

In this study, Chen et al. provide novel insight into Wilms tumor by investigating the mechanisms by which mutations in the microRNA biogenesis machinery drive tumorigenesis. They show that the transcription factor PLAG1 is a microRNA target gene that is overexpressed in Wilms tumors with mutations in microRNA processing genes and that PLAG1 transactivates IGF2, a key Wilms tumor oncogene, and drives mTORC1 signaling.
Keywords: IGF2, PLAG1, Wilms tumor, kidney, microRNA processing, pediatric cancer
Abstract
Many childhood Wilms tumors are driven by mutations in the microRNA biogenesis machinery, but the mechanism by which these mutations drive tumorigenesis is unknown. Here we show that the transcription factor pleomorphic adenoma gene 1 (PLAG1) is a microRNA target gene that is overexpressed in Wilms tumors with mutations in microRNA processing genes. Wilms tumors can also overexpress PLAG1 through copy number alterations, and PLAG1 expression correlates with prognosis in Wilms tumors. PLAG1 overexpression accelerates growth of Wilms tumor cells in vitro and induces neoplastic growth in the developing mouse kidney in vivo. In both settings, PLAG1 transactivates insulin-like growth factor 2 (IGF2), a key Wilms tumor oncogene, and drives mammalian target of rapamycin complex 1 (mTORC1) signaling. These data link microRNA impairment to the PLAG1–IGF2 pathway, providing new insight into the manner in which common Wilms tumor mutations drive disease pathogenesis.
Wilms tumor, the most common pediatric kidney cancer, is treated with surgery, radiation, and chemotherapy (Rivera and Haber 2005). This combination is effective for many patients, but a cure remains difficult for those with metastatic or anaplastic disease (Dome et al. 2006; Chu et al. 2010). The combination of vincristine and dactinomycin, first used in the 1960s, remains the primary treatment backbone used today (James et al. 1966). There are no therapeutically targetable mutations in Wilms tumor. Recent Wilms tumor sequencing studies have classified driver mutations into two main categories. Mutations affecting nephron differentiation (including WT1, CTNNB1, AMER1, MLLT1, SIX1, and SIX2) account for ∼40% of Wilms tumors, while mutations affecting microRNA (miRNA) production (DROSHA, DGCR8, DICER1, and XPO5) account for another ∼20% (Rakheja et al. 2014; Torrezan et al. 2014; Walz et al. 2015; Wegert et al. 2015; Gadd et al. 2017). With the lack of rational, targeted therapies, current trials are focused on refining risk stratification based on high-risk copy number changes such as loss of heterozygosity (LOH) of chromosome arms 1p and 16q (Dome et al. 2015).
Insulin-like growth factor 2 (IGF2) overexpression contributes to many cancers, and the IGF2 locus on chromosome 11p15 is the most commonly altered genetic location in Wilms tumor. Loss of imprinting (LOI) or LOH at 11p15 occurs in >70% of tumors (Ogawa et al. 1993; Moulton et al. 1994; Steenman et al. 1994). Under normal imprinting, IGF2 is expressed only from the paternal 11p15 allele, but LOI leads to biallelic expression. LOI at 11p15 is hypothesized to be an early event in tumorigenesis and is commonly seen in nephrogenic rests, the precursor lesion for Wilms tumor (Ogawa et al. 1993; Charles et al. 1998; Rump et al. 2005). However, IGF2 levels in Wilms tumor are much higher than in neighboring adjacent kidney tissue, which can also have 11p15 LOI (Wang et al. 1996). Thus, transformation may require cooperating mutations that further increase IGF2 transcription, but no cooperating mutations have been shown to augment IGF2 expression in Wilms tumor (Maiti et al. 2000; Ruteshouser et al. 2008; Wegert et al. 2015).
We and others recently described a unique molecular subset of Wilms tumors driven by recurrent mutations at “hot spot” residues in genes of the miRNA processing pathway (Rakheja et al. 2014; Walz et al. 2015; Wegert et al. 2015; Gadd et al. 2017). IGF2 LOI is significantly enriched in tumors with these mutations (Walz et al. 2015). miRNAs arise through two successive cleavage events. Primary transcripts form a stem–loop structure and are cleaved first by DROSHA and its cofactor, DGCR8, in the nucleus to release a pre-miRNA hairpin, which is transported to the cytoplasm by XPO5. In the cytoplasm, a second cleavage by DICER1 generates a miRNA duplex. One strand of this duplex, the mature miRNA, directs an Argonaute protein to target transcripts for degradation and translational repression.
We showed previously that oncogenic mutations in DROSHA and DICER1 have different functional effects. Specifically, DICER1 hot spot mutations specifically impair processing of miRNAs derived from the 5′ arms of pre-miRNA hairpins (referred to here as 5p miRNAs), while DROSHA mutations result in globally reduced miRNA processing (Rakheja et al. 2014). Thus, loss of 5p miRNAs is a common downstream effect of both types of mutations, while loss of 3p miRNAs occurs only in DROSHA mutant tumors. For this reason, we hypothesized that 5p miRNAs are the key tumor suppressors, while loss of 3p miRNAs can be tolerated but does not drive tumorigenesis; that is, we hypothesized that derepression of 5p miRNA gene targets drives tumor formation. LIN28B is one key target of the 5p miRNA let-7, and, indeed, overexpression of LIN28B in the developing mouse kidney produces Wilms-like tumors (Urbach et al. 2014). However, the contribution of other 5p miRNAs and the derepression of their target genes has not been investigated in Wilms tumor.
Here we show that pleomorphic adenoma gene 1 (PLAG1) is one the of most overexpressed genes in Wilms tumors with miRNA-impairing mutations. PLAG1 is a developmental transcription factor that normally drives IGF2 expression. We demonstrate that PLAG1 can be repressed by miR-16 and miR-34a, two 5p miRNAs severely impacted by Wilms tumor miRNA processing mutations. Ectopic PLAG1 expression in Wilms tumor cells accelerates proliferation in vitro, and ectopic PLAG1 expression in the developing mouse kidney produces neoplastic lesions. Last, specific copy number changes can also lead to increased PLAG1 expression, and Wilms tumors with high PLAG1 expression are clinically more aggressive. These data provide new insights into the pathogenesis of common Wilms tumor mutations.
Results
Wilms tumors with miRNA processing mutations overexpress PLAG1
To identify genes overexpressed in tumors with miRNA processing mutations, we examined publicly accessible microarray data from 75 relapsed favorable histology Wilms tumors. These data were collected and published by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative (Walz et al. 2015). Of these 75 tumors, 14 tumors had mutations in miRNA processing genes. When we compared gene expression profiles of tumors with and without mutations in miRNA processing genes, we found 31 genes that were overexpressed in these tumors (Fig. 1A).
Figure 1.
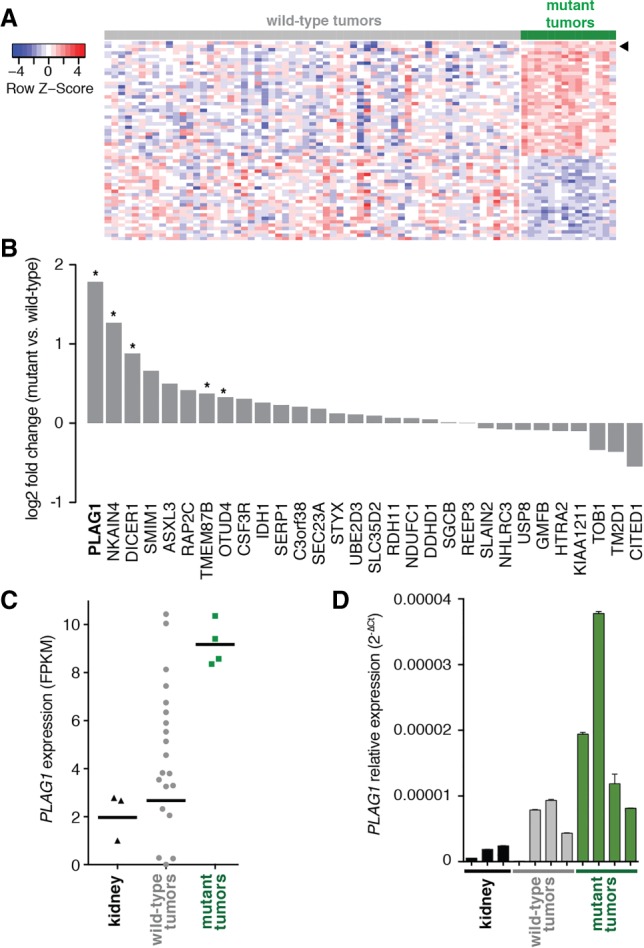
Wilms tumors with miRNA processing mutations express high levels of PLAG1. (A) Heat map of genes differentially expressed between Wilms tumors with and without mutations in the miRNA processing pathway (“wild-type” and “mutant,” respectively) in TARGET microarray data (false discovery rate <0.1). The arrowhead marks PLAG1. (B) Log2 fold change of expression for 31 genes overexpressed in the microarray in mutant tumors versus wild-type tumors, as measured by whole-transcriptome sequencing. (*) P < 0.05. (C) PLAG1 levels in individual samples, as measured by whole-transcriptome sequencing (FPKM [fragments per kilobase per million mapped reads]). Horizontal bars mark the geometric mean. (D) PLAG1 levels in individual samples, as measured by quantitative RT–PCR (qRT–PCR). Values shown are mean ± SD from three technical replicates.
We next performed RNA sequencing (RNA-seq) on an independent set of 24 tumors, which we analyzed previously by exome sequencing (Rakheja et al. 2014). Four of these 24 tumors had mutations in miRNA processing genes: DROSHA (two), DICER1 (one), or DGCR8 (one). Of the 31 genes that we identified as overexpressed in the TARGET microarray, five were significantly overexpressed in our independent RNA-seq (Fig. 1B). The most overexpressed gene was the transcription factor PLAG1. Expression of PLAG1 was uniformly high in tumors with miRNA processing mutations but varied more widely in tumors without these mutations (Fig. 1C,D). Adjacent normal kidney tissue expressed low levels of PLAG1.
PLAG1 was originally described as a frequently translocated gene in pleomorphic adenoma of the salivary glands (Kas et al. 1997). In the most common rearrangement, the promoters of PLAG1 and CTNNB1 are switched, resulting in PLAG1 overexpression. PLAG1 is oncogenic in NIH3T3 cells, and mice that overexpress PLAG1 in their salivary glands develop adenomas mimicking the human disease (Hensen et al. 2002; Declercq et al. 2005). PLAG1 is a zinc finger transcription factor expressed early in embryonic development, and inherited variants in PLAG1 determine organism size (Hensen et al. 2004; Karim et al. 2011; Rubin et al. 2012). In the contexts of both cancer and development, its most well-characterized target gene is IGF2 (Voz et al. 2000, 2004; Declercq et al. 2008b). Because IGF2 is highly expressed in Wilms tumors, we hypothesized that PLAG1 overexpression augments IGF2 expression in Wilms tumor.
Kidney-specific PLAG1 overexpression drives neoplasia
To explore the impact of PLAG1 overexpression on kidney development in vivo, we obtained mice carrying a latent lox-stop-lox-PLAG1 transgene (Declercq et al. 2005). These mice develop pleomorphic adenomas when bred to Cre driver lines active in the salivary gland. We recombined this transgene using Wt1Cre, which is expressed throughout the metanephric tissue as early as embryonic day 10.5 (E10.5) (Wilm and Munoz-Chapuli 2016). These Wt1Cre;LSL-PLAG1 mice develop severely enlarged kidneys by 3 wk of age and do not survive to weaning (Fig. 2A,B). As expected, the Igf2 transcript is elevated in PLAG1-overexpressing kidneys throughout development (Fig. 2C; Supplemental Fig. S1A).
Figure 2.

Kidney-specific PLAG1 overexpression drives neoplasia. (A) Gross dissection of a Wt1Cre;LSL-PLAG1 mouse and a wild-type littermate at 24 d of age. (B) Survival curve of Wt1Cre;LSL-PLAG1 mice versus Wt1Cre-only and LSL-PLAG1-only littermates. P < 0.05, log-rank test. (C) Relative expression of Igf2 taken from whole kidneys in Wt1Cre;LSL-PLAG1 mice versus LSL-PLAG1-only at postnatal day 8 (P8). Values are presented as mean ± SD from four technical replicates. (*) P < 0.05 by two-sided t-test comparing mean expression levels in each group of kidneys. (D) Kidneys from Wt1Cre;LSL-PLAG1, wild-type littermate, and Six2Cre;LSL-PLAG1 mice at P8. Bar, 500 µm. (E) High-power view of a Wt1Cre;LSL-PLAG1 kidney at P8. (SC) Simple cyst; (AC) atypical cyst; (D) dysplastic nodule; (g) glomerulus.
We next examined these kidneys histologically and found that they exhibit preneoplastic features. At E15.5, PLAG1-overexpressing kidneys are indistinguishable from their wild-type littermates (data not shown). However, by postnatal day 1 (P1), these mice possess fewer glomeruli than their wild-type littermates (Supplemental Fig. S1B,C). In addition, cysts have begun to form at the corticomedullary junction. By the end of the first week of life, these cysts have expanded to efface the entire renal parenchyma (Fig. 2D). As they develop, PLAG1-overexpressing kidneys progressively acquire neoplastic characteristics. The cysts are initially lined by a simple monolayer of cuboidal epithelium (Fig. 2E). Over a subsequent 2-wk period, the kidney parenchyma becomes completely replaced by expanding cysts with developing neoplastic characteristics with progressive cytologic and architectural atypia/dysplasia. The cyst epithelia develop micropapillae and multilayering, with more than one layer of cytologically atypical cells with enlarged nuclei, anisonucleosis, nuclear prominence, and occasional cytoplasmic clearing or vacuolation (atypical cysts). Similar atypical cells are noted in solid dysplastic nodules.
Wilms tumors are thought to arise from nephron progenitors; Wt1Cre marks both the nephron progenitor and stromal progenitor populations of the developing kidney. To further delineate the cell type responsible for these phenotypes, we next activated the PLAG1 transgene in specific compartments of the developing kidney. We found that PLAG1 activation using Six2Cre, which is specific to nephron progenitors, produced a phenotype identical to Wt1Cre (Fig. 2D). On the other hand, recombination with Foxd1Cre, which is specific to stromal precursors, produced normal kidneys (Supplemental Fig. S1D). These findings imply that the defects seen in Wt1Cre;LSL-PLAG1 mice arise from the nephron progenitor pool. This is in contrast to LIN28-driven kidney tumors, which arose under Wt1Cre but not Six2Cre, potentially implicating a separate cell of origin (Urbach et al. 2014).
miR-16 and miR-34 repress PLAG1
Because PLAG1 was expressed at high levels in tumors with mutations in miRNA processing genes, we next examined its expression in the absence of miRNAs. For this experiment, we knocked out DICER1 in WiT49 cells. This cell line was derived from an anaplastic Wilms tumor metastasis and bears no mutations in DROSHA, DGCR8, XPO5, or DICER1 (Alami et al. 2003). Specifically, we transiently expressed Cas9 with a guide RNA targeting the second coding exon of DICER1 (Supplemental Fig. S2A). This generated three independent clones with biallelic frameshift mutations. We confirmed loss of DICER1 protein in these clones by Western blot and confirmed miRNA depletion by quantitative RT–PCR (qRT–PCR) (Supplemental Fig. S2B,C). When we examined PLAG1 expression in these clones, we found that all three of these clones overexpress PLAG1 and IGF2 compared with parental cells (Supplemental Fig. S2D).
Having shown that global miRNA loss leads to an increase in PLAG1 expression, we next asked which individual miRNAs could be responsible for regulating PLAG1. We used the TargetScan algorithm, which predicts potential miRNA–mRNA interactions using two complementary computations: context+ score and probability of conserved targeting (PCT) (Lewis et al. 2003; Agarwal et al. 2015). The context+ score predicts miRNA–target interaction based on sequence characteristics alone, while PCT predicts biological significance based on evolutionary conservation. When the same miRNA family can target multiple sites of a 3′ untranslated region (UTR), the values at each of these sites are combined to produce a “total context+ score” and an “aggregate PCT” for that miRNA. Based on these computations, the top two miRNA families predicted to target the PLAG1 3′ UTR are the miR-16 and miR-34 families (Fig. 3A). These two miRNAs are 5p-derived and function as tumor suppressors in other contexts (Calin et al. 2002; Chang et al. 2007; He et al. 2007). Additionally, their expression is impaired in Wilms tumors with miRNA processing mutations (Fig. 3A, right). The PLAG1 sequence contains one miR-34a-binding site and two miR-16-binding sites, and, at all three sites, the entire 8-mer seed sequence is conserved from humans to Xenopus (Fig. 3B).
Figure 3.
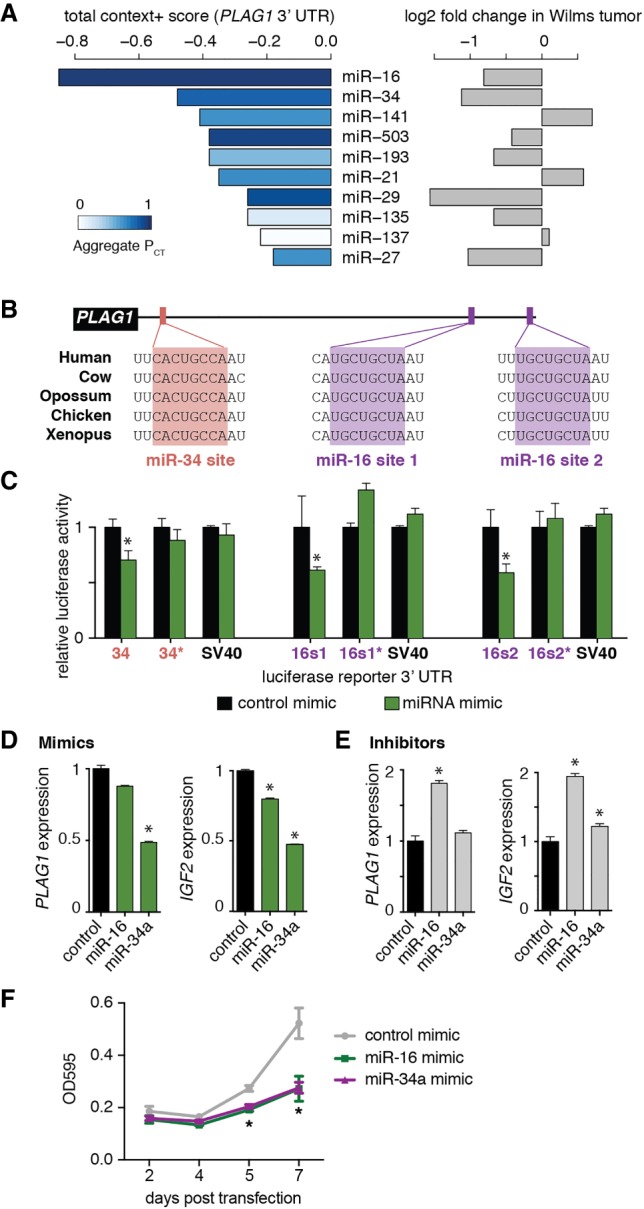
miR-16 and miR-34 repress PLAG1. (A) TargetScan (version 6.2) predictions of miRNA families targeting PLAG1 transcript, ranked by total context+ score (more negative values indicate stronger predicted interaction). Bars are colored by aggregate PCT (darker bars indicate more likely biological importance). Relative expression (log2 fold change) of each miRNA in Wilms tumors with miRNA processing mutations is shown at the right. (B) Conservation of binding sites for the miR-16 and miR-34 families in the PLAG1 3′ UTR. (C) Luciferase reporter assay confirming that miR-16 and miR-34a repress sequences from the PLAG1 3′ UTR in a sequence-specific manner. The sequence surrounding each site was cloned downstream from a luciferase reporter, and the seed sequence was mutated (marked by an asterisk). An SV40 polyadenylation sequence was used as a negative control. Values are presented as mean ± SD from five replicates. (*) P ≤ 0.05. (D,E) Expression of PLAG1 and IGF2 in WiT49 in response to mimics (D) or inhibitors (E) of miR-16 and miR-34a. Values are presented as mean ± SD from three technical replicates. (*) P ≤ 0.05. (F) The density of WiT49 cells after transfection of miRNA mimics, measured by crystal violet staining. Values are presented as mean ± SD from four technical replicates. (*) P < 0.05.
Using luciferase reporter assays, we confirmed that each of the binding sites in the PLAG1 3′ UTR responds to miR-16 and miR-34a in a sequence-dependent manner (Fig. 3C). These effect sizes are in line with the 10%–50% repression typical for interactions between a miRNA and a target mRNA (Baek et al. 2008; Guo et al. 2010). In addition, we examined the expression of PLAG1 in WiT49 cells in response to manipulations of miR-16 and miR-34a. Transfection of miR-16 or miR-34a mimics decreased PLAG1 and IGF2 levels in both parental WiT49 and DICER1 knockout cells (Fig. 3D; Supplemental Fig. S2E). Conversely, transfection of a miR-16 inhibitor increased endogenous PLAG1 and IGF2 (Fig. 3E). WiT49 cells do not express miR-34a (data not shown). This is consistent with observations that WiT49 is a p53 mutant anaplastic Wilms tumor, as miR-34a expression is dependent on p53 (Alami et al. 2003; Chang et al. 2007; He et al. 2007). As expected, the miR-34a inhibitor showed little effect on PLAG1 or IGF2 expression. Furthermore, transfection of these two mimics into WiT49 cells impaired growth, implying that they may act as tumor suppressors in Wilms tumor (Fig. 3F). Together, these results show that miR-16 and miR-34 repress PLAG1 in Wilms tumor cells.
PLAG1 expression augments IGF2 levels
Next, we examined the effects of PLAG1 overexpression on Wilms tumor cells in vitro. WiT49 cells express low levels of PLAG1 at baseline, so we stably overexpressed PLAG1 in WiT49 cells using a lentiviral vector (Fig. 4A). This led to more rapid proliferation. We then performed whole-transcriptome sequencing in these cells to identify PLAG1 transcriptional targets that might be accelerating proliferation in an unbiased manner. Very few individual genes were significantly overexpressed in PLAG1-overexpressing cells, and IGF2 was the only one whose promoter contains PLAG1-binding sites (Fig. 4B,C).
Figure 4.

PLAG1 expression enhances proliferation. (A) Immunoblot of WiT49 with lentivirally overexpressed PLAG1 and the cell density of WiT49 with or without ectopic PLAG1 expression, measured by crystal violet staining. Values are presented as mean ± SD from four technical replicates, (*) P < 0.05. (B) Differentially expressed genes measured by whole-transcriptome sequencing of WiT49 cells with or without ectopic PLAG1 expression in two replicates each. FPKM in parental cells is shown on the X-axis. Comparison of values in PLAG1-overexpressing cells versus parental cells is shown as −log10 of the P-value along the Y-axis. Predicted PLAG1 target genes are highlighted in red. (C) qPCR for IGF2 in PLAG1-overexpressing WiT49. Values are presented as mean ± SD from three technical replicates. (*) P < 0.05. (D) PLAG1 consensus binding sites (GRGGCN6-8GGG) aligned with IGF2 transcripts, marked by the transcription start site (P0–P4). (E) ChIP-qPCR (chromatin immunoprecipitation [ChIP] combined with qPCR) using Flag antibody in WiT49 cells transfected with empty vector versus PLAG1-3xFlag to detect binding to the human IGF2 P3 promoter region. Values are presented as mean ± SD from four technical replicates. (*) P < 0.05. (F) ChIP-qPCR using PLAG1 antibody in whole mouse kidneys at P4 to detect binding to the mouse Igf2 P2 promoter region (which is homologous to the human P3 promoter). Values are presented as mean ± SD from three technical replicates. (*) P < 0.05. (G) IGF2 transcript levels in WiT49 with or without PLAG1 overexpression, as measured by RNA-seq. Values are presented as mean ± SEM, taken from two replicates. (*) P < 0.05. (H) IGF2 transcript levels in Wilms tumors and normal kidneys, segregated by the transcription start site (P0–P4), as measured by RNA-seq.
IGF2 can be transcribed from five different promoters in the human genome, named P0–P4 (Fig. 4D). Transcriptional activity at these promoters is tightly controlled by developmental timing and cellular context (Brouwer-Visser and Huang 2015). PLAG1 recognizes the binding motif GRGGCN6-8GGG (Hensen et al. 2002; Voz et al. 2004), and the region upstream of the P3 and P4 transcription start sites contains a high concentration of PLAG1-binding sites (Fig. 4D). To confirm that exogenous PLAG1 binds to the P3 promoter, we performed chromatin immunoprecipitation (ChIP) for PLAG1-3xFlag in WiT49. Using an antibody against the Flag tag, we confirmed that exogenous PLAG1 was enriched at the IGF2 P3 promoter (Fig. 4E). Similarly, we also performed ChIP in whole transgenic mouse kidneys at P4. We used primers to detect binding to the mouse Igf2 P2 promoter, which is homologous to the human P3 promoter. As the mouse transgene does not have a Flag tag, we used a PLAG1 antibody, which also recognizes endogenous mouse Plag1 and Plagl1 protein. Perhaps due to this cross-reactivity, we detected enrichment in control kidneys that is not statistically significant (Fig. 4F). In PLAG1-overexpressing kidneys, however, enrichment at the P2 promoter is greater and is statistically significant.
Last, we also confirmed that IGF2 transcript levels reflect this promoter bias. In WiT49, PLAG1 overexpression specifically increased IGF2 transcripts arising from the P3 and P4 promoters (Fig. 4G). In human Wilms tumors, the P3 and P4 promoters are the most active IGF2 promoters (Fig. 4H).
PLAG1 overexpression drives mTOR activity
To further compare these two conditions, we next performed a gene set enrichment analysis (GSEA) using the “hallmark” gene sets in the molecular signatures database (Subramanian et al. 2005; Liberzon et al. 2015). The most significantly enriched gene set in PLAG1-overexpressing WiT49 cells was mammalian target of rapamycin complex 1 (mTORC1) signaling (Fig. 5A,B), a known downstream effector of IGF2. In parallel, we also performed a reverse-phase protein array to identify signaling pathways differentially regulated in PLAG1-overexpressing WiT49 cells in an unbiased manner. Markers of mTORC1 activity, especially the phosphorylated form of ribosomal protein S6, were among the most differentially active signals on the array (Fig. 5C). We confirmed mTORC1 activity by Western blot for phospho-S6 (Fig. 5D).
Figure 5.

PLAG1 expression drives mTOR activity. (A) The most highly enriched “hallmark” gene sets identified using GSEA in WiT49 cells with versus without PLAG1 overexpression. (NES) Normalized enrichment score. (B) GSEA of the “hallmark_mtorc1_signaling” gene set. (C) Reverse-phase protein array (190 validated antibodies only) in WiT49 cells with versus without PLAG1 overexpression. mTORC1 pathway phospho-proteins are highlighted in red. (D) Western blots assaying mTORC1 activity of WiT49 cells with PLAG1 overexpression with and without IGF2 knockdown. (E) Western blot assaying mTORC1 activity in WiT49 cells with and without PLAG1 overexpression, treated with NVP-AEW451 at 5 µM for 3 or 6 h. (F,G) Immunostaining of Wt1Cre;LSL-PLAG1 kidneys at E18.5 for PLAG1 (F) and phospho-S6 (G). Bar, 100 µm.
Next, we tested whether this PLAG1-induced increase in mTORC1 signaling was indeed dependent on IGF2. We tested this by silencing IGF2 with an siRNA or inhibiting IGF2 signaling with NVP-AEW541, a small molecule IGF1R inhibitor. This drug previously has shown activity against WiT49 xenografts (Bielen et al. 2012). We found that PLAG1 overexpression did not significantly affect sensitivity to NVP-AEW451 (Supplemental Fig. S3A). Nevertheless, with either knockdown or drug, IGF2 inhibition led to a significant reduction in S6 phosphorylation (Fig. 5D,E). Thus, through multiple approaches, we found that PLAG1 overexpression leads to an IGF2-dependent increase in mTORC1 signaling in WiT49 cells.
Last, we next interrogated mTORC1 signaling activity in PLAG1-overexpressing mouse kidneys. The cysts and solid nodules stain positive for both PLAG1 (nuclear) and phospho-S6 (cytoplasmic), while the surrounding kidney parenchyma is negative, reinforcing that these lesions arise cell-autonomously (Fig. 5F,G; Supplemental Fig. S3B,C). This is not the case in control kidneys (Supplemental Fig. S3D,E). Furthermore, we performed RNA-seq in P8 kidneys to examine the effect of PLAG1 overexpression on the transcriptome in vivo. Corroborating what we found in WiT49, RNA-seq in PLAG1-overexpressing kidneys showed that Igf2 was elevated and that the mTORC1 signaling gene set was enriched (Supplemental Fig. S3F,G).
Human Wilms tumors also overexpress PLAG1 through copy number changes
Given these findings on the effects of PLAG1 overexpression both in vitro and in vivo, we returned to the observation that some tumors express high PLAG1 levels even in the absence of mutations impairing miRNA processing. Specifically, we looked at the correlation between copy number changes and PLAG1 overexpression in the TARGET cohort. We noted that while many Wilms tumors are aneuploid, those with miRNA processing mutations are rarely aneuploid (Supplemental Fig. S4). In fact, the only aneuploid tumor in this subset actually has a germline DROSHA p.P82T mutation (Walz et al. 2015). This mutation is outside of the active RNase III domains and thus is of unclear significance. In particular, several of the tumors with miRNA processing mutations show a gain of chromosome 1q or a loss of chromosome 16q (six of 14 or eight of 14 tumors, respectively). These two features have been associated previously with high-risk disease, although loss of 16q was not validated in other recent studies. However, only rarely do these tumors show a third high-risk copy number change: loss of chromosome 1p (Bown et al. 2002).
In fact, the PLAG1 regulator miR-34a is encoded on chromosome 1p36. In addition, chromosome 8q, where PLAG1 is encoded, is gained in many relapsed Wilms tumors, often as a whole chromosome or chromosome arm in the background of an aneuploid tumor (Natrajan et al. 2007). These two copy number changes are largely mutually exclusive of mutations in miRNA processing genes (Fig. 6A). Tumors with any of these three changes express high PLAG1 levels, although many other tumors express relatively high PLAG1 levels due to other mechanisms (Fig. 6A,B). Last, we examined whether PLAG1 expression correlated with outcome in this data set. Regardless of mechanism, we found that high PLAG1 expression significantly correlates with earlier relapse in this cohort (Fig. 6C).
Figure 6.

Human Wilms tumors overexpress PLAG1 through multiple mechanisms, resulting in poorer survival. (A) Genomic aberrations in TARGET Wilms tumors, correlated with expression of PLAG1 by microarray. Copy number gains are shown in red, while copy number losses are shown in blue. (B) PLAG1 expression in tumors with gain of 8q12, where PLAG1 is encoded; loss of 1p, where MIR34A is encoded; or mutations in the miRNA processing pathway by microarray. (*) P < 0.05, unpaired t-test versus “none.” (C) Event-free survival in relapsed favorable histology Wilms tumor patients based on PLAG1 expression (“high” is defined as PLAG1 expression intensity >6). (**) P ≤ 0.01, log-rank test.
Discussion
Previous studies of miRNA target genes in Wilms tumor have focused on let-7 and its target gene, LIN28B (Urbach et al. 2014; Gadd et al. 2017). However, the nearly global impairment of miRNA processing seen in some Wilms tumors led us to search for other miRNA target genes that could contribute to Wilms tumor formation. Here, using a combination of in vitro, in vivo, and human tumor data, we demonstrate that PLAG1 is another important mediator of Wilms tumor pathogenesis. It can be up-regulated by impairment of miRNA processing or by copy number changes. PLAG1 overexpression enhances IGF2 levels, increases mTORC1 activity, induces dysplasia, and correlates with poor prognosis in human Wilms tumors. The modest increases that we observed in IGF2 are comparable with the increases caused by LOI in human Wilms tumors and in mouse models of Wilms tumor (Hu et al. 2011; Huang et al. 2016).
PLAG1 was originally described as an oncogene in salivary gland pleomorphic adenomas but recently has also been implicated in other cancers. Hepatoblastomas (which, like Wilms tumor, commonly harbor IGF2 LOI) are also reported to recurrently amplify the PLAG1 locus, leading to IGF2 overexpression (Zatkova et al. 2004). Like pleomorphic adenoma, translocations resulting in PLAG1 activation and promoter switching are also seen in other tumors, such as lipoblastoma, leiomyoma, and soft tissue myoepithelioma (Hibbard et al. 2000; Antonescu et al. 2013). It cooperates with core-binding factor to induce acute myeloid leukemia (Landrette et al. 2005). PLAG1 overexpression in various mouse tissues can produce salivary gland tumors, cavernous angiomatosis, or mammary gland adenomyoepitheliomas (Declercq et al. 2005, 2008a; Van Dyck et al. 2008). Chronic lymphocytic leukemia is commonly driven by deletion of the miR-15a/16 cluster, and PLAG1 is often overexpressed in this disease (Pallasch et al. 2009). However, to our knowledge, this is the first report connecting PLAG1 to Wilms tumor.
Although our work focuses on miR-16 and miR-34, other miRNAs are also predicted to regulate PLAG1 (Fig. 3A). Further work may elucidate how much other miRNAs contribute to PLAG1 regulation. Likewise, although we focus on IGF2 here, PLAG1 is also reported to regulate the transcription of Wnt pathway members, cell cycle regulators, and immunomodulatory factors (Voz et al. 2004; Declercq et al. 2008b). Future studies may distinguish how much other PLAG1 target genes contribute to the phenotypes that we described here. Last, regulation of IGF2 transcription is complex and likely involves other important factors besides PLAG1.
The genes on chromosomes 1p, 1q, and 16q that are responsible for driving poor prognosis are unknown. We saw that Wilms tumors with mutated miRNA processing genes rarely develop loss of 1p. We speculate that gain of 1q or loss of 16q may cooperate with miRNA depletion, while loss of 1p may be dispensable for these tumors because it contains miR-34a. The finding that miR-34a can be down-regulated by either miRNA processing mutation, p53 mutation, or copy number loss potentially makes miR-34a a central tumor suppressor in Wilms tumor.
Because PLAG1 is expressed at low levels in postnatal tissues, it could be an attractive target for novel Wilms tumor therapies. However, its relative importance with respect to LIN28 and other miRNA target genes is yet to be seen. Testing whether PLAG1 is required for tumor growth in the setting of miRNA-impairing mutations may require an in vivo model of miRNA impairment-driven Wilms tumor. In addition, zinc finger transcription factors traditionally have been difficult to target with small molecules. Both inhibitors against IGF2 signaling and small molecule MEK inhibitors have shown limited efficacy in Wilms tumor (Flores et al. 2013; Weigel et al. 2014). Our work suggests that inhibitors of the mTORC1 pathway could have activity in Wilms tumor.
Alternatively, therapeutic miRNA reintroduction may be particularly well suited to tumors driven by mutations in the miRNA processing pathway. In fact, miR-34a-based therapies have been tested in adult hepatocellular cancer, and miR-16-based therapies have shown promising results in early studies of adult cancer patients (van Zandwijk et al. 2017). Prior studies have focused on let-7, but our work suggests that miR-16 and miR-34 may be attractive therapeutic agents in Wilms tumor.
Materials and methods
Cell lines and cell culture
WiT49 cells were a gift from Dr. Sharon Plon's laboratory. The HEK293T line was obtained from American Type Culture Collection. Both lines were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS) and 1× antibiotic antimycotic (Gibco, 15240096) and cultured at 37°C with 5% CO2.
Animal models
The LSL-PLAG1 mouse line (previously also known as “PTMS1”) was a gift from Dr. Wim van de Ven's laboratory. These mice were maintained on a FVB/NJ background. Wt1Cre, Six2Cre, and Foxd1Cre mouse lines were obtained from the University of Texas Southwestern O'Brien Kidney Research Animal Models Core, and we propagated them on a C57BL6/J background. All animal studies were performed in accordance with University of Texas Southwestern Institutional Animal Care and Use Committee protocols 2015-101096 and 2016-101824.
Clinical samples
Wilms tumor clinical samples were obtained at Children's Medical Center under approval of the University of Texas Southwestern Institutional Review Board. Tumors were reviewed by a pediatric pathologist (D. Rakheja) before RNA was extracted from frozen specimens.
TARGET data analysis
Open access microarray data (generated on Affymetrix U133 Plus 2), copy number data (generated on Affymetrix SNP 6.0), mRNA-seq data (gene-level RPKM [reads per kilobase per million mapped reads] quantification tables), and clinical metadata were downloaded from the TARGET data matrix for Wilms tumors (https://ocg.cancer.gov/programs/target/data-matrix). Published whole-exome sequencing results were then used to categorize these tumors by mutational status.
For microarray data, microarray signals were log2 transformed and then compared between groups by two-sided Student's t-test, and the resulting P-values were corrected for multiple hypothesis testing using the Bonferroni-Hochberg correction. For copy number data, preprocessed log2 segment mean values for the PLAG1 and MIR34A loci were extracted. Copy number gain was defined as log2 segment mean value of >0.3 (i.e., >2.5 copies in a diploid background), and copy number loss was defined as log2 segment mean value of less than −0.4 (i.e., <1.5 copies in a diploid background).
RNA-seq and small RNA-seq from Wilms tumors
RNA was extracted from frozen specimens using the mirVana miRNA isolation kit (Thermo Fisher). Whole-transcriptome sequencing was performed at the University of Texas Southwestern McDermott Center Next-Generation Sequencing Core on the Illumina HiSeq2000 platform with 100-base-pair (bp) paired-end reads according to the manufacturer. Between 66 million and 193 million reads were obtained from each sample. After adapter removal and quality filtering, the reads were mapped to human reference genome (hg19) by Bowtie2 (Langmead and Salzberg 2012) and TopHat2 (Kim et al. 2013). Cufflinks (Trapnell et al. 2012) was used to assemble and estimate the relative abundances of transcripts at the gene and isoform levels. IGF2 transcripts were segregated by transcription start site by counting the number of reads spanning the first divergent exon to the last common exon.
Small RNA-seq multiplexed miRNA libraries for sequencing were prepared according to the Illumina TruSeq small RNA sample preparation protocol (Illumina). Sequencing was conducted on an Illumina HiSeq2000 according to the manufacturer's protocol (Illumina) in the McDermott Next-Generation Sequencing Core (University of Texas Southwestern). Samples were demultiplexed, and adapter sequences were removed. Next, miRNAs were identified, and read counts were generated by aligning the reads to human miRBase (version 20) based on the Smith-Waterman algorithm.
DICER1 knockout in WiT49
The single-guide RNA sequence 5′-ACCATCGTCTGTTTAAACAC-3′ was cloned into the pX458 plasmid (Addgene, 48138), which contains coding sequences for both SpCas9 and GFP. Seventy-two hours after this plasmid was transiently transfected into WiT49, the ∼25% brightest cells were isolated by fluorescence-activated cell sorting. The following day, the sorted cells were seeded into 96-well plates by limiting dilution to isolate monoclonal populations. After these clones had reached a sufficient size, they were genotyped by loss of the PmeI site (GTTTAAAC). Biallelic frameshift insertions/deletions (indels) were confirmed by TA cloning.
qRT–PCR
RNA was extracted using Trizol reagent (Thermo Fisher) according to the manufacturer's protocol. Complementary DNA was reverse-transcribed using the RT2 HT first strand kit (Qiagen). qPCR was then performed with the iTaq Universal SYBR Green Supermix (Bio-Rad) using the primers listed in Supplemental Table 1 in three replicates per condition except where otherwise noted. Data were analyzed using the 2−ΔΔCt method by normalizing to the expression of 18s RNA.
Luciferase reporter assay, miRNA mimic/inhibitor, and siRNA transfection
An ∼500-bp region surrounding each miRNA-binding site was cloned from human genomic DNA into the XbaI site of the pGL3 control plasmid using the primers listed below. The 8-bp miRNA seed sequence in each case was then mutated into a NotI site by site-directed mutagenesis to serve as a seed mutant control. These plasmids were transfected into HEK293 cells in 48-well format, with a Renilla luciferase plasmid as a transfection control at a 50:1 ratio using Lipofectamine RNAiMax transfection reagent (Thermo Fisher). miRIDIAN miRNA mimics or control mimics (4 pmol per well) were cotransfected simultaneously (Dharmacon). This was performed in five replicates per condition, and luciferase activity was assayed 48 h after transfection using the dual-luciferase reporter assay system (Promega).
Parental WiT49 cells were transfected with control, miR-16, or miR-34a miRIDIAN miRNA mimics or inhibitors. Cells were seeded at 5 × 105 cells per well in a six-well plate and transfected with 60 pmol of mimic or 50 pmol of inhibitor per well using RNAiMAX. DICER1 knockout clones were seeded at 1.5 × 105 cells per well in a 12-well plate and transfected with 30 pmol of each miRNA mimic per well. Transfected cells were harvested for RNA after 48 h. RNA was then prepared using TRIzol reagent (Thermo Fisher Scientific) and used for qRT–PCR.
Both parental and PLAG1-overexpressing WiT49 cells were transfected with control or IGF2 targeting Silencer Select siRNA (Thermo Fisher Scientific) using RNAiMAX. Cells were seeded at 2.5 × 105 cells per well in a six-well plate and transfected with 60 pmol of siRNA. After 48 h, cells were harvested for RNA or protein lysate and used for immunoblotting.
PLAG1 ectopic expression
The PLAG1-coding sequence was amplified from human cDNA using the oligos listed below to add a Flag tag. It was then cloned into pLX303 plasmid (Addgene, 25897)—into which cyan fluorescent protein had been inserted adjacent to the blasticidin selection gene—by gateway cloning. WiT49 was exposed to lentivirus packaged with this plasmid, and transduced cells were selected in blasticidin. For the purposes of ChIP, the PLAG1 lentiviral vector was modified to change the C terminus to contain a 3xFlag tag prior to infection.
Growth curves were calculated by seeding 15,000 cells per well into 24-well plates in four replicates per condition. Over subsequent days, individual plates were stained with crystal violet solubilized in acetic acid before absorbance at 595 nm was detected.
RNA-seq from WiT49
RNA was isolated using TRIzol reagent (Thermo Fisher) according to the manufacturer's protocol from two biological replicates of WiT49 and WiT49 + PLAG1 each. RNA-seq was performed by DNA Link. cDNA libraries were constructed by using KAPA stranded RNA-seq kit with RiboErase (HMR) (Kapa Biosystems). Amplified cDNA was validated and quantified on an Agilent Bioanalyzer. The purified libraries were normalized, pooled together, denatured, and diluted, followed by one 75-bp sequencing on a NextSeq500 (Illumina). From each sample, we obtained ∼25 million reads.
For gene expression analysis, reads were aligned to the reference genome (GRCh38) using TopHat version 2.0.14 and Bowtie version 2.10. The distribution of alignments was analyzed using Cufflinks version 2.2.1, and FPKM (fragments per kilobase per million mapped reads) values were quantile-normalized. Differential expression testing was performed using Cuffdiff version 2.2.1. PLAG1 target genes were defined as protein-coding genes whose promoters (from 1000 bp upstream of to 100 bp downstream from each transcription start site in hg19) contain a sequence with a >90% match for the JASPAR_CORE-PLAG1-MA0163.1-binding motif using the MotifDb package (version 1.12.1).
RNA-seq from mouse kidneys
RNA was isolated from mouse pup kidneys at specified ages using the TRIzol reagent. Total RNA-seq was performed by DNA Link on the Illumina NextSeq 500 platform with 75-bp single-end reads. Between 23 million and 32 million reads were obtained from each sample before demultiplexing and quality filtering. The mm10 mouse reference genome was modified manually to add the human PLAG1-coding sequence that is expressed in the LSL-PLAG1 mouse model. RNA-seq reads were mapped to this modified reference genome using HISAT2 (Kim et al. 2015). Stringtie was used to assemble transcripts and estimate the relative expression of transcripts at the gene and isoform levels (Pertea et al. 2016).
ChIP
ChIP was performed using both cell lines and mouse kidney tissue. Cells were grown to 80% confluency in multiple 15-cm dishes and fixed in formaldehyde. Whole kidneys were dissected from mouse pups at P4, dissociated using TrypLE Select (Gibco), incubated for 90 min at 37°C with intermittent mixing to achieve single-cell suspension, and then rescued with DMEM with 10% FBS before fixation.
Cells from both preparations were fixed using formaldehyde at a final concentration of 1% for 10 min. Formaldehyde was quenched using glycine at a final concentration of 125 mM for 1 min. Nuclei were then isolated in Farnham lysis buffer (5 mM HEPES, 85 mM KCl, 0.5% NP-40, 1× protease inhibitors) by passing the solution through a 20-gauge needle and sonicated in RIPA buffer in TPX microtubes using the Bioruptor UCD-200 (Diagenode). WiT49 chromatin was sonicated for four cycles of 10 min (30 sec on/30 sec off), while mouse kidney chromatin was sonicated for three cycles of 10 min (30 sec on/30 sec off). Remaining cellular debris was pelleted, and the soluble chromatin was diluted in dilution buffer (2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl at pH 8) and used for immunoprecipitation.
Sonicated chromatin was rotated overnight at 4°C with 5 µg of either anti-PLAG1 monoclonal antibody (Sigma, WH0005324M2), anti-Flag M2 monoclonal antibody (Sigma, F1804), or mouse IgG isotype control (Invitrogen, 10400C). Protein G Dynabeads (Invitrogen, 10003D) were washed and blocked in 5% BSA in 1× PBS by rotating overnight at 4°C. The next day, the blocked Dynabeads were pelleted on the DynaMag-2 rack (Thermo Fisher Scientific), resuspended in dilution buffer, and added to each immunoprecipitation reaction before rotating for 4 h at 4°C. The beads bound to chromatin–protein immunocomplexes were pelleted and then washed in a series of buffers by incubating for 15 min on ice: TSE I (0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris-HCl, 150 mM NaCl), TSE II (0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris-HCl, 500 mM NaCl), TSE III (0.25 M LiCl, 1% Igepal-630, 1 mM EDTA, 10 mM Tris-HCl, 1% deoxycholate), and TE (10 mM Tris, 1 mM EDTA). Chromatin–protein complexes were eluted off of the beads by incubating twice in elution buffer (1% SDS, 0.75% NaHCO3) for 15 min at 55°C. The eluted chromatin was then decross-linked overnight at 65°C. All remaining RNA and protein were digested by 20 µg of RNase A for 30 min at 37°C followed by 20 µg of proteinase K for 30 min at 55°C. The chromatin was then column-purified using the QIAquick PCR purification kit (Qiagen). Purified chromatin was used for qPCR.
Reverse-phase protein array
Two biological replicate cell pellets of WiT49 and WiT49 + PLAG1 were sent to the Reverse Phase Protein Array Core Facility of the M.D. Anderson Cancer Center, Houston, TX. Normalized median-centered log2 values were averaged within replicates, and the difference of these averages was plotted.
NVP-AEW451
To assess the effect of NVP-AEW451 on cell viability, parental and PLAG1-overexpressing WiT49 cells were seeded in four replicate wells of a 96-well plate at 3 × 103 cells per well. The next day, medium containing NVP-AEW541 (Selleck Chemicals) was added at concentrations ranging from 125 µM to 2 nM. After 72 h, the number of viable cells was quantified using CellTiter-Glo 2.0 (Promega). The IC50 was calculated using a nonlinear fit with variable slope (GraphPad Prism). Data are presented as IC50 in micromolar (95% confidence interval).
To assess the effect of NVP-AEW451 on S6 phosphorylation, parental WiT49- and PLAG1L-overexpressing cells were each seeded in three wells of a six-well plate at 2.5 × 105 cells per well. The next day, cells were treated with either 0.02% DMSO for 6 h or 5 µM NVP-AEW541 (Selleck Chemicals) for 3 or 6 h. The cells were then harvested for protein lysate and used for immunoblotting.
Immunohistochemistry
Immunohistochemical staining for PLAG1 and phospho-S6 was performed using standard immunoperoxidase techniques with hematoxylin counterstaining. Prior to staining, heat-based antigen retrieval was performed using Trilogy pretreatment solution (Cell Marque). A monoclonal antibody against PLAG1 raised in mice was used at a 1:1000 dilution (clone 3B7, Sigma-Aldrich), while a polyclonal antibody against phospho-S6 was used at a 1:400 dilution (Cell Signaling Technology, 2211).
Supplementary Material
Acknowledgments
We thank Barrett Updegraff for sharing plasmids, and Dr. Wim J.M. Van de Ven for sharing the PLAG1 mouse line with us. We also thank Thomas Carroll for his assistance and advice and for sharing the Wt1Cre, Foxd1Cre, and Six2Cre lines. K.S.C. is funded by an Alex's Lemonade Stand Foundation Young Investigator grant and a Damon Runyon-Sohn Pediatric Cancer Research Fellowship (DRSG 4P-13). This work was supported by National Institutes of Health grants K08CA207849 to K.S.C., P50CA196516 to J.T.M. and J.F.A., and R35CA197311 to J.T.M. J.T.M. is an Investigator of the Howard Hughes Medical Institute.
Author contributions: K.S.C. conceived and performed experiments, wrote the manuscript, and secured funding. E.K.S., A.B., D.R., D.N.-V., L.X., S.H.S., A.A.S., and C.F. performed the experiments. J.T.M. and J.F.A. provided expertise, supervision, funding, data interpretation, and manuscript editing.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.313783.118.
References
- Agarwal V, Bell GW, Nam JW, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alami J, Williams BR, Yeger H. 2003. Derivation and characterization of a Wilms’ tumour cell line, WiT 49. Int J Cancer 107: 365–374. [DOI] [PubMed] [Google Scholar]
- Antonescu CR, Zhang L, Shao SY, Mosquera JM, Weinreb I, Katabi N, Fletcher CD. 2013. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer 52: 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. 2008. The impact of microRNAs on protein output. Nature 455: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielen A, Box G, Perryman L, Bjerke L, Popov S, Jamin Y, Jury A, Valenti M, Brandon Ade H, Martins V, et al. 2012. Dependence of Wilms tumor cells on signaling through insulin-like growth factor 1 in an orthotopic xenograft model targetable by specific receptor inhibition. Proc Natl Acad Sci 109: E1267–E1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bown N, Cotterill SJ, Roberts P, Griffiths M, Larkins S, Hibbert S, Middleton H, Kelsey A, Tritton D, Mitchell C. 2002. Cytogenetic abnormalities and clinical outcome in Wilms tumor: a study by the U.K. cancer cytogenetics group and the U.K. Children's Cancer Study Group. Med Pediatr Oncol 38: 11–21. [DOI] [PubMed] [Google Scholar]
- Brouwer-Visser J, Huang GS. 2015. IGF2 signaling and regulation in cancer. Cytokine Growth Factor Rev 26: 371–377. [DOI] [PubMed] [Google Scholar]
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al. 2002. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci 99: 15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. 2007. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26: 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles AK, Brown KW, Berry PJ. 1998. Microdissecting the genetic events in nephrogenic rests and Wilms’ tumor development. Am J Pathol 153: 991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A, Heck JE, Ribeiro KB, Brennan P, Boffetta P, Buffler P, Hung RJ. 2010. Wilms’ tumour: a systematic review of risk factors and meta-analysis. Paediatr Perinat Epidemiol 24: 449–469. [DOI] [PubMed] [Google Scholar]
- Declercq J, Van Dyck F, Braem CV, Van Valckenborgh IC, Voz M, Wassef M, Schoonjans L, Van Damme B, Fiette L, Van de Ven WJ. 2005. Salivary gland tumors in transgenic mice with targeted PLAG1 proto-oncogene overexpression. Cancer Res 65: 4544–4553. [DOI] [PubMed] [Google Scholar]
- Declercq J, Skaland I, Van Dyck F, Janssen EA, Baak JP, Drijkoningen M, Van de Ven WJ. 2008a. Adenomyoepitheliomatous lesions of the mammary glands in transgenic mice with targeted PLAG1 overexpression. Int J Cancer 123: 1593–1600. [DOI] [PubMed] [Google Scholar]
- Declercq J, Van Dyck F, Van Damme B, Van de Ven WJ. 2008b. Upregulation of Igf and Wnt signalling associated genes in pleomorphic adenomas of the salivary glands in PLAG1 transgenic mice. Int J Oncol 32: 1041–1047. [PubMed] [Google Scholar]
- Dome JS, Cotton CA, Perlman EJ, Breslow NE, Kalapurakal JA, Ritchey ML, Grundy PE, Malogolowkin M, Beckwith JB, Shamberger RC, et al. 2006. Treatment of anaplastic histology Wilms’ tumor: results from the fifth National Wilms’ Tumor Study. J Clin Oncol 24: 2352–2358. [DOI] [PubMed] [Google Scholar]
- Dome JS, Graf N, Geller JI, Fernandez CV, Mullen EA, Spreafico F, Van den Heuvel-Eibrink M, Pritchard-Jones K. 2015. Advances in Wilms tumor treatment and biology: progress through international collaboration. J Clin Oncol 33: 2999–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores LG II, Yeh HH, Soghomonyan S, Young D, Bankson J, Hu Q, Alauddin M, Huff V, Gelovani JG. 2013. Monitoring therapy with MEK inhibitor U0126 in a novel Wilms tumor model in Wt1 knockout Igf2 transgenic mice using 18F-FDG PET with dual-contrast enhanced CT and MRI: early metabolic response without inhibition of tumor growth. Mol Imaging Biol 15: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S, Huff V, Walz AL, Ooms A, Armstrong AE, Gerhard DS, Smith MA, Auvil JMG, Meerzaman D, Chen QR, et al. 2017. A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49: 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. 2007. A microRNA component of the p53 tumour suppressor network. Nature 447: 1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensen K, Van Valckenborgh IC, Kas K, Van de Ven WJ, Voz ML. 2002. The tumorigenic diversity of the three PLAG family members is associated with different DNA binding capacities. Cancer Res 62: 1510–1517. [PubMed] [Google Scholar]
- Hensen K, Braem C, Declercq J, Van Dyck F, Dewerchin M, Fiette L, Denef C, Van de Ven WJ. 2004. Targeted disruption of the murine Plag1 proto-oncogene causes growth retardation and reduced fertility. Dev Growth Differ 46: 459–470. [DOI] [PubMed] [Google Scholar]
- Hibbard MK, Kozakewich HP, Dal Cin P, Sciot R, Tan X, Xiao S, Fletcher JA. 2000. PLAG1 fusion oncogenes in lipoblastoma. Cancer Res 60: 4869–4872. [PubMed] [Google Scholar]
- Hu Q, Gao F, Tian W, Ruteshouser EC, Wang Y, Lazar A, Stewart J, Strong LC, Behringer RR, Huff V. 2011. Wt1 ablation and Igf2 upregulation in mice result in Wilms tumors with elevated ERK1/2 phosphorylation. J Clin Invest 121: 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Mokkapati S, Hu Q, Ruteshouser EC, Hicks MJ, Huff V. 2016. Nephron progenitor but not stromal progenitor cells give rise to Wilms tumors in mouse models with β-catenin activation or Wt1 ablation and Igf2 upregulation. Neoplasia 18: 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James DH Jr, Hustu O, Wrenn EL Jr, Johnson WW. 1966. Childhood malignant tumors. Concurrent chemotherapy with dactinomycin and vincristine sulfate. JAMA 197: 1043–1045. [DOI] [PubMed] [Google Scholar]
- Karim L, Takeda H, Lin L, Druet T, Arias JAC, Baurain D, Cambisano N, Davis SR, Farnir F, Grisart B, et al. 2011. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet 43: 405–413. [DOI] [PubMed] [Google Scholar]
- Kas K, Voz ML, Roijer E, Astrom AK, Meyen E, Stenman G, Van de Ven WJ. 1997. Promoter swapping between the genes for a novel zinc finger protein and β-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat Genet 15: 170–174. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrette SF, Kuo YH, Hensen K, Barjesteh van Waalwijk van Doorn-Khosrovani S, Perrat PN, Van de Ven WJ, Delwel R, Castilla LH. 2005. Plag1 and Plagl2 are oncogenes that induce acute myeloid leukemia in cooperation with Cbfb-MYH11. Blood 105: 2900–2907. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. 2015. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti S, Alam R, Amos CI, Huff V. 2000. Frequent association of β-catenin and WT1 mutations in Wilms tumors. Cancer Res 60: 6288–6292. [PubMed] [Google Scholar]
- Moulton T, Crenshaw T, Hao Y, Moosikasuwan J, Lin N, Dembitzer F, Hensle T, Weiss L, McMorrow L, Loew T, et al. 1994. Epigenetic lesions at the H19 locus in Wilms’ tumour patients. Nat Genet 7: 440–447. [DOI] [PubMed] [Google Scholar]
- Natrajan R, Little SE, Sodha N, Reis-Filho JS, Mackay A, Fenwick K, Ashworth A, Perlman EJ, Dome JS, Grundy PE, et al. 2007. Analysis by array CGH of genomic changes associated with the progression or relapse of Wilms’ tumour. J Pathol 211: 52–59. [DOI] [PubMed] [Google Scholar]
- Ogawa O, Eccles MR, Szeto J, McNoe LA, Yun K, Maw MA, Smith PJ, Reeve AE. 1993. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms’ tumour. Nature 362: 749–751. [DOI] [PubMed] [Google Scholar]
- Pallasch CP, Patz M, Park YJ, Hagist S, Eggle D, Claus R, Debey-Pascher S, Schulz A, Frenzel LP, Claasen J, et al. 2009. miRNA deregulation by epigenetic silencing disrupts suppression of the oncogene PLAG1 in chronic lymphocytic leukemia. Blood 114: 3255–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, Khokhar S, Wickiser JE, Karandikar NJ, Malter JS, et al. 2014. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera MN, Haber DA. 2005. Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer 5: 699–712. [DOI] [PubMed] [Google Scholar]
- Rubin CJ, Megens HJ, Martinez Barrio A, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, et al. 2012. Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci 109: 19529–19536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rump P, Zeegers MP, van Essen AJ. 2005. Tumor risk in Beckwith-Wiedemann syndrome: a review and meta-analysis. Am J Med Genet A 136: 95–104. [DOI] [PubMed] [Google Scholar]
- Ruteshouser EC, Robinson SM, Huff V. 2008. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47: 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenman MJ, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP. 1994. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour. Nat Genet 7: 433–439. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrezan GT, Ferreira EN, Nakahata AM, Barros BD, Castro MT, Correa BR, Krepischi AC, Olivieri EH, Cunha IW, Tabori U, et al. 2014. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun 5: 4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Yermalovich A, Zhang J, Spina CS, Zhu H, Perez-Atayde AR, Shukrun R, Charlton J, Sebire N, Mifsud W, et al. 2014. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes Dev 28: 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck F, Scroyen I, Declercq J, Sciot R, Kahn B, Lijnen R, Van de Ven WJ. 2008. aP2-Cre-mediated expression activation of an oncogenic PLAG1 transgene results in cavernous angiomatosis in mice. Int J Oncol 32: 33–40. [PubMed] [Google Scholar]
- van Zandwijk N, Pavlakis N, Kao SC, Linton A, Boyer MJ, Clarke S, Huynh Y, Chrzanowska A, Fulham MJ, Bailey DL, et al. 2017. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol 18: 1386–1396. [DOI] [PubMed] [Google Scholar]
- Voz ML, Agten NS, Van de Ven WJ, Kas K. 2000. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res 60: 106–113. [PubMed] [Google Scholar]
- Voz ML, Mathys J, Hensen K, Pendeville H, Van Valckenborgh I, Van Huffel C, Chavez M, Van Damme B, De Moor B, Moreau Y, et al. 2004. Microarray screening for target genes of the proto-oncogene PLAG1. Oncogene 23: 179–191. [DOI] [PubMed] [Google Scholar]
- Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, Chen QR, Hsu CH, Yan C, et al. 2015. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WH, Duan JX, Vu TH, Hoffman AR. 1996. Increased expression of the insulin-like growth factor-II gene in Wilms’ tumor is not dependent on loss of genomic imprinting or loss of heterozygosity. J Biol Chem 271: 27863–27870. [DOI] [PubMed] [Google Scholar]
- Wegert J, Ishaque N, Vardapour R, Georg C, Gu Z, Bieg M, Ziegler B, Bausenwein S, Nourkami N, Ludwig N, et al. 2015. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27: 298–311. [DOI] [PubMed] [Google Scholar]
- Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, Krailo M, Villaluna D, Adamson PC, Blaney SM. 2014. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatr Blood Cancer 61: 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm B, Munoz-Chapuli R. 2016. The role of WT1 in embryonic development and normal organ homeostasis. Methods Mol Biol 1467: 23–39. [DOI] [PubMed] [Google Scholar]
- Zatkova A, Rouillard JM, Hartmann W, Lamb BJ, Kuick R, Eckart M, von Schweinitz D, Koch A, Fonatsch C, Pietsch T, et al. 2004. Amplification and overexpression of the IGF2 regulator PLAG1 in hepatoblastoma. Genes Chromosomes Cancer 39: 126–137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.