

Nelson et al. report that peritoneal macrophage respiration is enhanced by rosiglitazone, an activating PPARγ ligand, in a PPARγ-dependent manner. PPARγ functions as a checkpoint, guarding against inflammation, and is permissive for alternative activation by facilitating glutamine metabolism.
Keywords: macrophage, alternative activation, PPARγ, glutamine, rosiglitazone, metabolism
Abstract
The nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) is known to regulate lipid metabolism in many tissues, including macrophages. Here we report that peritoneal macrophage respiration is enhanced by rosiglitazone, an activating PPARγ ligand, in a PPARγ-dependent manner. Moreover, PPARγ is required for macrophage respiration even in the absence of exogenous ligand. Unexpectedly, the absence of PPARγ dramatically affects the oxidation of glutamine. Both glutamine and PPARγ have been implicated in alternative activation (AA) of macrophages, and PPARγ was required for interleukin 4 (IL4)-dependent gene expression and stimulation of macrophage respiration. Indeed, unstimulated macrophages lacking PPARγ contained elevated levels of the inflammation-associated metabolite itaconate and express a proinflammatory transcriptome that, remarkably, phenocopied that of macrophages depleted of glutamine. Thus, PPARγ functions as a checkpoint, guarding against inflammation, and is permissive for AA by facilitating glutamine metabolism. However, PPARγ expression is itself markedly increased by IL4. This suggests that PPARγ functions at the center of a feed-forward loop that is central to AA of macrophages.
The nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) is the master regulator of adipogenesis and fat cell function (Rosen et al. 1999; Rangwala and Lazar 2004) and the target of anti-diabetic thiazolidinedione drugs (Lehmann et al. 1995). Although its expression is predominant in adipocytes, PPARγ is also expressed in macrophages (Ricote et al. 1998; Chawla et al. 2001a; Lefterova et al. 2010). Indeed, mice lacking macrophage PPARγ are prone to atherosclerosis (Chawla et al. 2001b; Babaev et al. 2005), and PPARγ ligands have anti-atherosclerotic properties (Nagy et al. 1998; Tontonoz et al. 1998; Akiyama et al. 2002).
In macrophages, PPARγ has been shown to play important roles in inflammation and metabolism (Hevener et al. 2007; Odegaard et al. 2007). PPARγ is induced by interleukin 4 (IL4), a canonical inducer of the anti-inflammatory macrophage alternative activation (AA) pathway (Ricote et al. 1998; Bouhlel et al. 2007), and PPARγ ligands have anti-inflammatory properties (Huang et al. 1999; Li et al. 2000a; Stienstra et al. 2008). Studies of its metabolic role have focused largely on lipid metabolism, and PPARγ has been shown to control lipid uptake (Chawla et al. 2001a; Odegaard et al. 2007), extrusion (Chawla et al. 2001b; Chinetti et al. 2001; Akiyama et al. 2002), and intracellular metabolism (Odegaard et al. 2007). These properties have been linked to the role of PPARγ in inflammation and prevention of metabolic diseases (Chawla 2010).
Here, studying the regulation of macrophage metabolism by the thiazolidinedione rosiglitazone (Rosi), we report an unexpected role for PPARγ in glutamine metabolism. Glutamine is required for macrophage AA (Jha et al. 2015; Davies et al. 2017; Liu et al. 2017), and there is a remarkable overlap between the effects of glutamine depletion and the loss of PPARγ, providing a molecular and metabolic basis for the requirement of PPARγ for IL4-stimulated AA. IL4 also induces PPARγ, creating a feed-forward loop controlling AA, with PPARγ as the nexus.
Results
Rosi activates a PPARγ-dependent gene program in macrophages
We generated homozygous macrophage-specific PPARγ knockout (MPKO) mice by crossing LysM-Cre mice to C57BL/6J PPARγ floxed mice. Quantification of mRNA isolated from thioglycollate-induced peritoneal macrophages showed almost complete depletion of PPARγ mRNA (Fig. 1A), and Western blotting confirmed loss of macrophage PPARγ protein (Fig. 1B). These cells were used to assess the role of PPARγ in the action of Rosi, a potent insulin sensitizer in the thiazolidinedione class that is activating ligands for PPARγ (Soccio et al. 2014) but has been suggested to have other cellular actions (Divakaruni et al. 2013). Upon treatment with 1 µM Rosi for 24 h, Rosi modulated the expression of hundreds of genes in control macrophages, yet, importantly, the MPKO macrophages were globally unresponsive to Rosi (Fig. 1C), demonstrating that PPARγ is responsible for the vast majority of Rosi effects on gene transcription.
Figure 1.
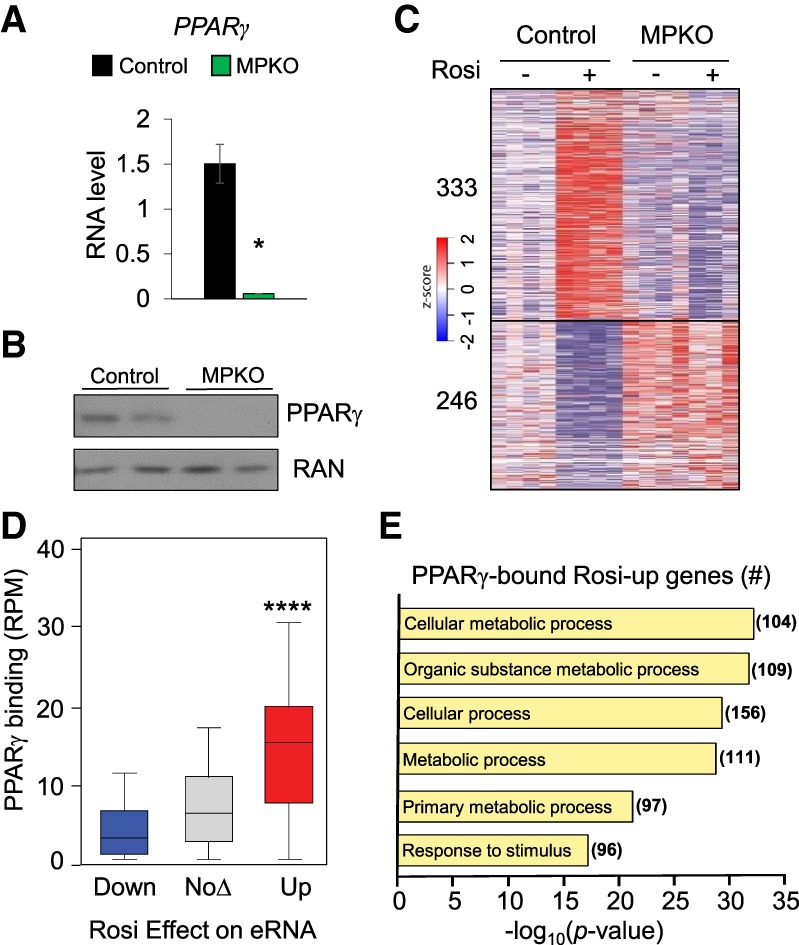
PPARγ binds at the genome to control macrophage enhancer RNA (eRNA) and gene transcription. (A) Confirmation of PPARγ knockout assessed with quantitative PCR (qPCR) analysis of PPARγ mRNA in control and MPKO macrophages. Data are shown as mean ± standard error. n = 6. (*) P < 0.05. (B) Western blot showing expression of PPARγ in control and MPKO macrophages with RAN as a loading control. (C) Heat map showing z-transformed RPKM (reads per kilobase per million mapped reads) values of all statistically significant (P < 0.05) Rosi-responsive genes in control macrophages treated with either vehicle or 1 µM Rosi and corresponding expression in MPKO macrophages. Data are shown as biological replicates. n = 3–4. (D) Total PPARγ ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) tag count in reads per million (RPM) within 2 kb of up-regulated, down-regulated, and unregulated eRNA sites identified by GRO-seq (global run-on sequencing) in response to Rosi treatment. (****) P < 0.0001. Differentially expressed eRNAs were defined with edgeR as having |fold change| > 1.5 and false discovery rate (FDR) < 0.05 in Rosi-treated macrophages as compared with the untreated control. (E) PANTHER gene ontology analysis of the Rosi-induced genes identified in red in D with significantly increased PPARγ occupancy at Rosi-induced eRNAs.
Comparison of sites of PPARγ binding from macrophage-specific ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) (Soccio et al. 2017) with Rosi-activated enhancers elucidated by macrophage-specific GRO-seq (global run-on sequencing) revealed markedly increased PPARγ binding at genes induced by Rosi (Fig. 1D). Enhancers down-regulated by Rosi had even less average PPARγ binding than unaffected genes (Fig. 1D), consistent with a coactivator redistribution mechanism for negative regulation that has also been observed in adipocytes and other systems (Step et al. 2014; Schmidt et al. 2016). Gene ontology analysis revealed that Rosi-induced genes with high PPARγ occupancy at Rosi-induced enhancer RNAs (eRNAs) exhibited a strong link to metabolism and metabolic processes (Fig. 1E).
PPARγ knockout macrophages have reduced respiration
We next investigated whether Rosi's activation of metabolic pathways altered macrophage respiration. Indeed, Rosi increased both basal and maximal oxygen consumption rates (OCRs) of control macrophages, and, consistent with the dependence of the Rosi-stimulated transcriptomes on PPARγ, this effect was abrogated in MPKO macrophages (Fig. 2A,B). Moreover, the baseline and maximal OCRs of MPKO macrophages were markedly attenuated relative to control even in the absence of exogenous PPARγ ligand (Fig. 2A,B). The striking effect of PPARγ depletion on macrophage respiration was independent of cell viability (Supplemental Fig. S1A) and mitochondrial density or number (Supplemental Fig. S1B).
Figure 2.
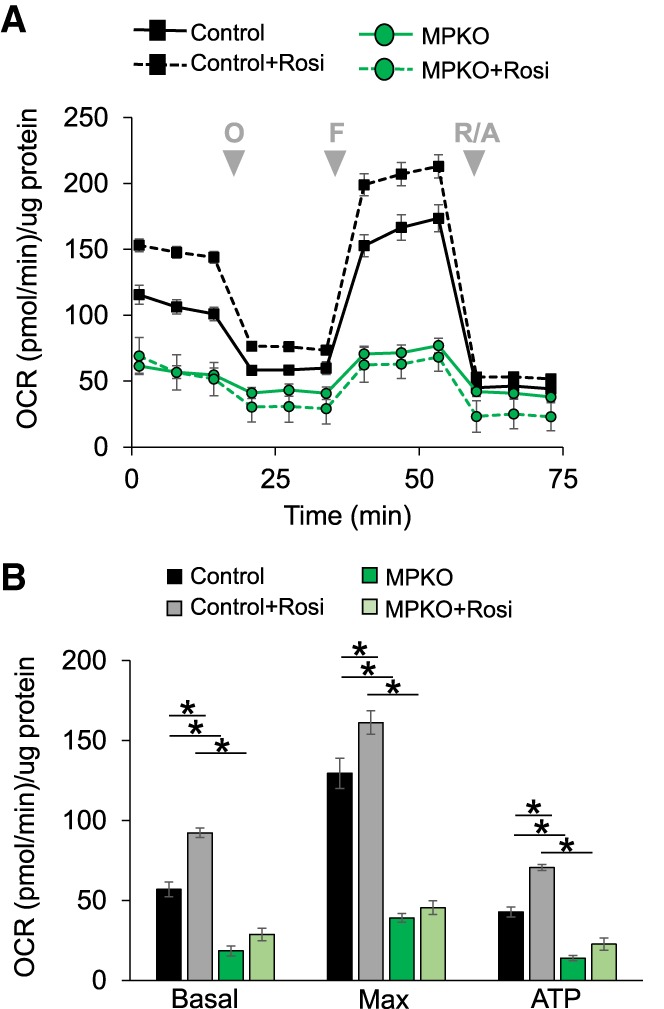
Loss of PPARγ reduces macrophage respiration and blocks the effects of Rosi. (A) Macrophage respiration as measured by OCR in DMEM containing 10 mM glucose and 2 mM glutamine in control and MPKO macrophages after 24 h of stimulation with 1 µM Rosi or DMSO vehicle control. During measurements, macrophages were treated with the complex V inhibitor oligomycin (O), the uncoupler fluoro-carbonyl cyanide phenylhyrazone (FCCP) (F), and the complex I and III inhibitors rotenone/antimycin A (R/A) at the times indicated (gray arrows). (B) Basal and maximal respiration and oxygen consumption from ATP production calculated from OCRs in control and MPKO macrophages. Calculations were corrected for nonmitochondrial respiration. Data are representative of at least three independent experiments. Data points are represented as mean ± SEM from n = 6–12 technical replicates. (*) P < 0.05.
PPARγ is not required for respiration from fatty acid or glucose
We next explored whether PPARγ was required for the use of specific energy sources for macrophage respiration. We first examined fatty acid oxidation, which has been reported to be defective in macrophages lacking PPARγ (Odegaard et al. 2007). Addition of palmitate to control macrophages in standard medium increased their respiration (Fig. 3A). Surprisingly, this was also the case for MPKO macrophages (Fig. 3A), whose defective basal and maximal OCRs were both largely rescued by palmitate (Fig. 3B).
Figure 3.
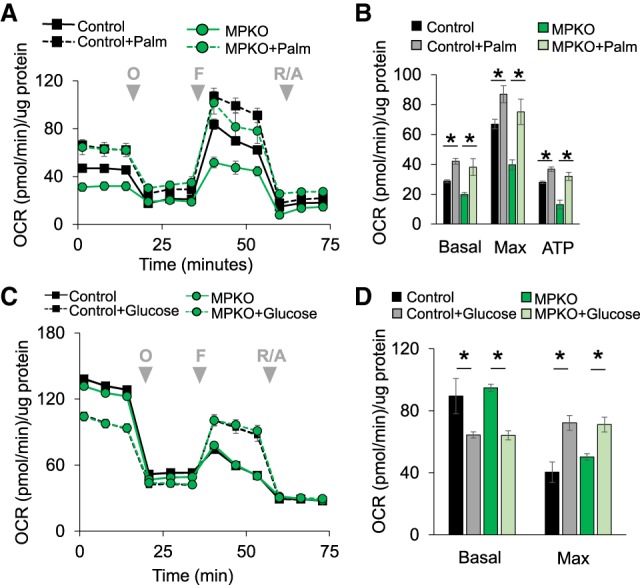
MPKO macrophages can use palmitate and glucose as fuel for respiration. (A) Macrophage respiration in control and MPKO macrophages measured in DMEM containing 10 mM glucose and 2 mM glutamine in the presence of either 10 nM free fatty acid as palmitate bound to BSA or an equal volume of BSA control added to the cells immediately before the assay and treated with oligomycin (O), FCCP (F), and rotenone/antimycin A (R/A) at the times indicated (gray arrowheads). (B) Quantification of basal and maximal respiration and OCR contributed by ATP production. (C) OCR in control and MPKO macrophages in nutrient-depleted medium with and without glucose. (D) Quantification of basal and maximum respiration from C. Data are representative of at least three independent experiments. Data points are represented as mean ± SEM from n = 6–12 technical replicates. (*) P < 0.05.
We next considered whether the MPKO macrophages would be defective in glucose utilization. However, glucose uptake was not impaired relative to control macrophages (Supplemental Fig. S2A). Moreover, control and MPKO macrophages respired similarly well with glucose as the only fuel added to nutrient-free medium; in both cases, glucose initially suppressed respiration fueled by internal catabolic processes (potentially by activating glycolytic ATP production and thus decreasing reliance on respiratory ATP) but enhanced respiration after addition of oligomycin and uncoupler (Fig. 3C,D). The extracellular acidification rate (ECAR) was also similar between control and MPKO macrophages using glucose as substrate, further indicating that PPARγ was not required for glycolysis or glucose utilization (Supplemental Fig. S2B).
PPARγ is required for respiration from glutamine
In addition to fatty acid oxidation and glycolysis, oxidative phosphorylation can be fueled by glutamine, the most abundant circulating amino acid in mammals and a major TCA substrate via its catabolism to α-ketoglutarate (αKG) (Newsholme et al. 1986; Fan et al. 2013; Curi et al. 2017). We found that, unlike control macrophages, MPKO macrophages were unable to effectively respire using glutamine as fuel (Fig. 4A). Likewise, the MPKO macrophages were deficient in respiration from dimethyl-αKG (DM-αKG), a cell-permeable analog of αKG (Fig. 4B). Since basal respiration varied from experiment to experiment, we performed a normalized comparison of substrate effects on MPKO OCR relative to control macrophages, which makes clear the extent to which MPKO respiration is fueled by the fatty acid and glucose substrates but not by glutamine or αKG (Fig. 4C). Note that these experiments used fluoro-carbonyl cyanide phenylhyrazone (FCCP) at 2 µM, which was sufficient to maximally induce respiration of control macrophages with either glucose or glutamine as the only fuel provided (Supplemental Fig. S3). Moreover, when respiration was attenuated by inhibition of fatty acid oxidation and glucose metabolism using etomoxir and UK5099, respectively, 10 mM glutamine largely restored oxidative capacity in control macrophages but had little effect on MPKO macrophages (Supplemental Fig. S4A,B), consistent with the conclusion that they are defective in glutamine oxidation.
Figure 4.
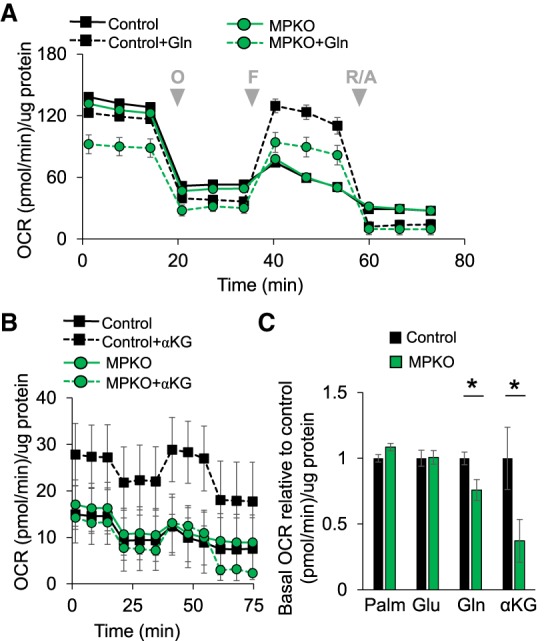
MPKO macrophages have impaired glutamine metabolism. (A) OCR in control and MPKO macrophages in nutrient-depleted medium with and without 2 mM glutamine as a sole fuel substrate. (B) OCR in control and MPKO macrophages in nutrient-depleted medium with only DM-αKG as a fuel substrate. (C) Basal OCR in the presence of only either 10 nM palmitate (Palm), 10 mM glucose (Glu), 2 mM glutamine (Gln), or 2 mM DM-αKG, shown as a percentage of basal respiration of nutrient-treated control OCR. Data are representative of at least three independent experiments. Data points are represented as mean ± SEM from n = 6–12 technical replicates. (*) P < 0.05.
PPARγ is required for IL4 function in macrophages
Our observation that PPARγ controls macrophage respiration from glutamine was intriguing because (1) glutamine is an important fuel source required for AA of macrophages (Jha et al. 2015; Liu et al. 2017); (2) PPARγ has been shown to be induced by IL4 (Huang et al. 1999), a classical inducer of AA; and (3) PPARγ has been reported to be required for AA (Bouhlel et al. 2007; Odegaard et al. 2007). We confirmed that PPARγ was induced by IL4 in control macrophages (Fig. 5A). Indeed, IL4 regulated hundreds of macrophage genes, including those involved in the AA response (such as Irf4, Ym1, and Ccl17) and genes critical to cell metabolism and the TCA cycle (such as Idh1 and Cs) (Fig. 5B). Remarkably, most of the IL4 effect was abrogated in the absence of PPARγ (Fig. 5B). Thus, although IL4 induces PPARγ expression, PPARγ is required for IL4 action.
Figure 5.
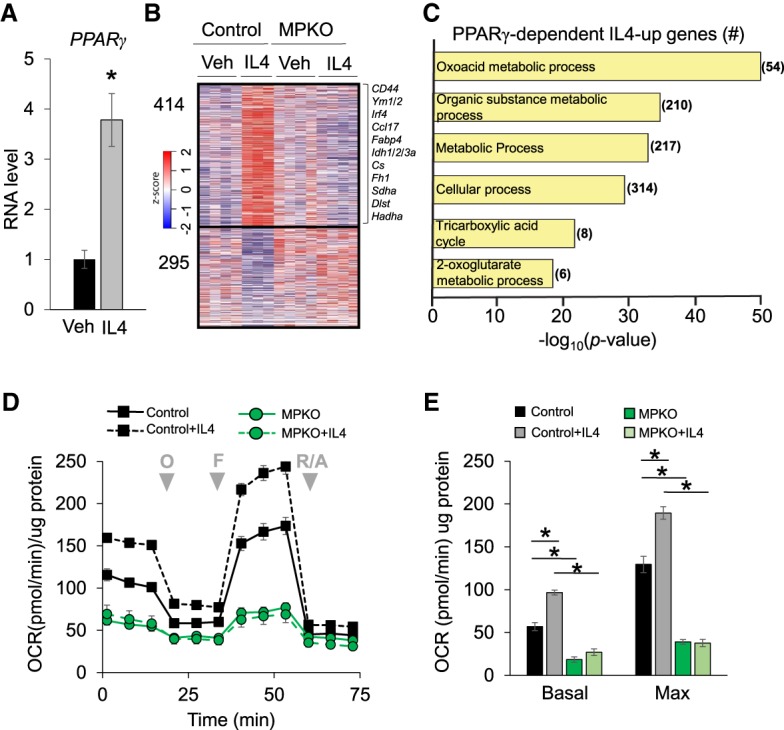
PPARγ directs an IL4-mediated metabolic gene program. (A) Expression of PPARγ mRNA measured by qPCR in control macrophages treated with IL4 for 24 h. n = 5 vehicle; n = 6 IL4-treated. (*) P < 0.05. (B) Heat map showing z-transformed RPKM values of all statistically significant (P < 0.05) IL4-responsive genes in control macrophages and corresponding expression in MPKO macrophages. Selected IL4-induced genes involved in immunity and metabolism are listed at the right. Data are shown as biological replicates. n = 3–4. (C) PANTHER gene ontology analysis of genes increased with IL4 treatment in control macrophages but not in MPKO macrophages. (D) OCR in control and MPKO macrophages treated with and without 20 ng/mL IL4 for 24 h prior to assay. (E) Quantification of basal and maximal respiration in IL4-treated macrophages. Data are representative of at least three independent experiments. OCR data points are represented as mean ± SEM from n = 6–12 technical replicates. (*) P < 0.05.
Transcripts induced by IL4 in control but not MPKO macrophages were strongly enriched for metabolism-related gene pathways, including those implicated in the TCA cycle and αKG metabolism (Fig. 5C). αKG is produced from glutamine and is critical for metabolism of AA macrophages (Palmieri et al. 2017). Indeed, IL4 stimulated the respiration of control macrophages but not MPKO macrophages (Fig. 5D), with PPARγ deletion impairing both basal and maximal respiration (Fig. 5E).
Unstimulated MPKO macrophages have an inflammatory character that phenocopies the effect of glutamine depletion
We next performed metabolic profiling on unstimulated control and MPKO macrophages. Itaconate stood out as the molecule with the largest and most statistically significant increase in the MPKO macrophages (Fig. 6A,B). Itaconate is a metabolite with anti-microbial and immunomodulatory properties generated from the TCA cycle intermediate aconitate by the enzyme IRG1 (Michelucci et al. 2013; Jha et al. 2015). Indeed, Irg1 expression was increased in MPKO macrophages (Fig. 6C). Moreover, expression of Idh1, which directs the conversion of isocitrate to αKG in the TCA cycle, was decreased in MPKOs (Fig. 6D). The increase in Irg1 and decrease in Idh1 in MPKO macrophages likely leads to catapleurotic removal of TCA intermediates. In addition, the accumulation of itaconate may also impair oxidative metabolism by inhibiting succinate dehydrogenase (Cordes et al. 2016).
Figure 6.
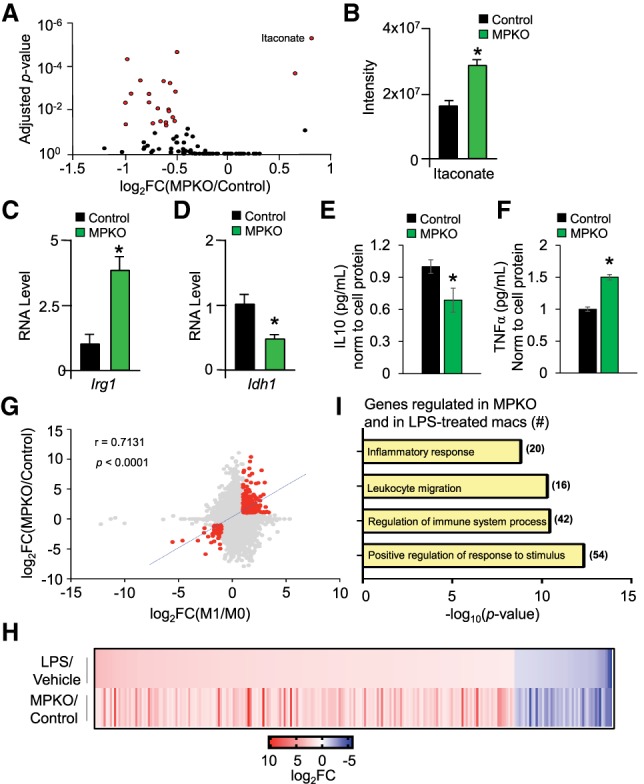
Unstimulated MPKO macrophages have an inflammatory phenotype. (A) Macrophage metabolites measured in cell lysate from control and MPKO macrophages cultured in DMEM supplemented with 10 mM glucose and 2 mM glutamine. Statistically significant metabolites are expressed in red and were determined using an unpaired t-test corrected for multiple comparisons using the Holm-Sidek method. (B) Quantitation of itaconate in cell lysate from control and MPKO macrophages cultured in Figure 3A. (C,D) Relative gene expression from RNA sequencing (RNA-seq) of Irg1 (C) and Idh1 (D) in control and MPKO macrophages as measured by normalized RPKM. n = 3–4 biological replicates each condition. (*) P < 0.05, Mann-Whitney t-test. (E) IL10 secreted into the supernatant of control and MPKO as measured by ELISA normalized to cell protein and shown relative to control. n = 6 each condition. (*) P <0.05. (F) TNFα secreted into the supernatant of control and MPKO macrophages normalized to cell protein and shown relative to control. n = 6 each condition. (*) P < 0.05. (G) Correlation scatter plots of gene expression (displayed in log2 fold change) between proinflammatory M1 lipopolysaccharide (LPS)-treated macrophages (compared with untreated M0 control; X-axis) and MPKOs (compared with wild-type control; Y-axis). A specific group of genes that met an arbitrary cutoff of Pearson's r > 0.5, |log2 fold change| > 1, and FDR < 0.05 were selected (highlighted in red with corresponding correlation values and best-fit line) for further functional investigation. (H) Heat map plotted as a function of log2 fold change values of genes identified in G to demonstrate the degree of similarity in gene expression between MPKO and LPS-treated macrophages. (I) PANTHER gene ontology analysis of genes identified in G showing significant enrichment for inflammatory and external stimulus response pathways.
Itaconate accumulation occurs during M1-type inflammatory activation of macrophages induced by the classic inflammatory stimulus lipopolysaccharide (LPS) (Cordes et al. 2016). Consistent with this, MPKO macrophages secreted less anti-inflammatory IL10 (Fig. 6E) and more proinflammatory TNFα (Fig. 6F) relative to controls, indicating that, in the basal state, MPKO macrophages have an inflammatory phenotype. Indeed, there was significant overlap between a subset of gene changes induced by loss of PPARγ in unstimulated macrophages and those caused by LPS-treated wild-type macrophages (Fig. 6G,H). The overlapping genes and pathways suggest that PPARγ serves as a metabolic checkpoint in unstimulated macrophages that inhibits the inflammatory phenotype and favors AA (Fig. 6I).
Gene set enrichment analysis (GSEA) independently revealed that PPARγ-dependent IL4-responsive genes were strongly enriched for TCA cycle and respiratory electron transport pathways (Fig. 7A), with the greatest effect on key TCA enzyme Idh1 (Fig. 7B). Indeed, there was a strong positive correlation globally between the up-regulation of gene expression by Rosi and IL4, including the TCA cycle genes (Fig. 7C). Since the respiratory defect in MPKO macrophages was due largely to the inability to oxidize glutamine and also because the MPKO macrophages are biased away from AA and toward inflammation, we compared the transcriptomes of IL4-stimulated glutamine-depleted macrophages (Jha et al. 2015), IL4-stimulated MPKO macrophages, and Rosi-stimulated MPKO macrophages to assess the potential of PPARγ in regulating AA-mediated glutamine metabolism. Remarkably, the effect of PPARγ knockout on the IL4 response was highly similar to that of glutamine depletion (Fig. 7D). Moreover, a similar transcriptional landscape, which includes the AA markers Irf4 and Ym1, was found in Rosi-treated MPKO macrophages, suggesting that PPARγ regulates a shared group of mediators required for not only IL4 but also Rosi response (Fig. 7D). These data suggest that the function of PPARγ to support glutamine metabolism is central to its role in macrophage AA.
Figure 7.
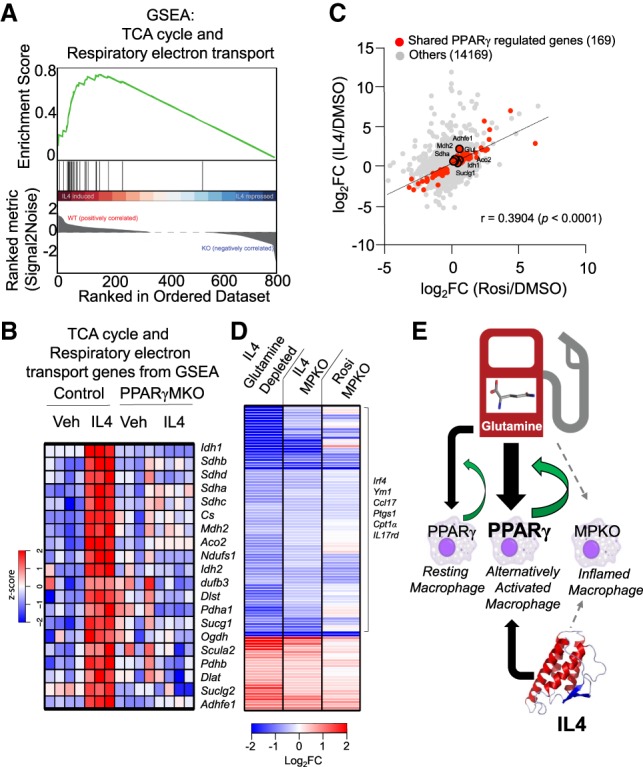
AA requires PPARγ for glutamine metabolism. (A) GSEA showing overrepresentation of TCA cycle and respiratory electron transport genes in IL4-treated controls and underrepresentation in MPKOs. (B) Heat map of z-transformed RPKM values of genes from GSEA in A in vehicle and IL4-treated control and MPKO macrophages ordered as a function of GSEA ranked metric score (high to low signal to noise ratio). (C) Scatter plot showing significant positive global correlation between Rosi-induced genes and IL4-induced genes in control macrophages. Genes in red are not induced with either Rosi or IL4 in MPKOs. (D) Heat map showing gene expression log2 fold changes of IL4-treated control macrophages in glutamine-depleted medium (vs. IL4-treated control macrophages in complete medium), IL4-treated MPKOs (vs. IL4-treated control macrophages), and Rosi-treated MPKOs (vs. Rosi-treated control macrophages). (E) PPARγ is required for glutamine-mediated AA, yet its expression is also induced by IL4. This places PPARγ at the center of a feed-forward loop for macrophage AA.
Discussion
We showed here that PPARγ controls macrophage glutamine metabolism, providing a link between transcription, AA, and metabolism. PPARγ is both required for IL4 stimulation of AA and amplified by IL4 signaling and thus serves as a nexus in a feed-forward pathway to AA (Fig. 7E).
The strong metabolic phenotype of the MPKO macrophages suggests that PPARγ is active in the absence of exogenous ligand either constitutively or due to endogenous ligands. Nonetheless, PPARγ-dependent gene regulation and metabolic activity are increased by Rosi treatment, indicating that the basal state is not fully active and amenable to pharmacological manipulation. These activities of PPARγ and Rosi likely play a major role in the anti-diabetic and anti-atherosclerotic activities of PPARγ per se (Huang et al. 1999; Li et al. 2000b; Akiyama et al. 2002; Babaev et al. 2005; Hevener et al. 2007) as well as the benefits of Rosi treatment (Chawla et al. 2001a,b; Odegaard et al. 2007; Stienstra et al. 2008).
Recently, it has been suggested that some of the effects of Rosi are mediated by cellular targets other than PPARγ (Nawrocki et al. 2006; Colca et al. 2013; Divakaruni et al. 2013). Although PPARγ has a critical role in adipocyte biology, it has not been possible to demonstrate that PPARγ is the target of Rosi in adipocytes because deletion of PPARγ causes the adipocytes to dedifferentiate. In contrast, macrophages are ideal because PPARγ is not required for maintenance of their differentiated state. The studies presented here leave no doubt that PPARγ is by far the main cellular target underlying the transcriptional and physiological effects of Rosi in macrophages. Our finding that PPARγ binds at Rosi-regulated enhancers near genes that are induced by the PPARγ ligand strongly suggests that PPARγ directly regulates the genes that it activates.
The overlapping transcriptomes of LPS-treated macrophages and unstimulated MPKO macrophages support a model in which PPARγ acts as an anti-inflammatory checkpoint. Macrophage itaconate has been identified as a LPS-responsive metabolite that is capable of initiating metabolic remodeling and limiting AA cytokine production (Strelko et al. 2011; Lampropoulou et al. 2016; Mills et al. 2018). This creates a break in the TCA cycle associated with the inflammatory phenotype and reduced oxidative respiration mirrored in the MPKO macrophages (Jha et al. 2015). The elevation of itaconate in unstimulated MPKO macrophages thus provides further evidence of a dual role for PPARγ as both anti-inflammatory and permissive for AA.
Our work confirms previous reports that IL4 increases macrophage respiration (Vats et al. 2006). Those studies of Balb/c macrophages chronically (96 h) treated with IL4 showed a role of PPARγ in fatty acid oxidation to promote oxidative phosphorylation in AA (Vats et al. 2006; Odegaard et al. 2007), while other studies suggest that fatty acid oxidation is not required for AA of C57Bl/6 macrophages (Nomura et al. 2016; Gonzalez-Hurtado et al. 2017). More recent studies demonstrate that glutamine metabolism is required for AA (Jha et al. 2015), likely via metabolism to αKG (Liu et al. 2017; Palmieri et al. 2017). In this regard, although much of the literature on PPARγ in macrophage metabolism has focused on lipids (Castrillo and Tontonoz 2004), the importance of PPARγ in controlling glutamine metabolism fits with its known importance in AA (Huang et al. 1999; Odegaard et al. 2007).
Macrophages use glutamine at high rates in part to provide energy for oxidative phosphorylation (Newsholme et al. 1986; Jha et al. 2015; Davies et al. 2017). Thus, the reduced expression of TCA enzymes in macrophages lacking PPARγ likely contributes to impaired glutamine metabolism. In particular, IDH1 was shown recently to strongly influence the activation state of the macrophage (Jha et al. 2015; O'Neill 2015). Decreased TCA cycle function in unstimulated MPKO macrophages directly opposes the metabolic reprogramming required for AA. The requirement of PPARγ for glutamine metabolism and AA as well as the normal increase in PPARγ upon AA stimulation place PPARγ at the center of a feed-forward loop required for IL4 to induce AA.
Materials and methods
Animals
PPARγf/f and LysMCre C57BL/6J mice were purchased from Jackson Laboratories. Mice were housed in a temperature-controlled specific pathogen-free facility under 12-h light/dark cycles. Adult male and female mice between the ages of 8 and 16 wk old were used in all experiments. Animal care and use procedures followed the guidelines of the Institutional Animal Care and Use Committee of the University of Pennsylvania in accordance with the guidelines of the National Institutes of Health.
Cell culture
Thioglycollate-elicited macrophages were harvested from the peritoneal cavity of mice. Four days after peritoneal injection with 3 mL of autoclaved 3% thioglycollate (Sigma-Aldrich), peritoneal macrophages were harvested from controls and MPKOs by lavage using Delbucco's phosphate-buffered saline (PBS) (Invitrogen) and cultured overnight in DMEM supplemented with 10% fetal bovine serum (Tissue Culture Biologics), 100 U/mL penicillin, and 100 µg/mL streptomycin (Invitrogen). When indicated, cultured macrophages were treated with DMSO vehicle, 1 µM Rosi (Cayman Chemical), or 20 ng/mL IL-4 (Peprotech) for 24 h. For metabolomics assays, control and MPKO macrophages were pooled from four mice and cultured in DMEM supplemented with 10% dialyzed fetal bovine serum (Tissue Culture Biologics), 100 U/mL penicillin, and 100 µg/mL streptomycin (Invitrogen).
Gene expression analysis
RNA was isolated from cells using TRIzol (Invitrogen) and purified with the Rneasy minikit (Qiagen). RT–PCR was performed using 500 ng–1 µg of RNA (Applied Biosystems) following the manufacturer's instructions. Quantitative PCR (qPCR) was performed on cultured macrophages using the primers PPARG (F: AGAAGCGGTGAACCACTGATATTC; R: AGAGGTCCACAGAGCTGATTCC) and 18s (F: AGTCCCTGCCCTTTGTACACA; R: CGATCCGAGGGCCTCACTA) with SYBR Green master mix (Applied Biosystems) on the QuantStudio 6 Flex real-time PCR system (Applied Biosytems). qPCR analysis was performed using the standard curve method, and all genes were normalized to the housekeeping gene 18s. For RNA sequencing (RNA-seq), RNA integrity was examined using an Agilent 2100 Bioanalyzer. RNA samples with RNA integrity number >7 were used for quantification with KAPA library quantification kit (Roche). RNA cleanup and library preparation were performed with the TRU-seq stranded total RNA library preparation kit according to the protocol provided by the manufacturer's instructions (Illumina).
RNA-seq data processing
High-throughput sequencing data were either generated internally through the Functional Genomics Core at the University of Pennsylvania or downloaded from published Gene Expression Omnibus data sets cited in the text. All publicly obtained raw data, including the IL4-treated glutamine-depleted and LPS-stimulated macrophages (Jha et al. 2015), were subjected to the same analysis pipeline to minimize processing variations. Sequencing reads were aligned to the University of California at Santa Cruz (UCSC) mm9 genome using TopHat (version 2.1.9) (Kim et al. 2013) with the following parameters: –g 1 --library-type fr-firststrand. Read counts were then obtained with featureCounts per the standard instructions (Liao et al. 2014). Differentially expressed genes (cutoff defined as fold change > 1.5, false discovery rate [FDR] < 0.05 > 0.5 RPKM [reads per kilobase per million mapped reads]) were identified using edgeR with design matrices normalized for variations in size sequencing, depth, and dispersion (Robinson et al. 2010). All gene ontology analyses were performed using PANTHER gene ontology enrichment analysis (Mi et al. 2013). Heat maps were generated in R with the “gplots” package heatmap 2 function (https://CRAN.R-project.org/package= gplots) by (1) identifying differentially expressed genes with the aforementioned cutoff and (2) mapping their corresponding z-transformed RPKM or log2 fold change values across all biological replicates (n = 3–4) and treatment conditions. The heat map color scheme was scaled accordingly for optimal graphical illustrations. Hierarchical clustering was applied unless specified otherwise.
Liquid chromatography-mass spectrometry (LC-MS) analysis
Extracts were prepared as described in Lu et al. (2017). In brief, cultured thioglycollate-elicited macrophages pooled from four control or four MPKO mice and further divided into four technical replicates were incubated on ice in a 40:40:20 acetonitrile:methanol:H20 + 0.5% formic acid solvent solution for 5 min before the cell lysates were collected and neutralized with a 15% NH4HCO3 in water solution for 15 min at −20°C. The lysates were spun at maximum speed for 15 min at 4°C, and the supernatant was frozen on dry ice and stored at −80°C before analysis. Cell extracts were analyzed using a quadrupole orbitrap mass spectrometer (Q Exactive, Thermo Fisher Scientific) coupled to hydrophilic interaction chromatography via electrospray ionization. LC separation was on a XBridge BEH amide column (2.1 mm × 150 mm, 2.5-µm particle size; Waters) using a gradient of solvent A (20 mM ammonium acetate, 20 mM ammonium hydroxide in 95:5 water:acetonitrile at pH 9.45) and solvent B (acetonitrile). Flow rate was 150 µL/min, column temperature was 25°C, autosampler temperature was 5°C, and injection volume was 10 µL. The LC gradient was 0 min and 90% B, 2 min and 85% B, 3 min and 75% B, 7 min and 75% B, 8 min and 70% B, 9 min and 70% B, 10 min and 50% B, 12 min and 50% B, 13 min and 25% B, 14 min and 25% B, 16 min and 0% B, 21 min and 0% B, 22 min and 90% B, and 25 min and 90% B. Autosampler temperature was 5°C, and injection volume was 3 µL. The mass spectrometer was operated in negative ion mode to scan from 70 to 1000 m/z at 1 Hz and a resolving power of 140,000 (Jang et al. 2018). Data were analyzed using MAVEN software (Melamud et al. 2010).
ELISAs
Cytokine secretion into the supernatant of thioglycollate-elicited cultured macrophages was measured with mouse Quantikine ELISA kits (TNFα [MTA00B] and IL10 [M1000B]) according to the manufacturer's instructions. Cytokine quantities in the supernatant were normalized to cell protein measured with BCA.
Immunoblotting
Primary antibodies for PPARγ (Santa Cruz Biotechnology) and RAN (BD Biosciences) were detected by a secondary horseradish peroxidase-conjugated antibody (Sigma) and an enhanced chemiluminescent substrate kit (PerkinElmer Western Lightning). Immunoblots were imaged with Odyssey Clx infrared imaging system (Li-Cor) and uniformly contrasted.
ChIP-seq
ChIP was performed as described previously (Steger et al. 2008) using 200 µg/0.1 mL PPARγ (anti-PPARγ [H100]; Santa Cruz Biotechnology, sc7196x, lot K1010) antibody. ChIP DNA was prepared for sequencing according to the amplification protocol provided by Illumina. Next-generation sequencing of ChIP-seq libraries was performed by the Functional Genomics Core at the University of Pennsylvania.
ChIP-seq data processing
Sequencing reads of PPARγ immunoprecipitated DNA of biological replicates for wild-type and PPARγ knockout macrophages (as input) were aligned to the UCSC mm9 genome using Bowtie1 (Langmead et al. 2009). Mapped reads were filtered for duplicates, biological replicates were pooled for read counts, and downstream peak calling and processing were done with HOMER version 4.6 per the instructions (Heinz et al. 2010). Peaks are defined as reads per million (RPM) > 1, fold change immunoprecipitation/input > 4, and FDR < 0.001.
GRO-seq
GRO-seq was performed using thioglycollate-elicited peritoneal macrophages pooled from 20 mice and treated with DMSO or 1 µM Rosi 24 h prior to the harvest. Cells were washed twice with ice-cold PBS and then swelled in cold swelling buffer (10 mM Tris at pH 7.5, 2 mM MgCl2, 3 mM CaCl2) for 5 min on ice. Cells were scraped off and centrifuged at 400g for 10 min. The nuclei were then extracted using lysis buffer (swelling buffer with 10% glycerol and 1% Igepal). After washing twice with lysis buffer, the nuclei were resuspended in freezing buffer (50 mM Tris at pH 8.3, 40% glycerol, 5 mM MgCl2, 0.1 mM EDTA). For each library, 1.5 × 107 nuclei were used. The nuclear run-on reaction was incubated in run-on buffer (10 mM Tris at pH 8.0; 5 mM MgCl2; 1 mM DTT; 300 mM KCl; 200 U/mL Superase-In; 1% sarkosyl; 500 mM ATP, GTP, and Br-UTP; 2 mM CTP) for 7 min at 30°C. Nascent transcripts were enriched with anti-BrUTP antibodies. After 10 min of RNA hydrolysis and reverse transcription, all products were used for GRO-seq library preparations in parallel to reduce batch effects as reported previously (Step et al. 2014).
GRO-seq data processing and eRNA analysis
FASTA file of trimmed (adapter and poly-A sequences) reads were generated prior to mapping. Bowtie1 was used for alignment to the UCSC mm9 genome with the following parameters: -n 1 -m 1. Downstream processing was done with HOMER version 4.6. Gene body transcription was calculated from processed .BED files by calculating tags in the gene body within a window of 2–12 kb from the transcription start site to capture acute changes without confounding the effects of paused signal noise at promoters. Tags aligned to noncoding RNAs, ribosomal RNA, small nucleolar RNA, small nuclear RNA, or transfer RNA were discarded to minimize false signal due to the overt abundance of these elements. Differential expression of Rosi-modulated genes was done with edgeR per the manufacturer's instructions (FDR < 0.05; RPKM > 0.5) eRNA analysis was done as described previously (Step et al. 2014). Briefly, HOMER's findPeaks function was used to identify putative transcript on both positive and negative strands. Start sites within 1 kb of each other are defined as bidirectional. eRNA tags were counted similarly to gene body transcription except for the application of a 2-kb window around known enhancers to account for both positive and negative tags.
Metabolic assays
Cells were uniformly plated in XF96 plates overnight before 24 h of stimulation with DMSO-vehicle, 1 µM Rosi, or 20 ng/mL IL4. OCR and ECAR were measured with the Seahorse XF96 extracellular flux analyzer (Agilent). Experiments were conducted in XF medium (nonbuffered DMEM containing 10 mM glucose [Invitrogen], 2 mM L-glutamine [Invitrogen], 2 mM sodium pyruvate [Invitrogen]), with OCR measured basally and in response to sequential addition of 2 µM oligomycin, 2 µM FCCP, and 0.5 µM rotenone + antimycin A. Fatty acid oxidation was measured in medium supplemented with a final concentration of 10 nM free palmitate bound to BSA or an equal volume of BSA control (Agilent). Glycolysis was measured as ECAR at baseline and after the addition of 0.2 µM oligomycin to impair respiration. Fuel preference was measured after 1 h of incubation in Seahorse XF base medium (Agilent) with no fuel substrates and then immediately before the assay supplemented with either 10 mM glucose, 4 mM glutamine, 4 mM DM-αKG (Sigma-Aldrich), or 10 nM free fatty acids in the form of palmitate BSA with unbound BSA as a control. All Seahorse XF data were normalized to total well protein quantified with Pierce BCA protein kit (Thermo Fisher). All calculations for respiration were corrected for nonmitochondrial respiration. Glucose uptake was measured with 1 µCi/mL 2-deoxy-D-[3H] glucose (Sigma-Aldrich) in cultured macrophages following 2 h of incubation in serum-free medium. 3H glucose uptake was detected with a PerkinElmer Tri-Carb 4910 TR scintillation counter.
Flow cytometry
Thioglycollate-elicited peritoneal macrophages from control or PPARγ knockout mice were FC-blocked with CD16/32 (2.4G2, 1:50; BD Bioscience) and rat IgG (1:50; Fisher Scientific) prior to staining with anti-mouse fluorochrome-conjugated monoclonal antibodies (clone and concentration shown; BD Bioscience, BioLegend, eBioscience) specific for CD11b (M1/70; 1:500) and F4/80 (BM8; 1:250). Cells were also stained with 100 nM MitoTracker red or 750 nM MitoTracker deep red (Thermo Fisher). Compensation was performed with cells or OneComp eBeads beads (Thermo Fisher). Cells were analyzed with a BD LSR II running DiVa software (BD Bioscience) and analyzed with FlowJo software (version 10.4, Tree Star).
Accession numbers
All sequencing data, including RNA-seq, ChIP-seq, and GRO-seq, were deposited to the Gene Expression Omnibus database under the superseries GSE111105, which includes RNA-seq data (GSE111102), ChIP-seq data (GSE111104), and GRO-seq data (GSE111103).
Supplementary Material
Acknowledgments
We thank Josh Rabinowitz (Princeton University) for helpful discussions. We thank the Penn Diabetes Research Center (National Institutes of Health DK19525) Metabolic Phenotypic Core (Will Quinn) for help with the glucose uptake assay, the Islet Cell Biology Core for access to the Seahorse bioanalyzer, the Functional Genomics Core for next-generation sequencing, and the Penn/Princeton Metabolomics Core for targeted metabolomics. This work was supported by National Institutes of Health grant DK49780 (to M.A.L.) and the Cox Research Institute. V.L.N. was supported by a post-doctoral fellowship from the American Heart Association (AHA 16POST29680002).
Author contributions: V.L.N. and H.C.B.N. were involved in project planning, experimental work, data analysis, and manuscript preparation. J.C.G.-C. performed experimental work and data analysis. E.R.B. was involved in project planning. W.Y.H. was involved in experimental work. J.R.D. was involved in project planning and experimental work. J.M.M. was involved in experimental work. D.A.H. was involved in project planning and experimental work. M.A.L was involved in project planning, data analysis, and manuscript preparation.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.312355.118.
References
- Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee Y-H, Ricote M, Glass CK, Brewer HB, et al. 2002. Conditional disruption of the peroxisome proliferator-activated receptor γ gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol 22: 2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF. 2005. Conditional knockout of macrophage PPARγ increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 25: 1647–1653. [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. 2007. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab 6: 137–143. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Tontonoz P. 2004. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu Rev Cell Dev Biol 20: 455–480. [DOI] [PubMed] [Google Scholar]
- Chawla A. 2010. Control of macrophage activation and function by PPARs. Circ Res 106: 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. 2001a. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 7: 48–52. [DOI] [PubMed] [Google Scholar]
- Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, et al. 2001b. A PPARγ–LXR–ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell 7: 161–171. [DOI] [PubMed] [Google Scholar]
- Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, Teissier E, Minnich A, Jaye M, Duverger N, et al. 2001. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med 7: 53–58. [DOI] [PubMed] [Google Scholar]
- Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell-Conrad AS, Wolfe CL, Wheeler JS, Coulter KR, Kilkuskie PM, et al. 2013. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)—relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One 8: e61551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, Koseki H, Cabrales P, Murphy AN, Hiller K, et al. 2016. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem 291: 14274–14284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curi R, de Siqueira Mendes R, de Campos Crispin LA, Norata GD, Sampaio SC, Newsholme P. 2017. A past and present overview of macrophage metabolism and functional outcomes. Clin Sci 131: 1329–1342. [DOI] [PubMed] [Google Scholar]
- Davies LC, Rice CM, Palmieri EM, Taylor PR, Kuhns DB, McVicar DW. 2017. Peritoneal tissue-resident macrophages are metabolically poised to engage microbes using tissue-niche fuels. Nat Commun 8: 2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, et al. 2013. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci 110: 5422–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD. 2013. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol 9: 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hurtado E, Lee J, Choi J, Selen Alpergin ES, Collins SL, Horton MR, Wolfgang MJ. 2017. Loss of macrophage fatty acid oxidation does not potentiate systemic metabolic dysfunction. Am J Physiol Endocrinol Metab 312, E381–E393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener AL, Olefsky JM, Reichart D, Nguyen MTA, Bandyopadyhay G, Leung H-Y, Watt MJ, Benner C, Febbraio MA, Nguyen A-K, et al. 2007. Macrophage PPARγ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest 117: 1658–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. 1999. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400: 378–382. [DOI] [PubMed] [Google Scholar]
- Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, Rabinowitz JD. 2018. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab 27: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SC-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. 2015. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42: 419–430. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC-C, Griss T, et al. 2016. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 24: 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterova MI, Steger DJ, Zhuo D, Qatanani M, Mullican SE, Tuteja G, Manduchi E, Grant GR, Lazar MA. 2010. Cell-specific determinants of peroxisome proliferator-activated receptor γ function in adipocytes and macrophages. Mol Cell Biol 30: 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. 1995. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J Biol Chem 270: 12953–12956. [DOI] [PubMed] [Google Scholar]
- Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. 2000a. Peroxisome proliferator-activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest 106: 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Pascual G, Glass CK. 2000b. Peroxisome proliferator-activated receptor γ-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol 20: 4699–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Liu P-S, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng W-C, Chou C-H, Vavakova M, et al. 2017. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 18: 985–994. [DOI] [PubMed] [Google Scholar]
- Lu W, Su X, Klein MS, Lewis IA, Fiehn O, Rabinowitz JD. 2017. Metabolite measurement: pitfalls to avoid and practices to follow. Annu Rev Biochem 86: 277–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD. 2010. Metabolomic analysis and visualization engine for LC-MS data. Anal Chem 82: 9818–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. 2013. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 8: 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, et al. 2013. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci 110: 7820–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. 2018. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Tontonoz P, Alvarez JGA, Chen H, Evans RM. 1998. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell 93: 229–240. [DOI] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, et al. 2006. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor γ agonists. J Biol Chem 281: 2654–2660. [DOI] [PubMed] [Google Scholar]
- Newsholme P, Curi R, Gordon S, Newsholme EA. 1986. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J 239: 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T. 2016. Fatty acid oxidation in macrophage polarization. Nat Immunol 17: 216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, et al. 2007. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LAJ. 2015. A broken krebs cycle in macrophages. Immunity 42: 393–394. [DOI] [PubMed] [Google Scholar]
- Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, Hooper DC, Lamers WH, Ghesquière B, McVicar DW, et al. 2017. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep 20: 1654–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala SM, Lazar MA. 2004. Peroxisome proliferator-activated receptor γ in diabetes and metabolism. Trends Pharmacol Sci 25: 331–336. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. 1998. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 391: 79–82. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. 1999. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 4: 611–617. [DOI] [PubMed] [Google Scholar]
- Schmidt SF, Larsen BD, Loft A, Mandrup S. 2016. Cofactor squelching: artifact or fact? BioEssays 38: 618–626. [DOI] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Lazar MA. 2014. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 20: 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Li Z, Chen ER, Foong YH, Benson KK, Dispirito JR, Mullican SE, Emmett MJ, Briggs ER, Peed LC, et al. 2017. Targeting PPARγ in the epigenome rescues genetic metabolic defects in mice. J Clin Invest 127: 1451–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim J-E, Chen J, Lazar MA, et al. 2008. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol 28: 2825–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Step SE, Lim H-W, Marinis JM, Prokesch A, Steger DJ, You S-H, Won K-J, Lazar MA. 2014. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARγ-driven enhancers. Genes Dev 28: 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienstra R, Duval C, Keshtkar S, van der Laak J, Kersten S, Müller M. 2008. Peroxisome proliferator-activated receptor γ activation promotes infiltration of alternatively activated macrophages into adipose tissue. J Biol Chem 283: 22620–22627. [DOI] [PubMed] [Google Scholar]
- Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, Roberts MF. 2011. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc 133: 16386–16389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. 1998. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93: 241–252. [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. 2006. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab 4: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.