

Abstract
Mitochondria undergo frequent fusion and fission events, which are essential to maintain a functional mitochondrial network. A disruption of these processes can cause severe neurodegeneration. Charcot–Marie–Tooth disease type 2A (CMT2A) is a neuropathy that is caused by mutations in the fusion factor Mfn2. It is generally assumed that impaired mitochondrial fusion causes CMT2A. However, the detailed molecular mechanism underlying the pathophysiology of CMT2A is only incompletely understood. In this issue of EMBO Reports, El Fissi et al established a fly model to analyze the consequence of frequently occurring MFN2 mutations on locomotor behavior, mitochondrial morphology, and function and find that some pathogenic mutants enhance fusion activity, indicating that increased mitochondrial fusion can drive CMT2A‐like pathology 1. Moreover, this novel assay system will be a useful tool to analyze CMT2A pathogenesis in vivo.
Subject Categories: Cancer, Signal Transduction
Mitochondria are essential organelles that not only produce the cell's energy in form of ATP through oxidative respiration but also participate in other processes such as the regulation of cell death or metabolism. Mitochondrial dysfunction causes a variety of diseases, such as neurodegeneration, heart failure, metabolic disorders, and aging 2. Mitochondria are highly dynamic and motile in the cytoplasm and frequently divide and fuse with each other 2, 3. Fusion and fission are regulated by highly conserved GTPase proteins. Dynamin‐related protein (Drp1, Dnm1 in budding yeast) is a fission‐stimulating GTPase, which is transported from the cytosol to mitochondrial fission sites on the outer membrane. Optic atrophy 1 (OPA1, Mgm1 in budding yeast) and mitofusin (Mfn)1 and Mfn2 (Fzo1 in budding yeast) are involved in mitochondrial inner and outer membrane fusion, respectively.
Through genetic analysis in yeast, it became clear that a finely tuned balance between fusion and fission is critical for mitochondrial morphology and function 3. Deletion of Fzo1 not only altered mitochondrial shape, but also impaired respiratory function and mitochondrial genome (mtDNA) maintenance. These defects could be rescued by the further depletion of the fission factor Dnm1 3. Studies in mammals confirmed these conclusions. While the knockout of either mitochondrial fusion or fission factors causes severe defects, such as neurodegeneration and heart failure 4, 5, the suppression of both fusion and fission factors simultaneously restores mitochondrial functions, at least to some extent 6. These results suggested that the appropriate balance of mitochondrial fusion and fission is important for mitochondrial homeostasis. These fission and fusion factors are not only essential to maintain mitochondrial and respiratory function, but also contribute to the regulation of mitophagy, a quality control system that degrades damaged mitochondria.
Both, mitochondrial fusion–fission and the mitochondrial quality control system are particularly important in neurons to avoid mitochondrial dysfunction resulting in neuronal death and neurodegeneration. Indeed, many neuropathologies are associated with mitochondrial dysfunction. Familial Parkinson's disease (PD) is caused by mutations in PINK1 and PARKIN, two proteins involved in mitophagy, the selective degradation of damaged mitochondria by autophagy. Autosomal dominant optic atrophy (ADOA) type I, a neurodegenerative disorder with optic nerve hypoplasia, is caused by dominant mutations in OPA1, and Charcot–Marie–Tooth neuropathy type 2A (CMT2A), a peripheral neural muscular atrophy, is caused by mutations in Mfn2 7.
The analysis of CMT2A patients resulted in the identification of many pathogenic variants of Mfn2. Among these variants, point mutations in the GTPase domain severely affect mitochondrial fusion, indicating that impaired fusion is causative of the neurodegeneration observed in these patients 8. It was recently reported that Mfn2 agonists could revert mitochondrial and neuronal defects of the GTPase domain mutation of Mfn2 (9). On the other hand, some mutants identified from CMT2A patients still maintain mitochondrial fusion capacity in cultured mammalian cells 8, and thus, the pathogenic mechanism induced by mutated Mfn2 remained incompletely understood.
In this issue of EMBO Reports, El Fissi et al established a fly model to analyze CMT2A pathogenesis in vivo 1. Marf is the ubiquitously expressed fly homolog of mitofusin. To study the pathogenicity of CMT2A alleles, the authors generated Marf alleles mimicking disease‐related mutations of human Mfn2 (Fig 1). These Marf variants were exogenously expressed in fly neurons, and all four Marf variants were found to dominantly impair locomotor activity. When analyzed by fluorescence microscopy, Marf mutants in the GTPase domain (such as R94Qlike) or mutants in the HB1 region (such as R364Wlike) induced large mitochondria‐containing structures in the neuronal soma. In both R94Qlike and R364Wlike mutant flies, mitochondrial respiration defects and ROS production were observed. However, electron microscopy revealed that the different Marf alleles caused distinct mitochondrial morphological changes; mitochondria expressing R94Qlike were smaller and aggregated in a part of the cytoplasm. Notably, mitochondrial delivery to neuromuscular junctions is only partially impaired in the mitochondrial fusion‐deficient cells. In contrast, expression of R364Wlike induced an elongated mitochondrial network, with bulb‐like shaped enlarged structures. The authors confirmed their findings in cultured mammalian cells, in which the GTPase domain mutant R94Q repressed mitochondrial fusion, as reported earlier 8. Similar to the fly model, the Mfn2‐R364W allele was not only functional in mitochondrial fusion, but actually accelerated fusion in living cells. Moreover, the bulb‐like mitochondria formed in R364Wlike‐expressing cells were similar to the mitochondrial morphology observed in Drp1‐deficient and thus fission‐deficient cells, further supporting the notion that they were formed by hyperfusion 10. Indeed, exogenous expression of Drp1 suppressed the phenotypes induced by R364Wlike. These data further support the conclusion that the R364Wlike mutant enhances mitochondrial fusion activity, and that mitochondrial hyperfusion can cause CMT2A pathogenesis, at least in fly neurons.
Figure 1. CMT2A pathology in the fly model and mitochondrial fusion activity of the pathogenic alleles.
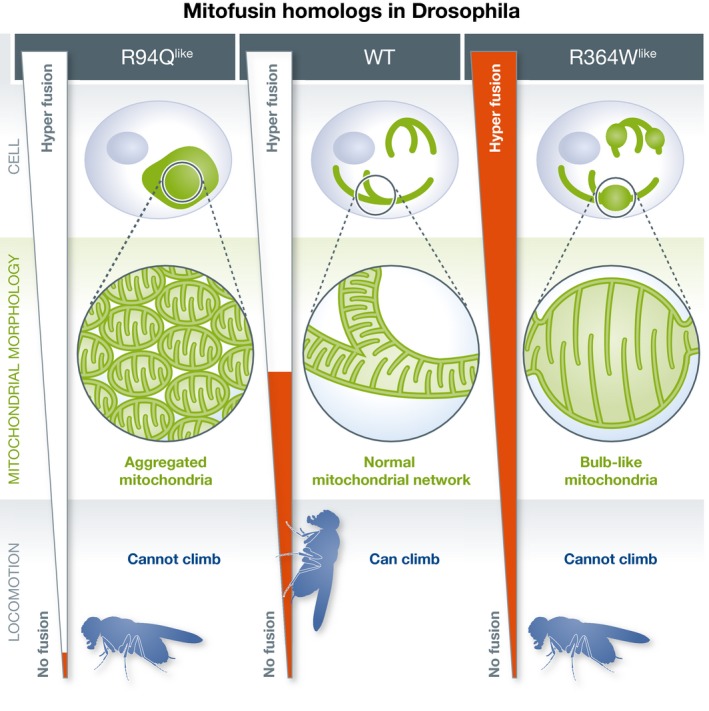
(Left panel) Inhibition of mitochondrial fusion activity in R94Qlike variant leads to aggregation of small mitochondria and impaired locomotor activity. (Middle panel) Appropriate mitochondrial fusion activity in wild‐type fly leads to normal mitochondrial network morphology and locomotor activity. (Right panel) Enhanced mitochondrial fusion activity in the R364Wlike variant leads to the formation of enlarged bulb‐like mitochondria and also impaired locomotor activity.
It was well known that mutations in the mitofusin GTPase domain severely block mitochondrial fusion. The paper by Fissi et al clearly showed that not only diminished but also accelerated fusion drives CMT2A pathology and neurodegeneration. Many recent studies focused on the structure and regulation of mitofusin with a particular focus on the role of mitofusin homo‐oligomerization to form membrane tethers before fusion 9. How mitofusin proteins mediate outer membrane fusion after membrane tethering is less well understood. The R364Wlike variant and its pathology in the fly model not only provide new insight on the pathogenesis of neurodegeneration that is also induced by hyper‐active mitochondrial fusion but might also provide a useful tool to study the mechanism of membrane fusion mediated by mitofusin proteins.
EMBO Reports (2018) 19: e46502
See also: https://doi.org/10.15252/embr.201745241 (August 2018)
References
- 1. Fissi NE, Rojo M, Aouane A et al (2018) EMBO Rep 19: e45241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nunnari J, Suomalainen A (2012) Cell 148: 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sesaki H, Jensen RE (1999) J Cell Biol 147: 699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen H, McCaffery JM, Chan DC (2007) Cell 130: 548–562 [DOI] [PubMed] [Google Scholar]
- 5. Ishihara N, Nomura M, Jofuku A et al (2009) Nat Cell Biol 11: 958–966 [DOI] [PubMed] [Google Scholar]
- 6. Chen H, Ren S, Clish C et al (2015) J Cell Biol 211: 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Züchner S, Mersiyanova IV, Muglia M et al (2004) Nat Genet 36: 449–451 [DOI] [PubMed] [Google Scholar]
- 8. Detmer SA, Chan DC (2007) J Cell Biol 176: 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rocha AG, Franco A, Krezel AM et al (2018) Science 360: 336–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ban‐Ishihara R, Ishihara T, Sasaki N et al (2013) Proc Natl Acad Sci USA 110: 11863–11868 [DOI] [PMC free article] [PubMed] [Google Scholar]