

Abstract
The synthesis of a library of nucleoside triphoshate mimetics is described where the Mg2+ chelated triphosphate sidechain is replaced by an uncharged methylenetriazole linked monosaccharide sidechain. The compounds have been evaluated as inhibitors of Bacillus anthracis pantothenate kinase and a competitive inhibitor has been identified with a Ki that is 3-fold lower than the Km value of ATP.
Background
Nucleoside triphosphates (NTPs) play a critical role in a vast range of biological processes ranging from protein phosphorylation and cell signalling to DNA replication. A major challenge for the design of NTP mimetics as inhibitors of NTP-dependent enzymes (eg. kinases, DNA/RNA polymerases) is the replacement of the tetra-anionic triphosphate sidechain with a neutral, or significantly less charged, motif that will facilitate cell permeability whilst ensuring they still bind to their biological target(s) with comparable or greater efficiency than the native substrate. Most kinases/polymerases accept their incoming NTP substrates pre-coordinated to a divalent magnesium cation which is essential for substrate binding and catalysis. The divalent cation facilitates substrate binding via coordination to both the triphosphate ligand and carboxylate sidechains (usually aspartates) in the NTP binding domain. The design of uncharged bioisosteres of the triphosphate sidechain has so far proven to be challenging.1,2 However, conformationally rigidified ATP mimetics have recently been prepared using a bicyclic (ethylene bridged) oxazolidine ring structure to mimic the enzyme-bound conformation of the Mg2+-complexed triphosphate sidechain.3 In this case, an inhibitor of human tyrosine kinase (HER-2) was developed with a Ki value of 88 μM (compared to K m(ATP) of 5 μM). NTPs adopt different conformations when bound to different enzymes. Molecules that mimic specific enzyme-bound conformation(s) of NTP-Mg2+ could help to confer inhibitor selectivity for individual (or sub-populations of) NTP-dependent enzymes.
In the sugar nucleotide arena, there have been numerous attempts to prepare uncharged pyrophosphate isosteres, but with limited success.4 One notable exception is lactosyl uridine 2, where the pyrophosphate-Mn2+ complex of the galactosyltransferase donor substrate, uridine-5′-diphosphogalactose (UDP-Gal) 1 is replaced with a bridging 1,4-β-D-glucose linkage (Fig. 1).5 Lactosyl uridine, 2, proved to be a competitive inhibitor of 1,4-GalT activity from leukemia ascites fluid with a Ki value comparable to Km of the native substrate 1 implying that the central glucose moiety of 2 is isosteric with a pyrophosphate-metal ion complex (Fig. 1). More recently, the inhibitory effect of 2 (500 μM) against bovine β-1,4-galactosyltransferase was shown to be <10% in the presence of Km concentrations of UDP-Gal (120 μM).6 This may be attributed to structural differences in GalTs from different species and in particular the gross binding conformation of their donor substrates.
Fig. 1.

Examples of pyrophosphate-M2+ mimicry by monosaccharides.
Pyrophosphate mimicry by hexoses is also observed in nature. The dolichol-pyrophosphate-α-D-GlcNAc-synthase inhibitor tunicamycin uses an L-rhamnose-configured monosaccharide to mimic the pyrophosphate-Mn2+ complex of the natural dolichol-pyrophospho-GlcNAc substrate (Fig. 1).7,8 The stereo-configuration and substitution patterns of the central sugar motifs in 2 and tunicamycin significantly differ (Fig. 1). Evidently, no single monosaccharide motif can operate as a generic pyrophosphate-metal ion isostere due to different ligand-binding requirements of sugar nucleotides with different enzymes. How a neutral monosaccharide effectively mimics a charged pyrophosphate-metal ion chelate could be a combination of appropriate spacing/orientation of the terminal nucleoside and sugar motifs and interactions of the bridging glucose motif with the pyrophosphate-binding pocket. Substrate-binding interactions between the pyrophosphate-metal ion and aspartate sidechains in the pyrophosphate binding pocket could perhaps be replaced by hydrogen bonding interactions with one or more hydroxyl groups of a monosaccharide. We were curious to see if any such examples of pyrophosphate-metal ion mimicry would translate into the design of NTP mimics by replacing the terminal β,γ-pyrophosphate motif with a neutral monosaccharide.
We recently reported the preparation of sugar nucleoside monophosphates (sugar-NMPs) such as 3 and 4, which could potentially serve as significantly less charged ATP mimics (Fig. 2).9 However, these sugar-NMPs still retain a charged and hydrolytically unstable glycosyl phosphate linkage. Replacing the remaining phosphate with a readily accessible O-methylenetriazole linker (Fig. 2) would resolve these polarity/stability concerns. The viability of the triazole group as a bioisostere for the phosphate group in the sugar-phosphate backbone of nucleic acids has recently been demonstrated.10 Herein, we describe the synthesis and evaluation of a parallel library of sugar-triazole-nucleosides (STNs) as inhibitors of the type-III pantothenate kinase from Bacillus anthracis (BaPanK). From this library, a competitive inhibitor is identified whose Ki is 3-fold lower than Km of the competing substrate (ATP).
Fig. 2.
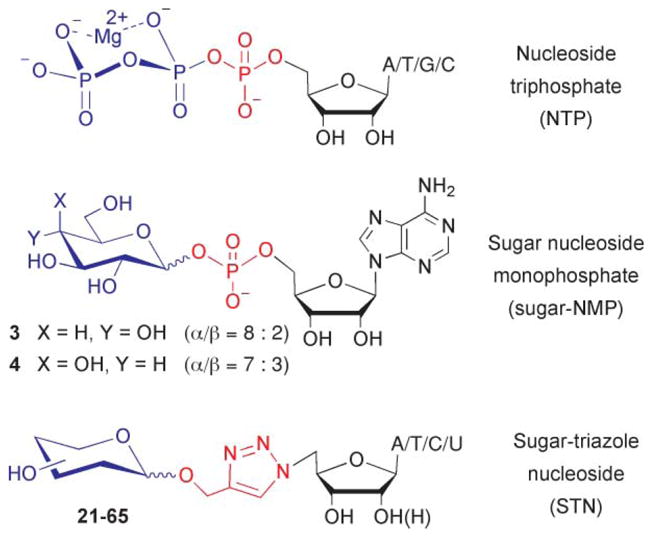
Proposed NTP mimics.
Results and discussion
Chemistry
AMP-glucose 3 and AMP-galactose 4 were prepared (as anomeric mixtures) using reported methods.9 For construction of the sugar-triazole-nucleoside (STN) library, 5′-azido uridine 7 and 5′-azido-2-deoxythymidine 8 were prepared, in good yield, by treatment of the free nucleosides, 5 and 6 with triphenylphosphine, carbon tetrabromide and lithium azide (Scheme 1).11
Scheme 1.
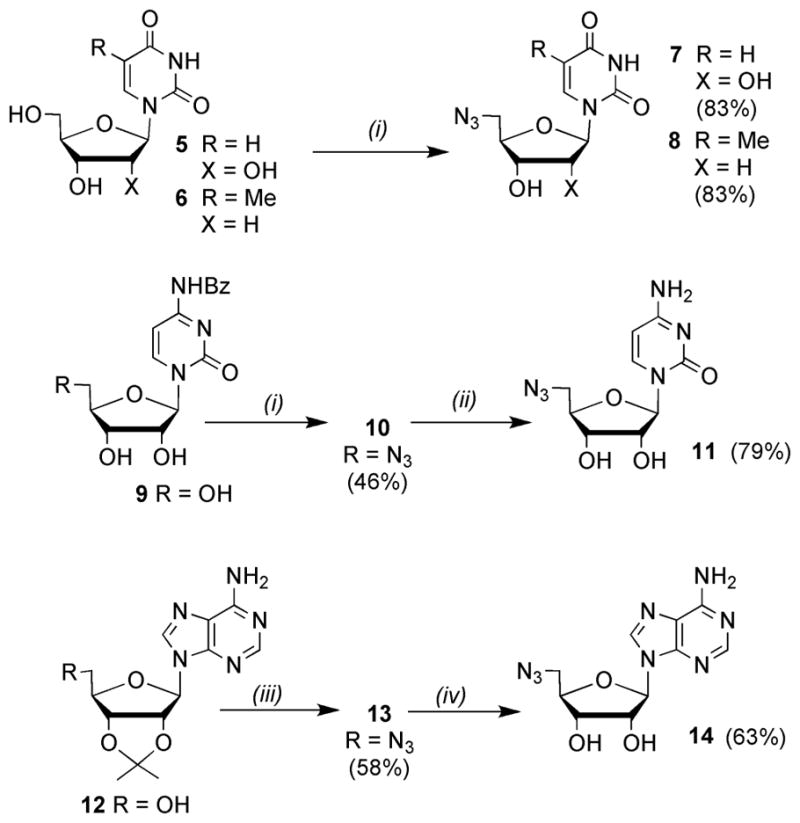
Reagents and conditions: (i) PPh3, LiN3, CBr4, DMF, r.t., 3 days; (ii) conc. NH4OH, MeOH, r.t., 6 days; (iii) PPh3, DIAD, DPPA, THF, r.t., 68 h; (iv) TFA/H2O (9:1), 0 °C–r.t., 25 min.
Similar procedures were use to access 5′-azido cytidine 11 from the N4-benzoyl protected derivative 9.11,12 The N-benzoyl protecting group was employed to prevent intermolecular alkylation of the nucleobase via reaction with the 5′-bromo nucleoside intermediate, which is generated in situ prior to reaction with lithium azide. 5′-Azido adenosine 14 has previously been prepared in five steps from N6-benzoyl adenosine.13 Herein, a more succinct route involved treatment of 2′,3′-O-isopropylidene adenosine 12 with diphenyl phosphoryl azide, diisopropylazodicarboxylate and triphenylphosphine to give 13.14 The isopropylidene protecting group served to enhance the solubility of 13, which facilitated purification by column chromatography prior to its removal under acidic conditions to give 14. A variety of methods were employed to access the different propargyl glycosides. Alpha-propargyl glycosides of D-Glc 15,15 D-GlcNAc 16,16 D-Man 1717 and D-Xyl 19 were all obtained using acid catalysed Fischer glycosylation conditions (Scheme 2). The same conditions were used to prepare O-propargyl derivatives of L-Ara 18 and D-Gal 20 as anomeric mixtures, which were inseparable by chromatography. However, for 20 the 4,6-O-benzylidene acetal derivatives, 21 and 22, were separable by chromatography enabling access to the desired α-galactoside 23.
Scheme 2.

Reagents and conditions: (i) propargyl alcohol, AcCl (cat.), 100–120 °C, 1–3 days; (ii) PhCH(OMe)2, TsOH, MeCN; (iii) AcOH(aq), 80 °C.
Treatement of peracetylated D-Rib 24, D-Gal 27a and D-Xyl 28a with propargyl alcohol in the presence of BF3·OEt18 was used to access the β-propargyl glycosides 26, 27c, 28c (Scheme 3).
Scheme 3.
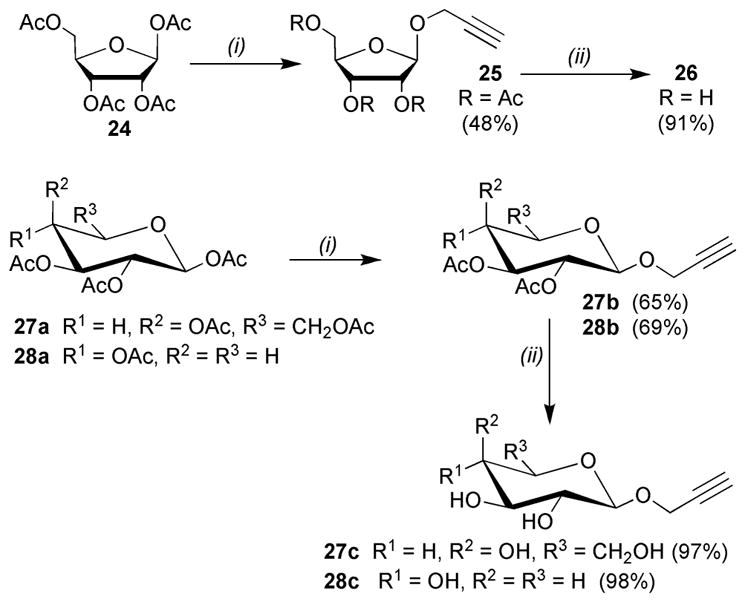
Reagents and conditions: (i) propargyl alcohol, BF3·OEt2, CH2Cl2, 0–20 °C; (ii) NaOMe, MeOH.
The β-propargyl glycoside of GlcNAc 31 was accessed via silver triflate promoted glycosylation of propargyl alcohol with glycosyl bromide 29 (Scheme 4a).19 Similar procedures were employed to access glucosamine derivative 35 (Scheme 4b). Preparation of 2-azido-2-deoxy-α-D-glucopyranosyl bromide 33 by treatment of peracetylated precursor 32 with HBr in glacial acetic acid was unsuccessful, providing a complex mixture of products (by TLC). An alternative treatment with titanium(IV) bromide20 gave the desired beta-anomer in 31% yield (40% of unreacted starting material was also recovered during chromatography). Reaction of 33 with propargyl alcohol in the presence of silver triflate provided propargyl glycoside 34 in good yield as an anomeric mixture (α:β ratio = 1:1.1). Subsequent deacetylation under Zemplen conditions in a sonicator21 rapidly proceeded to completion in just three minutes.
Scheme 4.
Reagents and conditions: (i) propargyl alcohol, AgOTf, 4Å molecular sieves, CH2Cl2, 0 °C, 2 h; (ii) NaOMe, MeOH; (iii) TiBr4, CH2Cl2/EtOAc; (iv) Zn dust, NH4Cl, EtOH/H2O, 90 °C.
Amino-sugar 36 could not be accessed by hydrogenation of azido-sugar 35 as this would also have reduced the alkyne, hence alternative methods were explored. Under Staudinger conditions (with tributylphosphine) the azide moiety of 35 was not reduced and only unreacted starting material was recovered. However treatment with zinc and ammonium chloride22 reduced the azide whilst leaving the alkyne aglycone intact.
The thymidine STNs 49–59 were the first to be prepared using the copper-(I)-catalysed [3 + 2] azide-alkyne cycloaddition (Cu-ACC) reaction between the aforementioned propargyl glycosides and azido-nucleoside 8 in the presence of copper sulfate and sodium ascorbate (Scheme 5, Table 1).23 Under these conditions, reactions took 1–3 days to reach completion (by TLC). For the adenosine, uridine and cytidine derivatives 37–48 and 60–81, alternative reaction conditions using copper(I) bromide, tris-(benzyltriazolylmethyl)amine (TBTA),24 under sonication21 gave improved yields and shorter reaction times (30–40 minutes).
Scheme 5.

Reagents and conditions: (i) 8, CuSO4 (5 mol %), sodium ascorbate (10 mol %), MeOH/H2O (3:1); (ii) 7 or 11 or 14, CuBr (5 mol %), sodium ascorbate (10 mol %), TBTA (5 mol %), MeOH/H2O/MeCN (1:1:1), r.t., sonication.
Table 1.
Compounds prepared and screened as inhibitors of BaPanK
|
|
|
|
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||||||
| Yield (%) | % enzyme activitya | Yield (%) | % enzyme activitya | Yield (%) | % enzyme activitya | Yield (%) | % enzyme activitya | |||||
| α-D-Glc | 37 | (72) | 93 | 49 | (61) | 98 | 6016 | (74) | 96 | 71 | (60) | 90 |
| β-D-Glc | 38 | (85) | 95 | 50 | (88) | 91 | 61 | (91) | 99 | 72 | (80) | 104 |
| α-D-Gal | 39 | (84) | 97 | 51 | (68) | 80b | 6216 | (81) | 105 | 73 | (67) | —c |
| β-D-Gal | 40 | (82) | 4 | 52 | (54) | —c | 63 | (87) | 105 | 74 | (70) | 86 |
| α-D-Man | 41 | (86) | 84 | 53 | (62) | 108 | 64 | (80) | 94 | 75 | (76) | 105 |
| α-D-Xyl | 42 | (88) | 90 | 54 | (60) | 102 | 65 | (94) | 91 | 76 | (82) | 102 |
| β-D-Xyl | 43 | (85) | 90 | 55 | (57) | 100 | 66 | (94) | 91 | 77 | (81) | 100 |
| α-D-GlcNAc | 44 | (80) | 95 | 56 | (82) | 92 | 6716 | (90) | 102 | 78 | (83) | 109 |
| β-D-GlcNAc | 45 | (53) | 87 | 57 | (49) | 98 | 68 | (76) | 110 | 79 | (68) | —c |
| L-Ara (α/β = 1:3) | 46 | (84) | 76 | 58 | (58) | 104 | 69 | (81) | —c | 80 | (80) | 91 |
| β-D-Rib | 47 | (71) | 95 | 59 | (81) | 100 | 70 | (81) | 83 | 81 | (70) | 107 |
| D-GlcN (α/β = 1.1:1) | 48 | (48) | 93 | |||||||||
Assays carried out in the presence of inhibitor (1 mM), ATP (125 μM) and pantothenate (250 μM). Percentage activity is relative to the same assay carried out in the absence of inhibitor.
No further decrease in activity was observed when inhibitor concentration was increased to 2 mM.
False positives (ie. inhibition of the PK/LDH coupled enzymes) in this assay.
Enzyme assays
The type-III pantothenate kinase from Bacillus anthracis (BaPanK) was used to study the NTP mimicry of all the STNs that were made. Pantothenate kinase (PanK) catalyses the first obligate step in Coenzyme A biosynthesis, namely the ATP-dependent phosphorylation of pantothenic acid (Scheme 6a). To date, the crystal structures of type-III PanK enzymes from Thermotoga maritima (TmPanK),25,26 Pseudomonas aeruginosa (PaPanK)27 and BaPanK28 have been reported, but co-crystallistaion of any of these Mg2+-dependent enzymes with ATP-Mg2+ has not yet been accomplished. Type-III PanK enzymes exhibit unusually high Km values of 3–10 mM29,25,27 compared to Km (ATP) values of <200 μM generally observed with PanK types I and II. Some type III panKs also display a broad tolerance for other NTPs as alternative phosphate donor substrates.27,29 For Type-III PanKs, the nucleobase end of the ATP binding site is open to the solvent and doesn’t support tight binding interactions with the adenosyl nucleobase, as shown by active site docking of the inert substrate mimic adenosine 5′-(β,γ-imido)triphosphate (AMPPNP) with PaPanK27 and the structure of the ADP:pantothenate ternary complex with TmPanK.26 For the studies reported herein, BaPanK activity/inhibition was monitored using a coupled assay as outlined in Scheme 6b.
Scheme 6.
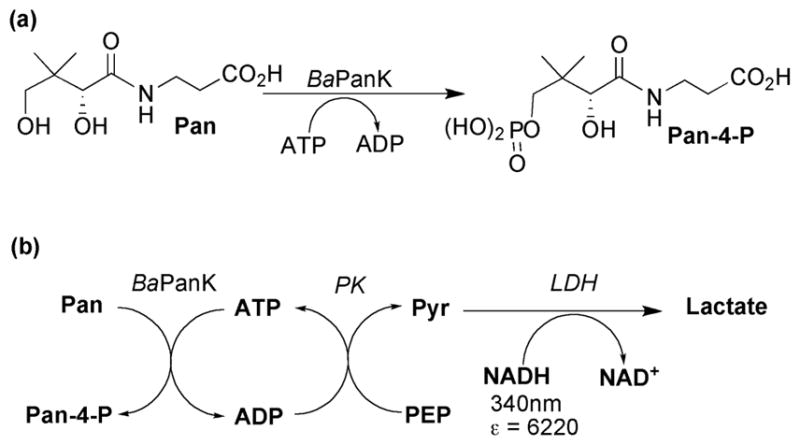
(a) BaPanK catalysed phosphorylation of pantothenate; (b) coupled assay used to monitor BaPanK activity; abbreviations: ADP = adenosine triphosphate, PK = pyruvate kinase, LDH = lactate dehydrogenase, PEP = phosphoenol pyruvate, Pyr = pyruvate.
BaPanK is able to utilise a range of NTPs as phosphate donors (Table 2). The catalytic efficiencies (kcat/K m) of the purine NTPs (ATP, dATP, dGTP) were 5–10-fold greater than their pyrimidine counterparts. This is primarily due to their lower Km values whereas the steady state turnover rates (kcat) of the pyrimidines were much closer to (and in some cases better than) those of the purine NTPs. The Km (ATP) for BaPanK (510 μM) is significantly lower than those reported for other type-III PanKs (3–10 mM).25–27,29 It is interesting to note that removal of the 2′-hydroxyl on the ribose ring leads to reductions in both Km and kcat, but with no significant overall reduction in catalytic efficiency (kcat/Km). The Km value for pantothenate is 275 (± 11) μM. All of the NTPs in Table 2 have previously been evaluated as substrates for PaPanK where a preference for purine NTPs was also observed.18 However, a direct qualitative comparison cannot be made because the NTP specificity of PaPanK was determined from relative enzyme activity measurements in the presence of a fixed concentration of the different NTPs (250 μM). Not all type-III enzymes share the same NTP substrate promiscuity. As well as ATP, HpPanK can also utilise GTP and CTP (but not UTP), whereas BsPanK cannot utilise any of these three alternative phosphate donors.20
Table 2.
NTP substrate kinetics with BaPanK
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (s−1mM−1) |
|---|---|---|---|
| ATP | 510 (± 19) | 1.5 (± 0.02) | 2.9 (± 0.2) |
| dATP | 138 (± 6) | 0.9 (± 0.01) | 6.6 (± 0.4) |
| dGTP | 117 (± 13) | 0.8 (± 0.02) | 6.5 (± 0.9) |
| CTP | 1759 (± 302) | 1.5 (± 0.15) | 0.9 (± 0.2) |
| dCTP | 839 (± 91) | 0.5 (± 0.02) | 0.6 (± 0.1) |
| dTTP | 723 (± 62) | 0.4 (± 0.01) | 0.5 (± 0.1) |
| UTP | 1715 (± 155) | 1.7 (± 0.06) | 1.0 (± 0.1) |
Compounds 37–81 were screened (at 1 mM concentrations) for BaPanK inhibition in the presence of ATP (125 μM) and pantothenate (250 μM). High inhibitor and low (<K m) ATP concentrations were chosen to ensure that even moderate ATP-competitive inhibitors wouldn’t go unnoticed. Under these conditions the majority of the STNs displayed no significant (ie. <20%) inhibition (Table 1). Compounds 52, 69, 73 and 79 proved to be false positives due to inhibition of the coupled enzymes (PK and/or LDH). Similar inhibitory effects on the coupled enzymes were also observed for the sugar-NMPs 3 and 4.
The β-galacto-configured adenosyl STN 40 displayed 96% inhibition of BaPanK activity under these assay conditions, but did not inhibit the PK/LDH coupled enzymes during control experiments. Detailed analysis showed 40 to be a competitive inhibitor of BaPanK (with respect to ATP) with a Ki of 164 μM (Fig. 3). This suggests that, for this enzyme, 40 operates as an ATP mimetic, which, despite lacking all of the negative charges of the parent triphosphate, has a Ki that is 3-fold lower than the Km value of the competing substrate (ATP). The anomeric configuration of the sugar-triazole sidechain is important as the α-galacto-configured adenosyl STN 39 shows no inhibitory activity. The axial configuration of the 4-hydroxyl of the galactose moiety is also of great importance as the gluco-configured epimer 38 was inactive. The anomeric mixture of L-arabino-configured ATP mimic 46 (with an axial 4-hydroxyl group) displayed 24% inhibition at 1 mM concentrations (Table 1). Based on the structure–activity data for 38–40, it is likely that the β-anomer is the active component of 46. The Km values for UTP, CTP and dTTP are, at most, only 3.5-fold greater than that of ATP (Table 2). Hence, the lack of any significant activity with the β-galacto-configured pyrimidines 52, 63, 74 (Table 1) suggests that the adenine nucleobase also makes a notable contribution to the efficacy of 40. In the absence of a type-III PanK structure co-crystallised with ATP, Mg2+ and the additional monovalent cation, reasons for inhibition by the β-galacto-configured STN 40 are not immediately clear. The structure of the ADP:pantothenate ternary complex with TmPanK shows how ATP-Mg2+ must bind in an extended conformation during the catalytic pathway.26 The loss of inhibitory activity going from the β-galactoside 40 to its α-anomer 39 could be the consequence of 40 being able to adopt such an extended conformation whereas its α-configured diastereoisomer 39 would be bent into a disfavourable orientation at the anomeric linkage. Evidently, the axial hydroxyl of the galactose portion of 40 plays a significant role in inhibitor recognition. Using Autodock (4.0.1),22 it has not been possible to predict a satisfactory binding orientation for 40 with the BaPanK apo-enzyme19 where inversion of the axial hydroxyl (ie. to 38) results in a significant loss of binding interactions and/or steric clashing that could plausibly account for the significant loss of inhibitor activity with 38 (data not shown). For future consideration, co-crystallisation studies of 40 with BaPanK would help establish the binding orientation of 40 as well as if/how the β-linked galacto-motif is able to mimic the β,γ-pyrophosphate portion of ATP with this enzyme.
Fig. 3.
Compound 40 as a competitive inhibitor of BaPanK. Double reciprocal plot of initial rate against ATP concentration in the presence of compound 40 at 0 μM (open triangles), 150 μM (closed circles) and 400 μM (open squares).
Conclusions
This study demonstrates how effective ATP mimics can be prepared where the charged triphosphate sidechain is replaced by an uncharged monosaccharide and that small variations in the monosaccharide stereoconfiguration lead to dramatic changes in inhibitor potency. It will be interesting to evaluate and compare the structure–activity relationship of this STN library with other NTP-dependent enzymes. More detailed mechanistic studies with BaPanK, and other kinases, are ongoing and will be reported in due course.
Experimental
General
1H and 13C NMR spectra were obtained using Bruker AV 300, DPX 300 or DRX 500 NMR spectrometers. Chemical shifts are quoted in parts per million (δ) using the residual solvent peak as an internal reference. IR spectra were recorded on a Perkin-Elmer RX I FT-IR system. Optical rotations ([α]D) were obtained with a Perkin-Elmer Model 341 Polarimeter, using the specified solvent and concentration, and are quoted in units of 10−1deg cm2 g−1. Thin layer chromatography (TLC) was carried out on Macherey-Nagel SIL G-25 UV254 glass-backed silica plates, which were visualised using a UV lamp, basic potassium permanganate solution or sulfuric acid (10% (v/v) in ethanol). Flash chromatography was carried out using Fluorochem silica gel for flash chromatography. Automated flash chromatography was performed on a Biotage Horizon or Biotage SP1 HPFC system using Si 12 + M or Si 40 + M pre-packed cartridges. Dry pyridine was purchased from Fluka, and other solvents were dried using a Braun 1 solvent purification system. Sonication-mediated reactions were carried out using a Branson model 2510 sonicator bath operating at a frequency of 40 kHz. TBTA was made as previously described.24 Propyl-2-ynyl-β-D-glucopyranoside was purchased from Aldrich. Recombinant BaPanK was overexpressed and purified as previously described.28 Rabbit muscle pyruvate kinase (PK) and rabbit muscle lactate dehydrogenase (LDH) were purchased from Sigma. Enzyme assays were carried out using either a PerkinElmer UV Lambda 25 spectrophotometer or a Tecan Saffire microplate reader. Compounds 7,11 8,11 11,11 1314 27a,18 27b,18 2919 and 3230 were prepared following published methods. Representative procedures are described below for the preparation of STNs 40, 46, 50, 65 and 78. Experimental methods and characterisation data for other STNs and all other synthetic intermediates are provided in the supplementary material.†
5′-Deoxy-5′-[4-(β-D-galactopyranosyloxymethyl)-1,2,3-triazol-1-yl]adenosine (40)
Prop-2-ynyl-β-D-galactopyranoside 27c (28.5 mg, 0.131 mmol) and 5′-azido-5′-deoxyadenosine 14 (42 mg, 0.144 mmol) were measured into a sample vial, followed by copper(I) bromide (1 mg, 6.6 μmol). Aqueous sodium ascorbate solution (40 mM, 332 μL, 13.3 μmol) and a solution of TBTA in acetonitrile (20 mM, 333 μL, 6.6 μmol) was added, followed by methanol (700 μL), acetonitrile (367 μL) and water (368 μL). The sample vial was sealed, the lid pierced with a syringe needle, and the reaction mixture was sonicated for 30 minutes. This was concentrated in vacuo and purified by automated chromatography (EtOAc/MeOH 7:3 as eluent) to give 40 (55 mg, 82%) as a white solid: [α]D20 = +2.3 (c = 1.48, DMSO); δH (500 MHz, CD3OD) 8.17 (s, 1H, H-2), 7.86, 7.85 (2 × s, 2H, H-8, H-5‴), 5.99 (d, 1H, J 4.5, H-1′), 4.96 (d, 1H, J 12.0, OCH AHB), 4.88 (dd, 1H, J 14.5, 4.5, H-5′a), 4.79 (dd, 1H, J 14.5, 3.5, H-5′b), 4.67 (d, 1H, J 12.0, OCHAH B), 4.44 (t, 1H, J 5.5, H-3′), 4.38 (m, 1H, H-4′), 4.34 (d, 1H, J 8.0, H-1″), 4.29 (t, 1H, J 5.0, H-2′), 3.84 (dd, 1H, J 3.5, 1.0, H-4″), 3.78 (dd, 1H, J 11.5, 7.0, H-6″a), 3.72 (dd, 1H, J 11.5, 5.0, H-6″b), 3.58 (dd, 1H, J 9.5, 7.5, H-2″), 3.55 (ddd, 1H, J 7.0, 4.5, 1.0, H-5″), 3.50 (dd, 1H, J 10.0, 3.5, H-3″); δC (125 MHz, CD3OD) 157.30 (C-6), 154.19 (C-2), 150.60 (C-4), 145.77 (C-4‴), 141.25 (C-8), 127.13 (C-5‴), 120.29 (C-5), 104.51 (C-1″), 90.10 (C-1′), 83.54 (C-4′), 76.93 (C-5″), 74.85, 74.81 (C-2′, C-3″), 72.39 (C-2″), 71.91 (C-3′), 70.57 (C-4″), 63.03 (CH2), 62.67 (C-6″), 52.01 (C-5′); HRMS (ES−) m/z calc. for C19H25N8O9 (M − H+): 509.1744; found 509.1758; m/z 509 (5%, M − H+).
5′-Deoxy-5′-[4-(L-arabinopyranosyloxymethyl)-1,2,3-triazol-1-yl]adenosine (46)
Was prepared from an anomeric mixture of prop-2-ynyl-α/β-L-arabinopyranoside 18 (27.4 mg, 0.146 mmol) and 5′-azido-5′-deoxyadenosine 14 (46.8 mg, 0.160 mmol) as described for 40. Following automated chromatography (EtOAc/MeOH 7:3 as eluent), 46 (59 mg, 84%) was obtained as a white solid with an α/β ratio of 1:3: [α]D20 = +92.2 (c = 1.19, DMSO); δH (500 MHz, DMSO-d 6) 8.15, 8.14, 8.06, 7.95, 7.92 (5 × s, 6H, H-2α, H-2β, H-8α, H-8β, H-5‴α, H-5‴β), 5.89 (d, 2H, H-1′α, H-1′β), 4.80–4.67 (m, 6H, H-1″β, H-5′aα, H-5′aβ, H-5′aα, H-5′bα, OCH AHBα), 4.57 (d, 1H, J 12.5, OCH AHBβ), 4.53 (t, 2H, J 5.0, H-2′α, H-2′β), 4.48 (d, 1H, J 12.5, OCHAH Bα), 4.42 (d, 1H, J 12.0, OCHAH Bβ), 4.30–4.20 (m, 4H, H-3′α, H-3′β, H-4′α, H-4′β), 4.17–4.13 (m, 1H, H-1″α), 3.72–3.50 (m, 6H, H-2″β, H-3″β, H-4″α, H-4″β, H-5″aα, H-5″β), 3.44–3.38 (m, 1H, H-5″bβ), 3.38–3.32 (m, 1H, H-5″bα), 3.32–3.28 (m, 2H, H-2″α, H-3″α); δC (125 MHz, DMSO-d 6) 156.06 (C-6), 153.10 (C-2), 149.47 (C-4α), 149.42 (C-4β), 143.99 (C-4‴β), 143.89 (C-4‴α), 140.16 (C-8α), 140.02 (C-8β), 125.34 (C-5‴), 119.09 (C-5), 102.56 (C-1″α), 99.17 (C-1″β), 87.97 (C-1′β), 87.85 (C-1′α), 82.56 (C-4′α), 82.41 (C-4′β), 72.85 (C-2′β), 72.76 (C-2′α), 72.53 (C-2″α), 70.94 (C-3′α), 70.90 (C-3′β), 70.56 (C-3″α), 69.02 (C-2″β), 68.64 (C-4″β), 68.44 (C-3″β), 67.71 (C-4″α), 65.55 (C-5″α), 63.37 (C-5″β), 61.14 (CH2α), 60.37 (CH2β), 51.49 (C-5′α), 51.36 (C-5′β); HRMS (ES+) m/z calc. for C18H25N8O8 (M + H+): 481.1795; found 481.1787; m/z 983 (44%, 2M + Na+), 961 (8%, 2M + H+), 503 (32%, M + Na+), 481 (100%, M + H+).
5′-Deoxy-5′-[4-(β-D-glucopyranosyloxymethyl)-1,2,3-triazol-1-yl]thymidine (50)
Prop-2-ynyl-β-D-glucopyranoside (23.5 mg, 0.108 mmol) and 5′-azido-5′-deoxythymidine 8 (31.7 mg, 0.119 mmol) were dissolved in MeOH (2 mL). Aqueous CuSO4 solution (0.05 M, 110 μL, 5.5 μmol) was added followed by aqueous sodium ascorbate solution (0.1 M, 110 μL, 11.0 μmol). This was stirred for 44 hrs then concentrated in vacuo and purified by automated chromatography (EtOAc/MeOH 3:1 as eluent) to give 50 (46 mg, 88%) as a white solid: [α]D20 = +23.7 (c = 0.99, MeOH); δH (500 MHz, CD3OD) 8.03 (s, 1H, H-5‴), 7.24 (d, 1H, J 1.0, H-6), 6.19 (t, 1H, J 7.0, H-1′), 4.97 (d, 1H, J 12.5, OCH AHB), 4.77 (d, 1H, J 12.5, OCHAH B), 4.78 (dd, 1H, J 14.5, 4.0, H-5′a), 4.70 (dd, 1H, J 14.5, 6.5, H-5′b), 4.41 (dt, 1H, J 6.0, 5.0, H-3′), 4.37 (d, 1H, J 7.5, H-1″), 4.15 (dt, 1H, J 7.0, 4.5, H-4′), 3.88 (dd, 1H, J 12.5, 2.0, H-6″a), 3.66 (dd, 1H, J 12.0, 6.0, H-6″b), 3.34 (t, 1H, J 9.0, H-3″), 3.29–3.26 (m, 2H, H-4″, H-5″), 3.20 (dd, 1H, J 9.0, 7.5, H-2″), 2.27 (dd, 2H, J 6.5, 6.0, H-2′), 1.89 (d, 3H, J 1.0, CH3); δC (125 MHz, CD3OD) 166.38 (C-4), 152.22 (C-2), 145.83 (C-4‴), 138.28 (C-6), 126.56 (C-5‴), 112.02 (C-5), 103.66 (C-1″), 87.00 (C-1′), 85.47 (C-4′), 78.11, 78.03 (C-3″, C-5″), 75.06 (C-2″), 72.40 (C-3′), 71.67 (C-4″), 63.03 (CH2), 62.92 (C-6″), 52.59 (C-5′), 39.56 (C-2′), 12.56 (CH3); HRMS (ES+) m/z calc. for C19H28N5O10 (M + H+): 486.1831; found 486.1833; m/z 524 (19%, M + K+), 508 (32%, M + Na+).
5′-Deoxy-5′-[4-(α-D-xylopyranosyloxymethyl)-1,2,3-triazol-1-yl]uridine (65)
Was prepared from prop-2-ynyl-α-D-xylopyranoside 19 (27.7 mg, 0.147 mmol) and 5′-azido-5′-deoxyuridine 7 (43.6 mg, 0.162 mmol) as described for 40. Following automated chromatography (EtOAc/MeOH 4:1 as eluent), 65 (63 mg, 94%) was obtained as a white solid: [α]D20 = +107.7 (c = 0.76, DMSO); δH (500 MHz, CD3OD) 8.04 (s, 1H, H-5″), 7.40 (d, 1H, J 8.0, H-6), 5.73–5.70 (m, 2H, H-1′, H-5), 4.86 (d, 1H, J 3.5, H-1″), 4.82 (dd, 1H, J 14.5, 3.5, H-5′a), 4.80 (d, 1H, J 12.5, OCH AHB), 4.73 (dd, 1H, J 15.0, 7.0, H-5′b), 4.64 (d, 1H, J 12.5, OCHAH B), 4.25 (dt, 1H, J 6.5, 3.5, H-4′), 4.19 (dd, 1H, J 5.5, 4.0, H-2′), 4.11 (t, 1H, J 6.0, H-3′), 3.59–3.53 (m, 2H, H-3″, H-5″a), 3.52–3.43 (m, 2H, H-4″, H-5″b), 3.38 (dd, 1H, J 9.0, 3.5, H-2″); δC (125 MHz, CD3OD) 166.15 (C-4), 152.08 (C-2), 145.72 (C-4‴), 143.44 (C-6), 126.62 (C-5‴), 103.12 (C-5), 100.06 (C-1″), 93.41 (C-1′), 83.00 (C-4′), 75.16 (C-3″), 74.19 (C-2′), 73.59 (C-2″), 71.95 (C-3′), 71.56 (C-4″), 63.34 (C-5″), 61.60 (CH2), 52.52 (C-5′); HRMS (ES−) m/z calc. for C17H22N5O10 (M-H+): 456.1367; found 456.1364; m/z 456 (100%, M − H+).
5′-Deoxy-5′-[4-(2-acetylamino-2-deoxy-α-D-glucopyranosy-loxymethyl)-1,2,3-triazol-1-yl]cytidine (78)
Was prepared from prop-2-ynyl-2-acetylamino-2-deoxy-α-D-glucoside 16 (31.2 mg, 0.120 mmol) and 5′-azido-5′-deoxycytidine 1 (35.5 mg, 0.132 mmol) as described for 40. Following automated chromatography (EtOAc/MeOH 7:3 as eluent), 78 (53 mg, 83%) was obtained as a white solid: δH (500 MHz, CD3OD) 8.02 (s, 1H, H-5‴), 7.35 (d, 1H, J 7.5, H-6), 5.87 (d, 1H, J 7.5, H-5), 5.71 (d, 1H, J 3.5, H-1′), 4.87 (d, 1H, J 3.5, H-1″), 4.84 (dd, 1H, J 14.5, 3.5, H-5′a), 4.81 (d, 1H, J 12.5, OCH AHB), 4.74 (dd, 1H, J 15.0, 7.0, H-5′b), 4.62 (d, 1H, J 12.5, OCHAH B), 4.26 (dt, 1H, J 7.0, 3.5, H-4′), 4.21 (dd, 1H, J 6.0, 3.5, H-2′), 4.08 (dd, 1H, J 7.0, 6.0, H-3′), 3.91 (dd, 1H, J 11.0, 3.5, H-2″), 3.83 (dd, 1H, J 12.0, 2.0, H-6″a), 3.69 (dd, 1H, J 12.0, 5.5, H-6″b), 3.66 (dd, 1H, J 10.5, 8.5, H-3″), 3.62 (ddd, 1H, J 9.5, 5.5, 2.0, H-5″), 3.36 (dd, 1H, J 10.0, 9.0, H-4″), 1.94 (s, 3H, CH3); δC (125 MHz, CD3OD) 173.79 (C=O), 167.78 (C-4), 158.15 (C-2), 145.42 (C-4‴), 143.67 (C-6), 126.50 (C-5‴), 98.06 (C-1″), 96.37 (C-5), 94.83 (C-1′), 82.76 (C-4′), 74.70 (C-2′), 74.30 (C-5″), 72.77 (C-3″), 72.38 (C-4″), 72.16 (C-3′), 62.81 (C-6″), 61.27 (CH2), 55.31 (C-2″), 52.62 (C-5′); HRMS (ES+) m/z calc. for C20H30N7O10 (M + H+): 528.2054; found 528.2051; m/z 550 (25%, M + Na+), 528 (100%, M + H+).
Enzyme assays
All enzyme assays were carried out at 37 °C in buffer containing HEPES (200 mM), MgCl2 (10 mM) and NH4Cl (60 mM), the pH of which was adjusted to 7.6 by careful addition of saturated NaOH solution.
Inhibition assays were carried out in a final volume of 1000 μL in disposable cuvettes containing NADH (150 μM), PEP (2 mM), pantothenate (250 μM), ATP (125 μM), BaPanK (7.5 μg), PK (7.5 units), LDH (15 units) plus inhibitor (1 mM). Reactions were initiated by the addition of BaPanK. Inhibitor stock solutions were prepared in DMSO and all assay mixtures contained a final quantity of 4% (v/v) DMSO. Compounds showing greater than 20% inhibition were further evaluated under the same assay conditions but using twice as much PK and LDH. Compounds which showed a decrease in % inhibition under these conditions were deemed false positives (ie. inhibitors of PK and/or LDH). For Ki determinations, assays were carried out at six ATP concentrations (0.5–2 × K m) in the presence of three different concentrations of inhibitor. Initial rates were measured from the linear region of product formation and Ki values were determined by weighted non-linear regression analysis of the hyperbola plot of rate against substrate concentration using the equation for linear competitive inhibition in Grafit version 5 (Erithacus Software Ltd).
NTP substrate kinetics were carried out in a final volume of 300 μL in a 96-well microplate. For the Km determination of ATP, each reaction mixture contained NADH (150 μM), PEP (2 mM), Pan (250 μM), BaPanK (2.5 μg), PK (2.5 units), LDH (5 units), and varying concentrations of ATP substrate. Reactions were initiated by addition of BaPanK. The same conditions were used for alternative NTP substrates except 12.5 units of PK was used to ensure that PK was not the rate limiting step in these coupled assays (as was observed when 2.5 units of PK were used). Kinetic data were analysed (by non-linear regression) using Grafit.
Supplementary Material
Acknowledgments
We are thankful for financial support from the Wellcome Trust (grant ref. 067525/Z/02/Z and VS/07/QUEB/A5) (WAW, CJH, NC), the Department of Employment and Learning (ASR), the National Institutes of Health (grant ref. GM-35394 (NIN, AC)) and a grant from the Southeast Regional Centre of Excellence for Biodefense and Emerging Infections. We also thank the Engineering and Physical Sciences Research Council (EPSRC) Mass Spectrometry Service Centre, Swansea, for invaluable support.
Footnotes
Electronic supplementary information (ESI) available: Additional experimental procedures for compound syntheses and characterisation data are provided in the supplementary information. See DOI: 10.1039/b909729e
Notes and references
- 1.Goldring AO, Balzarini J, Gilbert IH. Bioorg Med Chem Lett. 1998;8:1211–1214. doi: 10.1016/s0960-894x(98)00189-9. [DOI] [PubMed] [Google Scholar]
- 2.Goldring AO, Gilbert IH, Mahmood N, Balzarini J. Bioorg Med Chem Lett. 1996;6:2411–2416. [Google Scholar]
- 3.Liu F, Johnson EF, Austin DJ, Anderson KS. Bioorg Med Chem Lett. 2003;13:3587–3592. doi: 10.1016/s0960-894x(03)00761-3. [DOI] [PubMed] [Google Scholar]
- 4.Compain P, Martin OR. Bioorg Med Chem. 2001;9:3077–3092. doi: 10.1016/s0968-0896(01)00176-6. [DOI] [PubMed] [Google Scholar]
- 5.Wang R, Steensma DH, Takaoka Y, Yun JW, Kajimoto T, Wong CH. Bioorg Med Chem. 1997;5:661–672. doi: 10.1016/s0968-0896(97)00005-9. [DOI] [PubMed] [Google Scholar]
- 6.Ballell L, Young RJ, Field RA. Org Biomol Chem. 2005;3:1109–1115. doi: 10.1039/b500418g. [DOI] [PubMed] [Google Scholar]
- 7.Heifetz A, Keenan RW, Elbein AD. Biochemistry. 1979;18:2186–2192. doi: 10.1021/bi00578a008. [DOI] [PubMed] [Google Scholar]
- 8.Kimura K, Bugg TDH. Nat Prod Rep. 2003;20:252–273. doi: 10.1039/b202149h. [DOI] [PubMed] [Google Scholar]
- 9.Wlassof AW, Finlay RMJ, Hamilton CJ. Synth Commun. 2007;37:2927–2934. [Google Scholar]
- 10.Isobe H, Fujino T, Yamazaki N, Guillot-Nieckowski M, Nakamura E. Org Lett. 2008;10:3729–3732. doi: 10.1021/ol801230k. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto I, Sekine M, Hata T. J Chem Soc, Perkin Trans. 1;1980:306–310. [Google Scholar]
- 12.Ludwig PS, Schwendener RA, Schott H. Synthesis. 2002:2387–2392. [Google Scholar]
- 13.Wang T, Lee HJ, Tosh DK, Kim HO, Pal S, Choi S, Lee Y, Moon HR, Zhao LX, Lee KM, Jeong LS. Bioorg Med Chem Lett. 2007;17:4456–4459. doi: 10.1016/j.bmcl.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 14.Comstock LR, Rajski SR. Tetrahedron. 2002;58:6019–6026. [Google Scholar]
- 15.Blinkovsky AM, Dordick JS. Tetrahedron: Asymmetry. 1993;4:1221–1228. [Google Scholar]
- 16.Yeoh KK, Butters TD, Wilkinson BL, Fairbanks AJ. Carbohydr Res. 2009;344:586–591. doi: 10.1016/j.carres.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Hasegawa T, Numata M, Okumura S, Kimura T, Sakurai K, Shinkai S. Org Biomol Chem. 2007;5:2404–2412. doi: 10.1039/b703720a. [DOI] [PubMed] [Google Scholar]
- 18.Mereyala HB, Gurrala SR. Carbohydr Res. 1998;307:351–354. [Google Scholar]
- 19.Gillard JW, Israel M. Tetrahedron Lett. 1981;22:513–516. [Google Scholar]
- 20.Krist P, Kuzma M, Pelyvas IF, Simerska P, Kren V. Coll Czech Chem Commun. 2003;68:801–811. [Google Scholar]
- 21.Deng SL, Gangadharmath U, Chang CWT. J Org Chem. 2006;71:5179–5185. doi: 10.1021/jo060374w. [DOI] [PubMed] [Google Scholar]
- 22.Lin WQ, Zhang XM, Ze H, Yi J, Gong LZ, Mi AQ. Synth Commun. 2002;32:3279–3284. [Google Scholar]
- 23.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 24.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 25.Yang K, Eyobo Y, Brand LA, Martynowski D, Tomchick D, Strauss E, Zhang H. J Bacteriol. 2006;188:5532–5540. doi: 10.1128/JB.00469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang K, Strauss E, Huerta C, Zhang H. Biochemistry. 2008;47:1369–1380. doi: 10.1021/bi7018578. [DOI] [PubMed] [Google Scholar]
- 27.Hong BS, Yun MK, Zhang YM, Chohnan S, Rock CO, White SW, Jackowski S, Park HW, Leonardi R. Structure. 2006;14:1251–1261. doi: 10.1016/j.str.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Nicely NI, Parsonage D, Paige C, Newton GL, Fahey RC, Leonardi R, Jackowski S, Mallett TC, Claiborne C. Biochemistry. 2007;46:3234–3245. doi: 10.1021/bi062299p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brand LA, Strauss E. J Biol Chem. 2005;280:20185–20188. doi: 10.1074/jbc.C500044200. [DOI] [PubMed] [Google Scholar]
- 30.Liu JJ, Numa MMD, Liu HT, Huang SJ, Sears P, Shikhman AR, Wong CH. J Org Chem. 2004;69:6273–6283. doi: 10.1021/jo049355h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.