

Abstract
Despite recent mass spectrometry (MS)‐based breakthroughs, comprehensive ADP‐ribose (ADPr)‐acceptor amino acid identification and ADPr‐site localization remain challenging. Here, we report the establishment of an unbiased, multistep ADP‐ribosylome data analysis workflow that led to the identification of tyrosine as a novel ARTD1/PARP1‐dependent in vivo ADPr‐acceptor amino acid. MS analyses of in vitro ADP‐ribosylated proteins confirmed tyrosine as an ADPr‐acceptor amino acid in RPS3A (Y155) and HPF1 (Y238) and demonstrated that trans‐modification of RPS3A is dependent on HPF1. We provide an ADPr‐site Localization Spectra Database (ADPr‐LSD), which contains 288 high‐quality ADPr‐modified peptide spectra, to serve as ADPr spectral references for correct ADPr‐site localizations.
Keywords: ADP‐ribosylation, ARTD1/PARP1, genotoxic stress, HPF1, tyrosine ADP‐ribosylation
Subject Categories: Methods & Resources; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Protein ADP‐ribosylation is a physiologically and pathologically important post‐translational modification (PTM) that regulates cellular activities by modifying proteins with a single (MARylation) or multiple (PARylation) ADP‐ribose (ADPr) units derived from NAD+ 1, 2. The human genome encodes 17 intracellular ADP‐ribosyltransferases (ARTDs), formerly called poly ADP‐ribose polymerases (PARPs), that are distributed in different cellular compartments 2, 3. While ARTDs are structurally homologous and resemble the diphtheria toxin, the molecular mechanisms by which they modify target proteins are not fully defined. It is not yet clear whether each ARTD can modify all or only a specific subset of the known ADPr‐acceptor amino acids or whether they are all capable of modifying the same amino acid(s) within a given target protein.
For many years, technical challenges have limited the identification of ARTD‐specific target proteins. But the development of MARylated‐peptide enrichment and/or ADPr labeling techniques prior to mass spectrometry (MS)‐based ADP‐ribosylome analysis 4, 5, 6, 7 drastically improved the identification of ADP‐ribosylated proteins 8, 9, 10, 11, 12. Our group and others have shown that genotoxic stress conditions specifically induce the ADP‐ribosylation of hundreds of proteins. These studies identified ARTD1/PARP1 and histones to be the main acceptors of ADPr under these conditions 11, 12, 13, 14. ARTD1 and ARTD2/PARP2 are considered the two main enzymes that “write” nuclear PARylation 2, 3, 9. Although these studies have significantly advanced our understanding of cellular ADP‐ribosylation, it still remains unclear what the bona fide ADP‐ribosylation targets of these ARTDs are in vivo.
Proteomics studies have identified several ADPr‐acceptor amino acids in mammalian cells, namely lysine (K), arginine (R), aspartic acid (D), glutamic acid (E), cysteine (C), and serine (S) 2, 5, 7, 8, 11, 15, 16. Using ARTD1 as the writer of in vitro ADP‐ribosylation modifications, a few of the described ADPr‐acceptor amino acids have been biochemically validated by site‐directed mutagenesis or Edman sequencing 2, 3, 5, 11. Unfortunately, accurate ADPr‐site localization remains challenging partially due to the fact that MS search engines require the user to predefine all possible ADPr‐acceptor amino acids. Indeed, earlier reports from our group identified K‐ADPr sites within a KS modification motif 4, 11. Re‐analysis of these data to include S as an ADPr‐acceptor amino acid led to the reassignment of most of the K‐ADPr modifications to S‐ADPr 4, 5. Following the identification of S‐ADP‐ribosylation on histones and ARTD1 15, Bonfiglio et al found that histone PARylation factor 1 (HPF1, previously C4orf27) 17 functions as a cofactor for ARTD1 and ARTD2 that directs ADP‐ribosylation specifically toward S‐ADPr‐modifications 5. Together, these findings suggested that new ADPr‐acceptor amino acids await discovery and that previously reported sites potentially require revision due to the fact that not all possible ADPr‐acceptor amino acids were considered when analyzing the MS data.
Results and Discussion
Revising Mascot‐based MS search parameters for ADPr variable modifications identifies tyrosine as a novel potential ADPr‐acceptor amino acid
To determine whether new ADPr‐acceptor amino acids could be identified within our ADPr MS datasets 4, revised Mascot searches were used. The data used here were acquired from biological replicate H2O2‐treated HeLa cell samples (n = 4) using an ADPr‐optimized HCD‐PP‐EThcD/HCD MS workflow (Fig 1A). For these searches, we opened the variable ADPr PTM search parameters to include all 20 amino acids as potential ADPr‐acceptor amino acids (Appendix Table S1). False discovery rates (FDRs) were estimated at every filtering step using a target‐decoy search strategy, which, when combined with previously established stringent filtering steps (Mascot scores > 20 and expectation values < 0.05) 18, ensured that only high‐quality spectra were assessed further. Using this strategy, no decoy hits were detected in the reported lists of ADP‐ribosylated peptides and only strong potential ADP‐ribosylated peptide candidates were analyzed further. The individual searches returned ~700 ADPr‐site localizations per search and ~500 ADPr modifications that localized to “new” potential ADPr‐acceptor amino acids (Appendix Table S2). An obvious disadvantage of the semi‐repetitive search parameters used here is that they returned several ADPr‐site localizations for the same spectra. Therefore, the spectra annotations were validated manually to determine which of the predicted ADPr‐sites were supported the most.
Figure 1. MS acquisition method and refined MS bioinformatics workflows.

- Schematic of the sample preparation and HCD‐PP‐EThcD/HCD MS acquisition method workflow employed by Bilan et al 4 to generate the EThcD and HCD datasets used here.
- Schematic of the bioinformatics workflow employed here. In the initial phase of these analyses, variable ADPr PTM search parameters were opened to include all 20 amino acids as potential ADPr acceptors. Ion scores were then calculated to help identify the best‐quality ADPr‐site localizations within modified peptides and peptides for manual validation of ADPr‐site localization. Based on these manual validations, the search parameters were refined to include S, D, E, R, K, and Y (SDERKY) or S, R, and Y (SRY) as ADPr‐acceptor amino acids and the workflow was re‐applied.
Prior to manual review, an additional ion score filter was applied to the Mascot score and expectation value filtered ADP‐ribosylated peptide candidates. The ion scores were calculated based on the number of observed peptide fragment ions in the b, y, c, and z ion series normalized over the maximum number of observable theoretical peptide fragment ions (Fig 1B). Only ADPr‐site localizations for unique peptide sequences (117 unique peptides) that had at least one spectrum with ion score > 0.5 (448 unique spectra), indicating that at least half of the theoretical ions within a given spectrum for the peptide of interest were observed, were manually validated. During this validation process, ADPr‐spectra and ADPr‐site localizations were confirmed based on the presence of ADPr‐site‐determining ions and/or a complete series of annotated c/b/z/y (EThcD spectra) or b/y (HCD spectra) fragment ions. This review process identified 288 high‐quality spectra (181 EThcD and 107 HCD spectra), which supported confident ADPr‐site localization. In agreement with previous studies 4, 11, 15, 18, all known ADPr acceptors (S, D, E, R, and K) were identified and S‐ADPr modifications were the most abundant identified in vivo (Figs 2A and EV1A). In addition, several previously unidentified ADPr‐acceptor amino acids were predicted including alanine (A), leucine (L), asparagine (N), glutamine (Q), valine (V), and tyrosine (Y) (Figs 2A and EV1A, and Dataset EV1). However, several of the side chains of these new potential ADPr‐acceptor amino acids are biochemically inert. Moreover, all but Y were only identified in HCD spectra (Fig EV1A). While high‐resolution HCD fragmentation methods successfully and confidently identify ADP‐ribosylated peptides, the inherent fragility of ADPr modifications under these fragmentation conditions severely limits confident ADPr‐site localization 4, 5, 11, 15, 19. The fact that only one‐third of the spectra within the ADPr‐LSD are HCD spectra confirms that HCD fragmentation methodologies further supports this. Together, these findings led us to reason that some of these new ADPr‐acceptor amino acids were incorrectly assigned.
Figure 2. Refining the variable ADPr PTM MS search parameters identifies tyrosine as a novel ADPr‐acceptor amino acid.
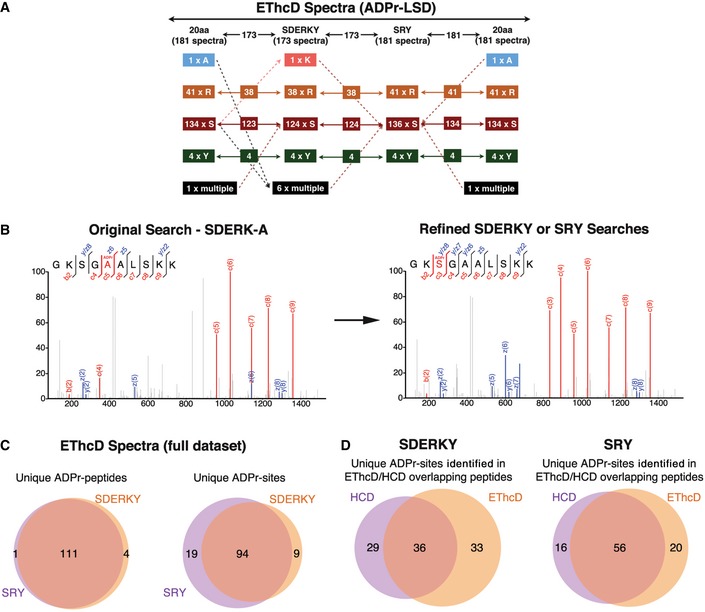
- The ADPr‐site localization results from the initial (20 amino acids) searches and refined searches were compared. The flowchart depicts how ADPr‐acceptor amino acid identifications changed for the 181 EThcD ADPr localization training spectra as the variable ADPr search parameters were refined. Importantly, these analyses identified novel tyrosine (Y) ADPr modifications at each phase of our analyses.
- The left panel depicts an EThcD fragmentation spectrum of an ARTD1 peptide in which the original search localized the ADPr modification to A5 of the peptide. The right panel is the same EThcD spectrum annotated after the data were searched using the refined ADPr search parameters developed here. The spectrum annotations on the right provide stronger evidence that this ADPr modification is on S3.
- Comparison of total number of unique ADP‐ribosylated peptides and unique ADPr sites identified (Mascot localization probability > 60%) in the SDERKY and SRY searches.
- Comparison of the ADPr‐site localizations (Mascot localization probability > 60%) for ADP‐ribosylated peptides that were identified by both EThcD and HCD fragmentation methods.
Figure EV1. HCD spectra ADPr‐site localizations show considerable movement as Mascot searches were refined.
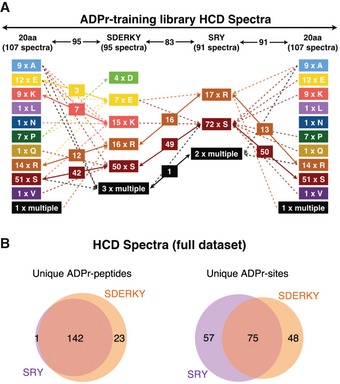
- ADPr‐site localization results from the initial (20 amino acids) searches and refined searches were compared. The flowchart depicts how ADPr‐acceptor amino acid localizations changed for the 107 HCD ADPr localization training spectra as the variable ADPr search parameters were refined.
- Comparison of total number of unique ADP‐ribosylated peptides and unique ADPr‐sites identified (Mascot localization probability > 60%) from HCD spectra in the SDERKY and SRY searches.
To tease this out, we used the manually curated 288 spectra (Dataset EV1) to build a spectra library for ADPr‐site localization training. The ADPr‐site Localization Spectra Database (ADPr‐LSD; Dataset EV2) was then used to iteratively refine the search parameters to confidently and accurately assign the ADPr‐site within modified peptides. Two search parameter refinement steps were carried out using Mascot where the ADPr‐acceptor amino acid groups were defined as SDERKY or SRY. These groups were defined by Y as the only new biochemically active potential ADPr‐acceptor amino acid in combination with either SDERK, representing all well‐established acceptor sites, or only SR as the two most abundant acceptor sites defined under the tested conditions. The refined peptide identification and ADPr‐site localization results were compared with each other and the original assignments (Figs 2A and EV1A). Throughout this process, we found that overall the ADPr‐site localization scores increased as the search parameters were refined to only include amino acids that could be confirmed by manual spectra annotation (Dataset EV1). Moreover, this step‐wise process bioinformatically and manually validated Y as a new potential ADPr‐acceptor amino acid. The improved spectra fragment ion annotations obtained using the refined search parameters also demonstrated that spectra previously assigned to other potential “new” ADPr‐acceptors (A, L, N, Q, and V) were indeed incorrectly localized (Fig 2B, Datasets EV1 and EV2). Nevertheless, our results also indicate that narrowing the search space too much can also result in the loss of ADPr‐PSM identifications (e.g., four spectra modified at D) when the “true” ADPr‐site is not queried in the search (e.g., see Dataset EV2; peptides PDPAKSAPAPK and PEPAKSAPAPK provide the best examples).
To assess the performance of our improved ADPr‐search parameters on the full dataset, rather than just the ADPr‐LSD spectra, we extended our analyses and compared our SDERKY and SRY results with each other. Similar to the results published by Bilan et al 4, the SDERKY search identified 197 unique ADPr‐peptides and the SRY search identified 179 unique ADPr‐peptides with perfect peptide identification overlaps (Figs 2C and EV1B, and Table 1, and Dataset EV3). Comparison of the ADPr‐site localizations returned by the SDERKY and SRY searches revealed > 80% ADPr‐site localization agreement for EThcD fragmentation‐based spectra (Fig 2C) and ~55% agreement for HCD spectra (Fig EV1B). Approximately one half of the ADPr‐peptides identified were found by both HCD and EThcD fragmentation methods. For these peptides, the ADPr‐site localizations were in agreement > 50% of the cases (Fig 2D). This ADPr‐site localization discrepancy highlights the abovementioned challenge of confidently assigning ADPr‐sites using HCD data alone. The majority of the spectra for the common EThcD and HCD ADPr‐PSMs contain KS/RS‐ADPr‐modification sites 4, 15 for which the ADPr‐sites are localized to K‐ADPr, R‐ADPr, or S‐ADPr using the HCD spectra while EThcD spectra localizations are almost exclusively S‐ADPr (Dataset EV3). Finally, and most intriguingly, these comparative analyses identified seven proteins that carry potential Y‐ADPr modifications (Table 2). Importantly, MaxQuant‐based analyses of these MS data using published ADPr search parameters with SDERKY or SRY as variable ADPr‐acceptor amino acids 11, 15 also identified Y‐ADPr modifications (data not shown). Importantly, the Mascot identified Y on RPS3A was also assigned by MaxQuant, further substantiating Y as a novel, albeit low abundant, in vivo ADPr‐acceptor amino acid. The total number of Y‐modified proteins identified in vivo was rather low compared to the S‐modified proteins. This is, however, similar to Y‐phosphorylation modifications, which are also low abundant compared to S/T‐phosphorylations. Studies have clearly demonstrated that Y‐phosphorylation modifications have very important functional roles in vivo 20, 21. Thus, it is possible that this is also the case for Y‐ADPr protein modifications, but additional investigations are required.
Table 1.
Summary of ADPr‐peptides and ADPr‐sites identified using different variable ADPr localization parameters
| SDERKY | SRY | |
|---|---|---|
| Unique ADPr‐peptides | ||
| HCD‐PP‐EThcD | 197 | 179 |
| Unique EThcD | 33 | 37 |
| Unique HCD | 82 | 67 |
| Overlap | 82 | 75 |
| Unique ADPr‐peptides with Mascot localization probability > 60% | ||
| HCD‐PP‐EThcD | 151 | 160 |
| EThcD | 35 | 39 |
| HCD | 58 | 54 |
| Overlap | 58 | 67 |
| Unique ADPr‐sites with Mascot localization probability > 60% | ||
| HCD‐PP‐EThcD | 190 | 189 |
| EThcD | 67 | 57 |
| HCD | 87 | 76 |
| Overlap | 36 | 56 |
Table 2.
Y‐ADPr‐modified proteins
| Gene name | Peptide sequence_ADPr‐site | Y‐ADPr‐site on protein | Spectra type |
|---|---|---|---|
| RPS3A | KTSYAQHQQVR_Y4 | Y155 | EThcD |
| HPF1 | TFHGAGLVVPVDKNDVGYRELPETDADLKR_Y18 | Y238 | EThcD/HCD |
| HNRNPA1 | NQGGYGGSSSSSSYGSGR_Y5 | Y252 | HCD |
| HNRNPA2B1 | NMGGPYGGGNYGPGGSGGSGGYGGR_Y6 | Y319 | HCD |
| HDGF | STANKYQVFFFGTHETAFLGPK_Y6 | Y38 | HCD |
| YY1 | KSYLSGGAGAAGGGGADPGNKKWEQK_Y3 | Y185 | HCD |
| P4HB | LAKVDATEESDLAQQYGVRGYPTIK_Y16 | Y94 | HCD |
ARTD1 is the main writer of genotoxic stress‐induced ADP‐ribosylome
To dissect the contributions of ARTD1 and ARTD2 on the observed H2O2‐induced ADP‐ribosylome, we knocked down their expression in HeLa cells via stable expression of short hairpin RNAs (shRNAs) targeting ARTD1 (shARTD1), ARTD2 (shARTD2), or control shRNAs. Knockdown efficiency was verified by Western blot (Fig EV2A). The engineered cell lines were treated in biological triplicate with H2O2 or left untreated, and the resulting ADP‐ribosylomes were defined using HCD‐based MS as previously described 11. These datasets were then analyzed for variable SDERKY or SRY ADP‐ribosylation using Mascot. In general, while the untreated ADP‐ribosylomes of each cell line were quite similar, specific and significant decreases in the total number of ADP‐ribosylated spectra and proteins identified were observed in H2O2‐treated shARTD1 cells (Figs 3A and EV2B, and Dataset EV4). In fact, shARTD1 ADP‐ribosylation levels were more similar to untreated shControl or untreated shARTD2 cells than those observed for H2O2‐treated shControl or shARTD2 cells (Fig EV2B). These findings confirmed that ARTD1 is the main driver of nuclear ADP‐ribosylation under the tested conditions 22. To address a potential crosstalk between ARTD1 and ARTD2, we quantified the identified ADPr‐sites using MS1‐based label‐free quantification (LFQ). While quantified relative ADPr‐peptide abundances were consistent among biological replicates (Spearman's rank correlation coefficients 0.71–0.83), poor correlation between the shARTD1 and the shControl or shARTD2 samples was observed (Fig EV2C and D). Moreover, our data demonstrate that ARTD1 is the main writer of genotoxic stress‐induced nuclear de novo ADP‐ribosylation and suggests that ARTD2 does not functionally complement ARTD1 in vivo. Finally, our findings also indicate that ARTD1 and ARTD2 only marginally contribute to the steady‐state (untreated) ADP‐ribosylome (Figs 3A and EV2B).
Figure EV2. Analysis of shControl, shARTD1, and shARTD2 knockdown HeLa cells.
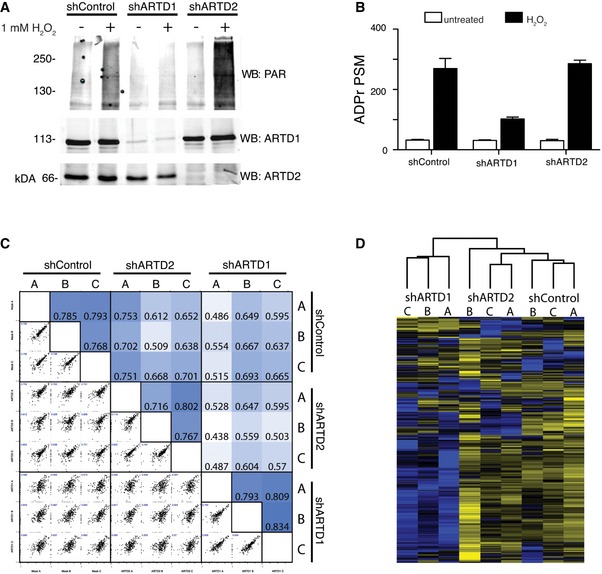
- α‐ARTD1, α‐ARTD2, and α‐PAR Western blot analyses of untreated and H2O2‐treated shControl, shARTD1, and shARTD2 HeLa cells lysates confirm specific knockdown of ARTD1 or ARTD2 and specific inhibition of H2O2‐induced PAR following loss of ARTD1.
- Number of ADPr‐PSMs identified in untreated and H2O2‐treated HeLa cells following ARTD1 or ARTD2 knockdown. Error bars represent standard deviations of the mean (n = 3).
- Heat map of Spearman's rank correlation coefficient and multi‐scatter plots showing reproducibility between cell line ADP‐ribosylomes.
- Heat map of unsupervised clustering of the z‐score of ADPr‐site abundance in the different cell lines.
Figure 3. ARTD1 mediates proteome‐wide S‐ and Y‐ADP‐ribosylation.
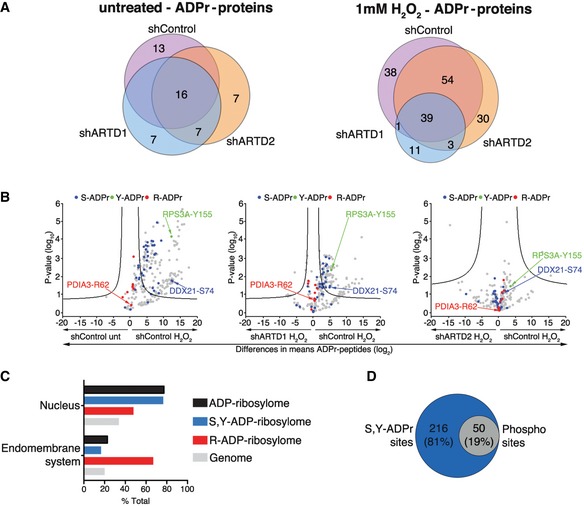
- Venn diagrams indicate the overlap of ADP‐ribosylated proteins identified in untreated and H2O2‐treated HeLa cells following ARTD1 or ARTD2 knockdown.
- Volcano plots comparing ADP‐ribosylated peptides identified in the different cell lines and conditions. ADP‐ribosylation sites confirmed by EThcD spectra are color‐coded for S‐ADPr (blue), Y‐ADPr (green), and R‐ADPr (red) modifications. Serine (DDX21), tyrosine (RPS3A), and arginine (PDIA3) modifications that were identified within our ADPr localization training spectra library are annotated. The black hyperbolic line represents a permutation‐based false discovery rate (FDR) of 5% and a minimal fold change of 2.
- Gene ontology cellular localization annotation of ADP‐ribosylated proteins following H2O2 treatment: Only SRY ADP‐ribosylated proteins with a Mascot localization score > 90% were considered.
- Comparison of the S‐ and Y‐ADP‐ribosylation identified here with the HeLa phosphoproteome identified by Sharma et al 20.
Previous studies observed PAR formation in ARTD1 knockout cells and suggested that ARTD2 may be responsible for these modifications 23. Here, we found that ARTD1 and ARTD2 do not modify the same ADP‐ribosylation target proteins in H2O2‐treated HeLa cells, indicating that ARTD2 is likely regulated by a different stress stimulus than ARTD1. Recent observations confirm that ARTD1 and ARTD2 regulate two independent, but intrinsically linked aspects of DNA base damage tolerance by promoting base excision repair directly 24. Nevertheless, ARTD1−/−/ARTD2−/− mice are not viable 25; thus, it is possible that ARTD1 and ARTD2 functionally compensate for each other indirectly by modifying different target proteins that function in the same cellular processes.
Proteome‐wide S‐ADP‐ribosylation and Y‐ADP‐ribosylation are mediated by ARTD1
To determine proteome‐wide ARTD1 ADPr‐acceptor amino acid specificity, comparative analysis of our optimized ADPr search parameters (SDERKY versus SRY) revealed a slight (~15%) increase in the number of unique peptides identified in the SRY search, while comparable numbers of ADPr‐spectra matches were identified (Fig EV3A and B). Overall, the increased search parameter (SDERKY) did not lead to a significant decrease in R‐ADPr, but reduced S‐ADPr assignments in favor of K‐ADPr, D‐ADPr, and E‐ADPr modifications (Fig EV3C and D). These searches demonstrated that R is the primary steady‐state ADPr‐acceptor amino acid. Unfortunately, the ART responsible for this modification is not yet known, but the cytoplasmic localization of proteins suggests that a cytoplasmic ART writes these modifications.
Figure EV3. Comparison of SRY and SDERKY ADPr searches following ARTD1 and ARTD2 knockdown reveals that ARTD1 is the writer of cellular Ser‐ADPr and Tyr‐ADPr modifications.
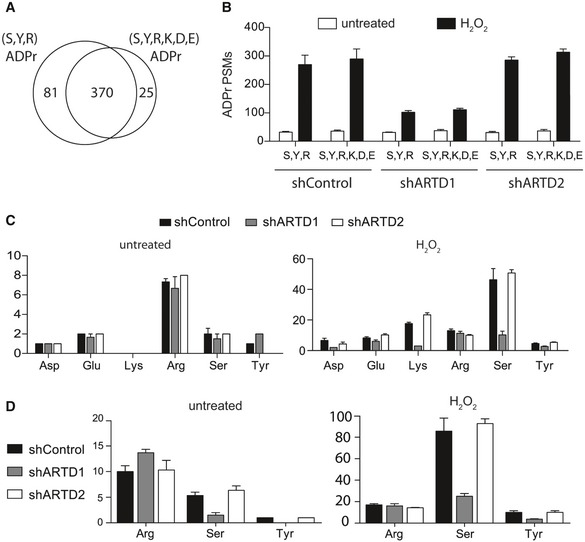
-
AVenn diagram depicts the overlap of the total number of unique ADPr‐peptides identified following H2O2 treatment of shControl, shARTD1, and shARTD2 cells by Mascot‐based searches that considered either S, R, and Y or S, D, E, R, K, and Y as variable ADPr‐acceptor amino acids. Note that ADPr‐site localization within the peptide was ignored for this analysis.
-
BTotal number of ADPr‐peptide spectrum matches (PSMs) identified in the different cell lines and treatment conditions for SRY and SDERKY searches. Error bars represent standard deviations of the mean (n = 3).
-
C, DTotal number of ADPr‐sites localized (Mascot localization score > 60%) in the SDERKY (c) and SRY (d) searches. Error bars represent standard deviations of the mean (n = 3).
Following H2O2 treatment, the major modification site identified was S. Interestingly, our data indicate that both S and Y modifications required ARTD1 (Fig EV3C and D). Quantification of the site‐specific ADP‐ribosylation changes in response to ARTD1 or ARTD2 knockdown, via volcano plot analyses and annotation of EThcD‐localized ADPr sites, further confirmed that S‐ADP‐ribosylation and Y‐ADP‐ribosylation were dependent on ARTD1 (Fig 3B). These findings are in agreement with earlier in vitro reports of ARTD1 using protein microarrays 26, 27 or purified histones 5. ARTD1 was among the main acceptors of ADP‐ribosylation under the tested conditions and was modified at S but not Y. This might be explained by the observation that, both in vivo and in vitro, ARTD1 is highly modified in its automodification domain (Ref. 5, 11 and this study), which spans amino acids 373–524 and does not contain a Y residue. We also demonstrate that R is the main steady‐state ADPr‐acceptor amino acid in HeLa cells.
Pathway analysis identifies links between the ADP‐ribosylome, cellular functions, and the phosphoproteome
GO analysis of the ADPr‐ribosylated proteins identified revealed that S‐ADP‐ribosylated and Y‐ADP‐ribosylated proteins are typically nuclear proteins that interact with RNA and/or DNA and play a role in chromatin organization (Fig 3C). In contrast, similar to our previous studies 22, R‐ADP‐ribosylated proteins localized to the endomembrane system and function in protein folding and/or endoplasmic reticulum (ER) stress‐associated processes. Studies have demonstrated that ~90% of the phosphorylation sites observed in mammalian cells localize to S, whereas less than 1% localize to Y 20. Given that our studies identified S and Y as the main H2O2‐induced ADPr‐acceptor amino acids, we investigated the potential link between protein ADP‐ribosylation and phosphorylation modifications. Comparison of our datasets with those recently published by Sharma et al 20 revealed that 19% of the S‐ADPr sites that we identified here have also been identified under different experimental conditions as phosphorylation sites in HeLa cells (Fig 3D and Dataset EV5), which suggests that crosstalk between phosphorylation and O‐linked ADP‐ribosylation may occur.
RPS3A‐Y155 is ADP‐ribosylated by ARTD1 in the presence of HPF‐1
Our extensive manual and bioinformatic analyses identified seven proteins that were potentially ADP‐ribosylated on Y residues in vivo (Table 2), and our LFQ‐based data indicated that their novel Y‐ADP‐ribosylation is modified by ARTD1 (Figs 3B and EV3C and D). The low abundance of these modifications, relative to S‐ADP‐ribosylation, and the uncertainty surrounding ADP‐ribosylation sites localized using HCD spectra 5 prompted us to independently validate Y as an ADPr‐acceptor amino acid using in vitro ADP‐ribosylation assays. For these studies, we proceeded with RPS3A, as several high‐quality EThcD spectra supporting Y155‐ADPr modification were identified at each phase of our bioinformatic analyses [Fig 4A and Dataset EV2 (KTSYAQHQQVR)]. Full‐length recombinant wild‐type RPS3A (RPS3A‐WT) and Y155F mutant (RPS3A‐Y155F) proteins were generated for these studies. Little or no [32P]‐ADPr was incorporated into RPS3A‐WT and RPS3A‐Y155F when the modification reactions were carried out with ARTD1 alone in the presence of physiological levels of NAD+ (Fig 4B). Recent studies have demonstrated that the in vitro enzymatic activity of ARTD1 can be augmented by HPF1 and that ARTD1 modification specificity shifts toward S residues 5, 17. We reasoned that Y‐ADP‐ribosylation should be biochemically similar to O‐linked S‐ADP‐ribosylation; thus, addition of HPF1 could provide ARTD1 the enzymatic specificity required to confirm Y‐ADP‐ribosylation. Based on the incorporation of [32P]‐ADPr, we found that HPF1 not only augmented ARTD1 automodification but also meditated very strong and, seemingly, specific trans‐modification of RPS3A‐WT relative to that observed for RPS3A‐Y155F (Fig 4B and C). Together, these data strongly indicate that RPS3A is ADP‐ribosylated by ARTD1 at Y155, confirming that Y can act as an ADPr‐acceptor amino acid and demonstrating that HPF1 functions to specifically tune the enzymatic activity of ARTD1 toward O‐linked S‐ and Y‐ADP‐ribosylation.
Figure 4. Biochemical validation of RPS3A‐Y155 ADPr modification.
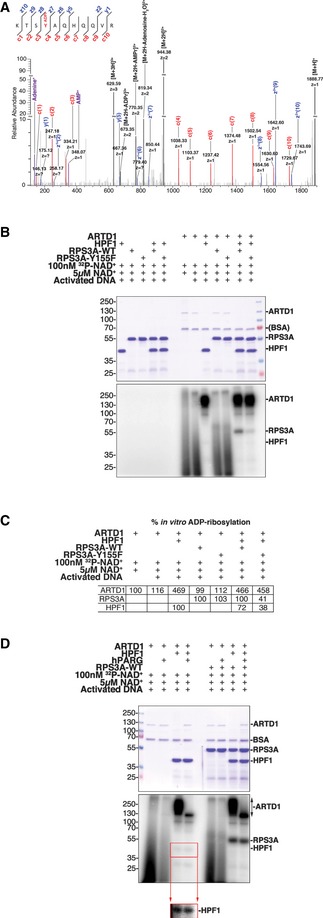
- High‐resolution EThcD fragmentation spectrum of an RPS3A peptide modified with ADP‐ribose on Y155.
- Recombinant RPS3A‐WT and RPS3A‐Y155F were in vitro ADP‐ribosylated with [32P]‐NAD+, activated DNA, and recombinant ARTD1 in the presence or absence of recombinant HPF1. Samples were resolved by SDS–PAGE and analyzed by autoradiography. Top: Coomassie blue‐stained polyacrylamide gel; bottom: autoradiograph with levels adjusted to enhance exposure.
- Relative percentages of the in vitro ADP‐ribosylation (radioactive signal) of ARTD1 (relative to radioactive signal with [32P]‐NAD+ and activated DNA alone), RPS3A‐WT, and RPS3A‐Y155F (relative to radioactive signal for RPS3A‐WT for each condition) are shown (n = 3).
- Recombinant ARTD1 and RPS3A‐WT were in vitro ADP‐ribosylated with [32P]‐NAD+ and activated DNA in the presence or absence of recombinant HPF1. The indicated samples were then treated with recombinant human PARG (hPARG) to reduce PAR modifications to MAR modifications. The samples were resolved by SDS–PAGE and analyzed by autoradiography. Top: Coomassie blue‐stained polyacrylamide gel; middle: autoradiograph with levels adjusted to enhance exposure; bottom inset: autoradiograph of HPF1 section only with enhanced exposure.
Tyrosine residues can be MARylated and likely PARylated
To elucidate whether the Y residue can be MARylated or PARylated, ARTD1 and ARTD1/RPS3A were in vitro‐modified in the presence or absence of HPF1. Following modification, each reaction was then treated with PARG to specifically remove the PAR chains from the modified proteins (Fig 4D). Very little protein‐associated in vitro‐modification was observed in the absence of HPF1, while HPF1 induced strong automodification of ARTD1, as well as trans‐modification of RPS3A and weak modification of HPF1. Interestingly, when these modified proteins were treated with PARG we observed a very strong reduction in ARTD1 [32P]‐ADPr signals clearly suggesting that most ARTD1 automodifications are PAR (Fig 4D). HPF1 was also ADP‐riboslylated under these conditions (Fig 4B and D), but to a lesser extent than to RPS3A. PARG treatment did not alter the modification of HPF1, suggesting that HPF1 is MARylated under these conditions. In contrast, we observed a slight decrease in the RPS3A [32P]‐ADPr signals and a small shift in the gel migration pattern for this protein following PARG treatment. Additionally, enrichment of H2O2 cell lysates with the WWE domain, known to interact only with PARylated proteins, and subsequent analysis of the modified peptides by MS revealed that RPS3A is PARylated following H2O2 treatment (Appendix Table S3). While our WWE enrichment and PARG ADPr reduction experiments clearly indicated that RPS3A is PARylated, analysis of our in vivo data revealed that RPS3A can also be ADP‐ribosylated at K152, S154, and Y155. Thus, these data do not allow us to clearly determine whether Y‐155 PARylation alone is responsible for the ADPr modification and protein migration changes observed. It is possible that K152 and/or S154 are PARylated alone or in conjunction with Y155. As a result, additional experiments using K152A, S154A, and Y155F RPS3A mutant proteins are required to determine which of the identified RPS3A ADPr‐acceptor amino acids are PARylated and whether tyrosine residues can be PARylated.
MS analysis of in vitro ADP‐ribosylation assays confirms that RPS3A and HPF1 are Y‐ADPr trans‐ADP‐ribosylation target proteins
While the findings presented in Fig 4 clearly demonstrate that RPS3A is ADP‐ribosylated at Y155, residual ADP‐ribosylation was observed in the RPS3A‐Y155F mutant protein, which indicated that other ADPr sites may also be present. Indeed, K152 and S154 of RPS3A were also identified as potential ADPr sites in the in vivo MS data presented above (Dataset EV3). To define the exact localization of the in vitro ADPr modifications on RPS3A, the ARTD1, HPF1, and RPS3A in vitro modification reactions were analyzed by MS using our ADPr‐optimized HCD‐PP‐EThcD/HCD workflow 4, 28. As our group and others have clearly demonstrated that ETD or ETD‐like fragmentation methods provide the most accurate data for localizing ADPr sites within a modified peptide 4, 19, only the resulting EThcD spectra were used to define the ADPr‐sites written on these proteins in vitro. MS analysis of RPS3A‐WT or RPS3A‐Y155F in vitro ADP‐ribosylated in the presence of ARTD1 alone failed to identify any ARTD1, RPS3A‐WT, or RPS3A‐Y155 ADP‐ribosylated peptides (data not shown). In line with the radioactivity assays presented above, addition of HPF1 to the reactions led to identification of several ARTD1 ADPr‐sites (S499, S504, S507, and S519). Importantly, S499, S507, and S519 were identified as the major genotoxic stress‐induced in vivo ADPr‐sites present in ARTD1 (Dataset EV3 and 4, 5), indicating that the in vitro modification conditions used here closely mimic those met in vivo following genotoxic stress.
Our in vivo analyses also identified a single ADP‐ribosylated peptide from HPF1 (TFHGAGLVVPVDKNDVGYRELPETDADLKR) where the ADPr modification was localized to Y238 (Fig EV4A). Interestingly, while the signals acquired for HPF1 ADP‐ribosylation in the [32P]‐NAD+ in vitro modification assays presented in Fig 4B and D were very weak, our MS analyses of the ARTD1/HPF1 in vitro ADPr modifications confirmed that Y238 of HPF1 is indeed ADP‐ribosylated (Fig EV4B). Together, these data clearly demonstrated that HPF1 is itself a trans‐ADP‐ribosylation target protein and that it is modified at Y238. Previous studies have demonstrated that both Y238 and R239 are required for its ADP‐ribosylation factor functions 17. Given the reported importance of Y238 in HPF1 functional activities, we wonder if not just the amino acid itself but ADP‐ribosylation of Y238 helps control HPF1 ADP‐ribosylation cofactor functions. Further experiments including genetic complementation of HPF1 knockout cells with HPF1 WT and modification‐deficient mutants are required.
Figure EV4. Spectral evidence supporting HPF1‐Y238 ADP‐ribosylation.

- High‐resolution EThcD fragmentation spectra of the HPF1 peptide TFHGAGLVVPVDKNDVGYRELPETDADLKRELPETDADLKR identified during re‐analysis of in vivo HeLa H2O2‐treated MS datasets. This spectrum supports ADP‐ribosylation of Y238.
- High‐resolution EThcD fragmentation spectra of HPF1 peptide TFHGAGLVVPVDKNDVGYRELPETDADLKR acquired during MS analysis of ARTD1/HPF1 in vitro ADP‐ribosylation assays. This spectrum also supports ADP‐ribosylation of Y238.
Similarly, addition of HPF1 to the ARTD1/RPS3A‐WT reaction enabled us to confirm that both Y155 and S154 function as ADPr sites on RPS3A, with Y155 functioning as the main acceptor site (eight EThcD spectral IDs) compared to S154 (three EThcD spectral IDs; Fig EV5A). These findings were confirmed by our MS analysis of ARTD1/HPF1/RPS3A‐Y155F in vitro modifications, which revealed that RPS3A‐Y155F mutant proteins only carry S154 ADPr modifications (Fig EV5B). Taken together, these findings demonstrate that Y serves as the main ADPr‐acceptor amino acid within HFP1 and RPS3A both in vivo and in vitro.
Figure EV5. Spectral evidence supporting RPS3A‐Y155 and ‐S154 in vitro ADP‐ribosylation.

- High‐resolution EThcD fragmentation spectra of RPS3A peptide KTSYAQHQQVR acquired during MS analysis of ARTD1/HPF1/RPS3A‐WT in vitro ADP‐ribosylation assays. Top spectrum supports ADPr localization on Y155, and the bottom spectrum supports ADPr localization on S154.
- High‐resolution EThcD fragmentation spectrum of RPS3A peptide KTSFAQHQQVR acquired during MS analysis of ARTD1/HPF1/RPS3A‐Y155F in vitro ADP‐ribosylation assays. Spectrum supports ADPr localization on S154.
We have made great strides toward overcoming the bioinformatic challenges associated with analyzing ADP‐ribosylome MS datasets, but improvements are still required. It will also be important to explore whether other MS search engines and data analysis pipelines identify more ADPr‐peptides and assign ADPr‐sites with greater confidence 29, 30, 31. Moreover, our ability to confidently assign only S‐ADPr, R‐ADPr, and Y‐ADPr modifications in vivo raises concerns as to why E‐ADPr and D‐ADPr were not robustly detected using the methodologies applied here; especially given that other studies provide strong evidence that these modifications are written in vivo and in vitro 8, 9, 12. It could simply be that the peptide fragmentation (EThcD versus CID 32, 33) methods used here limit D‐ADPr and E‐ADPr modification identifications. T‐ADP‐ribosylation modifications were not identified, suggesting that T is not at all ADP‐ribosylated by a mammalian ART or to an undetectable extent under the conditions tested. Additional studies are required to clarify these potential biases.
For proteomic researchers embarking on ADP‐ribosylome studies, we think that this study has clarified some of the bioinformatic challenges associated with this PTM and hope they take advantage of the insights gained. We recommend that whenever possible ETD/EThcD fragmentation be used for confident ADPr‐site localizations while HCD confidently identify ADP‐ribosylated peptides. Moreover, we recommend that ETD/EThCD spectra be searched in duplicate with variable ADPr modifications on S, D, E, R, K, and Y and on just S, R, and Y. Comparative analyses of these results will identify ADPr sites that are common to both searches and those that differ. ADPr‐site localizations that differ between these searches require manual validation of the spectra annotations. Finally, the ADPr‐site Localization Spectra Database (ADPr‐LSD) included here provides spectral references for the ADPr sites identified within and can be used by researchers as a training resource to improve high‐quality ADPr‐spectra identifications and ADPr‐site localizations.
Materials and Methods
Generation of stable shARTD1 and shARTD2 cell lines
Virus generation and HeLa cell transduction were carried out as previously described 34. Briefly, short hairpin RNAs (shRNAs), targeting the mRNA coding sequences ARTD1 (CCGAGAAATCTCTTACCTCAA) or ARTD2 (TCTGAATCCAGATGGTTATA), or a scramble shRNA (CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG) were cloned into pRDI292. These vectors were then used to generate replication incompetent retroviruses in HEK‐293 cells. HeLa cells (Kyoto) were infected with the resulting retroviruses for 8 h, cultured for 48 h without selection, and placed under selective growth conditions (2 μg/ml puromycin) to select for transduced cells. ARTD1 and ARTD2 knockdowns were confirmed on the mRNA and protein levels via qPCR and Western blot analyses, respectively.
Cell culture conditions and ADPr‐peptide enrichment
shControl, shARTD1, and shARTD2 HeLa cells (Kyoto) were cultured in Dulbecco's modified Eagle's medium [DMEM, supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptavidin] at 37°C with 5% CO2. Cells were either untreated or treated in triplicate with 1 mM H2O2 in PBS containing 1 mM MgCl2. Cells were lysed and processed for MS analysis as previously described using either GST‐Af1521 or GST‐WWE recombinant proteins 4, 11.
Liquid chromatography and mass spectrometry analysis
The ADP‐ribosylomes of shControl, shARTD1, and shARTD2 HeLa cells were identified using an Orbitrap Q Exactive HF mass spectrometer (Thermo Fisher Scientific) coupled to a nano EasyLC 1000 (Thermo Fisher Scientific). The peptides were loaded into the MS using a reverse‐phase C18 (ReproSil‐Pur 120 C18‐AQ, 1.9 μm, Dr. Maisch GmbH) packed self‐made column (75 μm × 150 mm) that was connected to an empty PicoTip emitter (New Objective, Woburn, MA). Peptides were injected into the MS at a flow rate of 300 nl/min and were separated using a 90‐min gradient of 2 to 25% buffer B. Solvent compositions of buffer A and buffer B were 0.1% formic acid and 0.1% formic acid/99.9% acetonitrile, respectively.
For mass spectrometer (MS) analysis, the MS was set to acquire full‐scan MS spectra (300–1,700 m/z) at a resolution of 60,000 after accumulation to an automated gain control (AGC) target value of 3 × 106. Charge‐state screening was enabled, and unassigned charge states and single charged precursors were excluded. Ions were isolated using a quadrupole mass filter with a 2 m/z isolation window. The maximum injection time was set to 240 ms, and HCD fragmentation was performed at 28% normalized collision energy (NCE). Finally, selected ions were dynamically excluded for 20 s.
MS data analysis
For the bioinformatic analyses aimed at identifying novel ADPr‐acceptor amino acids, raw files from Af1521‐enriched H2O2‐treated HeLa samples measured by the HCD‐PP‐EThcD/HCD method 4 were used. These files can be accessed via ProteomeXchange (PXD004676). MS and MS/MS spectra were converted to Mascot generic format (MGF) using Proteome Discoverer, v2.1 (Thermo Fisher Scientific, Bremen, Germany). Separate MGF files were created from the raw file for each type of fragmentation, and the files were processed further as previously described 4.
The resulting MGF files were searched against UniProtKB human database (taxonomy 9606, version 20140422) using Mascot 2.5.1.3 (Matrix Science) as previously described 4 (Appendix Table S1). For HCD fragmentation, ADP‐ribosylation was defined as previously described by Rosenthal et al 18. For EThcD fragmentation, the modification was defined similarly, but marker ions at m/z 428.0372, 348.0709, 250.0940, 136.0623 were ignored for scoring. For initial analysis, the files were searched with fixed carbamidomethylation (C), except for the search with C as a potential ADPr‐acceptor amino acid where C carbamidomethylation was set as variable, and variable oxidation (M). ADP‐ribosylation was set as a variable modification, and 14 separate searches for each MGF file were performed to cover all 20 amino acid acceptors: Individual searches allowed ADPr modifications to be localized on the five experimentally validated ADPr‐acceptor amino acids (S, D, E, R and K) and on one additional amino acid. Upon completion, all search results were integrated and the spectra were filtered based on the presence of at least two of the known ADPr marker ions 35.
Peptides were considered correctly identified when a Mascot score > 20 and an expectation value < 0.05 were obtained. Prior to manual spectra validation, we applied an ion score filter to reduce the spectra for manual validation to properly fragmented peptides. The ion score was based on observed peptide fragment ions in the b, y, c, and z ion series normalized to the maximum number of observable theoretical ions. We manually curated ADPr‐peptide spectra with an ion score > 0.5.
Using this filter, we identified 448 unique spectra in which the ADPr‐site localization was validated manually. Following manual validation, the files were searched again using Mascot with fixed carbamidomethylation (C), variable oxidation (M) and variable ADP‐ribosylation on S, D, E, R, K, and Y (SDERKY) or S, R, and Y (SRY). All ADPr‐site localizations mentioned in the text had Mascot localization scores > 60%, unless otherwise stated.
Label‐free quantification MS analysis of shControl, shARTD1, and shARTD2 ADP‐ribosylomes
Progenesis QI software (v. 3.0.6039.34628, Nonlinear Dynamics, Durham, NC) was used for MS1 precursor‐based label‐free quantification of the shControl, shARTD1, and shARTD2 ADP‐ribosylomes. For these analyses, the raw data were imported into Progenesis and aligned based on MS1 peak retention time. Sample loading variations were normalized based on total signal intensity, and the results obtained were exported as MGFs. These MGFs were then searched for variable ADPr modifications (SDERKY and SRY) using Mascot as described above. The Mascot search results were then imported into Scaffold software (v.4.7.2) and filtered for protein and peptide false discovery rates (FDRs) ≤ 0.01. When multiple charge‐state precursors were sequenced for the same peptide, total peptide amounts were calculated by summing individual charge‐state values. To confidently localize ADPr sites within the identified ADPr‐peptides, the HCD ADPr‐peptides were compared to the EThcD ADPr‐peptides identified in the Bilan et al dataset presented above. All identified ADPr‐peptides were included in this analysis, but only ADPr‐peptides for which we had corresponding high‐quality EThcD fragmentation spectra were assigned a modification site. Volcano plot analysis of the quantified ADPr‐peptides and their non‐modified counterparts was performed using two‐sample testing in Perseus, with permutation‐based FDRs of 5% and minimal fold change of 2 36. Gene ontology analysis was performed using the PANTHER database 37.
Cloning and protein purification
Human HPF1 (pET21a), human RPS3A‐WT (pGEX6P‐3), and human RPS3A‐Y155F (pGEX6P‐3) bacterial expression vectors were constructed by GenScript (Piscataway, NJ, USA). Plasmids were transformed into BL21 Escherichia coli, and protein expression was induced by adding 1 mM IPTG at OD600 0.4–0.6 for 3 h at 30°C. Batch purification of GST‐tagged or His‐tagged proteins was carried out using glutathione sepharose 4B beads (GE Healthcare) or ProBond™ Nickel‐Chelating Resin (Thermo Fisher Scientific) according to the manufacturer's manual. Expression and purification of all recombinant proteins were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) followed by Coomassie staining.
In vitro ADP‐ribosylation assay with recombinant proteins
In vitro ADP‐ribosylation assays were performed based on previously described methods 5, 17. Briefly, recombinant RPS3A (50 pmol) and HPF1 (50 pmol) proteins were mixed as indicated with ARTD1 (2.5 pmol; Trevigen, Gaithersburg, MD, USA) in 1× PARP1 buffer (Trevigen). Activated DNA (Trevigen), cold NAD+, and [32P]NAD+ (6.25 μM, 0.3 μCi/μl; Perkin Elmer) were then added to the reactions at final concentrations of 40 ng/μl, 5 μM, and 100 nM, respectively. Reactions were incubated at 25°C for 20 min. Reactions were stopped via addition of SDS–PAGE sample buffer to 1× final concentration and heat denaturation at 95°C for 10 min. The reaction volume for all in vitro ADP‐ribosylation assays was 25 μl. Samples were resolved by SDS–PAGE and analyzed by autoradiography.
For PARG assays, ARTD1, HPF1, and RSP3A‐WT were ADP‐ribosylated as described above, and the reactions were stopped via addition of PJ34 to a final concentration of 10 μM. The MgCl2 reaction buffer concentration was adjusted to 15 mM, and recombinant human PARG (2.5 pmol) was added to the reactions. PAR digestion was carried out at 37°C for 15 min. Samples were resolved by SDS–PAGE and analyzed by autoradiography.
For mass spectrometry analyses, the reactions were scaled accordingly to allow in vitro modification of 100 μg of RPS3A‐WT or RPS3A‐Y155F. The reactions were carried out as described above and stopped via addition of PJ34 to a final concentration of 10 μM. These samples were then processed for MS analysis as previously described 4, 28, 38.
Western blot analysis
For Western blot analyses, protein samples were resolved using standard SDS–PAGE methods and transferred to nitrocellulose membranes (Merck Millipore). Membranes were then blocked for 3 h at room temperature with blocking buffer (25 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Tween‐20, and 3% bovine serum albumin) and probed overnight with primary antibodies, diluted in blocking buffer, at 4°C. Membranes were then washed with TBS‐T (25 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Tween‐20) and incubated at room temperature for with IRDye‐conjugated secondary antibodies (1:15,000, LI‐COR). Blots were visualized using the Odyssey infrared imaging system (LI‐COR). Dilutions used for the primary antibodies were as follows: 1:1,000 α‐ARTD1 (H‐250 sc‐7150, Santa Cruz); 1:1,000 α‐ARTD2 (Active Motif); 1:1,000 α‐PAR 10H (in‐house).
Visualization of MS spectra and manual validation
Candidate spectra were visualized using lorikeet, a JavaScript‐based viewer 39. Using a custom python script, one HTML document was created per candidate peptide, and each peptide page displays all collected spectra (regardless of the source file) in a vertically stacked fashion to allow for fast comparison.
Data availability
The mass spectrometry proteomics data for the shControl, shARTD1, and shARTD2 HeLa cells as well as the MS analysis of in vitro ADP‐ribosylated ARTD1, HFP1, RPS3A‐WT, and RPS3A‐Y155F recombinant proteins including the RAW files, peak list files (MGFs), and result files (mzIdent) have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009948 (https://doi.org/10.6019/pxd009948). The WWE enrichment data that support the findings of this study are available from the corresponding author on request.
Author contributions
DMLP, ML, VB, KN, KG, LM, and MOH conceived the project and performed data analysis. DMLP, EF, and KN expressed and purified recombinant proteins, and/or performed biochemical assays. ML performed sample preparation and mass spectrometry analysis. LM provided bioinformatics support and compiled the ADPr‐LSD. RI generated HPF1 mutant clones. DMLP, ML, and MOH prepared the manuscript. MOH directed and supervised all aspects of the study. All authors critically reviewed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Acknowledgements
We would like to thank Tobias Suter (University of Zurich) for helpful discussions and for providing editorial assistance. We would also like to thank Patrick Pedrioli (ETH Zürich) for his valuable advice and helpful discussions. We also thank Paolo Nanni, Christian Panse, Peter Gehrig, and Jonas Grossmann from the Functional Genomics Center (FGCZ) of the University of Zurich for discussions and technical advice. We would like to thank Prof. Michael L. Nielsen (University of Copenhagen) for valuable help with MS searches and helpful discussions. ML is supported by the Forschungskredit (FK‐15‐052) from the University of Zurich. KG is supported by the Swiss Cancer Research Foundation (KFS‐3740‐08‐2015‐R). ADP‐ribosylation research in the laboratory of MOH is funded by the Canton of Zurich and the Swiss National Science Foundation Grants (SNF 310030_157019, 31003A_176177).
EMBO Reports (2018) 19: e45310
References
- 1. Barkauskaite E, Jankevicius G, Ahel I (2015) Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP‐dependent protein ADP‐ribosylation. Mol Cell 58: 935–946 [DOI] [PubMed] [Google Scholar]
- 2. Hottiger MO (2015) Nuclear ADP‐ribosylation and its role in chromatin plasticity, cell differentiation, and epigenetics. Annu Rev Biochem 84: 227–263 [DOI] [PubMed] [Google Scholar]
- 3. Luo X, Kraus WL (2012) On PAR with PARP: cellular stress signaling through poly(ADP‐ribose) and PARP‐1. Genes Dev 26: 417–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bilan V, Leutert M, Nanni P, Panse C, Hottiger MO (2017) Combining HCD and EThcD fragmentation in a product dependent‐manner confidently assigns proteome‐wide ADP‐ribose acceptor sites. Anal Chem 89: 1523–1530 [DOI] [PubMed] [Google Scholar]
- 5. Bonfiglio JJ, Fontana P, Zhang Q, Colby T, Gibbs‐Seymour I, Atanassov I, Bartlett E, Zaja R, Ahel I, Matic I (2017) Serine ADP‐ribosylation depends on HPF1. Mol Cell 65: 932–940 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chapman JD, Gagne JP, Poirier GG, Goodlett DR (2013) Mapping PARP‐1 auto‐ADP‐ribosylation sites by liquid chromatography‐tandem mass spectrometry. J Proteome Res 12: 1868–1880 [DOI] [PubMed] [Google Scholar]
- 7. Rosenthal F, Hottiger MO (2014) Identification of ADP‐ribosylated peptides and ADP‐ribose acceptor sites. Front Biosci (Landmark Ed) 19: 1041–1056 [DOI] [PubMed] [Google Scholar]
- 8. Daniels CM, Ong SE, Leung AK (2014) Phosphoproteomic approach to characterize protein mono‐ and poly(ADP‐ribosyl)ation sites from cells. J Proteome Res 13: 3510–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, Kraus WL (2016) Chemical genetic discovery of PARP targets reveals a role for PARP‐1 in transcription elongation. Science 353: 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML (2013) Proteome‐wide identification of poly(ADP‐Ribosyl)ation targets in different genotoxic stress responses. Mol Cell 52: 272–285 [DOI] [PubMed] [Google Scholar]
- 11. Martello R, Leutert M, Jungmichel S, Bilan V, Larsen SC, Young C, Hottiger MO, Nielsen ML (2016) Proteome‐wide identification of the endogenous ADP‐ribosylome of mammalian cells and tissue. Nat Commun 7: 12917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Wang J, Ding M, Yu Y (2013) Site‐specific characterization of the Asp‐ and Glu‐ADP‐ribosylated proteome. Nat Methods 10: 981–984 [DOI] [PubMed] [Google Scholar]
- 13. Andersson A, Bluwstein A, Kumar N, Teloni F, Traenkle J, Baudis M, Altmeyer M, Hottiger MO (2016) PKCalpha and HMGB1 antagonistically control hydrogen peroxide‐induced poly‐ADP‐ribose formation. Nucleic Acids Res 44: 7630–7645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rank L, Veith S, Gwosch EC, Demgenski J, Ganz M, Jongmans MC, Vogel C, Fischbach A, Buerger S, Fischer JM et al (2016) Analyzing structure‐function relationships of artificial and cancer‐associated PARP1 variants by reconstituting TALEN‐generated HeLa PARP1 knock‐out cells. Nucleic Acids Res 44: 10386–10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, Matic I (2016) Serine is a new target residue for endogenous ADP‐ribosylation on histones. Nat Chem Biol 12: 998–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharifi R, Morra R, Denise Appel C, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B et al (2013) Deficiency of terminal ADP‐ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J 32: 1225–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gibbs‐Seymour I, Fontana P, Rack JG, Ahel I (2016) HPF1/C4orf27 is a PARP‐1‐interacting protein that regulates PARP‐1 ADP‐ribosylation activity. Mol Cell 62: 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenthal F, Nanni P, Barkow‐Oesterreicher S, Hottiger MO (2015) Optimization of LTQ‐Orbitrap mass spectrometer parameters for the identification of ADP‐ribosylation sites. J Proteome Res 14: 4072–4079 [DOI] [PubMed] [Google Scholar]
- 19. Bonfiglio JJ, Colby T, Matic I (2017) Mass spectrometry for serine ADP‐ribosylation? Think o‐glycosylation!. Nucleic Acids Res 45: 6259–6264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sharma K, D'Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr‐based signaling. Cell Rep 8: 1583–1594 [DOI] [PubMed] [Google Scholar]
- 21. Hunter T (2009) Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol 21: 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bilan V, Selevsek N, Kistemaker HAV, Abplanalp J, Feurer R, Filippov DV, Hottiger MO (2017) New quantitative mass spectrometry approaches reveal different ADP‐ribosylation phases dependent on the levels of oxidative stress. Mol Cell Proteomics 16: 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Althaus FR, Kleczkowska HE, Malanga M, Muntener CR, Pleschke JM, Ebner M, Auer B (1999) Poly ADP‐ribosylation: a DNA break signal mechanism. Mol Cell Biochem 193: 5–11 [PubMed] [Google Scholar]
- 24. Ronson GE, Piberger AL, Higgs MR, Olsen AL, Stewart GS, McHugh PJ, Petermann E, Lakin ND (2018) PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1‐dependent Rad51 regulation. Nat Commun 9: 746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boehler C, Gauthier L, Yelamos J, Noll A, Schreiber V, Dantzer F (2011) Phenotypic characterization of Parp‐1 and Parp‐2 deficient mice and cells. Methods Mol Biol 780: 313–336 [DOI] [PubMed] [Google Scholar]
- 26. Feijs KL, Kleine H, Braczynski A, Forst AH, Herzog N, Verheugd P, Linzen U, Kremmer E, Luscher B (2013) ARTD10 substrate identification on protein microarrays: regulation of GSK3beta by mono‐ADP‐ribosylation. Cell Commun Signal 11: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Troiani S, Lupi R, Perego R, Depaolini SR, Thieffine S, Bosotti R, Rusconi L (2011) Identification of candidate substrates for poly(ADP‐ribose) polymerase‐2 (PARP2) in the absence of DNA damage using high‐density protein microarrays. FEBS J 278: 3676–3687 [DOI] [PubMed] [Google Scholar]
- 28. Leutert M, Bilan V, Gehrig P, Hottiger MO (2017) Identification of ADP‐ribose acceptor sites on in vitro modified proteins by liquid chromatography‐tandem mass spectrometry. Methods Mol Biol 1608: 137–148 [DOI] [PubMed] [Google Scholar]
- 29. Codrea MC, Nahnsen S (2016) Platforms and pipelines for proteomics data analysis and management. Adv Exp Med Biol 919: 203–215 [DOI] [PubMed] [Google Scholar]
- 30. Eng JK, Jahan TA, Hoopmann MR (2013) Comet: an open‐source MS/MS sequence database search tool. Proteomics 13: 22–24 [DOI] [PubMed] [Google Scholar]
- 31. Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI (2017) MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry‐based proteomics. Nat Methods 14: 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haag F, Buck F (2015) Identification and analysis of ADP‐ribosylated proteins. Curr Top Microbiol Immunol 384: 33–50 [DOI] [PubMed] [Google Scholar]
- 33. Rood JE, Leung AK, Chang P (2011) Methods for purification of proteins associated with cellular poly(ADP‐ribose) and PARP‐specific poly(ADP‐ribose). Methods Mol Biol 780: 153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ariumi Y, Turelli P, Masutani M, Trono D (2005) DNA damage sensors ATM, ATR, DNA‐PKcs, and PARP‐1 are dispensable for human immunodeficiency virus type 1 integration. J Virol 79: 2973–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hengel SM, Goodlett DR (2012) A review of tandem mass spectrometry characterization of adenosine diphosphate‐ribosylated peptides. Int J Mass Spectrom 312: 114–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- 37. Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Larsen SC, Leutert M, Bilan V, Martello R, Jungmichel S, Young C, Hottiger MO, Nielsen ML (2017) Proteome‐wide identification of in vivo ADP‐ribose acceptor sites by liquid chromatography‐tandem mass spectrometry. Methods Mol Biol 1608: 149–162 [DOI] [PubMed] [Google Scholar]
- 39. Riffle M, Merrihew GE, Jaschob D, Sharma V, Davis TN, Noble WS, MacCoss MJ (2015) Visualization and dissemination of multidimensional proteomics data comparing protein abundance during Caenorhabditis elegans development. J Am Soc Mass Spectrom 26: 1827–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Data Availability Statement
The mass spectrometry proteomics data for the shControl, shARTD1, and shARTD2 HeLa cells as well as the MS analysis of in vitro ADP‐ribosylated ARTD1, HFP1, RPS3A‐WT, and RPS3A‐Y155F recombinant proteins including the RAW files, peak list files (MGFs), and result files (mzIdent) have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009948 (https://doi.org/10.6019/pxd009948). The WWE enrichment data that support the findings of this study are available from the corresponding author on request.