

Abstract
Motivation
Network biology is widely used to elucidate mechanisms of disease and biological processes. The ability to interact with biological networks is important for hypothesis generation and to give researchers an intuitive understanding of the data. We present visJS2jupyter, a tool designed to embed interactive networks in Jupyter notebooks to streamline network analysis and to promote reproducible research.
Results
The tool provides functions for performing and visualizing useful network operations in biology, including network overlap, network propagation around a focal set of genes, and co-localization of two sets of seed genes. visJS2jupyter uses the JavaScript library vis.js to create interactive networks displayed within Jupyter notebook cells with features including drag, click, hover, and zoom. We demonstrate the functionality of visJS2jupyter applied to a biological question, by creating a network propagation visualization to prioritize risk-related genes in autism.
Availability and implementation
The visJS2jupyter package is distributed under the MIT License. The source code, documentation and installation instructions are freely available on GitHub at https://github.com/ucsd-ccbb/visJS2jupyter. The package can be downloaded at https://pypi.python.org/pypi/visJS2jupyter.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Networks are ubiquitous in biology, from protein–protein interactions to metabolic, neuronal and signaling networks (Barabasi et al., 2004, Kreeger and Lauffenburger, 2010). Interacting with networks in real time can help generate hypotheses, reveal underlying biological mechanisms, and provide an intuitive understanding of data that static forms cannot. Here, we present a light-weight tool, visJS2jupyter, which integrates the flexibility and aesthetic benefits of networks rendered in vis.js (visjs.org) directly into Jupyter notebook cells (Perez and Granger, 2007). In contrast to the static networks produced by existing Python network visualization tools, such as NetworkX (Schult and Swart, 2008), visJS2jupyter networks are interactive. The tool also provides options to export networks for further analysis and biological interpretation in Cytoscape (Shannon et al., 2003), or Cytoscape.js (Franz et al., 2015). We thus enable a seamless transition from the powerful and reproducible development and data analysis environment of Jupyter notebooks to interactive network analysis and visualization, within the same coding environment.
2 Main features
Drawing basic networks
Drawing basic but easily adaptable interactive networks in Jupyter notebook cells is a main functionality of visJS2jupyter. Users provide as input a network in NetworkX format, and a list of node-specific and edge-specific attributes to display, including label, position, color, size and shape. The NetworkX graph object flexibly stores nodes (represented by any Python object), and node attributes, along with edges and edge attributes which link the nodes. These attributes may be passed along to visJS2jupyter for visualization. Other optional arguments apply general styles to the graph, such as edge styles, highlight colors, and physics properties. We also provide functionality to map scalar NetworkX node or edge attributes to any Python colormap, along with options for scaling and transforming the attribute. See the supplemental information for a short programming example.
Network visualizations
The visualizations module builds on basic drawing functions in order to perform operations on graphs and to visualize their results. The module includes three functions. The first of these, ‘draw_graph_overlap’, displays nodes and edges which are shared between two networks, along with the nodes and edges unique to a single network. The second, ‘draw_heat_prop’, allows for investigation of the local network neighborhood of a set of seed genes by simulating a network propagation simulation (Vanunu et al., 2010) (alternatively referred to as heat propagation). Finally, ‘draw_colocalization’, enables examination of the shared network neighborhood between two sets of seed genes by running a network propagation simulation from each set of seed genes and displaying the product of the resulting heat vectors. Genes which are nearby to both sets of seed genes will have higher combined heat values. This method is similar to that proposed in (Paull et al., 2013). Resulting networks can also be exported to Cytoscape for further visualization and analysis.
Implementation
The open-source visJS2jupyter package is written in Python and is specifically designed to be used within a Python-kernel Jupyter notebook. It uses the vis.js (http://visjs.org/) browser-based visualization library written in JavaScript to provide interactive network graphics. vis.js was selected because it was more easily integrated into the Jupyter framework than other browser-based javascript libraries, such as cytoscape.js. Network and data manipulations are implemented using NetworkX, NumPy and Pandas libraries. Its functionality operates on graphs in the standard NetworkX format.
3 Example use case
Network propagation methods (e.g. Vanunu et al., 2010) allow for exploration of the genetic landscape of diseases by prioritizing likely disease-related genes and identifying possible drug-repurposing candidates (Novarino et al., 2014). We demonstrate the network propagation functionality of visJS2jupyter by prioritizing genes of interest in autism (see supplemental information for the Jupyter notebook to reproduce this analysis). A comparison of network propagation to other gene prioritization methods may be found in Börnigen et al. (2012) and Guala and Sonnhammer (2017).
The function draw_heat_prop simulates heat propagating from a set of seed nodes on a background interaction network. Beginning with the list of all known autism disease risk genes (859 genes) from the SFARI database (Banerjee-Basu and Packer, 2010), and using the STRING database (Szklarczyk et al., 2014) as a background interaction network converted to NetworkX format, we randomly selected 25 of these genes to use as seed nodes. We then ran draw_heat_prop (Fig. 1A) to identify potentially related genes and create a fully interactive network visualization of the autism sub-network. After propagation, we measured how many of the left-out set are recovered. On average, 23 (out of 751 autism risk genes contained in the STRING interactome) known disease risk genes are recovered in the top 200 highest ranking genes when the simulation is seeded with 25 known disease genes (Fig. 1B). To establish a baseline for comparison, we also ran a control condition in which we measured the number of disease risk genes which were recovered in N genes randomly selected from the interactome and compared to the number recovered in the N highest ranking network propagation genes. This experiment was repeated k = 100 times for each value of N (100 < N < 2000, Fig. 1B). Consistently, we recovered many more autism genes from network propagation than from the control condition (p = 1E-32, top 200 genes, rank-sum test), demonstrating that the network propagation method successfully prioritizes disease-related genes.
Fig. 1.
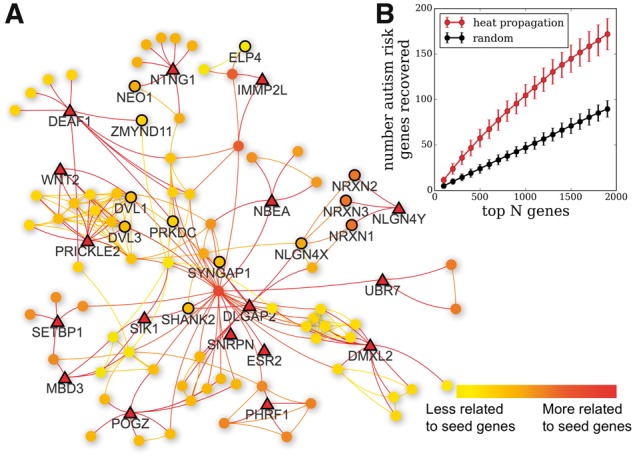
Network propagation visualization used to prioritize autism risk genes. (A) Autism sub-network, created by randomly selecting 25 known autism risk genes (triangles) as seeds for the heat propagation simulation. Here we display the largest connected component of the autism sub-network. Top 100 related genes are shown as circles, color-coded with decreasing propagated heat value (color online). Nodes with bold outlines are recovered autism risk genes. (B) Validation of heat propagation as method of gene prioritization. Top curve shows average number of autism risk genes in a set that are recovered in the top N genes based on propagation from 25 randomly selected genes in that set, while the bottom curve denotes the number of autism risk genes recovered in N randomly selected genes (Color version of this figure is available at Bioinformatics online.)
4 Conclusions
This tool brings the processes of building, interacting with and analyzing networks together in Jupyter notebooks, thus streamlining network analysis workflows. Together with flexible drawing of networks, the more advanced network functions enable interpretation of biological network data. Further examples of how the functions of visJS2jupyter may be used to produce interactive networks with layered and integrated biological attributes are provided in the visJS2jupyter GitHub repository.
Funding
This work was partially supported by the National Institute of Health [UL1TR001442] of CTSA.
Conflict of Interest: none declared.
Supplementary Material
References
- Banerjee-Basu S., Packer A. (2010) SFARI Gene: an evolving database for the autism research community. Dis. Models Mech., 3, 133–135. [DOI] [PubMed] [Google Scholar]
- Barabasi A. et al. (2004) Network biology: understanding the cell’s functional organization. Nat. Rev. Genet., 5, 101–113. [DOI] [PubMed] [Google Scholar]
- Börnigen D. et al. (2012) An unbiased evaluation of gene prioritization tools. Bioinformatics, 28, 3081–3088. [DOI] [PubMed] [Google Scholar]
- Franz M. et al. (2015) Cytoscape.js: a graph theory library for visualization and analysis. Bioinformatics, 32, 309–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guala D., Sonnhammer E. (2017) A large-scale benchmark of gene prioritization methods. Sci. Rep., 7, 46598.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreeger P., Lauffenburger D. (2010) Cancer systems biology: a network modeling perspective. Carcinogenesis, 31, 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarino G. et al. (2014) Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science, 343, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez F., Granger B. (2007) IPython: a system for interactive scientific computing. Comput. Sci. Eng., 9, 21–29. [Google Scholar]
- Paull E.O. et al. (2013) Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion Through Interacting Events (TieDIE). Bioinformatics, 29, 2757–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schult D., Swart P. (2008) Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conferences (SciPy 2008), pp. 11–16.
- Shannon P. et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res., 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D. et al. (2014) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res., 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanunu O. et al. (2010) Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol., 6, e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.