

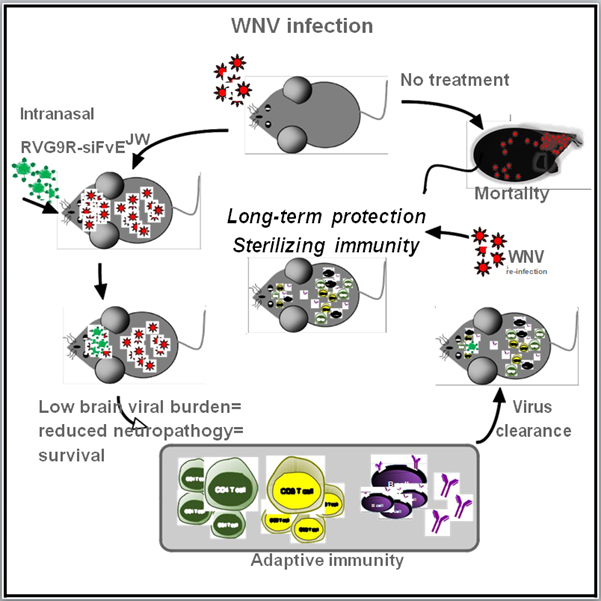
Summary
No vaccines or therapeutics are licensed for West Nile virus (WNV), a mosquito-transmitted neuroencephalitic flavivirus. The siRNA, siFvEJW, targets a conserved sequence within the WNV E protein and limits virus infection. Using a Rabies virus-derived neuron-targeting peptide (RVG9R) and an intranasal route for delivering siFvEJW to the CNS, we demonstrate that treatment of WNV-infected mice at late stages of neuroinvasive disease results in recovery. Selectively targeting virus in the CNS lowers viral burdens in the brain, reduces neuropathology, and results in a 90% survival rate at 5-6 days post-infection (when viral titers peak in the CNS), while placebo-treated mice succumb by day 9-10. Importantly, CNS virus clearance is achieved by humoral and cell-mediated immune responses to WNV infection in peripheral tissues, which also engender sterilizing immunity against subsequent WNV infection. These results indicate that intranasal RVG9R-siRNA treatment offers efficient late-stage therapy and facilitates natural long-term immunity against neuroinvasive flaviviruses.
Keywords: West Nile Virus, therapeutics, natural immunity, CNS delivery, flavivirus, siRNA, Rabies virus glycoprotein
eTOC Blurb
No vaccines or therapies exist for WNV disease. Beloor, et.al., show that intranasal treatment with a neuron-penetrating peptide complexed to antiflaviviral siRNAs is therapeutic for neuroinvasive WNV disease. The approach reduces CNS viral burden allowing development of immune responses to WNV. This enables recovery and protects against future WNV infection.
Introduction
Infectious flaviviruses like the West Nile (WNV) and Zika have exhibited unprecedented activity and geographic expansion in recent years (Salimi et al., 2016). Human WNV infection is a febrile illness that can progress to meningitis and encephalitis; the elderly are at a 20-fold higher risk for severe neurological manifestations and mortality (Diamond, 2009). No vaccines or therapies exist for WNV disease and current treatment is supportive care. Naturally-acquired immunity is believed to protect lifelong against WNV and humoral immunity, a critical mediator of this immune protection (Nicolle, 2003; Suthar et al., 2013). WNV vaccines may induce long-term immunity, however their clinical development has been stymied by sporadic disease incidence impeding field efficacy studies and poor cost-effectiveness of mass vaccination (Amanna and Slifka, 2014).
As meningoencephalitis is the most common (50-84%) diagnosis in hospitalized WNV patients (Hayes et al., 2005; Jehi and Sila, 2009), therapeutics that improve clinical and virologic outcome after virus dissemination in the CNS hold great value. We previously demonstrated the utility of small interfering RNA (siRNA) in the prophylactic treatment of flaviviral encephalitis by employing an RVG9R peptide for siRNA delivery in vivo (Kumar et al., 2007). RVG9R, a chimeric peptide derived from the Rabies virus glycoprotein (RVG) fused to nona-arginine (9R), targets nicotinic acetylcholine receptors (nAchR) expressed on neuronal cells and mediated transvascular delivery of siRNA into the CNS. Intravenous (i.v.) treatment with antiflaviviral siRNA complexed to RVG9R protected mice from lethal challenge with Japanese encephalitis virus (JEV), an encephalitic flavivirus closely related to WNV. Here, through a nose-to-brain route, which maximized siRNA payloads to the brain, RVG9R-siRNA substantially reduced brain viral burden and rescued mice from fatal WNV encephalitis when administered even at late stages of infection after virus had invaded the CNS. Importantly, siRNA treatment delayed mortality enabling natural immune responses to peripheral virus replication, which mediated virus clearance during primary infection and sterilizing immunity upon re-exposure to WNV.
Results
Intranasal administration of RVG9R-siRNA elicits widespread gene silencing in the mouse brain
The most severe manifestations of WNV infection are a result of neuropathology and brain injury that positively correlate with, and are in part caused by high viral burden in the CNS (Diamond, 2009; Shrestha et al., 2003). C3H mice, an established animal model for WNV infection, display a high mortality rate to WNV and virus entry into the CNS occurs by day 2 post-infection (p.i.) (Brown et al., 2007). When C3H mice were challenged with 100LD50 WNV (causes 100% mortality at day 9-10 p.i.), significant viral burden was detected in brain tissues by day 5 p.i. (Figure S1A). However, despite high viral burden (~6 log10 pfu/g tissue by day 6), mice exhibited morbidity by days 8-9 and mortality at days 9-10 following a dramatic increase in TUNEL-positivity (Figures S1B and S1C), implicating severe neuronal injury as the determinant of lethality (Shrestha et al., 2003). This revealed a time frame after virus invasion of the CNS that might be exploitable for therapeutic benefit. We therefore evaluated RVG9R-siRNA under therapeutic settings.
To maximize delivery to the brain, we explored the intranasal (i.n.) nose-to-brain route that can provide direct access to the CNS from the olfactory region bypassing the blood brain barrier (BBB) (Dhuria et al., 2010; Lochhead and Thorne, 2012; Miyake and Bleier, 2015). Using a positioning device for placement of mice in the head down and forward position (Dhuria et al., 2010), we performed preliminary biodistribution experiments in Balb/c mice. i.n. administration of RVG9R (or a control 9R peptide) complexed to FITC-labeled siRNA (siFITC, 5 µg) resulted in strong fluorescent signal in the brain and paranasal sinus indicating trafficking of siRNA from the upper olfactory region to neural tissues (Figures 1A and S2A). Notably, this procedure resulted in minimal delivery to the lungs. It also maximized brain delivery of siRNA as i.v. administration at the same dose did not result in appreciable brain signal (Figure S2B) or mRNA knockdown (see below). A single i.n. administration of RVG9R-siRNA targeting murine superoxide dismutase-1 (siSOD1, 13.5 µg) resulted in a significant decrease in target mRNA levels indicating functional delivery of siRNA (Figure S2C). mRNA knockdown was documented in multiple regions of the brain, including those distal to the sinus/brain interface, known to harbor virus during WNV infection (Lim et al., 2011). Increasing siRNA dosing frequency to 4 times over a 48 h period resulted in ~50% knockdown in SOD-1 mRNA throughout the brain (Figure 1B) while comparable knockdown via the i.v. route required increasing the siRNA dose to ~5 times more per injection (Figure S2D). No silencing occurred when siRNA was complexed to the control peptide 9R (Figure S2E), despite some brain localization (Figure 1A) reconfirming that RVG9R-mediated siRNA delivery required engaging a cellular receptor (Zeller et al., 2015).
Figure 1. I.n. RVG9R-siRNA elicits widespread gene silencing in the brain.

(A) The indicated organs were examined for fluorescence 24 h after i.n. administration of PBS (mock), and 9R or RVG9R-complexed siFITC. Left- representative fluorescent images overlaid with their bright field images; Right- cumulative data from three mice depicting mean fluorescence intensities (MFIs) in arbitrary pixel values ± standard error (SE). (B) qPCR for SOD1 mRNA in mouse brain regions at 24 h after the last i.n. treatment with RVG9R-siSOD1 (solid bars) and RVG9R-siNT (open bars). Data are mean percent gene expression (± SD) relative to GAPDH after normalizing with corresponding data of mock-treated cohort. n = 3 mice per treatment per time point. NT-non-targeting. See also Figure S2.
I.n. RVG9R-siFvEJW is therapeutic for WNV in mice with active CNS infection
We explored if mice could be treated therapeutically with RVG9R complexed to siFvEJW, an siRNA targeting a conserved sequence on the single-stranded RNA genome corresponding to the fusion-loop region of the WNV E protein (Kumar et al., 2006). Mice pre-infected with 100LD50 WNV and i.n. administered RVG9R-siFvEJW (9.1 μg siRNA) on days 3 & 4 or 4 & 5 p.i. were completely rescued from WNV-induced mortality (Figure 2A). Treatment with RVG9R complexed to siRNA targeting luciferase (siNT) or a scrambled siRNA (siScr) did not improve survival. Initiating the first i.n. treatment as late as day 5 or 6 p.i., when brain WNV titers reached 4-6 log10 pfu per gram tissue (Figure S1A), was also remarkably therapeutic resulting in corresponding survival rates of 90 and 50% (Figure 2A). This was accompanied by >1000 and >10-fold reduction respectively in WNV brain burden at day 9 p.i. (Figure 2B). Accordingly, glial fibrillary acidic protein (GFAP)- a marker for injury-induced astrogliosis, TUNEL-positive apoptotic neurons as well as viral antigen were substantially reduced in brain tissue (Figures 2C, 2D and S3A). Interestingly, WNV-challenged mice treated on days 3 & 4 via the i.v. route, succumbed by day 10, illustrating the markedly enhanced CNS bioavailability of i.n. delivered siRNA. Importantly, WNV infection resolved and virus titers became undetectable in the brains of surviving mice at 30 days p.i. (Figure 2B). At this time point, viremia, viral antigen and/or viral RNA were also not detected in peripheral organs including spleen, kidneys, lung and liver (not shown). Examination of the brains of surviving mice for pathologic abnormalities by H&E staining revealed no indications of neuropathologic damage (not shown). Thus, i.n. administration of RVG9R-siFvEJW to mice with active CNS WNV infection resulted in reduced neuropathology, survival and eventual virologic cure.
Figure 2. I.n. RVG9R-siFvEJW is therapeutic for WNV infection.

(A) Kaplan-Meier survival curves of C3H mice treated with RVG9R-siRNA formulations at the indicated days p.i. with 100LD50 WNV. Data are from 3 experiments with mouse cohorts of atleast n = 5 mice per group per treatment. Survival statistics in all test cohorts (except treatment at days 6 & 7) achieved significance by the log-rank test when compared to mock- or siNT-treated mice. siNT = siLuc, siScr= scrambled siRNA for siFvEJW. (B) Mean WNV titers per gram of brain tissue at the indicated days p.i. Each data point represents one mouse. The dotted line depicts the assay’s limit of detection. ND- not detectable. (C) Representative images of mouse brain tissue sections analyzed by fluorescent immunohistochemistry at day 9 p.i. Scale bar = 30 µm. (D) Graphical presentation of data in (C) depicting MFI of GFAP-positive cells and numbers of TUNEL-positive cells per high power field (n=10) combined from 4 mice per treatment condition. See also Figure S3.
Antiviral immune responses are critical for recovery in RVG9R-siFvEJW treated mice
Cell-mediated immune responses orchestrate WNV clearance from the CNS (Diamond, 2009; Netland and Bevan, 2013). To assess if siRNA mechanisms were sufficient for virus elimination, we treated WNV-infected immunodeficient NSG mice i.n. with RVG9R-siFvEJW at days 4 & 5 p.i. (Figure 3A). Although the median survival time increased from 10 to 12 days (P = 0.0047), the treatment did not impact mortality signifying an important role for immune responses in recovery. As predictable from the brain-specific profile of siRNA biodistribution (Figure 1), WNV replication in peripheral tissues of C3H mice was not curtailed after i.n. treatment and splenic WNV burden was comparable between the RVG9R-siFvEJW and control cohorts at day 8 p.i. becoming undetectable by day 11 p.i. (Figure S3B). Thus, we looked for germinal center responses to WNV infection. We identified T follicular helper cells (TFH, CD4+PD1+CXCR5+) and germinal center B cells (GC B, BB220+Fas+GL7+) in the spleens of RVG9R-siFvEJW treated C3H mice at 8 days p.i. (Figure 3B). Sera from mice surviving WNV challenge at 11 and 30 days p.i. revealed significant titers of virus-neutralizing antibodies (Mean PRNT90=84.5±24.8 and 144.8±41.6; P = 0.0034; Figure 3C).
Figure 3. Immune responses mediate recovery in RVG9R-siFvEJW treated WNV-infected mice.

(A) Kaplan-Meier survival curves of immunodeficient NSG mice treated i.n. with RVG9R-siFvEJW on days 4 & 5 p.i. with 100LD50 WNV. n = 6 mice per group. (B) Flow cytometric detection of TFH (CD4+, PD1+ and CXCR5+) and GC B (B220+, Fas+ and GL7+) cells in spleens of RVG9R-siFvEJW treated C3H mice on day 8 p.i. Left- representative plots; Right- cumulative data. Each data point represents one mouse. Bars are geometric mean. (C) Serum PRNT90 of mice treated with RVG9R-siFvEJW at the indicated days p.i. with WNV. (D) Kaplan-Meier survival curves of C3H recipients that received splenocyte populations (w/w/o immune serum) at day 5 p.i. with WNV from donor RVG9R-siFvEJW treated mice that survived WNV challenge. n = 3-4 mice per group. See also Figure S3.
T cells (particularly CD8+) elicited during WNV infection and recruited to the CNS by chemokines secreted from infected neurons aid in WNV clearance and limit disease severity while antibodies control peripheral viremia (Netland and Bevan, 2013; Shrestha et al., 2008; Sitati and Diamond, 2006). To delineate the relative contribution of T cells and humoral responses in recovery from neuroinvasive WNV disease, we performed adoptive transfer experiments using RVG9R-siFvEJW treated mice that survived WNV infection as immune donors. Splenocytes isolated on day 12 after viral challenge (when splenic virus titers were absent) from multiple animals were pooled and depleted of CD8+ T cells and used as the source of immune cells or after replenishing CD8+ T cells. Transfer of pooled 12-day immune serum (individual WNV-specific PRNT90>100) or splenocytes replete with CD8+ T cells into naive C3H mice 1 day prior to WNV challenge protected from mortality (Figure S3C and data not shown). Transfer of splenocytes lacking CD8+ T cells along with immune serum or after repleting CD8+ T cells into WNV-challenged C3H mice at day 5 p.i., when the virus is in the CNS, significantly delayed mortality (14 and 16 days, P = 0.0455 and 0.0023 respectively); however recipient mice eventually succumbed to WNV infection (Figure 3D). A combination of the treatments i.e.- splenocytes replete with CD8+ T cells and immune serum, in contrast, completely rescued mice from lethal encephalitis. As splenocytes are composed mainly of B cells (45%), CD4 T cells (30%) and other immune cell types besides CD8 T cells (20%) (Diamond et al., 2003), these data illustrate that an intact, coordinated adaptive immune response with both humoral and cell-mediated immunity is essential for viral clearance at late stages p.i. Furthermore, adoptive transfer of splenocytes with immune serum was not protective beyond day 5 p.i (Figure S3C) unless supplemented with i.n. siRNA, again pointing to the existence of therapeutic time window when an intervention strategy is needed to quickly reduce viral burden in the CNS. These data suggest that for effective immune-mediated viral clearance, CNS viral loads need to be contained below a critical threshold to prevent irreparable neuronal injury and lethality. Thus, interventions like RVG9R-siFvEJW that reduce, rather than abrogate, virus replication in the CNS improve chances for recovery in overt WNV disease.
Long-term sterilizing immunity in RVG9R-siFvEJW treated mice that survive WNV disease
Neutralizing antibodies are recognized as the primary mechanism of resistance to WNV in virus endemic regions (Shrestha et al., 2008). We therefore assessed if the immune responses in RVG9R-siFvEJW treated mice could protect against a second challenge with WNV (Fig. 4A, schematic). Rechallenge of mice that survived primary infection a month later with 100LD50 of WNV resulted in 100% survival regardless of initial magnitude of neutralizing antibody titer (Figures 4B and 3C). Post-rechallenge PRNT titers were comparable to those in mice immunized and boosted with H2O2-inactivated WNV (Figure 4C). Notably, no viremia or viral RNA was detected in the serum, spleen or brain at any time after rechallenge signifying sterilizing immunity to WNV (Figure 4D, Figure S4A and data not shown). Rechallenge via the intracranial (i.c.) route however, resulted in mortality indicating that virus control was achieved primarily in peripheral tissues preventing CNS dissemination and that siRNA effects in the brain were limited to the first round of infection (Figure S4B). Durable immunity is a hallmark of natural WNV infection (Suthar et al., 2013), and serum neutralizing antibody titers persisted at 6 months after primary challenge (Figure 4C, red symbols). Accordingly, rechallenge at this time also protected 100% of the mice (Figure 4B, red symbols). Thus, therapeutic and long-lasting protective immunity to WNV can be engendered by a brain-targeting strategy that employs small interfering RNA to control WNV infection selectively in the CNS.
Figure 4. I.n. RVG9R-siFvEJW treatment induces long-lasting sterilizing immunity to WNV.

(A) Experimental timeline for re-challenge studies. (B) Kaplan-Meier survival curves of RVG9R-siFvEJW treated C3H mice challenged for a second time with 100LD50 WNV either 1 or 6 months after primary challenge. n= 5-8 mice per group. (C) Serum PRNT90 of C3H mice on day 21 after the second WNV challenge (Post 2°), im munization with H2O2-inactivated WNV (WNVinact) or at 6 months after primary challenge (Pre 2°). Each data point represents one mouse. Bars are geometric mean values. ns- non-significant. (D) Data depicting proportion of mice positive for the specified clinical or virologic parameter. siFvEJW refers to mice treated with i.n. RVG-9R-siFvEJW at days 5 and 6 p.i., a,b-Positive for WNV RNA on day 4 or 8 after challenge; c,d,e-Positive for infectious WNV on day 4, 8 or 30 after challenge, nd- not determined. See also Figure S4.
Discussion
Timely control of CNS viral burden in WNV encephalitis is essential to minimize neuronal injury and prevent lethal or permanently damaging pathologies. Most experimental WNV therapeutics are effective prior to or soon after infection in animals (Acharya and Bai, 2016; Diamond, 2009). Neutralizing antibodies have exhibited some degree of potency in encephalitic WNV patients (Ben-Nathan et al., 2009; Haley et al., 2003; Shimoni et al., 2001) and in animal studies after virus dissemination into the CNS (Gould et al., 2005; Morrey et al., 2006; Oliphant et al., 2005). However, cost-efficacy issues and the risk of antibody-dependent enhancement of infection with heterologous flaviviruses (Acosta and Bartenschlager, 2016) raise concerns of a therapy that solely relies on antibodies. Here we describe a mortality-reversing post-exposure approach that utilized RVG9R for intranasally delivering antiviral siRNAs into the CNS bypassing the BBB and lowered brain viral burden and neuropathology at late stages of WNV infection. This allowed the host to develop natural humoral and cell-mediated immunity against WNV, that are critical in preventing and resolving infection in WNV-endemic populations.
The olfactory and trigeminal nerves penetrate the nasal mucosal lining permitting direct access to the CNS bypassing the BBB (Dhuria et al., 2010; Lochhead and Thorne, 2012; Miyake and Bleier, 2015). While multiple mechanisms are postulated for nose-to-brain transport (Quintana et al., 2016), trafficking of RVG9R to sites distal from the olfactory area was perhaps through long-range retrograde trans/poly-synaptic transport, which is also a feature of viral vectors pseudotyped with RVG or RVG fusion proteins (Beier et al., 2013; Mazarakis et al., 2001; Schoderboeck et al., 2015). Live imaging of neuronal cells in vitro indicate that cellular entry might occur at the site of receptor aggregation by temporary delocalization of the cell membrane or from within endosomes (Zeller et al., 2015). Nevertheless, the extremely favorable dynamics of siRNA delivery to the CNS resulted in significant target knockdown as early as 12 h after treatment at a 5-times lower dose at half the dosing frequency used in i.v. and i.c. protocols (Kumar et al., 2006; Kumar et al., 2007).
Despite widespread brain distribution of RVG9R-siFvEJW, complete elimination of viral RNA did not occur likely because of excessive brain viral burden and perhaps, vesicular sequestration of virus replication complexes from the cytoplasmic RNAi machinery (Geiss et al., 2005). Nevertheless, the decrease in brain virus titers was sufficient to vastly reduce neuronal damage, the basis for morbidity and mortality. Limb paralysis and tremors in hamsters are directly associated with injury and apoptosis of anterior horn motor neurons of the spinal cord (Morrey et al., 2008). Caspase 3-deficiency also decreases neuronal injury and increases resistance to lethal WNV infection despite high brain viral burden (Samuel et al., 2007). As RVG9R can transduce microglia and astrocytes (Kim et al., 2010) that are dominant sources of proinflammatory and neurotoxic mediators in WNV encephalomyelitis (van Marle et al., 2007), reducing virus infection in these cells would also reduce neuropathology.
As i.n. treatment excluded systemic circulation, it facilitated immune responses to peripheral WNV spread that were critical for recovery from primary infection as well as protecting against re-infection. CD8 T cells, via mechanisms of direct cytotoxicity and antiviral cytokine secretion, mediate clearance of WNV-infected cells while CD4 T cells sustain antibody production and effector CD8 T cells in the CNS (Netland and Bevan, 2013; Shrestha et al., 2008; Sitati and Diamond, 2006). Humoral immunity is more linked to control and clearance of peripheral WNV. Our studies, however, established an indispensable role for both humoral and cell-mediated immune responses in recovery from neuroinvasive WNV infection. Adoptive transfer of WNV-immune splenocytes depleted of CD8+ T cells but with immune serum for immediate antiviral activity was insufficient to reverse mortality. Adoptive transfer of splenocytes (with CD8+ T cells) significantly delayed time to death, however immune serum was necessary for survival. As prophylactic adoptive transfer splenocytes without immune serum protected mice, a critical role for antibodies in the timely control of virus dissemination within the CNS and perhaps, clearance through Fc-effector mechanisms is apparent. Interestingly, these immune responses did not protect against i.c. WNV challenge as in another study (Shrestha et al., 2008), probably because C3H mice display high viral burden (Brown et al., 2007). This nonetheless highlights the potency of the approach from a preclinical standpoint given the therapeutic success in an animal model with accelerated CNS infection and high mortality.
Despite several successful preclinical animal studies and some promising human trials, the i.n. route is yet to be clinically exploited. I.n. delivery to the CNS is heavily influenced by the anatomy of the human nose, which unlike in rodents, has a highly reduced mucosal surface area within the nasal vestibule. However, with newer approaches like mucosal flap reconstruction and pressurized olfactory devices for drug deposition at sites beyond the nasal valve where the absorptive mucosal epithelium is located, nose-to-brain delivery of therapeutics such as RVG9R-siFvEJW is achievable in the near future (Djupesland et al., 2006; Miyake and Bleier, 2015). RVG9R-siFvEJW was not immunogenic or inflammatory in mice (Kumar et al., 2007). However, given potential concerns of off-target toxicity (Garber, 2017), siRNA therapeutics appear best suited for short-term application. Unlike in hepatocytes where siRNA persists for months (Garber, 2017), siRNA activity in neurons appears to have a shorter duration (~3 weeks) (Omi et al., 2004) and RVG9R-siFvEJW treated animals succumbed to intracerebrally injected WNV a month after treatment. Thus, an effective siRNA delivery system for neuronal cells coupled with the minimally-invasive i.n. regimen with reduced systemic exposure, can be transformative for treating acute flaviviral neuroencephalitis, particularly in the elderly. The approach also offers better scope for strategic clinical testing in a targeted human population with confirmed WNV disease in contrast to vaccine studies that recruit more loosely defined ‘at-risk’ populations. The fusion loop-encoding sequence in domain II of the viral Env gene targeted by siFvEJW is well-conserved (Kumar et al., 2006) and may cross-target multiple flaviviruses. Nevertheless, a cocktail of siRNAs targeting multiple conserved sequences in the flaviviral genome can better tackle quasispecies diversity adding to the potency of adaptive immune responses. Thus, i.n. RVG9R-siRNA formulations can represent a versatile broad-spectrum antiflaviviral that blocks viral replication and minimizes neuropathology while allowing development of natural immune responses for therapy and durable immunity against encephalitic flaviviruses.
STAR Methods
Contact for Reagents and Resource Sharing
Request for reagents will be fulfilled by the corresponding author Priti Kumar (priti.kumar@yale.edu).
Experimental Model and Subject Details
Mice
All animal experiments were performed according to protocols approved by the Institutional Review Board and the Institutional Animal Care and Use Committee (IACUC) of Yale University. 6-8 week old female C3H/HeNcrl (C3H) (Charles river laboratories, Wilmington, MA) and immunodeficient NOD.Cg-PrkdcSCID Il2rgtm1Wjl/SzJ (NSG) (Jackson Laboratory, Farmington, CT) mice were purchased. Female Balb/c (6-8 weeks old) mice were obtained from Orient Bio, Seoul, South Korea.
Cells, Virus and Reagents
The African Green Monkey kidney epithelial cell line, Vero (female) from American Type Culture Collection were cultured in complete DMEM (Life technologies, Grand Island, NY) at 37°C in a 5% CO2 atmosphere. WNV strain CT 2741 was obtained from Dr. Erol Fikrig, Yale University. WNV stocks were propagated in Vero cells and titrated by plaque assay in Vero cells.
The RVG9R peptide was synthesized at the Tufts University Peptide Synthesis Core Facility. Sequences for siLuc, siCD4, siSOD-1 and siFveJW are in Table S1. FITC-labeled siLuc and siRNA targeting human CD4 (siCD4) were used as non-targeting (NT) control siRNA in biodistribution and functional target gene knockdown studies. siLuc was the NT control in WNV-challenge experiments. All siRNAs were purchased from Dharmacon (Thermo Fisher Scientific, Lafayette, CO). RVG-siRNA complexes were formulated in a 25:1 peptide:sirna molar ratio in 5% dextrose.
Method Details
Intranasal siRNA Delivery
RVG9R-siRNA complexes (formulated at a 25:1 peptide:siRNA molar ratio in 25 µl of PBS containing 5% glucose) were administered i.n. to mice (anesthetized i.p. with ketamine/xylazine hydrochloride) placed in a head down-and-forward position using a mouse positioning device (Signet Biotech). Solutions were applied drop-wise (1-2 μl per drop) using a micropipette over 30 min per animal alternating between each nare. Mouth and the alternate nare were closed during application to ensure uptake. siRNA biodistribution and analysis of gene silencing was performed in Balb/c mice after i.n. treatment with RVG9R-siRNA.
siRNA Biodistribution and Knockdown Analysis
Balb/c mice were i.n. treated with RVG9R-siRNA. Mouse organs were imaged 24 h later using a Kodak imaging station (Kodak IS440CF, Rochester, NY) and fluorescence analyzed by the Kodak Digital Science Image Analysis Software. To assess gene silencing, RNA from brain tissue collected in RNAiso (TaKaRa Bio, Shiga, Japan) was reverse-transcribed into cDNA using iScript TM cDNA synthesis kit (Bio-rad, Hercules, CA). qPCR was performed with primer pairs for murine SOD-1 (forward 5’CCAGTGCAGGACCTCATTTT3’; reverse 5’CACCTTTGCCCAAGTCATCT3’) and murine GAPDH (forward 5’AACTTTGGCATTGTGGAAGG3’; reverse 5’GGAGACAACCTGGTC CTCAG3’) using the SYBR premix Ex Taq perfect real-time (TaKaRa Bio, Shiga, Japan) on a 7500 Real Time PCR System.
Animal Studies
C3H mice were i.p. infected with 100 pfu (100 times the 50% lethal dose [LD50]) of WNV, strain CT 2741. Infection at LD50 resulted in 100% mortality. Mice were randomly divided into groups and treated twice with RVG9R-siRNA at 24 h intervals i.v. through the tail vein or i.n. on days 3 & 4, 4 & 5, 5 & 6, or 6 & 7 p.i. Mock-infected and mock-treated (RVG9R-siLuc or PBS on days 5 & 6) mice were controls. n ≥ 5 in each group. Mice were monitored daily for signs of morbidity (defined by clinical signs including ruffled fur and ataxia for at least 1 day and/or > 5% weight loss or hind limb paralysis), mortalility and survival upto 30 days. Experiments were conducted in parallel for tissue analyses. Mice that survived primary challenge were re-infected either 1 or 6 months later with 100LD50 of WNV i.p. or 50LD50 i.c. Here, 3-9 month old age-matched wild-type C3H mice were used in control groups. Mice immunized and boosted with 6.5 log10 pfu equivalents of H2O2-inactivated WNV were positive control. In other experiments, 6-8 week old immunodeficient NOD/SCIDIL2Rγ−/− mice were i.p. challenged with 100LD50 WNV.
Detection of Infectious Virus
Viral titers were determined by an Immunoperoxidase Monolayer assay (IPMA) (Durbin et al., 2001). WNV-infected Vero cells were overlaid with 1% methylcellulose (EMD Chemicals, Gibbstown, NJ) and infectious foci detected with the anti-flavivirus 4G2 monoclonal antibody and peroxidase staining.
For PRNT assays, heat-inactivated immune mouse sera (56°C, 30 minutes) serially diluted in DMEM were incubated for 1 hour at 37°C after mixing with 100 PFU WNV. No loss in neutralizing activity occurred after heat inactivation. The mixtures were used in IPMA and PRNT90 was calculated by linear regression by plotting the log dilutions of test sera against the percent reduction in viral foci. The reciprocal of the highest dilution of the test sera required to neutralize 90% of the virus was defined as PRNT90.
Viral RNA was isolated from mouse tissue homogenates using the RNAII Spin Kit (Clontech, Mountain View, CA) and detected by SYBR Green qRT-PCR with WNV env-specific (5′-TCACGCATCTCTCCACCAAAG-3′ and 5′-GGGTCAGCACGTTTGTCATTG-3′) (Papin et al., 2004).
Fluorescent Immunostaining
Mouse brain tissue fixed in 10% neutral-buffered formalin (Sigma, St. Louis, MO) for 72 h was paraffin embedded, and sectioned. TUNEL staining was performed using the In Situ Cell Death Detection kit-fluorescein (Roche, Indianapolis, IN) followed by immediate overnight staining at 4°C for WNV antigen with anti-WNV hyperimmune ascitic fluid and a TRITC-labeled anti-mouse secondary antibody. For GFAP staining, antigens were unmasked with Target Retrieval Solution (Dako North America, Inc., Carpenteria, CA) followed by overnight incubation at 4°C with anti-GFAP (Dako North America, Inc., Carpenteria, CA) followed by an anti-rabbit secondary FITC-labeled antibody. All sections were counterstained with DAPI. Cells were visualized at 400X magnification with a Nikon Eclipse fluorescent microscope.
Adoptive and Passive Transfer Experiments
RVG9R-siFvEJW treated C3H mice that survived primary infection were euthanized on D14 p.i. and CD8+ T cells purified by MACS (Miltenyi Biotech, Auburn, CA) (>96% purity) from splenocytes pooled from multiple donors. Recipient and control mice were sub-lethally irradiated with 300 rad 4 h prior to WNV infection, which does not affect time to death (Webb and Jagelman, 1979).
For pre-challenge adoptive transfer, splenocytes depleted of CD8+ T cells (10 × 106 cells/mouse) or reconstituted with purified CD8+ T cells (additional 4.6 ×106 CD8+T cells/mouse) were i.v. injected followed by i.p challenge with 100LD50 WNV in an hour. For post-challenge adoptive transfer, recipient mice were i.v. injected 5 days p.i. as above with splenocytes depleted of or reconstituted with purified CD8 T-cells along with 0.2 ml of heat inactivated immune serum pooled from donor mice.
Germinal Center Analysis
Splenocytes isolated at day 8 p.i. were treated with Fc receptor blocker (Innovex Biosciences, Richmond, CA) and stained with anti-mouse B220-PerCP, GL7-FITC and FAS-PE or anti-mouse CD4-A647, PD-1-PE and CXCR5-PerCP (BioLegend, San Diego, CA) prior to flow cytometry on a FACS Calibur (BD). The data were analyzed by FlowJo software (Tristar Inc., San Carlos, CA).
Quantification and Statistical Analysis
Statistical analyses were performed with the Prism software (GraphPad Prism). Grouped data were analyzed by one- or two-way ANOVA followed by multiple comparisons (Tukey or Sidak post-hoc tests). Non-grouped analyses was performed using the Mann-Whitney test. Kaplan-Meier survival curves were analyzed by Log-Rank (Mantel-Cox) test. Statistical significance was computed at P ≤ 0.5. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; and ****P ≤ 0.0001. Number of animals and replicates for each experiment are indicated in the figure legends.
Supplementary Material
Highlights.
Intranasal treatment with RVG9R-siFvEJW is therapeutic for neuroinvasive WNV.
Viral burden is lowered in the CNS reducing neuropathology and mortality.
The approach allowed development of natural immunity to WNV.
Immune responses mediated recovery and protection against WNV.
Acknowledgements
We thank Michel Ledizet (L2 Diagnostics) for the 4G2 monoclonal antibody, Dr. John Anderson (Connecticut Agricultural Experiment Station) for hyperimmune ascitic fluid, Dr. Choukri Ben Mamoun (Yale University), for the use of the Nikon fluorescent microscope. This work was funded in part by NIH grants R01AI112443 and R21AI122384 (P.K), T32AI055403 and ASM Robert D. Watkins Graduate Research Fellowship (N.M.), and Korea Ministry of Health & Welfare grant HI17C1046 and NRF 2009-0079989 (S.K.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
P.K., S.K.L, J.B., and N. M. conceived and performed experiments and wrote the manuscript, P.K., and S.K.L, secured funding and provided resources, I.U. and A.J. performed experiments. P.D.U. helped with experiments and edited the manuscript. E.F. provided some resources.
Declaration of Interest
P.K. and S.K.L. are cofounders of Signet Biotech. P.K and S.K.L are listed as co-inventors for patent PCT/US07/12152, which includes in part claims relating to RVG9R. P.K, S.K.L, and I.U. are listed as co-inventors for a patent (PCT/KR2016/014220) submitted, which includes claims relating to a mouse positioning device.
References
- Acharya D, and Bai F (2016). An Overview of Current Approaches Toward the Treatment and Prevention of West Nile Virus Infection, Vol 1435 (Humana Press; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta EG, and Bartenschlager R (2016). Paradoxical role of antibodies in dengue virus infections: considerations for prophylactic vaccine development. Expert review of vaccines 15, 467–482. [DOI] [PubMed] [Google Scholar]
- Amanna IJ, and Slifka MK (2014). Current trends in West Nile virus vaccine development. Expert review of vaccines 13, 589–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Saunders AB, Oldenburg IA, Sabatini BL, and Cepko CL (2013). Vesicular stomatitis virus with the rabies virus glycoprotein directs retrograde transsynaptic transport among neurons in vivo. Front Neural Circuits 7, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nathan D, Gershoni-Yahalom O, Samina I, Khinich Y, Nur I, Laub O, Gottreich A, Simanov M, Porgador A, Rager-Zisman B, et al. (2009). Using high titer West Nile intravenous immunoglobulin from selected Israeli donors for treatment of West Nile virus infection. BMC infectious diseases 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AN, Kent KA, Bennett CJ, and Bernard KA (2007). Tissue tropism and neuroinvasion of West Nile virus do not differ for two mouse strains with different survival rates. Virology 368, 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhuria SV, Hanson LR, and Frey WH, 2nd (2010). Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci 99, 1654–1673. [DOI] [PubMed] [Google Scholar]
- Diamond MS (2009). Progress on the development of therapeutics against West Nile virus. Antiviral Res 83, 214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Shrestha B, Marri A, Mahan D, and Engle M (2003). B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol 77, 2578–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djupesland PG, Skretting A, Winderen M, and Holand T (2006). Breath actuated device improves delivery to target sites beyond the nasal valve. Laryngoscope 116, 466–472. [DOI] [PubMed] [Google Scholar]
- Durbin AP, Karron RA, Sun W, Vaughn DW, Reynolds MJ, Perreault JR, Thumar B, Men R, Lai CJ, Elkins WR, et al. (2001). Attenuation and immunogenicity in humans of a live dengue virus type-4 vaccine candidate with a 30 nucleotide deletion in its 3’-untranslated region. Am J Trop Med Hyg 65, 405–413. [DOI] [PubMed] [Google Scholar]
- Garber K (2017). Worth the RISC? Nat Biotechnol 35, 198–202. [DOI] [PubMed] [Google Scholar]
- Geiss BJ, Pierson TC, and Diamond MS (2005). Actively replicating West Nile virus is resistant to cytoplasmic delivery of siRNA. Virol J 2, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould LH, Sui J, Foellmer H, Oliphant T, Wang T, Ledizet M, Murakami A, Noonan K, Lambeth C, Kar K, et al. (2005). Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J Virol 79, 14606–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley M, Retter AS, Fowler D, Gea-Banacloche J, and O’Grady NP (2003). The role for intravenous immunoglobulin in the treatment of West Nile virus encephalitis. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 37, e88–90. [DOI] [PubMed] [Google Scholar]
- Hayes EB, Sejvar JJ, Zaki SR, Lanciotti RS, Bode AV, and Campbell GL (2005). Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis 11, 1174–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehi L, and Sila CA (2009). Neurologic complications of West Nile virus. In Disease Management (Cleveland Clinic). [Google Scholar]
- Kim SS, Ye C, Kumar P, Chiu I, Subramanya S, Wu H, Shankar P, and Manjunath N (2010). Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol Ther 18, 993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Lee SK, Shankar P, and Manjunath N (2006). A single siRNA suppresses fatal encephalitis induced by two different flaviviruses. PLoS Med 3, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung K-E, Hee Kim M, Davidson BL, Kyung Lee S, Shankar P, and Manjunath N (2007). Transvascular delivery of small interfering RNA to the central nervous system. Nature 448, 39–43. [DOI] [PubMed] [Google Scholar]
- Lim SM, Koraka P, Osterhaus AD, and Martina BE (2011). West Nile virus: immunity and pathogenesis. Viruses 3, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, and Thorne RG (2012). Intranasal delivery of biologics to the central nervous system. Adv drug deliv rev 64, 614–628. [DOI] [PubMed] [Google Scholar]
- Mazarakis ND, Azzouz M, Rohll JB, Ellard FM, Wilkes FJ, Olsen AL, Carter EE, Barber RD, Baban DF, Kingsman SM, et al. (2001). Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum Mol Genet 10, 2109–2121. [DOI] [PubMed] [Google Scholar]
- Miyake MM, and Bleier BS (2015). The blood-brain barrier and nasal drug delivery to the central nervous system. Am J Rhinol Allergy 29, 124–127. [DOI] [PubMed] [Google Scholar]
- Morrey JD, Siddharthan V, Olsen AL, Roper GY, Wang H, Baldwin TJ, Koenig S, Johnson S, Nordstrom JL, and Diamond MS (2006). Humanized monoclonal antibody against West Nile virus envelope protein administered after neuronal infection protects against lethal encephalitis in hamsters. J Infect Dis 194, 1300–1308. [DOI] [PubMed] [Google Scholar]
- Morrey JD, Siddharthan V, Wang H, Hall JO, Skirpstunas RT, Olsen AL, Nordstrom JL, Koenig S, Johnson S, and Diamond MS (2008). West Nile virus-induced acute flaccid paralysis is prevented by monoclonal antibody treatment when administered after infection of spinal cord neurons. J Neurovirol 14, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netland J, and Bevan MJ (2013). CD8 and CD4 T cells in west nile virus immunity and pathogenesis. Viruses 5, 2573–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolle L (2003). Experiencing West Nile virus. Can J Infect Dis 14, 75–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, et al. (2005). Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nature medicine 11, 522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omi K, Tokunaga K, and Hohjoh H (2004). Long-lasting RNAi activity in mammalian neurons. FEBS Lett 558, 89–95. [DOI] [PubMed] [Google Scholar]
- Papin JF, Vahrson W, and Dittmer DP (2004). SYBR green-based real-time quantitative PCR assay for detection of West Nile Virus circumvents false-negative results due to strain variability. J Clin Microbiol 42, 1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana DS, Guastella AJ, Westlye LT, and Andreassen OA (2016). The promise and pitfalls of intranasally administering psychopharmacological agents for the treatment of psychiatric disorders. Mol Psychiatry 21, 29–38. [DOI] [PubMed] [Google Scholar]
- Salimi H, Cain MD, and Klein RS (2016). Encephalitic Arboviruses: Emergence, Clinical Presentation, and Neuropathogenesis. Neurotherapeutics 13, 514–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Morrey JD, and Diamond MS (2007). Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J Virol 81, 2614–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoderboeck L, Riad S, Bokor AM, Wicky HE, Strauss M, Bostina M, Oswald MJ, Empson RM, and Hughes SM (2015). Chimeric rabies SADB19-VSVg-pseudotyped lentiviral vectors mediate long-range retrograde transduction from the mouse spinal cord. Gene Ther 22, 357–364. [DOI] [PubMed] [Google Scholar]
- Shimoni Z, Niven MJ, Pitlick S, and Bulvik S (2001). Treatment of West Nile virus encephalitis with intravenous immunoglobulin. Emerg Infect Dis 7, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Gottlieb D, and Diamond MS (2003). Infection and injury of neurons by West Nile encephalitis virus. J Virol 77, 13203–13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Ng T, Chu HJ, Noll M, and Diamond MS (2008). The relative contribution of antibody and CD8+ T cells to vaccine immunity against West Nile encephalitis virus. Vaccine 26, 2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati EM, and Diamond MS (2006). CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J Virol 80, 12060–12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Diamond MS, and Gale M Jr. (2013). West Nile virus infection and immunity. Nat Rev Microbiol 11, 115–128. [DOI] [PubMed] [Google Scholar]
- van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, et al. (2007). West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J Virol 81, 10933–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb HE, and Jagelman S (1979). Viral infections of the CNS in the immune compromised host with special reference to certain paraneoplastic syndromes In CNS complications of malignant disease, Whitehouse J, and H Kay, eds. (Palgrave Macmillan; ), pp. 258–280. [Google Scholar]
- Zeller S, Choi CS, Uchil PD, Ban HS, Siefert A, Fahmy TM, Mothes W, Lee SK, and Kumar P (2015). Attachment of cell-binding ligands to arginine-rich cell-penetrating peptides enables cytosolic translocation of complexed siRNA. Chem Biol 22, 50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.