profound clinical profile of HCM.4, 5, 8, 9, 10, 11, 12, 13
Case report
A 39‐year‐old man presented in 1998 with shortness of breath during exertion. Nine years earlier, in 1989, he was treated for myocarditis after an upper respiratory tract infection presumably due to a self‐limited virus infection. No test was performed then for any specific viruses. The echocardiogram (ECHO) showed a mildly reduced septal movement of the left ventricle. A year later, during follow‐up, a new ECHO showed an entirely ordinary function of the left ventricle, but the left ventricular wall was at the upper reference thickness limit. In the following 8 years, he did not have any heart problems. There was no family history of heart disease.
In 1998, the index patient experienced heart symptoms including breathlessness and therefore electrocardiography (ECG) (Figure 1 ) and ECHO (Figure 2 ) were performed. He had a regular sinus rhythm. Auscultation revealed a systolic murmur, heard best at the left parasternal region. The blood pressure was normal. The ECHO showed a thick interventricular septum at about 26 mm, and the posterior wall of the left ventricle was more than 16 mm thick. There were signs of outflow obstruction with a maximal flow velocity of 2.8 m/s at rest. An exercise test, where he cycled up to 130 W (55% of reference), was stopped because of a hypotension episode. He was initially treated with beta‐blockers. As no positive effect had been observed, he had a pacemaker (PM) implanted in 1999 with short A‐V interval. He was feeling better after PM implantation, and objectively, he was in a better condition.
Figure 1.
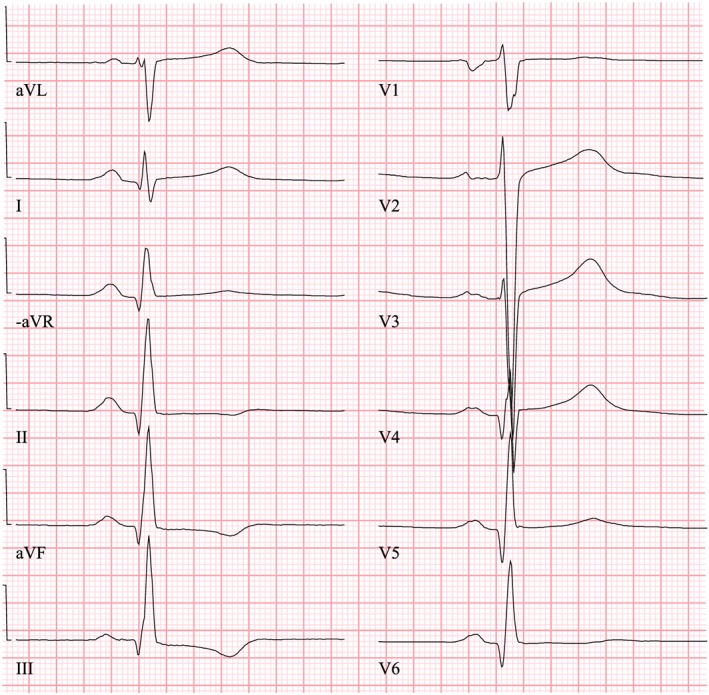
The electrocardiography (ECG) of the index patient taken in 1998, when he was seeking medical advice for breathlessness. The ECG figure showed sinus rhythm with increased R‐wave amplitude in the left‐sided ECG leads (V4–V6) and increased S‐wave depth in the right‐sided leads (V1–V3), which are typical signs of hypertrophy of the left ventricle.
Figure 2.
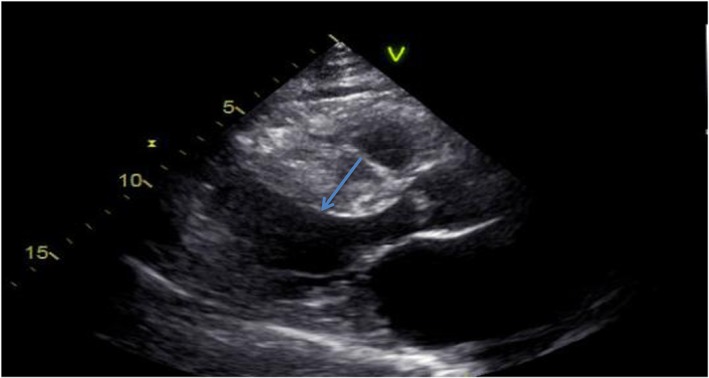
Echocardiogram of the index patient performed in 1998. The echocardiogram showed a thick septum (arrow) at about 26 mm, and the posterior wall of the left ventricle was more than 16 mm thick.
He experienced several problems with PM pocket infections that resulted in surgical extraction of the whole PM system in 2010. His clinical condition remained stable until 2013. Then he was admitted because of severe retrosternal chest pain that was due to a heart attack. Primary coronary angiography showed that the third marginal branch of the circumflex artery was occluded. The left anterior descending artery showed only slight atherosclerosis. The coronary circulation was ‘right dominant’ as the posterior descending artery was supplied by the right coronary artery, without any essential narrowing. He was treated with a drug‐eluting stent with a good result. As regards risk factors, he did not have any history of coronary artery disease and had no known thrombophilia. He was not obese, but he was smoking, which he stopped doing after the heart attack. He had slightly elevated cholesterol levels: total cholesterol of 6.8 mmol/L and LDL of 4.6 mmol/L. The ECHO showed a vigorous hypertrophied left ventricle with a septum thickness of 27 mm and at least moderate systolic dysfunction with a generally restrained wall motion. The worst abnormal movement was seen apically. No outflow obstruction was seen. The left atrium was moderately dilated. The pressure in the lung circulation was about 50 mmHg. The heart rate monitoring revealed short episodes of non‐sustained ventricular tachycardia, and this, together with the hypertrophied septum, prompted an implantable cardioverter defibrillator implantation. A year later, he suffered from atrial fibrillation attacks. During these episodes, he was extremely limited in his physical condition with severe dyspnoea, fatigue, and lack of energy, precluding any activity. A spirometry was also performed, showing a normal FEV1/VC ratio of 81% and normal total lung capacity. He had an optimal medical heart failure therapy with full‐dose beta‐blockers, angiotensin‐converting enzyme inhibitor and aldosterone antagonist. He was not a candidate for cardiac resynchronization therapy as he did not have a width‐increased QRS complex. Ablation of his atrial fibrillation would not have had any profound effect because of the severe impairment of the ventricle systolic function.
In 2015, ECHO showed a slightly dilated (about 80 mL/m2), asymmetrically hypertrophied left ventricle with severe impaired systolic and diastolic functions. VO2 max cycling in 2016 showed that he had a severely reduced maximum oxygen uptake of 1.2 L/min, equal to 14 mL/kg/min. A heart catheterization was performed. At rest, there was hypokinetic circulation with a cardiac output of 3.6 L/min and a systolic system pressure around 105 mmHg. End‐diastolic left ventricular pressure was moderately elevated to 24 mmHg with a corresponding rise in the pulmonary capillary wedge pressure, and secondary to this, there was a moderate pressure increase in the pulmonary circulation (pulmonary pressure: 56/27 mmHg). Pulmonary vascular resistance was slightly higher, 2.7 mmHg/L/min. The filling pressure of the left ventricle rose even when he lifted his feet to the bike pedals at rest. During the exercise test load, the end‐diastolic left ventricular pressure was severely elevated up to 38 mmHg, corresponding to a rise in pulmonary capillary wedge pressure and secondary elevated pressure in the pulmonary circulation with systolic levels of 70 mmHg. Magnetic resonance imaging of the heart was not performed, but the patient underwent a myocardial biopsy that showed moderate interstitial fibrosis, focal cell hypertrophy, and even atrophy, with no signs of storage/infiltrative disease. His severe clinical deterioration as well as the ominous results of the above examinations prompted a new heart to be transplanted, and he has recently undergone the operation well.
Genetic method
Next‐generation sequencing with a heart cardiomyopathy gene panel, including 84 genes (Table 1), was performed. The index patient was found to be homozygous for the MYBPC3 variant NM_000256.3:c.2618C>A,p.(Pro873His). Multiplex ligation‐dependent probe amplification was used to confirm that the MYBPC3 c.2618 G>A substitution was in true homozygous state. With American College of Medical Genetics and Genomics criteria14 for predicting pathogenicity of genetic variants, MYBPC3 c.2618 C>A was predicted to be ‘likely pathogenic’ (Class 2) in the heterozygous state.
Table 1.
Table of the genes included in the panel, as well as coverage and reference sequences used for testing the proband
| Gene | NCBI reference sequence | Coverage (%) |
|---|---|---|
| ABCC9 | NM_005691.3 | 99.18 |
| ACTA2 | NM_001613.2 | 100.00 |
| ACTC1 | NM_005159.4 | 97.18 |
| ACTN2 | NM_001103.2 | 100.00 |
| AKAP9 | NM_005751.4 | 99.35 |
| ANK2 | NM_001148.4 | 99.97 |
| ANKRD1 | NM_014391.2 | 100.00 |
| APOB | NM_000384.2 | 100.00 |
| BAG3 | NM_004281.3 | 100.00 |
| CACNA1C | NM_000719.6 | 100.00 |
| CACNA2D1 | NM_000722.2 | 97.59 |
| CACNB2 | NM_201590.2 | 97.31 |
| CASQ2 | NM_001232.3 | 96.17 |
| CAV3 | NM_033337.2 | 100.00 |
| CBS | NM_001178009.1 | 100.00 |
| COL3A1 | NM_000090.3 | 99.39 |
| COL5A1 | NM_000093.3 | 100.00 |
| COL5A2 | NM_000393.3 | 99.33 |
| CSRP3 | NM_003476.4 | 100.00 |
| DES | NM_001927.3 | 100.00 |
| DMD | NM_004006.2 | 99.37 |
| DSC2 | NM_024422.3 | 100.00 |
| DSG2 | NM_001943.3 | 99.26 |
| DSP | NM_004415.2 | 99.92 |
| EMD | NM_000117.2 | 100.00 |
| FBN1 | NM_000138.4 | 100.00 |
| FBN2 | NM_001999.3 | 99.95 |
| FHL1 | NM_001449.4 | 100.00 |
| FHL2 | NM_201555.1 | 100.00 |
| GLA | NM_000169.2 | 99.69 |
| GPD1L | NM_015141.3 | 100.00 |
| JUP | NM_002230.2 | 100.00 |
| KCNE1 | NM_000219.4 | 100.00 |
| KCNE2 | NM_172201.1 | 100.00 |
| KCNE3 | NM_005472.4 | 100.00 |
| KCNH2 | NM_000238.3 | 99.86 |
| KCNJ2 | NM_000891.2 | 100.00 |
| KCNJ5 | NM_000890.3 | 100.00 |
| KCNJ8 | NM_004982.3 | 100.00 |
| KCNQ1 | NM_000218.2 | 98.52 |
| LAMP2 | NM_002294.2 | 99.19 |
| LDB3 | NM_001080116.1 | 100.00 |
| LDLR | NM_000527.4 | 100.00 |
| LDLRAP1 | NM_015627.2 | 100.00 |
| LMNA | NM_170707.3 | 100.00 |
| MYBPC3 | NM_000256.3 | 100.00 |
| MYH11 | NM_002474.2 | 99.53 |
| MYH6 | NM_002471.3 | 99.5 |
| MYH7 | NM_000257.2 | 100.00 |
| MYL2 | NM_000432.3 | 91.42 |
| MYL3 | NM_000258.2 | 100.00 |
| MYLK | NM_053025.3 | 99.86 |
| MYLK2 | NM_033118.3 | 100.00 |
| MYOZ2 | NM_016599.4 | 100.00 |
| NEBL | NM_006393.2 | 100.00 |
| NEXN | NM_144573.3 | 100.00 |
| PCSK9 | NM_174936.3 | 99.62 |
| PKP2 | NM_004572.3 | 100.00 |
| PLN | NM_002667.3 | 100.00 |
| PRKAG2 | NM_016203.3 | 100.00 |
| RBM20 | NM_001134363.1 | 99.46 |
| RYR2 | NM_001035.2 | 99.46 |
| SCN1B | NM_001037.4 | 100.00 |
| SCN3B | NM_018400.3 | 100.00 |
| SCN4B | NM_174934.3 | 100.00 |
| SCN5A | NM_198056.2 | 100.00 |
| SLC2A10 | NM_030777.3 | 100.00 |
| SMAD3 | NM_005902.3 | 99.77 |
| SNTA1 | NM_003098.2 | 100.00 |
| TAZ | NM_000116.3 | 100.00 |
| TCAP | NM_003673.3 | 100.00 |
| TGFB2 | NM_001135599.2 | 100.00 |
| TGFB3 | NM_003239.2 | 100.00 |
| TGFBR1 | NM_004612.2 | 96.83 |
| TGFBR2 | NM_003242.5 | 100.00 |
| TMEM43 | NM_024334.2 | 100.00 |
| TMPO | NM_003276.2 | 100.00 |
| TNNC1 | NM_003280.2 | 100.00 |
| TNNI3 | NM_000363.4 | 100.00 |
| TNNT2 | NM_001001430.2 | 99.31 |
| TPM1 | NM_001018005.1 | 99.53 |
| TTN | NM_133378.4 | 99.86 |
| TTR | NM_000371.3 | 100.00 |
| VCL | NM_014000.2 | 100.00 |
Clinical and genetic follow‐up of relatives
Cascade genetic screening using Sanger sequencing was offered to family members, primarily to first‐degree relatives (Figures 3 and 4 ). The patient's parents were not alive and could not be genetically tested. There was no known consanguinity. So far, 12 family members have been analysed for the MYBPC3 c.2618 C>A variant. One brother to our index patient also had the mutation in the homozygous form and seven first‐degree or second‐degree relatives were shown to be heterozygous carriers of the variant. All the relatives who have the variant in heterozygous form have a normal ECG, a normal ECHO, and no heart problems so far.
Figure 3.
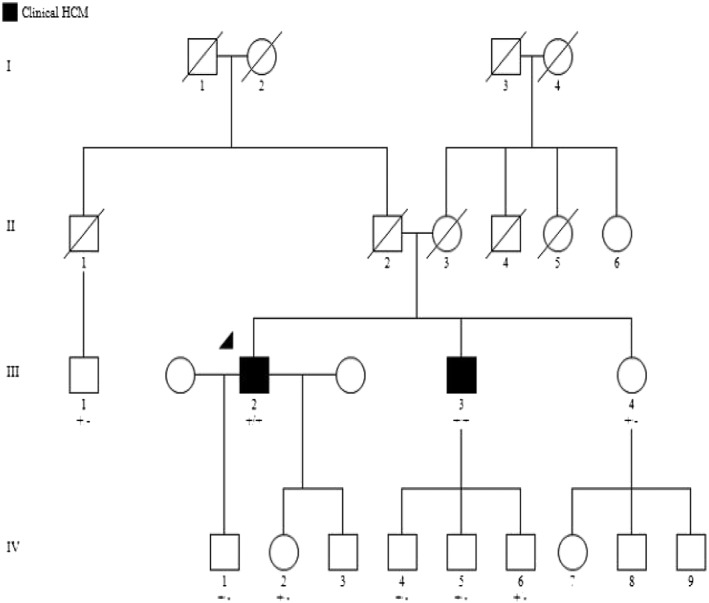
Pedigree analysis of the family in this study. Circles in the pedigree denote female, squares male. A crossed‐over symbol indicates that this particular individual has died. The arrow points out the index patient. The genetic variant MYBPC3 c.2618 C>A is indicated +/+ if homozygous and +/− if heterozygous. The individuals 7, 8, and 9 in the IV generation are also being offered genetic testing, but the results are not yet known.
Figure 4.
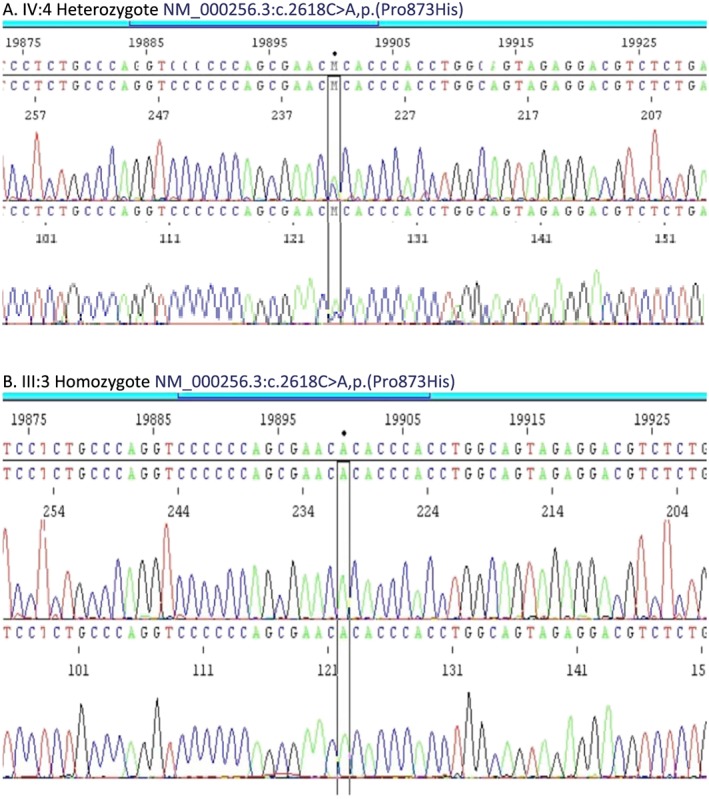
Sanger sequencing results showing (A) heterozygote (IV: 4 in Figure 3 ) and (B) homozygote (III: 3 in Figure 3 ) for NM_000256.3:c.2618C>A,p.(Pro873His). The variant nucleotide position is indicated by parallel vertical lines.
Our patient (III: 2 in the pedigree) has three children, one daughter and two sons. The youngest son (IV: 3 in the pedigree) has not been properly evaluated yet. The eldest son (IV: 1 in the pedigree), who is 29 years old, has the variant in heterozygous form. He has a normal ECG, a normal ECHO, and no heart problems. Our patient's daughter (IV: 2 in the pedigree) is 25 years old. She also has the variant in heterozygous form. She does not have any cardiopulmonary symptoms either. ECHO in 2016 showed a normal‐sized left ventricle with normal systolic and diastolic functions.
The patient's sister (III: 4 in the pedigree), who is 43 years old, has the variant in heterozygous form. She does not experience any heart problems. Her ECG shows sinus tachycardia, with 105 b.p.m. and slight ST‐T changes inferiorly. The ultrasonography of the heart has been completely normal without any signs of cardiomyopathy. She has normal wall thickness of the left ventricle with normal systolic and diastolic functions. A cardiac stress test showed normal work capacity; she cycled up to 143 W (88% of reference) and stopped because of general fatigue. She has three children, two sons who are 16 and 10 years old (IV: 8 and 9 in the pedigree) and an 18‐year‐old daughter (IV: 7 in the pedigree). All of them have been offered genetic testing, and they are still waiting for the results. None of them have experienced any heart symptoms.
The patient's 54‐year‐old brother (III: 3 in the pedigree) who has the variant in homozygous form cycled 220 W (corresponding to 95% of reference) during an exercise test. No arrhythmia and no evidence of underlying coronary insufficiency were seen. He can sometimes experience a chest discomfort even at rest but no pain, dizziness, fainting, or shortness of breath. The ECHO showed a normal‐sized left ventricle, regionally increased wall thickness, moderately increased septum (16 mm), slightly increased over the posterior wall (13 mm) and a normal systolic function (ejection fraction of about 56%). The ECG showed sinus rhythm with increased R‐wave amplitude in the left‐sided ECG leads, as seen in ventricular hypertrophy. This man has three sons (10, 16, and 20 years old; IV: 4, 5, and 6 in the pedigree). All of them are heterozygous for the variant and have normal ECGs and ECHOs. A cousin to our index patient (III: 1 in the pedigree) who is 52 years old also carries the variant in heterozygous form. He also has a normal ECHO and ECG and does not have any cardiac symptoms.
Discussion
In the majority of patients with familial cardiomyopathy owing to a variant in one of the genes encoding sarcomeric proteins, a single autosomal‐dominant pathogenic mutation is found. MYBPC3 encodes for the thick filament‐associated protein cardiac myosin‐binding protein C (cMyBP‐C), a signalling node in cardiac myocytes that contributes to the maintenance of sarcomeric structure and regulation of contraction and relaxation. MYBPC3 is the most frequently mutated gene in HCM, representing 40–50% of all HCM mutations. The majority of MYBPC3‐associated HCM mutations are heterozygous, and patients often have a late disease onset with a benign disease progression.15 Studies have also reported that 70% of the mutations in MYBPC3 are truncating variants, which cause a more severe HCM phenotype than those associated with missense and in‐frame deletions.16 In contrast to heterozygous pathogenic mutations, homozygous or compound heterozygous truncating pathogenic MYBPC3 variants cause severe neonatal cardiomyopathy, which leads to heart failure and death within the first year of life.13 Furthermore, several studies have demonstrated that HCM patients carrying multiple pathogenic gene variants have poorer prognosis in terms of earlier disease onset, increased left ventricular hypertrophy, and increased frequency of heart failure and sudden cardiac death, when compared with those carrying a single mutation.5, 6, 11, 16 Thus, the present case supports previous claims that rare homozygous variants may aggravate the clinical severity of HCM. Our HCM patient with a homozygous missense variant in MYPBC3 needed a heart transplantation, while his brother who also has the variant in homozygosis and the clinical phenotype of HCM will require specific attention at follow‐up as there is the possibility of more adverse manifestations of HCM later. None of the heterozygotes in the family has so far demonstrated any sign of HCM.
The MYBPC3 Pro873His variant described here has been reported in the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/), at a frequency of 0.00006781 (14/206 446 in the entire studied population, 13/97 288 in the European non‐Finnish population).
One fundamental question pertains to the nature of the gene product derived from the mutant allele. Certain mutant proteins may be more or less stable or incorporate more or less efficiently into the sarcomere. Wijnker et al.17 compared the pathomechanisms of a truncating mutation and a missense mutation in MYBPC3 in engineered heart tissues. The widely accepted hypothesis is that truncating MYBPC3 mutations causes haploinsufficiency, as opposed to missense mutations, which incorporate into the sarcomere and act in a dominant‐negative fashion. Some studies18, 19 provided evidence that truncated cMyBP‐Cs are not detectable in human patient samples, as they seem to be susceptible to degradation by nonsense‐mediated RNA decay. In contrast to truncating mutations, missense mutations lead to stable mutant cMyBP‐Cs that exert a more potent effect in disrupting sarcomere function.16 The MYBPC3 variant described here presumptively leads to the substitution of a non‐polar residue (Pro) with a positively charged residue (His). It occurs in the C7 domain of the protein and could interfere with the protein incorporation in the A‐band of the sarcomere. The ‘poison peptide’ hypothesis proposes that mutant sarcomeric proteins incorporate into myofibrils and act as dominant‐negative proteins.20
Evidently, conflicting interpretations of pathogenicity have been obtained for heterozygous missense and compound heterozygous missense MYBPC3 variants. For example, another case was presented with the MYBPC3 Pro873His variant associated with HCM where one additional MYBPC3 (Asp745Gly) variant of uncertain significance was also observed. The patient was diagnosed with HCM at 18 years of age. He had asymmetric hypertrophy with a maximal wall thickness of 30 mm, and he had received appropriate therapy from his implantable cardioverter defibrillator.10 Predictions from in silico tools were supportive of a deleterious effect. However, because of co‐occurrence with an additional MYBPC3 variant, limited familial data, and insufficient evidence, the MYBPC3 Pro873His variant found in that patient must be classified as one of ‘uncertain significance’ according to American College of Medical Genetic and Genomics criteria.14
In addition, the MYBPC3 Pro873His variant has been identified previously in an HCM individual who was also homozygous for the variant.21 In that study, while trying to identify genes that are consistently associated to HCM progressing to dilated cardiomyopathy, the authors found a homozygous MYBPC3 Pro873His mutation in a man from a gipsy family, where consanguinity between the parents was very likely. That male patient was clinically evaluated at the age of 27. He had a moderate/severe hypertrophy (interventricular septum: 27 mm; left ventricular outflow tract obstruction: 50 mmHg) and showed a non‐sustained ventricular tachycardia at Holter monitoring. As in our case, the patient's hypertrophied heart progressed to a slightly dilated left ventricle with severe systolic impairment.
The question of whether the myocarditis event that our index patient had experienced before he was clinically diagnosed with HCM might have contributed, as an external factor, to the clinical appearance of the disease, by triggering the underlying genetic substrate, arises. It is also possible that the heart attack that he faced might have worsened his ventricular function. However, the occlusion of the branch of the circumflex artery cannot explain the severe impairment of the systolic function. Thus, homozygosity of the MYBPC3 Pro873His variant seems to have caused a cumulative deleterious effect on the clinical course of our HCM patient who progressed to end‐stage heart failure. Given the important clinical issue of risk stratification for determining which HCM patients are at highest risk of heart failure and sudden cardiac death, the presence of bi‐allelic MYBPC3 missense variants in an individual may be a major factor in this cardiac event risk algorithm.
In conclusion, allelic balance may influence genotype–phenotype correlations and should be an important consideration in the risk stratification of patients with HCM and their family members. Our data show that homozygosity for the MYBPC3 Pro873His variant very likely increases the risk of HCM pathogenesis and may contribute a cumulative effect, leading to earlier disease onset and adverse cardiac events. Interestingly, none of the heterozygote carriers in the present study have so far developed any hypertrophic features, suggesting that this variant as a heterozygote trait has a low penetrance and might be insufficient to cause a strong HCM phenotype. Therefore, homozygous and compound heterozygous MYBPC3 variants should prove to be clinically useful prognostic markers. However, the current evidence is preliminary, and more studies and case reports are required before the potential for translating the basic genetic information into clinical decision making concerning longitudinal evaluation and monitoring of HCM patients can be fully assessed.
Conflict of interest
None declared.
Funding
This work was supported by Region Östergötland (ALF) under grant LIO‐609681 and by FORSS (Medical Research Council of Southeast Sweden) under grant FORSS/572421.
Ethics approval
Regional Ethics Committee in Linköping, Dnr 2016/389‐31. Participants in the present study have signed an informed consent to participate in the study.
Data accessibility
The datasets generated and/or analysed during the current study are not publicly available but are available from the corresponding author on reasonable request. The individuals in the present study have not given their permission for publishing data on an individual basis.
Acknowledgement
We are very grateful to the patient and relatives included in this study.
Kissopoulou, A. , Trinks, C. , Green, A. , Karlsson, J.‐E. , Jonasson, J. , and Gunnarsson, C. (2018) Homozygous missense MYBPC3 Pro873His mutation associated with increased risk for heart failure development in hypertrophic cardiomyopathy. ESC Heart Failure, 5: 716–723. 10.1002/ehf2.12288.
References
- 1. Baxi AJ, Restrepo CS, Vargas D, Marmol‐Velez A, Ocazionez D, Murillo H. Hypertrophic cardiomyopathy from A to Z: genetics, pathophysiology, imaging, and management. Radiographics: a review publication of the Radiological Society of North America, Inc 2016; 36: 335–354. [DOI] [PubMed] [Google Scholar]
- 2. Brito D, Cepeda B. Hypertrophic Cardiomyopathy. Treasure Island (FL): StatPearls; 2017. [Google Scholar]
- 3. Hebl VB, Miranda WR, Ong KC, Hodge DO, Bos JM, Gentile F, Klarich KW, Nishimura RA, Ackerman MJ, Gersh BJ, Ommen SR, Geske JB. The natural history of nonobstructive hypertrophic cardiomyopathy. Mayo Clin Proc 2016; 91: 279–287. [DOI] [PubMed] [Google Scholar]
- 4. Alpert NR, Mohiddin SA, Tripodi D, Jacobson‐Hatzell J, Vaughn‐Whitley K, Brosseau C, Warshaw DM, Fananapazir L. Molecular and phenotypic effects of heterozygous, homozygous, and compound heterozygote myosin heavy‐chain mutations. Am J Physiol Heart Circ Physiol 2005; 288: H1097–H1102. [DOI] [PubMed] [Google Scholar]
- 5. Fourey D, Care M, Siminovitch KA, Weissler‐Snir A, Hindieh W, Chan RH, Gollob MH, Rakowski H, Adler A. Prevalence and clinical implication of double mutations in hypertrophic cardiomyopathy: revisiting the gene–dose effect. Circ Cardiovasc Genet 2017; 10: e001685. [DOI] [PubMed] [Google Scholar]
- 6. Mohiddin SA, Begley DA, McLam E, Cardoso JP, Winkler JB, Sellers JR, Fananapazir L. Utility of genetic screening in hypertrophic cardiomyopathy: prevalence and significance of novel and double (homozygous and heterozygous) beta‐myosin mutations. Genet Test 2003; 7: 21–27. [DOI] [PubMed] [Google Scholar]
- 7. van Spaendonck‐Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM, Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, de Boer RA, Jongbloed JD, Pinto YM, van Tintelen JP. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years' experience. Eur J Heart Fail 2013; 15: 628–636. [DOI] [PubMed] [Google Scholar]
- 8. Garcia‐Castro M, Reguero JR, Alvarez V, Batalla A, Soto MI, Albaladejo V, Coto E. Hypertrophic cardiomyopathy linked to homozygosity for a new mutation in the myosin‐binding protein C gene (A627V) suggests a dosage effect. Int J Cardiol 2005; 102: 501–507. [DOI] [PubMed] [Google Scholar]
- 9. Gray B, Yeates L, Medi C, Ingles J, Semsarian C. Homozygous mutation in the cardiac troponin I gene: clinical heterogeneity in hypertrophic cardiomyopathy. Int J Cardiol 2013; 168: 1530–1531. [DOI] [PubMed] [Google Scholar]
- 10. Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet 2005; 42: e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maron BJ, Maron MS, Semsarian C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm 2012; 9: 57–63. [DOI] [PubMed] [Google Scholar]
- 12. Richard P, Charron P, Leclercq C, Ledeuil C, Carrier L, Dubourg O, Desnos M, Bouhour JB, Schwartz K, Daubert JC, Komajda M, Hainque B. Homozygotes for a R869G mutation in the beta‐myosin heavy chain gene have a severe form of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 2000; 32: 1575–1583. [DOI] [PubMed] [Google Scholar]
- 13. Wessels MW, Herkert JC, Frohn‐Mulder IM, Dalinghaus M, van den Wijngaard A, de Krijger RR, Michels M, de Coo IFM, Hoedemaekers YM, Dooijes D. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. EJHG 2015; 23: 922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin‐binding protein C (MYBPC3) in cardiac pathophysiology. Gene 2015; 573: 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017; 121: 749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wijnker PJ, Friedrich FW, Dutsch A, Reischmann S, Eder A, Mannhardt I, Mearini G, Eschenhagen T, van der Velden J, Carrier L. Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue. J Mol Cell Cardiol 2016; 97: 82–92. [DOI] [PubMed] [Google Scholar]
- 18. Helms AS, Davis FM, Coleman D, Bartolone SN, Glazier AA, Pagani F, Yob JM, Sadayappan S, Pedersen E, Lyons R, Westfall MV, Jones R, Russell MW, Day SM. Sarcomere mutation‐specific expression patterns in human hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2014; 7: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mohamed IA, Krishnamoorthy NT, Nasrallah GK, Da'as SI. The role of cardiac myosin binding protein C3 in hypertrophic cardiomyopathy‐progress and novel therapeutic opportunities. J Cell Physiol 2017; 232: 1650–1659. [DOI] [PubMed] [Google Scholar]
- 20. Geisterfer‐Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990; 62: 999–1006. [DOI] [PubMed] [Google Scholar]
- 21. Nanni L, Pieroni M, Chimenti C, Simionati B, Zimbello R, Maseri A, Frustaci A, Lanfranchi G. Hypertrophic cardiomyopathy: two homozygous cases with ‘typical’ hypertrophic cardiomyopathy and three new mutations in cases with progression to dilated cardiomyopathy. Biochem Biophys Res Commun 2003; 309: 391–398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analysed during the current study are not publicly available but are available from the corresponding author on reasonable request. The individuals in the present study have not given their permission for publishing data on an individual basis.