

Abstract
Genetic and genomic studies, including genome-wide association studies (GWAS) have accelerated the discovery of genes contributing to glaucoma, the leading cause of irreversible blindness world-wide. Glaucoma can occur at all ages, with Mendelian inheritance typical for the rare early onset disease (before age 40) and complex inheritance evident in common adult-onset forms of disease. Recent studies have suggested possible therapeutic targets for some patients with early-onset glaucoma based on the molecular and cellular events caused by MYOC, OPTN and TBK1 mutations. Diagnostic genetic tests using early-onset glaucoma genes are also proving useful for pre-symptomatic disease detection and genetic counseling. Recent GWAS completed for three types of common adult-onset glaucoma have identified novel loci for POAG (primary-open-angle glaucoma) (ABCA1, AFAP1, GMDS, PMM2, TGFBR3, FNDC3B, ARHGEF12, GAS7, FOXC1, ATXN2, TXNRD2); PACG (primary angle-closure glaucoma (EPDR1, CHAT, GLIS3, FERMT2, DPM2-FAM102); and exfoliation syndrome (XFS) glaucoma (CACNA1A). In total sixteen genomic regions have been associated with POAG (including the normal tension glaucoma (NTG) subgroup), 8 with PACG and 2 with XFS. These studies are defining important biological pathways and processes that contribute to disease pathogenesis.
Introduction
Glaucoma is a term used to describe a group of disorders that have in common progressive degeneration of the optic nerve causing visual compromise and eventually blindness. Collectively, glaucoma is the leading cause of irreversible blindness world-wide. Elevated intraocular pressure (IOP) is a major risk factor for most types of glaucoma. Fluid formed by the ciliary body (aqueous humor) is removed by the trabecular meshwork and Schlemm’s canal located in the ocular ‘angle’ that forms at the junction of the cornea and iris (Fig. 1). The IOP level is dependent on the rate of fluid removal, which is decreased in all types of glaucoma with elevated IOP. Glaucoma subgroups are defined as ‘open-angle’ or ‘closed-angle’ depending on the position of the ocular lens and iris relative to the trabecular meshwork (Fig. 1). Open angle glaucoma is further divided into subgroups defined by the ocular features that characterize them. For example exfoliation syndrome (XFS) and the related glaucoma (XFG) are defined by the accumulation of a characteristic fibrillar material on the ocular lens and trabecular meshwork (Fig. 2). Primary open angle glaucoma (POAG), a type of glaucoma defined by anatomically normal structures in the absence of any secondary cause of glaucoma, such as XFS, also includes the normal tension glaucoma (NTG) subgroup patients who develop glaucomatous optic neuropathy in the absence of abnormally elevated IOP.
Figure 1.
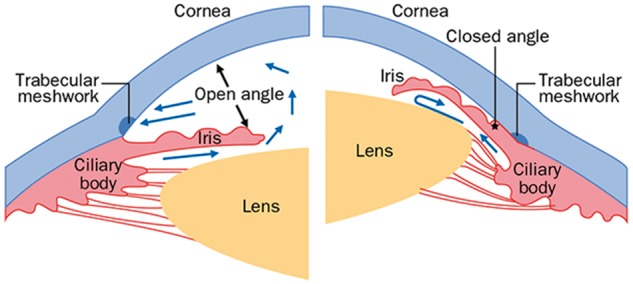
Schematic diagram of the ocular anterior segment in open-angle and closed-angle glaucoma. Under normal conditions (open angle) the aqueous humor formed by the ciliary body flows around the lens and iris (blue arrows) and exits the eye through the trabecular meshwork, through Schlemm’s canal and empties into aqueous veins and the episcleral venous system. In the closed angle, the iris and lens are positioned anteriorly causing an obstruction of aqueous flow through the trabecular meshwork.
Figure 2.
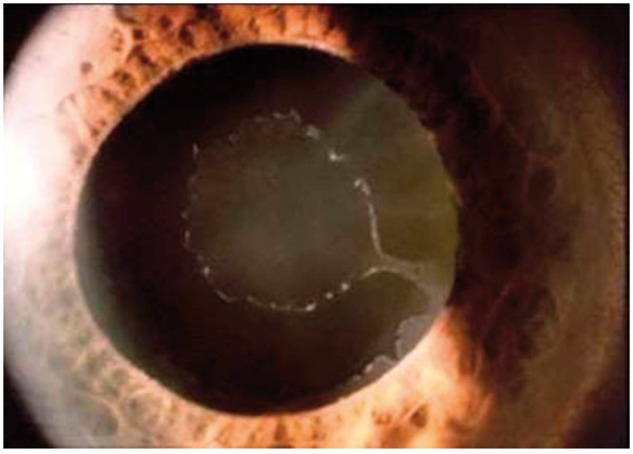
Image of an eye with exfoliation syndrome (XFS). The exfoliation material is evident as white fibrillar material on the lens and pupil margin.
Glaucoma can occur at all ages, with early onset disease (before the age of 40) exhibiting Mendelian inheritance and adult onset forms (developing after age 40) inherited as complex traits (1). Generally, mutations in genes causing early onset glaucoma are rare with large biological effects, while variants contributing to the adult-onset glaucomas are common with smaller effects (2). Genome-wide association studies (GWAS) have successfully identified genomic loci for POAG (3–11) (Table 1), NTG (5), XFS (12,13) and PACG (primary angle closure glaucoma) (14,15). These advances will be discussed in this review, as well as new information regarding genes responsible for Mendelian forms of open angle glaucoma. While the function of these genes and genomic loci in health and disease are not completely understood, these findings are providing new insights into the underlying molecular mechanisms responsible for glaucoma that could eventually lead to novel gene-based therapies, including strategies for neuroprotection. Additionally, gene-based diagnostic testing for mutations responsible for early-onset Mendelian glaucoma can detect individuals at the earliest stages of disease when therapy is most effective.
Table 1.
Primary open angle glaucoma (POAG) loci discovered by GWAS
| Population | Case/control (N) |
New Loci | Reference | |
|---|---|---|---|---|
| Discovery | Replication | |||
| Iceland | 1263/34,877 | 2175/2064 (European) 299/1607 (Chinese) | CAV1/CAV2 | Thorliefsson et al., Nat. Genet. 2010. (3) |
| Australian (ANZRAG) | 615/3956 | 892/4582 (Australian) | CDKN2BAS, TMCO1 | Burdon et al., Nat. Genet. 2011. (4) |
| US European ancestry (NEIGHBOR) | 2170/2347 | 976/1140 (GLAUGEN) | SIX6, 8q22 (NTG) | Wiggs et al., PLoS Genet., 2012. (5) |
| Japanese | 1394/6599 | 1802/7212 | CDKN2BAS, SIX6 | Osman et al., Hum. Mol. Genet., 2012. (6) |
| Australian (ANZRAG) | 1,155/1992 | 3548/9496 (Australian and US European) | ABCA1, AFAP1, GMDS | Gharahkhani et al., Nat. Genet., 2014. (7) |
| Chinese | 1007/1009 | 1899/4965 (Chinese and Singaporean Chinese) | ABCA1, PMM2 | Chen et al., Nat. Genet., 2014. (8) |
| Multi-ethnic (Illumina exome array) | 3504/9746 | 9173/26,780 (multi-ethnic) | TGFBR3, FNDC3B | Li et al., Hum. Mol. Genet., 2015. (9) |
| European (Rotterdam) | 8105 (population)* | 7471 population1225 POAG cases and 4117 controls | ARHGEF12 | Springelkamp et al., Hum. Mol. Genet., 2015. (10) |
| US European ancestry (NEIGHBORHOOD) | 3,853/33,480 | 3164/9242 (Australian, European, Singaporean Chinese) | TXNRD2, ATXN2, FOXC1, GAS7 | Cooke Bailey et al., Nat. Genet., 2016. (11) |
The reference number in this review is listed in parentheses after the abbreviated reference.
The discovery analysis for this study was a population-based study for IOP (N = 8105). Replication was done in a second population based analysis of 7471 individuals. The top SNPs after replication were assessed for association with POAG in 1225 cases and 4117 controls.
Early-Onset Open Angle Glaucoma
Early onset glaucoma with Mendelian inheritance can involve ocular developmental abnormalities that will be covered in another review in this issue (Fingert). Some types of early onset glaucoma, however, are associated with developmentally normal ocular structures including juvenile open angle glaucoma (JOAG) and familial normal tension glaucoma, to be discussed in the following sections.
Familial normal tension glaucoma (NTG)
Rare mutations involving two genes, an OPTN (optineurin) missense mutation (E50K) and CNVs (copy number variations) involving TBK1 (Tank-binding protein 1), cause early-onset familial NTG with autosomal dominant inheritance (16–20). Individuals affected by NTG caused by OPTN and TBK1 mutations develop severe disease prior to age 40 (17,18). Together mutations in these two genes account for approximately 2-3% of NTG (20). OPTN and TBK1 have important roles in critical cellular processes, notably autophagy and NF-κB signaling (20,21), and the encoded proteins are known to interact (22,23). Optineurin normally negatively regulates NF-κB, a process that is modulated by TBK1 (21). The E50K OPTN mutation enhances TBK1-OPTN binding (24) potentially increasing NF-κB activity and promoting cell death (25). Phosphorylation of OPTN by TKB1 also promotes the recruitment of microtubule-associated protein 1 light chain 3 beta (MAP1LC3B, LC3B) an important step in the initiation of autophagy (26), and this interaction is also being enhanced by the OPTN E50K missense mutation (23,27).
Recently, OPTN and TBK1 mutations have been identified in patients with ALS (amyotrophic lateral sclerosis) (28) and another ALS associated protein ATXN2 (29), has also been associated with POAG (see below) (11). These results suggest that genes encoding other ALS-related proteins, or proteins involved in autophagy and or NF-κB signaling could also contribute to NTG. However, one such protein, sequestosome (SQSTM1) that encodes an autophagy receptor that is a target of TBK1 phosphorylation, does not appear to contribute to NTG (30).
Juvenile open angle glaucoma (JOAG)
JOAG is defined as onset of open angle glaucoma before age 40. Typically, patients affected by JOAG develop a severe form of glaucoma characterized by very high IOP that can be difficult to control by current therapies. Existing therapies are most effective at early disease stages, however, patients may be asymptomatic and not seek medical attention early in the disease, limiting the opportunity for early intervention (31). The identification of genes responsible for JOAG and other early-onset forms of glaucoma would make gene-based diagnostic testing possible, providing for early detection of at-risk individuals before irreversible vision loss occurs.
MYOC (myocilin) mutations are an important cause of JOAG with dominant inheritance (32). Most disease-causing mutations are missense alleles located in the third exon that codes for the olfactomedin domain (33). A relatively common nonsense mutation (GLN368X) is associated with the mildest MYOC-related disease (34), while many missense mutations (notably PRO370LEU and TYR347HIS) cause the most severe phenotype. Overall MYOC mutations account for 8-36% of JOAG (35,36) and 2-4% of adult-onset POAG (35,37) depending on the ethnicity of the population. Deletions involving MYOC or nonsense mutations near the N-terminal do not cause disease suggesting that the underlying genetic mechanism is not loss of function but dominant negative or gain of function (38,39). Recently MYOC mutations have been shown to cause protein misfolding and protein aggregation causing ER stress (40). Sodium 4-phenylbutyrate, a molecular chaperone known to relieve the misfolded protein response in urea cycle disorders, also relieved ER stress and lowered IOP in a transgenic MYOC mouse (41,42), identifying a new opportunity for novel gene-based therapies for MYOC mutation carriers. Since absence of Myocilin does not result in ocular or systemic diseases (38), other strategies to eliminate or reduce MYOC expression could also be developed as effective therapeutics.
Common Adult-Onset Glaucoma with Complex Inheritance
Primary open-angle glaucoma
Primary open-angle glaucoma is the most common form of glaucoma in most populations worldwide (43). Patients with POAG have glaucoma despite anatomically normal ocular structures including open angles. Like other forms of glaucoma, IOP elevation is an important risk factor for POAG, however, up to one-third of POAG patients with optic nerve degeneration have IOP in the normal range, defining the normal tension glaucoma (NTG) POAG subgroup (44). Patients with NTG may have increased susceptibility to optic nerve degeneration compared to POAG overall (5).
Recent genome-wide association studies (GWAS) completed for POAG and NTG in European Caucasian and Asian populations have identified ABCA1, AFAP1, GMDS, PMM2, TGFBR3, FNDC3B, ARHGEF12, GAS7, FOXC1, ATXN2, and TXNRD2 (7–11) bringing the total number of genes/loci significantly associated with disease to 16 (Table 1). Several loci have been associated at the genome-wide level in both Asian and European Caucasian populations including CDKN2BAS, SIX6, and ABCA1 (6,8). GWAS in African-American or other African populations have not yet been done, an important area of future research considering the high disease prevalence in these populations (43).
Current SNPs associated with POAG are common (minor allele frequencies > 0.3) and have odds ratios (ORs) ranging from 1.4 (CDKN2BAS, rs7866783; SIX6, rs33912345) to 1.17 (FOXC1, rs2745572 and TXNRD2, rs35934224) (11). Interestingly, some disease-associated SNPs have stronger evidence for association in specific phenotypic subgroups supporting the genetic and phenotypic heterogeneity of the disease. For example, association of CDKN2BAS SNPs is stronger for NTG compared to POAG overall (OR = 1.6 for NTG compared to 1.4 for POAG overall) (11) and the association of CAV1/CAV2 SNPs is stronger for the POAG subgroup with paracentral visual field loss (OR = 1.57 compared with 1.26 for POAG overall) (45).
POAG associated loci involve diverse biological processes including cytokine signaling (CDKN2BAS, TGFBR2, FNDC3B), lipid metabolism (ABCA1, CAV1/CAV2, ARHGEF12), membrane biology (CAV1/CAV2), extracellular matrix (AFAP1), fucose and mannose metabolism (GMDS, PMM2), cell division (CDKN2BAS, TMCO1, GAS7) and ocular development (SIX6, FOXC1) (2). Several pathways are particularly interesting. Interactions among ABCA1, CAV1/CAV2, and ARHGEF12 can influence lipid and cholesterol metabolism (46,10), and recent studies suggest that statin treatment for hypercholesterolemia may be protective for glaucoma (47). Further studies investigating statin use in individuals with associated risk variants in these loci could reveal clinically relevant pharmacogenetic relationships. Recent studies are also identifying important contributions of mitochondria to glaucoma pathogenesis (48). TXNRD2, significantly associated with POAG in a recent GWAS (11), codes for thioredoxin reductase 2, a mitochondrial protein necessary for reducing damaging reactive oxygen species generated by oxidative phosphorylation and other mitochondrial functions (49). The mitochondria-rich retinal ganglion cells damaged in glaucoma are known to be susceptible to oxidative stress (50) suggesting that reduction in reactive oxygen species could be neuroprotective. The importance of mitochondrial function is also evident from gene-set analyses using mitochondrial protein-encoding genes that show an association with POAG and in particular NTG (51).
Primary angle-closure glaucoma (PACG)
PACG is a major cause of irreversible blindness, especially in Asia. Patients with PACG can have acute, subacute or chronic presentations. Regardless of symptoms, PACG patients develop elevated IOP secondary to apposition of the peripheral iris and trabecular meshwork that creates a barrier to fluid flowing out of the eye (Fig. 1). PACG can result in very high IOP causing optic nerve degeneration (52).
Various studies suggest that PACG has a genetic component (53–55), however, no environmental risk factors have been identified. While familial PACG has been reported in Basset Hounds (56), in humans, familial aggregation of early-onset angle-closure glaucoma phenotypically is within the spectrum of nanophthalmos, an extreme form of hyperopia that can cause closure of the angles due to age-related enlargement of the lens in the small hyperopic eye. Nanopthalmos can be inherited as an autosomal dominant or recessive trait and mutations in MFRP (57) and TMEM98 (58) have been identified in recessive and dominant forms of the disease, respectively. In addition, there is an autosomal recessive form of retinal degeneration termed bestrophinopathy that is commonly accompanied by angle closure glaucoma and produced by mutations in BEST1 (59).
Genome wide association studies have identified 8 genes/loci for the common adult-onset form of PACG: PLEKHA7, COL11A1, PCMTD1-ST18, EPDR1, CHAT, GLIS3, FERMT2, and DPM2-FAM102 (14,15). These loci were identified in large case-control sets mostly from Asia. The effect sizes for these variants are between ∼1.2-1.4 and only explain <2% of the genetic variance in PACG. None of these PACG loci were associated with primary open-angle glaucoma (POAG) in a Singaporean Chinese sample consisting of 986 cases and 3916 controls while only 2 POAG loci (rs2226035, ARHGEF12 and rs12150284, GAS7) were nominally associated with PACG, suggesting there is little etiological overlap between these two major forms of glaucoma (15).
Common variants in genes that cause inherited forms of nanophthalmos have been ruled out as candidate genes for the common forms of PACG with complex inheritance. The concept that PACG is directly correlated with eye size is not entirely supported by genomic data, as variants identified for axial length (AxL) in a genome-wide quantitative-linked trait (QLT) analysis (60) were not related to PACG (15). However, genes that are related to the depth of the anterior chamber (ACD) can be associated with disease. For instance the intergenic PACG susceptibility locus between PCMTD1 and ST18 (rs1015213) was associated with smaller ACD but not AxL in a European sample of 986 subjects (61). It should be noted that the LD block for rs1015213 includes PCMTD1 but not ST18 and that PCMTD1 is expressed in the anterior segment while ST18 has more limited ocular expression (15). Furthermore a QLT analysis identified ABCC5 associated with ACD in a dataset consisting 4276 PACG cases and 18,801 controls, this variant was also associated with PACG (OR = 1.13; 95%CI: 1.06-1.22; P = 0.00046) (62).
Eyes with angle closure glaucoma have a thicker retinal choroid than normal eyes and eyes affected by POAG even after adjusting for AxL (63), suggesting that cell-cell adhesion in the vascular uveal tract represents an important attribute of PACG. Several PACG genetic loci (EPDR1, FERMT2 and PLEKHA7) are involved in cell adhesion (15). Interestingly, anticholinergics are known to precipitate acute PACG attacks and one PACG susceptibility locus (CHAT) encodes an enzyme involved in generating acetylcholine (15) implicating acetylcholine metabolism in PACG pathogenesis, and suggesting a potential target for therapeutic prevention in high-risk populations.
Exfoliation syndrome and glaucoma
In exfoliation syndrome (XFS) there is an accumulation of fibrillar material in the anterior ocular segment, most conspicuously at the pupillary margin and on the lens surface (Fig. 2). These deposits lodge in the trabecular meshwork and contribute to elevated IOP, optic nerve degeneration and glaucoma. Similar deposits with unclear clinical significance have been detected in non-ocular tissues (64). Overall, XFS appears to be a form of deleterious ocular aging that is also associated with premature cataract formation, cataract surgery complications and retinal venous occlusive disease (65). The mechanisms underlying formation of the disease-related extracellular deposits remain unknown but the condition appears to have both genetic (66) and environmental components (67).
There are no familial aggregation or candidate gene studies that have identified significant genetic risk factors for XFS. Remarkably, a genome-wide association study using a discovery set consisting of only 75 unrelated cases and 14,470 population-based controls from Iceland identified LOXL1 (lysyl oxidase like 1) SNPs (rs3825942; rs1048661 and rs2165241) significantly associated with XFS (12). The top SNP effect size is amongst the highest observed for common complex disease in the GWAS era (∼20-fold for rs3825942) and suggests that LOXL1 has an important role in XFS pathogenesis. LOXL1 is involved in elastogenesis and collagen crosslinking which could impact XFS development by modulating extracelluar matrix stability. The co-occurrence of systemic (68,69) and ocular vasculopathies (70) with XFS, and an association between pelvic organ prolapse and XFS, collectively suggest that LOXL1 maintenance of elastin and collagen is altered in XFS (71). In European Caucasians and most populations world-wide the disease-associated variants (rs3825942; rs1048661 and rs2165241) are the common alleles present in up to 99% of cases and up to 80% of controls (72). However, in some Asian and African populations the common variants associated with disease are flipped compared to the European Caucasians (73). Collectively, these observations suggest that LOXL1 is necessary but not sufficient for disease development and that other genetic variants and also environmental factors are likely to contribute to the disease development.
A second XFS GWAS using meta-analyses of multi-ethnic populations identified CACNA1A as an additional locus for XFS (13). CACNA1A codes for a P/Q voltage dependent calcium channel. The observation that XFS disease burden does not correlate with LOXL1 risk variant frequencies in world-wide populations (72,74) also prompted a search for environmental risk factors for the condition. Interestingly multivariable analyses indicated that XFS disease burden increases in extra-equatorial regions (75) and environmental risk factors that could explain this trend were sought. Higher coffee consumption and lower dietary folate intake exhibit these trends and were found to be associated with increased risk of XFS (76). Furthermore, more time spent outdoors appears to be a strong risk factor for XFS (77) implicating ocular UV exposure, which is known to up-regulate LOXL1 activity (78). While genetic and environmental studies have shed light on XFS, further research and the identification of additional genetic loci and environmental risk factors will be needed to gain a better understanding of disease pathogenesis.
Conclusion
Genetic and genomic studies are finding important genes contributing to glaucoma. Glaucoma-related genes are beginning to define relevant biological pathways and processes that could be targets for novel gene-based therapies. Gene discovery is also enabling the development of gene-based tests capable of identifying individuals at risk before irreversible blindness occurs. Further research will be required to fully define the genetic architecture of glaucoma, a necessary step before comprehensive genetic testing and targeted gene-based therapy can be achieved.
Acknowledgements
LRP and JLW are supported by the Harvard Glaucoma Center of Excellence and Research to Prevent Blindness and the Margolis fund (Boston, MA, USA). The Arthur Ashley Foundation also supported LRP. JLW and LRP were supported by NIH/NEI.
Conflict of Interest statement. None declared.
Funding
Harvard Glaucoma Center of Excellence, Research to Prevent Blindness and the Margolis Fund, The Arthur Ashley Foundation, NIH/NEI grants R01 EY022305, R01 EY020928 and R01 EY015473.
References
- 1. Allen K.F., Gaier E.D., Wiggs J.L. (2015) Genetics of primary inherited disorders of the optic nerve: clinical applications. Cold Spring Harb. Perspect. Med., 5, a017277.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wiggs J.L. (2015) Glaucoma genes and mechanisms. Prog. Mol. Biol. Transl. Sci., 134, 315–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thorleifsson G., Walters G.B., Hewitt A.W., Masson G., Helgason A., DeWan A., Sigurdsson A., Jonasdottir A., Gudjonsson S.A., Magnusson K.P.. et al. (2010) Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat. Genet., 42, 906–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burdon K.P., Macgregor S., Hewitt A.W., Sharma S., Chidlow G., Mills R.A., Danoy P., Casson R., Viswanathan A.C., Liu J.Z.. et al. (2011) Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat. Genet., 43, 574–578. [DOI] [PubMed] [Google Scholar]
- 5. Wiggs J.L., Yaspan B.L., Hauser M.A., Kang J.H., Allingham R.R., Olson L.M., Abdrabou W., Fan B.J., Wang D.Y., Brodeur W.. et al. (2012) Common variants at 9p21 and 8q22 are associated with increased susceptibility to optic nerve degeneration in glaucoma. PLoS Genet., 8, e1002654.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Osman W., Low S.K., Takahashi A., Kubo M., Nakamura Y. (2012) A genome-wide association study in the Japanese population confirms 9p21 and 14q23 as susceptibility loci for primary open angle glaucoma. Hum. Mol. Genet., 21, 2836–2842. [DOI] [PubMed] [Google Scholar]
- 7. Gharahkhani P., Burdon K.P., Fogarty R., Sharma S., Hewitt A.W., Martin S., Law M.H., Cremin K., Bailey J.N., Loomis S.J.. et al. (2014) Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat. Genet., 46, 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y., Lin Y., Vithana E.N., Jia L., Zuo X., Wong T.Y., Chen L.J., Zhu X., Tam P.O., Gong B.. et al. (2014) Common variants near ABCA1 and in PMM2 are associated with primary open-angle glaucoma. Nat. Genet., 46, 1115–1119. [DOI] [PubMed] [Google Scholar]
- 9. Li Z., Allingham R.R., Nakano M., Jia L., Chen Y., Ikeda Y., Mani B., Chen L.J., Kee C., Garway-Heath D.F.. et al. (2015) A common variant near TGFBR3 is associated with primary open angle glaucoma. Hum. Mol. Genet., 24, 3880–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Springelkamp H., Iglesias A.I., Cuellar-Partida G., Amin N., Burdon K.P., van Leeuwen E.M., Gharahkhani P., Mishra A., van der Lee S.J., Hewitt A.W.. et al. (2015) ARHGEF12 influences the risk of glaucoma by increasing intraocular pressure. Hum. Mol. Genet., 24, 2689–2699. [DOI] [PubMed] [Google Scholar]
- 11. Bailey J.N., Loomis S.J., Kang J.H., Allingham R.R., Gharahkhani P., Khor C.C., Burdon K.P., Aschard H., Chasman D.I., Igo R.P. Jr. et al. (2016) Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat. Genet., 48, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thorleifsson G., Magnusson K.P., Sulem P., Walters G.B., Gudbjartsson D.F., Stefansson H., Jonsson T., Jonasdottir A., Jonasdottir A., Stefansdottir G.. et al. (2007) Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science, 317, 1397–1400. [DOI] [PubMed] [Google Scholar]
- 13. Aung T., Ozaki M., Mizoguchi T., Allingham R.R., Li Z., Haripriya A., Nakano S., Uebe S., Harder J.M., Chan A.S.. et al. (2015) A common variant mapping to CACNA1A is associated with susceptibility to exfoliation syndrome. Nat. Genet., 47, 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vithana E.N., Khor C.C., Qiao C., Nongpiur M.E., George R., Chen L.J., Do T., Abu-Amero K., Huang C.K., Low S.. et al. (2012) Genome-wide association analyses identify three new susceptibility loci for primary angle closure glaucoma. Nat. Genet., 44, 1142–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khor C.C., Do T., Jia H., Nakano M., George R., Abu-Amero K., Duvesh R., Chen L.J., Li Z., Nongpiur M.E.. et al. (2016) Genome-wide association study identifies five new susceptibility loci for primary angle closure glaucoma. Nat. Genet., 48, 556–562. [DOI] [PubMed] [Google Scholar]
- 16. Rezaie T., Child A., Hitchings R., Brice G., Miller L., Coca-Prados M., Héon E., Krupin T., Ritch R., Kreutzer D., Crick R.P., Sarfarazi M. (2002) Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science, 295, 1077–1079. [DOI] [PubMed] [Google Scholar]
- 17. Aung T., Rezaie T., Okada K., Viswanathan A.C., Child A.H., Brice G., Bhattacharya S.S., Lehmann O.J., Sarfarazi M., Hitchings R.A. (2005) Clinical features and course of patients with glaucoma with the E50K mutation in the optineurin gene. Invest. Ophthalmol. Vis Sci., 46, 2816–2822. [DOI] [PubMed] [Google Scholar]
- 18. Hauser M.A., Sena D.F., Flor J., Walter J., Auguste J., Larocque-Abramson K., Graham F., Delbono E., Haines J.L., Pericak-Vance M.A.. et al. (2006) Distribution of optineurin sequence variations in an ethnically diverse population of low-tension glaucoma patients from the United States. J. Glaucoma, 15, 358–363. [DOI] [PubMed] [Google Scholar]
- 19. Fingert J.H., Robin A.L., Stone J.L., Roos B.R., Davis L.K., Scheetz T.E., Bennett S.R., Wassink T.H., Kwon Y.H., Alward W.L.. et al. (2011) Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum. Mol. Genet., 20, 2482–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fingert J.H., Robin A.L., Scheetz T.E., Kwon Y.H., Liebmann J.M., Ritch R., Alward W.L. (2016) Tank-binding kinase 1 (TBK1) gene and open-angle glaucomas (An American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc., 114, T6. [PMC free article] [PubMed] [Google Scholar]
- 21. Sudhakar C., Nagabhushana A., Jain N., Swarup G. (2009) NF-kappaB mediates tumor necrosis factor alpha-induced expression of optineurin, a negative regulator of NF-kappaB. PLoS One, 4, e5114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richter B., Sliter D.A., Herhaus L., Stolz A., Wang C., Beli P., Zaffagnini G., Wild P., Martens S., Wagner S.A.. et al. (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl Acad. Sci. U S A, 113, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Minegishi Y., Iejima D., Kobayashi H., Chi Z.L., Kawase K., Yamamoto T., Seki T., Yuasa S., Fukuda K., Iwata T. (2013) Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum. Mol. Genet., 22, 3559–3567. [DOI] [PubMed] [Google Scholar]
- 24. Morton S., Hesson L., Peggie M., Cohen P. (2008) Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett., 582, 997–1002. [DOI] [PubMed] [Google Scholar]
- 25. Ying H., Yue B.Y. (2012) Cellular and molecular biology of optineurin. Int. Rev. Cell Mol. Biol., 294, 223–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rogov V.V., Suzuki H., Fiskin E., Wild P., Kniss A., Rozenknop A., Kato R., Kawasaki M., McEwan D.G., Löhr F.. et al. (2013) Structural basis for phosphorylation-triggered autophagic clearance of Salmonella. Biochem. J., 454, 459–466. [DOI] [PubMed] [Google Scholar]
- 27. Sirohi K., Swarup G. (2016) Defects in autophagy caused by glaucoma-associated mutations in optineurin. Exp. Eye Res., 144, 54–63. [DOI] [PubMed] [Google Scholar]
- 28. Cirulli E.T., Lasseigne B.N., Petrovski S., Sapp P.C., Dion P.A., Leblond C.S., Couthouis J., Lu Y.F., Wang Q., Krueger B.J.. et al. (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science, 347, 1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elden A.C., Kim H.J., Hart M.P., Chen-Plotkin A.S., Johnson B.S., Fang X., Armakola M., Geser F., Greene R., Lu M.M.. et al. (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature, 466, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scheetz T.E., Roos B.R., Solivan-Timpe F., Miller K., DeLuca A.P., Stone E.M., Kwon Y.H., Alward W.L., Wang K.. et al. (2016) SQSTM1 mutations and glaucoma. PLoS One, 11, e0156001.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Souzeau E., Tram K.H., Witney M., Ruddle J.B., Graham S.L., Healey P.R., Goldberg I., Mackey D.A., Hewitt A.W., Burdon K.P.. et al. (2017) Myocilin predictive genetic testing for primary open-angle glaucoma leads to early identification of at-risk individuals. Ophthalmology, 124, 303–309. [DOI] [PubMed] [Google Scholar]
- 32. Turalba A.V., Chen T.C. (2008) Clinical and genetic characteristics of primary juvenile-onset open-angle glaucoma (JOAG) Semin. Ophthalmol. 23, 19–25. [DOI] [PubMed] [Google Scholar]
- 33. Hewitt A.W., Craig J.E., Mackey D.A. (2006) Complex genetics of complex traits: the case of primary open-angle glaucoma. Clin. Exp. Ophthalmol., 34, 472–484. [DOI] [PubMed] [Google Scholar]
- 34. Nag A., Lu H., Arno M., Iglesias A.I., Bonnemaijer P., Broer L., Uitterlinden A.G., Klaver C.C., van Duijn C., Hysi P.G.. et al. (2017) Evaluation of the Myocilin Mutation Gln368Stop Demonstrates Reduced Penetrance for Glaucoma in European Populations. Ophthalmology, 124, 547–553. [DOI] [PubMed] [Google Scholar]
- 35. Wiggs J.L., Allingham R.R., Vollrath D., Jones K.H., De La Paz M., Kern J., Patterson K., Babb V.L., Del Bono E.A., Broomer B.W.. et al. (1998) Prevalence of mutations in TIGR/Myocilin in patients with adult and juvenile primary open-angle glaucoma. Am. J. Hum. Genet., 63, 1549–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Souzeau E., Burdon K.P., Dubowsky A., Grist S., Usher B., Fitzgerald J.T., Crawford A., Hewitt A.W., Goldberg I., Mills R.A.. et al. (2013) Higher prevalence of myocilin mutations in advanced glaucoma in comparison with less advanced disease in an Australasian disease registry. Ophthalmology, 120, 1135–1143. [DOI] [PubMed] [Google Scholar]
- 37. Fingert J.H., Héon E., Liebmann J.M., Yamamoto T., Craig J.E., Rait J., Kawase K., Hoh S.T., Buys Y.M., Dickinson J.. et al. (1999) Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum. Mol. Genet., 8, 899–905. [DOI] [PubMed] [Google Scholar]
- 38. Lam D.S., Leung Y.F., Chua J.K., Baum L., Fan D.S., Choy K.W., Pang C.P. (2000) Truncations in the TIGR gene in individuals with and without primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci., 41, 1386–1391. [PubMed] [Google Scholar]
- 39. Wiggs J.L., Vollrath D. (2001) Molecular and clinical evaluation of a patient hemizygous for TIGR/MYOC. Arch. Ophthalmol., 119, 1674–1678. [DOI] [PubMed] [Google Scholar]
- 40. Donegan R.K., Hill S.E., Freeman D.M., Nguyen E., Orwig S.D., Turnage K.C., Lieberman R.L. (2015) Structural basis for misfolding in myocilin-associated glaucoma. Hum. Mol. Genet., 24, 2111–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zode G.S., Kuehn M.H., Nishimura D.Y., Searby C.C., Mohan K., Grozdanic S.D., Bugge K., Anderson M.G., Clark A.F., Stone E.M.. et al. (2011) Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J. Clin. Invest., 121, 3542–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zode G.S., Bugge K.E., Mohan K., Grozdanic S.D., Peters J.C., Koehn D.R., Anderson M.G., Kardon R.H., Stone E.M., Sheffield V.C. (2012) Topical ocular sodium 4-phenylbutyrate rescues glaucoma in a myocilin mouse model of primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci., 53, 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tham Y.C., Li X., Wong T.Y., Quigley H.A., Aung T., Cheng C.Y. (2014) Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology, 121, 2081–2090. [DOI] [PubMed] [Google Scholar]
- 44. Anderson D.R., Drance S.M., Schulzer M.. Collaborative Normal-Tension Glaucoma Study Group. (2001) Natural history of normal-tension glaucoma. Ophthalmology, 108, 247–253. [DOI] [PubMed] [Google Scholar]
- 45. Loomis S.J., Kang J.H., Weinreb R.N., Yaspan B.L., Cooke Bailey J.N., Gaasterland D., Gaasterland T., Lee R.K., Lichter P.R., Budenz D.L.. et al. (2014) Association of CAV1/CAV2 genomic variants with primary open-angle glaucoma overall and by gender and pattern of visual field loss. Ophthalmology, 121, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu R., Tsuboi T., Okumura-Noji K., Iwamoto N., Yokoyama S. (2016) Caveolin-1 facilitates internalization and degradation of ABCA1 and probucol oxidative products interfere with this reaction to increase HDL biogenesis. Atherosclerosis, 253, 54–60. [DOI] [PubMed] [Google Scholar]
- 47. Talwar N., Musch D.C., Stein J.D. (2017) Association of Daily Dosage and Type of Statin Agent With Risk of Open-Angle Glaucoma. JAMA Ophthalmol., 135, 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davis C.H., Kim K.Y., Bushong E.A., Mills E.A., Boassa D., Shih T., Kinebuchi M., Phan S., Zhou Y., Bihlmeyer N.A.. et al. (2014) Transcellular degradation of axonal mitochondria. Proc. Natl Acad. Sci. U S A, 111, 9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen Y., Cai J., Jones D.P. (2006) Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett., 580, 6596–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin W.J., Kuang H.Y. (2014) Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy, 10, 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Khawaja A.P., Cooke Bailey J.N., Kang J.H., Allingham R.R., Hauser M.A., Brilliant M., Budenz D.L., Christen W.G., Fingert J., Gaasterland D.. et al. (2016) Assessing the association of mitochondrial genetic variation with primary open-angle glaucoma using gene-set analyses. Invest. Ophthalmol. Vis. Sci., 57, 5046–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Foster P.J.1., Buhrmann R., Quigley H.A., Johnson G.J. (2002) The definition and classification of glaucoma in prevalence surveys. Br. J. Ophthalmol., 86, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kong X., Chen Y., Chen X., Sun X. (2011) Influence of family history as a risk factor on primary angle closure and primary open angle glaucoma in a Chinese population. Ophthalmic Epidemiol., 18, 226–232. [DOI] [PubMed] [Google Scholar]
- 54. Amerasinghe N., Zhang J., Thalamuthu A., He M., Vithana E.N., Viswanathan A., Wong T.Y., Foster P.J., Aung T. (2011) The heritability and sibling risk of angle closure in Asians. Ophthalmology, 118, 480–485. [DOI] [PubMed] [Google Scholar]
- 55. Kavitha S., Zebardast N., Palaniswamy K., Wojciechowski R., Chan E.S., Friedman D.S., Venkatesh R., Ramulu P.Y. (2014) Family history is a strong risk factor for prevalent angle closure in a South Indian population. Ophthalmology, 121, 2091–2097. [DOI] [PubMed] [Google Scholar]
- 56. Ahram D.F., Grozdanic S.D., Kecova H., Henkes A., Collin R.W., Kuehn M.H. (2015) Variants in nebulin (NEB) are linked to the development of familial primary angle closure glaucoma in Basset Hounds. PLoS One, 10, e0126660.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sundin O.H., Leppert G.S., Silva E.D., Yang J.M., Dharmaraj S., Maumenee I.H., Santos L.C., Parsa C.F., Traboulsi E.I., Broman K.W.. et al. (2005) Extreme hyperopia is the result of null mutations in MFRP, which encodes a frizzled-related protein. Proc. Natl. Acad. Sci. U S A, 102, 9553–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Awadalla M.S., Burdon K.P., Souzeau E., Landers J., Hewitt A.W., Sharma S., Craig J.E. (2014) Mutation in TMEM98 in large white kindred with autosomal dominant nanophthalmos linked to 17p12-q12. JAMA Ophthalmol., 132, 970–977. [DOI] [PubMed] [Google Scholar]
- 59. Crowley C., Paterson R., Lamey T., McLaren T., De Roach J., Chelva E., Khan J. (2014) Autosomal recessive bestrophinopathy associated with angle closure glaucoma. Doc. Ophthalmol., 129, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cheng C.Y., Schache M., Ikram M.K., Young T.L., Guggenheim J.A., Vitart V., MacGregor S., Verhoeven V.J., Barathi V.A., Liao J.. et al. (2013) Nine loci for ocular axial length identified through genome-wide association studies, including shared loci with refractive error. Am. J. Hum. Genet., 93, 264–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Day A.C., Luben R., Khawaja A.P., Low S., Hayat S., Dalzell N., Wareham N.J., Khaw K.T., Foster P.J. (2013) Genotype-phenotype analysis of SNPs associated with primary angle closure glaucoma (rs1015213, rs3753841 and rs11024102) and ocular biometry in the EPIC-Norfolk Eye Study. Br. J. Ophthalmol., 97, 704–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nongpiur M.E., Khor C.C., Jia H., Cornes B.K., Chen L.J., Qiao C., Nair K.S., Cheng C.Y., Xu L., George R.. et al. (2014) ABCC5, a gene that influences the anterior chamber depth, is associated with primary angle closure glaucoma. PLoS Genet., 10, e1004089.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arora K.S., Jefferys J.L., Maul E.A., Quigley H.A. (2012) The choroid is thicker in angle closure than in open angle and control eyes. Invest. Ophthalmol. Vis. Sci., 53, 7813–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Streeten B.W., Li Z.Y., Wallace R.N., Eagle R.C. Jr, Keshgegian A.A. (1992) Pseudoexfoliative fibrillopathy in visceral organs of a patient with pseudoexfoliation syndrome. Arch. Ophthalmol., 110, 1757–1762. [DOI] [PubMed] [Google Scholar]
- 65. Pasquale L.R., Borrás T., Fingert J.H., Wiggs J.L., Ritch R. (2016) Exfoliation syndrome: assembling the puzzle pieces. Acta Ophthalmol., 94, e505–e512. [DOI] [PubMed] [Google Scholar]
- 66. Damji K.F., Bains H.S., Amjadi K., Dohadwala A.A., Valberg J.D., Chevrier R., Gould L.F., Zackon D.H., Addison D.J. (1999) Familial occurrence of pseudoexfoliation in Canada. Can. J. Ophthalmol., 34, 257–265. [PubMed] [Google Scholar]
- 67. Taylor H.R. (1979) Pseudoexfoliation, an environmental disease? Trans. Ophthalmol. Soc. UK, 99, 302–307. [PubMed] [Google Scholar]
- 68. Cousins C.C., Kang J.H., Bovee C., Wang J., Greenstein S.H., Turalba A., Shen L.Q., Brauner S., Boumenna T., Blum S.. et al. (2017) Nailfold capillary morphology in exfoliation syndrome. Eye (Lond), Jan 13 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Praveen M.R., Shah S.K., Vasavada A.R., Diwan R.P., Shah S.M., Zumkhawala B.R., Thomas R. (2011) Pseudoexfoliation as a risk factor for peripheral vascular disease: a case-control study. Eye, 25, 174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brooks A.M., Gillies W.E. (1987) The development of microneovascular changes in the iris in pseudoexfoliation of the lens capsule. Ophthalmology, 94, 1090–1097. [DOI] [PubMed] [Google Scholar]
- 71. Wirostko B.M., Curtin K., Ritch R., Thomas S., Allen-Brady K., Smith K.R., Hageman G.S., Allingham R.R. (2016) Risk for exfoliation syndrome in women with pelvic organ prolapse: A Utah project on exfoliation syndrome (UPEXS) study. JAMA Ophthalmol., 134, 1255–1262. [DOI] [PubMed] [Google Scholar]
- 72. Fan B.J., Pasquale L.R., Rhee D., Li T., Haines J.L., Wiggs J.L. (2011) LOXL1 promoter haplotypes are associated with exfoliation syndrome in a U.S. Caucasian population. Invest. Ophthalmol. Vis. Sci., 52, 2372–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Founti P., Haidich A.B., Chatzikyriakidou A., Salonikiou A., Anastasopoulos E., Pappas T., Lambropoulos A., Viswanathan A.C., Topouzis F. (2015) Ethnicity-based differences in the association of LOXL1 polymorphisms with pseudoexfoliation/pseudoexfoliative glaucoma: a meta-analysis. Ann. Hum. Genet., 79, 431–450. [DOI] [PubMed] [Google Scholar]
- 74. Hewitt A.W., Sharma S., Burdon K.P., Wang J.J., Baird P.N., Dimasi D.P., Mackey D.A., Mitchell P., Craig J.E. (2008) Ancestral LOXL1 variants are associated with pseudoexfoliation in Caucasian Australians but with markedly lower penetrance than in Nordic people. Hum. Mol. Genet., 17, 710–716. [DOI] [PubMed] [Google Scholar]
- 75. Kang J.H., Loomis S., Wiggs J.L., Stein J.D., Pasquale L.R. (2012) Demographic and geographical features of exfoliation glaucoma in two United States-based prospective cohorts. Ophthalmology, 119, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dewundara S., Pasquale L.R. (2015) Exfoliation syndrome: a disease with an environmental component. Curr. Opin. Ophthalmol., 26, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kang J.H., Wiggs J.L., Pasquale L.R. (2014) Relation between time spent outdoors and exfoliation glaucoma or exfoliation glaucoma suspect. Am. J. Ophthalmol., 158, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zenkel M., Krysta A., Pasutto F., Juenemann A., Kruse F.E., Schlötzer-Schrehardt U. (2011) Regulation of lysyl oxidase-like 1 (LOXL1) and elastin-related genes by pathogenic factors associated with pseudoexfoliation syndrome. Invest. Ophthalmol. Vis. Sci., 52, 8488–8495. [DOI] [PubMed] [Google Scholar]