

Van Alstyne et al. show that loss of SMN-dependent regulation of Mdm2 and Mdm4 alternative splicing underlies p53-mediated death of motor neurons in SMA, establishing a causal link between snRNP dysfunction and neurodegeneration.
Keywords: spinal muscular atrophy (SMA), survival motor neuron (SMN), small nuclear ribonucleoprotein (snRNP), p53, Mdm2, Mdm4
Abstract
Ubiquitous deficiency in the survival motor neuron (SMN) protein causes death of motor neurons—a hallmark of the neurodegenerative disease spinal muscular atrophy (SMA)—through poorly understood mechanisms. Here, we show that the function of SMN in the assembly of spliceosomal small nuclear ribonucleoproteins (snRNPs) regulates alternative splicing of Mdm2 and Mdm4, two nonredundant repressors of p53. Decreased inclusion of critical Mdm2 and Mdm4 exons is most prominent in SMA motor neurons and correlates with both snRNP reduction and p53 activation in vivo. Importantly, increased skipping of Mdm2 and Mdm4 exons regulated by SMN is necessary and sufficient to synergistically elicit robust p53 activation in wild-type mice. Conversely, restoration of full-length Mdm2 and Mdm4 suppresses p53 induction and motor neuron degeneration in SMA mice. These findings reveal that loss of SMN-dependent regulation of Mdm2 and Mdm4 alternative splicing underlies p53-mediated death of motor neurons in SMA, establishing a causal link between snRNP dysfunction and neurodegeneration.
Perturbation of RNA homeostasis is a common theme of many neurodegenerative disorders in which genetic mutations are associated with the direct or indirect impairment of RNA-binding proteins or factors controlling RNA processing at multiple levels (Cooper et al. 2009; Li et al. 2014; Conlon and Manley 2017). Remarkably, while disease-linked proteins are ubiquitously expressed and carry out essential biological processes, pathology is characterized by selective degeneration of specific neuronal populations. Furthermore, these proteins are often multifunctional and involved in the regulation of diverse RNA pathways, making it difficult to discern specific disease-relevant events among many transcriptome abnormalities. These complexities have made it remarkably challenging in most instances to establish causal links between RNA dysregulation and disease etiology. Nonetheless, dissecting the RNA-mediated mechanisms underlying the dysfunction and death of select neurons is necessary to uncover the molecular basis of human disease and identify drivers of neurodegeneration, which may also represent key targets for therapeutic intervention.
One prominent example of a neurodegenerative disease featuring RNA dysfunction is spinal muscular atrophy (SMA), a leading genetic cause of death in infancy that results from ubiquitous deficiency in the survival motor neuron (SMN) protein (Burghes and Beattie 2009; Tisdale and Pellizzoni 2015). SMN is part of a macromolecular complex that mediates assembly of Sm-class small nuclear ribonucleoproteins (snRNPs) of the pre-mRNA splicing machinery (Meister et al. 2001; Pellizzoni et al. 2002) as well as U7 snRNPs that carry out histone mRNA 3′ end processing (Pillai et al. 2003; Tisdale et al. 2013). SMN has also been implicated in the assembly of other RNA-binding proteins with a variety of coding and noncoding RNAs (Li et al. 2014), the most characterized of which is the formation of specific mRNPs for axonal transport and localized translation in neurons (Donlin-Asp et al. 2017). Reflecting the emerging multifaceted role of SMN in RNP assembly and post-transcriptional gene regulation, disruption of several distinct SMN-dependent RNA pathways has been proposed to contribute to SMA pathogenesis (Li et al. 2014; Donlin-Asp et al. 2016). However, proving clear mechanistic links between select SMN-dependent RNA pathways and specific disease-relevant features of SMA remains an outstanding challenge.
Here, we sought to address this issue by focusing on the molecular mechanisms of motor neuron death in SMA. The classical hallmark of SMA in both patients and mouse models is the progressive loss of spinal motor neurons, leading to skeletal muscle atrophy (Burghes and Beattie 2009; Tisdale and Pellizzoni 2015). Studies in SMA mice have shown that motor neuron death occurs cell-autonomously (Gogliotti et al. 2012; Martinez et al. 2012; McGovern et al. 2015; Fletcher et al. 2017), and distinct motor neuron pools display differential vulnerability to SMN deficiency (Mentis et al. 2011; Fletcher et al. 2017; Simon et al. 2017)—a feature reflected in the varying degree of denervation of the respective target muscles (Ling et al. 2012). This faithfully recapitulates clinical characteristics of the disease in which axial and proximal muscles are more affected than distal ones (Oskoui et al. 2017). Importantly, degeneration of vulnerable motor neurons in a mouse model of severe SMA is driven by a p53-dependent death pathway (Simon et al. 2017), and p53 induction has also been documented in motor neurons from milder mouse models of the disease (Murray et al. 2015; Jangi et al. 2017) as well as post-mortem spinal cords of SMA patients (Simic et al. 2000). However, the molecular mechanisms linking SMN deficiency to p53 activation and motor neuron death in SMA are unknown.
Under normal conditions, p53 expression is maintained at low levels by Mdm2 and Mdm4—two nonredundant negative regulators that act in concert to prevent unwarranted induction of the p53 pathway, which can lead to cell cycle arrest or cell death in a context-dependent manner (Vousden and Prives 2009). Mdm2 mainly serves as an E3 ubiquitin ligase that targets p53 for degradation, while Mdm4 inhibits p53 transcriptional activity in addition to enhancing Mdm2 function (Marine et al. 2006; Shadfan et al. 2012). We hypothesized that p53 activation and motor neuron degeneration in SMA could involve dysregulation of the repressive activity of Mdm2 and Mdm4 (i.e., p53 anti-repression) through splicing dysfunction induced by SMN deficiency. Consistent with this possibility, previous studies implicated alternative splicing of critical exons of Mdm2 and Mdm4 in the regulation of p53. Mdm2 transcripts that exclude exon 3 produce a truncated form of the protein that induces p53 stabilization through multiple mechanisms (Saucedo et al. 1999; Perry et al. 2000; Giglio et al. 2010). Skipping of exon 7 in Mdm4 introduces an early stop codon that induces nonsense-mediated decay or production of an inactive protein, leading to p53 activation through loss of function (Bezzi et al. 2013; Bardot et al. 2015; Dewaele et al. 2016).
In this study, we identify Mdm2 exon 3 and Mdm4 exon 7 as critical downstream targets whose coordinated alternative splicing is regulated by SMN through its function in snRNP biogenesis. Importantly, we show that defective splicing of these key regulatory exons induced by SMN deficiency, which occurs early and is most pronounced in vulnerable motor neurons, acts as a biological switch governing initiation of the p53 response in SMA mice. Through selective induction of exon skipping in wild-type mice, we demonstrate that perturbation of Mdm2 and Mdm4 alternative splicing synergistically induces p53 anti-repression such that both splicing events, but neither alone, are necessary and sufficient to elicit robust activation of p53 in vivo. Conversely, correction of either deficit by restoration of full-length Mdm2 or Mdm4 is sufficient to suppress p53 induction and prevent motor neuron degeneration in SMA. Thus, while SMN deficiency induces widespread transcriptome perturbations with the potential to contribute to SMA pathology (Zhang et al. 2008; Li et al. 2014), we show that it is the selective dysregulation of Mdm2 and Mdm4 alternative splicing that underlies p53 anti-repression and motor neuron death in a mouse model of the disease. These findings provide a direct mechanistic link between the disruption of SMN's function in the assembly of the snRNP constituents of the splicing machinery and the molecular basis of neurodegeneration in SMA.
Results
SMN deficiency disrupts Mdm2 and Mdm4 alternative splicing in SMA motor neurons
To determine the mechanisms by which SMN deficiency induces p53 in SMA, we investigated whether SMN regulates alternative splicing of Mdm2 exon 3 and Mdm4 exon 7, changes in which have been linked to p53 activation (Perry et al. 2000; Giglio et al. 2010; Bardot et al. 2015; Dewaele et al. 2016). In previously established NIH3T3-SmnRNAi fibroblasts with doxycycline (Dox)-inducible RNAi knockdown of Smn to ∼10% of normal levels (Lotti et al. 2012; Ruggiu et al. 2012; Tisdale et al. 2013), RT–PCR analysis revealed that the inclusion of both Mdm2 exon 3 and Mdm4 exon 7 was significantly reduced upon Smn deficiency (Fig. 1A–D), while splicing of other Mdm2 and Mdm4 exons was not affected (Supplemental Fig. S1). Moreover, these splicing changes were reversed by expression of RNAi-resistant human SMN in NIH3T3-SMN/SmnRNAi cells (Fig. 1A–D; Lotti et al. 2012; Ruggiu et al. 2012; Tisdale et al. 2013), highlighting the specificity of the effects for SMN depletion. These results demonstrate that SMN regulates the alternative splicing of specific exons of Mdm2 and Mdm4 mRNAs in vitro.
Figure 1.

SMN deficiency disrupts Mdm2 and Mdm4 alternative splicing in SMA motor neurons. (A) RT–PCR analysis of Mdm2 exon 3 splicing in NIH3T3-SmnRNAi and NIH3T3-SMN/SmnRNAi cells cultured with or without Dox using primers in exon 2 (E2) and exon 5 (E5). A schematic of the alternatively spliced exon is shown at the top. Arrowheads indicate the locations of primers. (B) RT–PCR analysis of Mdm4 exon 7 splicing in NIH3T3-SmnRNAi and NIH3T3-SMN/SmnRNAi cells cultured with or without Dox using primers in exon 5 (E5) and exon 8 (E8). A schematic of the alternatively spliced exon is shown at the top. Arrowheads indicate the locations of primers. (C) The percentage of Mdm2 exon 3 inclusion from RT–PCR data in A. Data represent mean and standard error of the mean (SEM). n = 3. (D) The percentage of Mdm4 exon 7 inclusion from RT–PCR data in B. Data represent mean and SEM. n = 3. (E) RT–PCR of Mdm2 exon 3 splicing in wild-type and SMA spinal cord at postnatal day 1 (P1), P6, and P11 using exon 2 and exon 5 primers. (F) RT–PCR of Mdm4 exon 7 splicing in wild-type and SMA spinal cords at P1, P6, and P11 using exon 5 and exon 8 primers. (G) The percentage of Mdm2 exon 3 inclusion in wild-type and SMA spinal cords from RT–PCR data in E. Data represent mean and SEM. n = 3. (H) The percentage of Mdm4 exon 7 inclusion in wild-type and SMA spinal cords from RT–PCR data in F. Data represent mean and SEM. n = 3. (I) Choline acetyltransferase (ChAT) and p53 immunostaining of L2 spinal segments from wild-type and SMA mice at P1, P6, and P11. Bar, 100 µm. (J) Schematic of the experimental approach used to isolate retrogradely labeled muscle-identified motor neurons from wild-type and SMA mice. (K) Bright-field and fluorescent images of a representative L1–L3 spinal segment used to isolate cholera toxin B subunit (CTb)-labeled motor neurons before and after laser capture microdissection (LCM). Bar, 200 µm. (L) RT–PCR analysis of Mdm2 exon 3 and Mdm4 exon 7 splicing in L1–L3 motor neurons from wild-type and SMA mice at P6. Exon 2 and exon 5 primers were used for Mdm2. Exon 5 and exon 8 primers were used for Mdm4. (M) The percentage of Mdm2 exon 3 and Mdm4 exon 7 inclusion in wild-type and SMA motor neurons from RT–PCR data in L. Data represent mean and SEM. n = 3. All statistics were performed with two-tailed unpaired Student's t-test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) no significance.
To assess whether SMN deficiency affects Mdm2 and Mdm4 splicing regulation in vivo, we used the well-established SMNΔ7 mouse model of SMA harboring homozygous knockout of mouse Smn, two copies of the human SMN2 gene, and transgenic SMNΔ7 cDNA (Le et al. 2005). We specifically monitored alternative splicing of Mdm2 and Mdm4 mRNAs by RT–PCR in the spinal cords of wild-type and SMA mice at presymptomatic (postnatal day 1 [P1]), early symptomatic (P6), and late symptomatic (P11) stages of the disease in this animal model. We found that SMN deficiency induces time-dependent progressive accumulation of Mdm2Δ3 and Mdm4Δ7 mRNAs in the spinal cords of SMA mice relative to control mice (Fig. 1E–H), revealing SMN-dependent dysregulation of Mdm2 and Mdm4 alternative splicing in vivo.
The accumulation of alternatively spliced Mdm2Δ3 and Mdm4Δ7 transcripts correlates with an increase in the number of cells displaying p53 activation induced by SMN deficiency over disease progression as revealed by immunohistochemistry analysis of spinal cords from wild-type and SMA mice with anti-p53 antibodies (Fig. 1I). As the onset of p53 activation occurs earliest in motor neurons before extending to other spinal neurons that do not degenerate in SMA mice (Fig. 1I; Simon et al. 2017), we next sought to determine the effects of SMN deficiency on Mdm2 and Mdm4 alternative splicing in vulnerable SMA motor neurons at an early stage of the disease. To do so, we retrogradely labeled vulnerable motor neurons that innervate the axial muscles iliopsoas (IL) and quadratus lumborum (QL) by intramuscular injection of fluorescently conjugated cholera toxin B subunit (CTb) in wild-type and SMA mice at P2 (Fig. 1J). We then isolated CTb-labeled motor neurons located in the lumbar segments L1–L3 by laser capture microdissection (LCM) from the spinal cords of injected mice at P6 (Fig. 1K)—a time point at which ∼60% of these motor neurons express p53 in SMA mice (see also Figs. 1I, 4A,B; Simon et al. 2017). Remarkably, RT–PCR analysis showed a severe reduction in the inclusion of Mdm2 exon 3 and Mdm4 exon 7 in vulnerable SMA motor neurons relative to wild-type motor neurons (Fig. 1L,M). Thus, SMN deficiency strongly affects the alternative splicing of Mdm2 and Mdm4 mRNAs in vulnerable SMA motor neurons. Moreover, consistent with a potential involvement in p53 activation and motor neuron degeneration, splicing dysregulation of these mRNAs occurs earlier and to a much greater extent in these disease-relevant neurons than in whole spinal cords of SMA mice.
Defective snRNP biogenesis induces Mdm2 and Mdm4 splicing dysregulation and p53 activation
Given SMN's essential function in the assembly of spliceosomal snRNPs (Meister et al. 2001; Pellizzoni et al. 2002), we investigated whether SMN-dependent effects on Mdm2 and Mdm4 splicing were mediated by deficits in snRNP biogenesis. To do so, we used a previously established cellular model in which Dox-dependent RNAi knockdown of SmB, a core protein component of spliceosomal snRNPs (Li et al. 2014), results in the selective impairment of snRNP biogenesis without altering SMN levels in NIH3T3-SmBRNAi fibroblasts (see also Fig. 2E,F; Ruggiu et al. 2012). Importantly, RT–PCR analysis showed that SmB knockdown strongly reduced inclusion of Mdm2 exon 3 and Mdm4 exon 7 in Dox-treated NIH3T3-SmBRNAi cells relative to untreated controls (Fig. 2A–D), mimicking the effects of SMN deficiency on the splicing of these mRNAs. Further characterization of NIH3T3-SmBRNAi cells revealed that SmB deficiency also led to marked up-regulation of p53 protein levels (Fig. 2E,F), nuclear accumulation of p53 (Fig. 2G), and robust mRNA up-regulation of p53 transcriptional targets (Fig. 2H). Thus, defective snRNP biogenesis dysregulates Mdm2 and Mdm4 alternative splicing and activates p53 in mammalian cells.
Figure 2.

Defective snRNP biogenesis dysregulates Mdm2 and Mdm4 alternative splicing and induces p53 activation. (A) RT–PCR analysis of Mdm2 exon 3 splicing in NIH3T3-SmBRNAi cells cultured with or without Dox using exon 2 (E2) and exon 5 (E5) primers. (B) RT–PCR analysis of Mdm4 exon 7 splicing in NIH3T3-SmBRNAi cells cultured with or without Dox using exon 5 (E5) and exon 8 (E8) primers. (C) The percentage of Mdm2 exon 3 inclusion from RT–PCR data in A. Data represent mean and SEM. n = 3. (D) The percentage of Mdm4 exon 7 inclusion from RT–PCR data in B. Data represent mean and SEM. n = 3. (E) RT-qPCR analysis of SmB mRNA levels in NIH3T3-SmBRNAi cells cultured with or without Dox. Data represent mean and SEM. n = 3. (F) Western blot analysis of NIH3T3-SmBRNAi cells cultured with and without Dox. (G) p53 and phospho-p53S18 immunostaining of NIH3T3-SmBRNAi cells cultured with or without Dox. Bar, 50 µm. (H) RT-qPCR analysis of mRNA levels of p53 transcriptional targets in NIH3T3-SmBRNAi cells cultured with or without Dox. Normalized data from Dox-treated cells relative to untreated cells are shown as mean and SEM. n = 3. (I) ChAT, SmB, and p53 immunostaining of L2 spinal segments from wild-type and SMA mice at P6. Bar, 25 µm. Arrowheads point to p53+ SMA motor neurons. (J) Normalized SmB fluorescence intensity in wild-type as well as p53+ and p53− SMA L2 motor neurons at P6 from experiments as in I. Each point represents SmB fluorescent intensity in a single L2 motor neuron, and data were collected from three mice. Statistics were performed with two-tailed unpaired Student's t-test (C,D,E,H) or one-way ANOVA with Tukey's post-hoc test (J). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
We showed previously that snRNP levels are reduced much more prominently in the nuclei of SMA motor neurons than other cells in the spinal cord of SMA mice (Ruggiu et al. 2012; Tisdale and Pellizzoni 2015). To investigate whether SMN-dependent snRNP reduction correlated with p53 activation in SMA motor neurons in vivo, we performed immunostaining experiments with antibodies against p53 and SmB using spinal cords from wild-type and SMA mice at P6 followed by quantification of the nuclear levels of SmB as a proxy for snRNP expression in vulnerable L2 motor neurons (Fig. 2I). We found that p53+ SMA motor neurons had significantly lower levels of nuclear SmB than either SMA or wild-type motor neurons that are p53− (Fig. 2I,J), indicating a correlation between snRNP reduction and p53 activation in SMA motor neurons.
Taken together, these findings mechanistically link snRNP dysfunction induced by SMN deficiency to dysregulation of Mdm2 and Mdm4 splicing and up-regulation of p53, consistent with a snRNP-mediated mechanism of p53 activation in SMA.
Skipping of SMN-regulated Mdm2 and Mdm4 exons synergistically activates p53
To determine whether accumulation of alternatively spliced Mdm2 and Mdm4 mRNAs induced by SMN deficiency is sufficient to induce p53 activation in vivo, we designed antisense morpholino oligonucleotides (MOs) complementary to the 5′ splice sites of mouse Mdm2 exon 3 and Mdm4 exon 7 (Fig. 3A), which are predicted to block U1 snRNP binding and promote exon skipping without altering endogenous mRNA levels. The Mdm2 and Mdm4 MOs were delivered by intracerebroventricular (ICV) injection in wild-type mice at P0 either alone or in combination in order to define the individual contribution of these splicing events to p53 activation. RT–PCR analysis of Mdm2 exon 3 and Mdm4 exon 7 splicing in the spinal cords of injected mice at P11 showed that Mdm2 and Mdm4 MOs acted selectively to induce skipping of their targeted exons (Fig. 3B). Moreover, injection of differing amounts of MOs demonstrated dose-dependent accumulation of alternatively spliced Mdm2Δ3 and Mdm4Δ7 mRNAs relative to wild-type mice injected with the highest dose of a nontargeting MO used as a control (Supplemental Fig. S2A). These experiments validated the specificity and efficacy of the designed MOs for inducing skipping of the intended alternatively spliced exons.
Figure 3.

Skipping of SMN-regulated Mdm2 and Mdm4 exons synergistically triggers p53 activation. (A) Schematic of splice-switching MOs targeting the 5′ splice sites of Mdm2 exon 3 and Mdm4 exon 7. (B) RT–PCR analysis of Mdm2 exon 3 and Mdm4 exon 7 splicing in the spinal cords of wild-type mice at P11 following injection of 400 µg of control MO or 200 µg of each splice-switching MO individually or together at P0. Exon 2 and exon 5 primers were used for Mdm2. Exon 5 and exon 8 primers were used for Mdm4. (C) ChAT and p53 immunostaining of L5 spinal segments at P11 from wild-type mice injected with 400 µg of control or 200 µg of each splice-switching MO at P0. Bar, 250 µm. (D) RT-qPCR analysis of p53 transcriptional targets in the spinal cords of wild-type mice at P11 following injection of 400 µg of control or 200 µg of each splice-switching MO at P0. Data represent mean and SEM. n = 3. (E) RT-qPCR analysis of p53 and Smn mRNA from the same groups as in D. Data represent mean and SEM. n = 3. (F) ChAT and p53 immunostaining of L5 spinal segments at P11 from wild-type mice injected with 200 µg of control or 100 µg of each splice-switching MO at P0. Insets show representative images of L5 medial motor column (MMC) motor neurons at higher magnification. Bar, 100 µm; bar in inset, 25 µm. (G) The percentage of p53+ L5 MMC motor neurons from experiments as in F. Data represent mean and SEM. n = 3. (H) ChAT and phospho-p53Ser18 immunostaining of L5 spinal segments from experiments as in F. Insets show representative images of L5 MMC motor neurons at higher magnification. Bar, 100 µm; bar in inset, 25 µm. (I) The total number of L5 MMC motor neurons from experiments as in H. Data represent mean and SEM. n = 3. Statistics were performed with one-way ANOVA with Tukey's post-hoc test (D,E) or two-tailed unpaired Student's t-test (G,I). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) no significance.
Next, we analyzed whether treatment with these splice-switching MOs had an effect on the p53 pathway. Strikingly, immunostaining experiments revealed that either MO alone induced little, if any, up-regulation of p53 in the mouse spinal cord at P11, while treatment with both MOs triggered robust and widespread p53 accumulation (Fig. 3C). Moreover, RT-qPCR analysis of p53 transcriptional targets, which are also induced in SMA mice (see also Fig. 4E; Murray et al. 2015; Jangi et al. 2017; Simon et al. 2017), showed relatively small increases upon treatment with either MO individually and strong induction when MOs were delivered in combination (Fig. 3D). Importantly, these synergistic effects were dose-dependent (Supplemental Fig. S2B,C), and activation of the p53 pathway induced by MOs targeting Mdm2 and Mdm4 occurred without changes in the expression of Smn or p53 mRNAs (Fig. 3E), consistent with a post-transcriptional mechanism involving impairment of p53 degradation with consequent protein stabilization. Together, these experiments reveal the remarkable potency with which p53 can be activated through concurrent perturbation of Mdm2 exon 3 and Mdm4 exon 7 splicing.
Figure 4.

Mdm2 and Mdm4 gene delivery suppresses p53 activation in SMA. (A) ChAT and p53 immunostaining of L2 spinal segments at P11 from uninjected wild-type mice and SMA mice injected at P0 with adeno-associated virus serotype 9 (AAV9)-GFP, AAV9-Mdm2, and AAV9-Mdm4 either alone or in combination as indicated. Bar, 50 µm. (B) The percentage of p53+ L2 motor neurons at P11 from the same groups as in A. Data represent mean and SEM. n = 3. (C) ChAT and p53 immunostaining of L5 spinal segments at P11 from the same groups as in A. Bar, 50 µm. (D) The percentage of p53+ L5 MMC motor neurons at P11 in the same groups as in A. Data represent mean and SEM. n = 3. (E) RT-qPCR analysis of p53 transcriptional targets in the spinal cords at P11 from uninjected wild-type mice and SMA mice injected at P0 with either AAV9-GFP or AAV9-Mdm2 and AAV9-Mdm4 together. Data represent mean and SEM. n = 6. (F) RT-qPCR analysis of the levels of aberrantly spliced Stasimon (Stas Aber) mRNA, 3′ end-extended H1c (H1c pre) mRNA, and chondrolectin (Chodl) mRNA in the spinal cords at P11 from the same groups as in E. Data represent mean and SEM. n = 6. All statistics were performed with one-way ANOVA with Tukey's post-hoc test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) no significance.
Last, we analyzed the effects in motor neurons and found that ∼15% of L5 medial motor column (MMC) motor neurons were p53+ following delivery of Mdm2 and Mdm4 MOs in wild-type mice (Fig. 3F,G), demonstrating that increased Mdm2 and Mdm4 skipping is sufficient to induce p53 in a motor neuron population that is vulnerable in SMA. We showed previously that SMA motor neuron death requires the convergence of p53 up-regulation with p53 phosphorylation (Simon et al. 2017). Accordingly, phosphorylation of Ser18 of mouse p53 (phospho-p53S18) is a specific marker of degenerating SMA motor neurons, and overexpression of unphosphorylated p53 is not sufficient to induce degeneration of wild-type motor neurons in vivo (Simon et al. 2017). We therefore performed motor neuron counts and immunostaining for phospho-p53S18 in the spinal cords of wild-type mice following coinjection of Mdm2 and Mdm4 MOs. We did not observe phospho-p53S18 expression in motor neurons or other spinal cells (Fig. 3H), and, as expected, in accordance with our previous findings, there was no change in the number of L5 MMC motor neurons relative to control mice (Fig. 3I). Thus, skipping of SMN-regulated Mdm2 and Mdm4 exons can act as an initiating step for p53 stabilization but not for phosphorylation of Ser18 in vivo.
Mdm2 and Mdm4 gene delivery inhibits p53 activation in SMA mice
To determine whether changes in Mdm2 and Mdm4 alternative splicing were responsible for p53 activation in SMA, we used adeno-associated virus serotype 9 (AAV9)-mediated gene delivery of full-length mouse Mdm2 and Mdm4 cDNAs in SMA mice by ICV injection at P0 (Supplemental Fig. S3A). SMA mice injected with AAV9-GFP and uninjected wild-type mice were used as controls. RT-qPCR analysis demonstrated robust expression of Mdm2 and Mdm4 mRNAs over endogenous levels in the spinal cords of mice injected with the corresponding AAV9 vectors relative to controls at P11 (Supplemental Fig. S3B,C). We also attempted to monitor Mdm2 and Mdm4 protein levels but failed to identify commercially available antibodies that detected endogenous or overexpressed levels of these proteins by either immunohistochemistry or Western blot. AAV9-Mdm2 and AAV9-Mdm4 did not alter the low levels of SMN produced by the SMN2 gene at the mRNA or protein level (Supplemental Fig. S3D,E). We then investigated whether AAV9-Mdm2 or AAV9-Mdm4 affected the expression of p53 in vulnerable motor neurons of SMA mice. In agreement with our previous study (Simon et al. 2017), we found that ∼60% of L2 motor neurons and ∼70% of L5 MMC motor neurons are p53+ in SMA mice injected with AAV-GFP at P11 (Fig. 4A–D), while no p53 expression is detectable in wild-type motor neurons. Importantly, injection of AAV9-Mdm2 and AAV9-Mdm4 either individually or in combination strongly reduced p53 expression in these SMA motor neurons (Fig. 4A–D), with p53 suppression being more effective upon coinjection of both vectors. The observation that overexpression of either Mdm2 or Mdm4 efficiently inhibits nuclear accumulation of p53 in SMA motor neurons is consistent with p53 induction requiring the synergistic effects of exon skipping in both Mdm2 and Mdm4, which are invariably muted when the full-length mRNA isoform of either gene is reinstated.
Next, to determine whether Mdm2 and Mdm4 overexpression selectively inhibits p53 transcriptional activity in SMA mice, we monitored by RT-qPCR the mRNA levels of several p53 target genes in the spinal cords of SMA mice relative to wild-type mice at P11. We found that coinjection of AAV9-Mdm2 and AAV9-Mdm4, but not AAV9-GFP, robustly decreased the mRNA levels of p53-regulated genes that are induced by SMN deficiency in the spinal cords of SMA mice (Fig. 4E), demonstrating functional inhibition of p53. As expected, the correction was not complete because AAV9 mainly transduces motor neurons but not many other spinal cells that express p53 at a late disease stage in SMA mice (see Fig. 1I; Simon et al. 2017). To establish the specificity of the effects of Mdm2 and Mdm4 for the suppression of p53-dependent gene changes in SMA, we monitored known RNA processing events in other SMN-regulated pathways disrupted in SMA. Specifically, we analyzed U12-dependent splicing of Stasimon (Lotti et al. 2012), U7-dependent 3′ end processing of histone mRNAs (Tisdale et al. 2013), and the expression of chondrolectin mRNA (Zhang et al. 2008; Bäumer et al. 2009), all of which were altered in the spinal cords of SMA mice relative to wild-type mice but not corrected by Mdm2 and Mdm4 overexpression (Fig. 4F). Together, these results demonstrate that overexpression of full-length Mdm2 and/or Mdm4 selectively suppresses the SMN-dependent activation of p53 in SMA mice.
Mdm2 and Mdm4 restoration inhibits motor neuron degeneration and improves motor function in SMA mice
Based on the link between SMN-dependent dysregulation of Mdm2 and Mdm4 alternative splicing and the induction of p53, we sought to determine the effect of overexpressing Mdm2 and Mdm4 on motor neuron survival in SMA mice. To do so, we delivered AAV9-Mdm2 and AAV9-Mdm4 either individually or in combination in SMA mice by ICV injection at P0 and then quantified the number of vulnerable L2 and L5 MMC motor neurons at P11, which is significantly decreased in SMA mice according to our previous studies (Mentis et al. 2011; Fletcher et al. 2017; Simon et al. 2017). Remarkably, we found that overexpression of either Mdm2 or Mdm4 alone strongly suppressed degeneration of L2 and L5 MMC SMA motor neurons with an efficacy comparable with that achieved with their coexpression (Fig. 5). Thus, restoration of either Mdm2 or Mdm4 full-length mRNAs is sufficient to prevent p53 up-regulation induced by SMN deficiency through the synergistic effects of Mdm2 and Mdm4 missplicing and the subsequent p53-dependent cascade leading to degeneration of SMA motor neurons.
Figure 5.

Full-length Mdm2 and Mdm4 overexpression prevents motor neuron degeneration in SMA mice. (A) ChAT immunostaining of L2 spinal segments at P11 from uninjected wild-type mice and SMA mice injected with AAV9-GFP, AAV9-Mdm2, and AAV9-Mdm4 either alone or in combination. Bar, 50 µm. (B) The total number of L2 motor neurons from the same groups as in A at P11. Data represent mean and SEM. n ≥ 3. (C) ChAT immunostaining of L5 spinal segments at P11 from the same groups as in A. Bar, 100 µm. (D) The total number of L5 MMC motor neurons from the same groups as in A at P11. Data represent mean and SEM. n ≥ 4. All statistics were performed with one-way ANOVA with Tukey's post-hoc test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
In addition to motor neuron degeneration, SMN deficiency leads to several deficits in the motor circuit (Tisdale and Pellizzoni 2015). To further assess the impact of Mdm2 and Mdm4 overexpression on the SMA neuromuscular phenotype, we investigated functional and morphological parameters of the sensory–motor circuit that are severely affected in the mouse model of the disease (Mentis et al. 2011; Fletcher et al. 2017). First, we analyzed changes in sensory–motor function by monitoring the spinal reflex in the L2 segment of the spinal cord at P11. We found that AAV-mediated Mdm2 and Mdm4 overexpression moderately but significantly increased the amplitude of the monosynaptic reflex that is strongly reduced in SMA mice injected with AAV9-GFP relative to wild-type mice (Fig. 6A,B). This partial functional recovery likely reflects the sparing of SMA motor neurons rather than specific benefits on the number and function of the proprioceptive (VGluT1+) synapses that impinge on motor neuron somata, the loss of which was not prevented by AAV9-Mdm2 and AAV9-Mdm4 injection in SMA mice (Fig. 6C,D). These experiments also indicate that SMA motor neurons rescued from death by Mdm2 and Mdm4 gene delivery are capable of eliciting functional responses (i.e., firing action potentials) following synaptic activation.
Figure 6.

Full-length Mdm2 and Mdm4 overexpression moderately improves spinal reflexes and motor behavior in SMA mice. (A) Traces of extracellular recordings of L2 ventral roots following L2 dorsal root stimulation from uninjected wild-type mice and SMA mice injected with either AAV9-GFP or AAV9-Mdm2 and AAV9-Mdm4 at P11. Arrowheads indicate the stimulus artifact. Arrows indicate the monosynaptic response. Bars, 5 msec and 0.5 mV. (B) Spinal reflex amplitudes from the same groups as in A. Data represent mean and SEM. n ≥ 3. (C) ChAT and VGluT1 immunostaining of L2 spinal segments from the same groups as in A at P11. Bar, 25 µm. (D) Total number of VGluT1+ synapses on L2 motor neuron somata from the same groups as in A at P11. Data represent mean and SEM. n ≥ 22 neurons from three mice per group. (E) Immunostaining of presynaptic (with NF-M and SYP antibodies) and post-synaptic (with BTX) terminals of neuromuscular junctions (NMJs) in the QL muscle from the same groups as in A at P11. Arrowheads indicate fully denervated NMJs. Bar, 25 µm. (F) The percentage of fully denervated NMJs in the QL muscle from the same groups as in A at P11. Data represent mean and SEM. n ≥ 3. (G) Compound muscle action potential (CMAP) traces recorded from the QL muscle following L2 ventral root stimulation in the same groups as in A at P11. Arrowheads indicate the stimulus artifact. Arrows indicate the peak of the M response. Bars, 5 msec and 0.5 mV. (H) The amplitude of the M response from the QL muscle in the same groups as in A at P11. Data represent mean and SEM. n ≥ 3. (I–K) Righting time (I), weight gain (J), and survival (K) of uninjected wild-type mice (n = 15) and wild-type and SMA mice injected with AAV9-GFP (n = 16) or AAV9-Mdm2 and AAV9-Mdm4 (n = 10) as indicated. Statistics were performed with one-way ANOVA with Tukey's post-hoc test (B,D,F,H) or two-way ANOVA with Bonferroni's post-hoc test (I). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) no significance.
We next analyzed the impact of Mdm2 and Mdm4 overexpression on the innervation and function of neuromuscular junctions (NMJs) in the QL muscle of SMA mice. AAV9-Mdm2 and AAV9-Mdm4 did not improve either the severe NMJ denervation (Fig. 6E,F) or the strong reduction in the amplitude of the compound muscle action potential (CMAP) (Fig. 6G,H) found in AAV9-GFP-injected SMA mice relative to wild-type mice. Nevertheless, overexpression of Mdm2 and Mdm4 transiently improved the righting time of SMA mice (Fig. 6I) and, while it had no toxic effects in normal mice, did not enhance weight gain or survival in SMA mice (Fig. 6I–K). These results are consistent with both the beneficial effects of genetic or pharmacological inhibition of p53 in the same model being limited to improved motor behavior (Simon et al. 2017) and the contribution of peripheral pathology to the growth and survival phenotype of SMA mice (Hua et al. 2011; Hamilton and Gillingwater 2013). They also indicate that Mdm2 and Mdm4 restoration improves motor function by acting on the p53-mediated pathway governing motor neuron degeneration but not on the mechanisms underlying synaptic loss in SMA mice.
Discussion
Establishing clear mechanistic links between the multiple roles of SMN in RNA regulation and specific downstream phenotypic effects relevant to SMA pathology is critical in understanding disease mechanisms. Since SMN deficiency can trigger widespread deficits in gene expression that can impact many downstream cellular processes (Li et al. 2014), an outstanding challenge in the field has been to identify which functions and specific targets of SMN are causally related to clinical features of SMA. Here, we addressed this issue by focusing on the molecular mechanisms underlying a hallmark of SMA (motor neuron degeneration) that have long remained elusive. Building on our recent discovery that p53 activation is central to the degenerative process of SMA motor neurons (Simon et al. 2017), we demonstrate that SMN function in snRNP biogenesis is required for coordinated regulation of Mdm2 and Mdm4 alternative splicing and that defective inclusion of a key regulatory exon in each of these genes acts synergistically to drive p53 anti-repression and motor neuron degeneration in a mouse model of SMA. Collectively, our findings establish a causal link between disrupted assembly of spliceosomal snRNPs and neurodegeneration and reveal the identity of the upstream molecular events that underlie this pathogenic process in SMA (Fig. 7).
Figure 7.
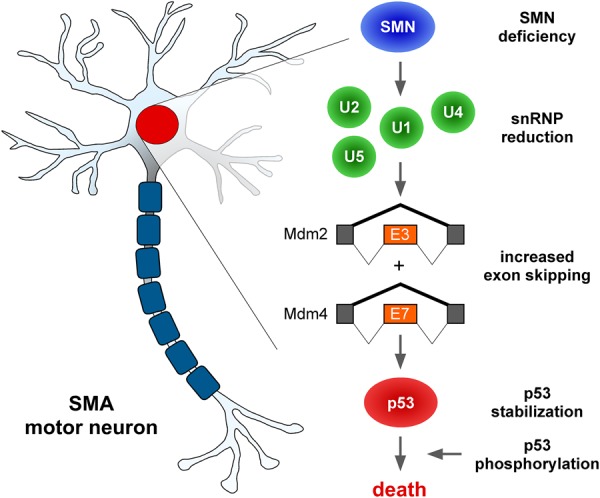
Splicing mechanisms of p53 anti-repression and motor neuron death in SMA. Schematic summary of the molecular cascade initiated by SMN deficiency, followed by reduction in the levels of spliceosomal snRNPs of the major spliceosome and consequent dysregulation of Mdm2 exon 3 and Mdm4 exon 7 alternative splicing, resulting in their increased skipping. Dysregulation of both splicing events is required to synergistically induce p53 stabilization, which is the initiating step in the death pathway leading to motor neuron loss in SMA mice. See the text for additional details.
The discovery that p53 activation drives motor neuron degeneration in SMA mice raised important mechanistic questions regarding the identity of the upstream triggers induced by SMN deficiency (Simon et al. 2017). Given that SMA motor neurons showed no evidence for DNA damage, which is a well-known inducer of p53 activation (Simon et al. 2017), we focused on other possible mechanisms. Using both cellular and animal models, we show that SMN deficiency disrupts the balance between inclusion and exclusion of key regulatory exons in Mdm2 and Mdm4 (two well-established effectors in the cellular control of p53) (Marine et al. 2006), leading to a switch in their biological activity from repression to activation of p53. The requirement of SMN for the regulation of Mdm2 exon 3 and Mdm4 exon 7 alternative splicing is demonstrated in mammalian cells in vitro using a well-controlled homogeneous model system (Lotti et al. 2012; Tisdale et al. 2013) as well as in vivo through the analysis of both the spinal cords and motor neurons from SMA mice (Fig. 1). Moreover, previous studies provide the basis to explain how increased levels of alternatively spliced Mdm2 and Mdm4 mRNAs and a corresponding reduction in the levels of full-length transcripts trigger p53 anti-repression through loss-of-function and dominant-negative effects on the activity of Mdm2 (Perry et al. 2000; Giglio et al. 2010) and Mdm4 (Bezzi et al. 2013; Bardot et al. 2015).
From a mechanistic standpoint, our findings indicate that Mdm2 and Mdm4 exon skipping induced by SMN deficiency is a direct consequence of impaired assembly of spliceosomal snRNPs, which is the best-characterized activity of SMN (Burghes and Beattie 2009; Li et al. 2014). First, the reduction of snRNP levels upon SMN depletion is well documented in both NIH3T3 cells (Lotti et al. 2012) and SMA motor neurons (Ruggiu et al. 2012; Tisdale and Pellizzoni 2015), in which we identified Mdm2 and Mdm4 splicing defects (Fig. 1). Second, the earliest and strongest effects of SMN depletion on alternative splicing of Mdm2 and Mdm4 mRNAs are found in disease-relevant vulnerable SMA motor neurons relative to other spinal cells (Fig. 1), coincident with more pronounced snRNP reduction and p53 up-regulation (Fig. 2). Third, selective impairment of snRNP biogenesis through SmB knockdown in NIH3T3 cells induces robust skipping of both Mdm2 exon 3 and Mdm4 exon 7 as well as p53 activation in the presence of normal SMN levels (Fig. 2), thereby directly linking dysregulation of these alternative splicing events to snRNP dysfunction. Potential mechanisms underlying motor neuron selectivity at early disease stages include the intrinsically low efficiency of SMN2 exon 7 splicing and activation of a negative feedback that specifically exacerbates SMN2 splicing defects and downstream effects of SMN deficiency through snRNP dysfunction in SMA motor neurons (Jodelka et al. 2010; Ruggiu et al. 2012). At later symptomatic stages, wider dysregulation of Mdm2 and Mdm4 splicing and accumulation of p53 in the spinal cords of SMA mice likely involves similar mechanisms induced by hypoxia and other stressors that decrease SMN2 splicing (Bebee et al. 2012; Somers et al. 2016). In agreement with regulation of Mdm2 and Mdm4 splicing by the snRNP biogenesis pathway, knockdown of protein arginine methyltransferase 5—a protein that carries out symmetric dimethylation of a subset of Sm proteins prior to their transfer to the SMN complex (Friesen et al. 2001; Meister et al. 2001)—has been shown to promote skipping of Mdm4 exon 7 and p53 activation (Bezzi et al. 2013). Expanding on this study in which SMN function and Mdm2 splicing were not investigated, our data indicate that coordinated regulation of Mdm2 and Mdm4 alternative splicing acts as a sensor of impaired snRNP biogenesis and splicing dysfunction to elicit p53-mediated biological responses and that this SMN-dependent regulatory axis is disrupted in SMA.
To assess directly the role of Mdm2 and Mdm4 alternative splicing in the regulation of p53, we used splice-switching oligonucleotides that specifically modify the inclusion of SMN-regulated exons. Remarkably, we found that exclusion of either Mdm2 exon 3 or Mdm4 exon 7 alone had mild effects on p53 activity, while concomitant induction of both Mdm2 and Mdm4 exon skipping events synergizes to elicit a much more robust p53 response, including widespread nuclear accumulation of p53 and up-regulation of its transcriptional targets in the spinal cords of wild-type mice (Fig. 3). Thus, coordinate skipping of SMN-regulated exons of Mdm2 and Mdm4 is both necessary and sufficient for robust p53 induction, likely through protein stabilization, based on the function of these factors in p53 regulation (Marine et al. 2006) and because mRNA levels of p53 were unchanged (Fig. 3E). However, these splicing events alone do not cause the death of wild-type motor neurons in vivo. This is consistent with our previous demonstration that p53 overexpression per se is not sufficient to induce degeneration of motor neurons but requires convergence of at least another activating event (N-terminal phosphorylation of p53, of which Ser18 phosphorylation is a marker) (Simon et al. 2017) that is independent from Mdm2 and Mdm4 exon skipping (Fig. 3). Importantly, functional analysis of the role of Mdm2 and Mdm4 splicing dysregulation in SMA revealed that overexpression of either target is sufficient to prevent p53 accumulation and rescue SMA motor neurons from degeneration to levels comparable with their coexpression in SMA mice (Figs. 4 and 5). This further highlights the requirement of both splicing events for synergistic induction of p53 and, conversely, the sufficiency of correcting either deficit for suppression of p53-mediated death of SMA motor neurons. Therefore, while future studies are needed to define the converging pathways responsible for N-terminal phosphorylation of p53, our results conclusively establish that dysregulated alternative splicing of two specific target genes induced by SMN deficiency is the initiating event that underlies synergistic activation of p53 and neurodegeneration in SMA (Fig. 7).
Our findings have potential therapeutic implications for human disease that include but are not limited to neurodegeneration in SMA. Mdm2 and Mdm4 are negative regulators of p53 that prevent its undue activation under normal conditions (Vousden and Prives 2009). However, they are also oncogenes, as their overexpression promotes uncontrolled cell proliferation through excess inhibition of the tumor suppressor activity of p53 in certain cancers (Wade et al. 2013). Moreover, enhanced skipping of Mdm4 exon 7 (exon 6 in human MDM4) has been proposed as a candidate therapeutic approach to activate the p53 response in specific cancers (such as melanomas and diffuse large B-cell lymphoma) that retain a functional p53 allele (Dewaele et al. 2016; Valianatos et al. 2017). Considering the strong synergistic effects that we demonstrated here, induction of both Mdm2 exon 3 and Mdm4 exon 7 skipping has the potential for much more pronounced activation of p53 and increased therapeutic benefit in cancer. Conversely, promoting the inclusion of either SMN-regulated exon in Mdm2 or Mdm4 mRNAs could represent a candidate approach to counteract the loss of SMA motor neurons in an SMN-independent manner. Our results indicate that selective blockade of motor neuron death results in partial functional recovery and improved motor ability early in the disease course of SMA mice (Fig. 6), while the overall benefit on the neuromuscular phenotype is limited by the progressive loss of central synapses and NMJ innervation, which are distinct pathogenic events. At a time when disease-modifying treatments are emerging but comprehensive understanding of what aspects of SMA pathology are insufficiently addressed in patients is lacking (Finkel et al. 2017; Mendell et al. 2017), there is increasing interest in the identification of combinatorial approaches that might complement SMN-based therapeutics (Van Alstyne and Pellizzoni 2016; Groen et al. 2018). Thus, despite the fact that rescue of motor neuron survival alone is expectedly not sufficient to reverse the severe SMA phenotype (Gogliotti et al. 2012; Martinez et al. 2012; McGovern et al. 2015; Fletcher et al. 2017), selectively targeting the motor neuron death pathway has the potential to enhance the clinical benefit of SMN-inducing therapeutics by preventing the irreplaceable loss of the most affected cell type in SMA and extending the window of opportunity for amelioration of synaptic pathology. Induction of p53 has also been documented in patient tissue and experimental models of several chronic and acute neurodegenerative diseases (Culmsee and Mattson 2005), including—but not limited to—Alzheimer's disease (de la Monte et al. 1997; Kitamura et al. 1997), Parkinson's disease (Duan et al. 2002; Mogi et al. 2007; Qi et al. 2016), ischemic stroke, and traumatic brain injury (Yang et al. 2016). In light of our results, it would be relevant to evaluate the involvement of Mdm2 and Mdm4 splicing dysregulation in neuronal loss in this diverse group of neurological disorders.
In conclusion, our findings mechanistically link dysregulation of alternative splicing induced by SMN deficiency with motor neuron death, the key hallmark of SMA. Furthermore, they identify decreased SMN function in snRNP biogenesis as the initial trigger and defective alternative splicing of Mdm2 and Mdm4 as the downstream effectors responsible for the up-regulation of p53 that underlies the neurodegenerative process in SMA. In addition to elucidating a fundamental pathogenic mechanism of SMA, the molecular cascade of events emerging from our study provides new entry points for exploring SMN-independent neuroprotective approaches for preventing motor neuron death that may also have broader implications in other neurodegenerative diseases associated with perturbations of RNA splicing and p53 regulation.
Materials and methods
DNA constructs
Plasmids containing cDNAs for full-length mouse Mdm2 and Mdm4 were purchased from Dharmacon (Mdm2 clone ID 6415937, accession no. BC050902; Mdm4 clone ID 3599906, accession no. BC003750). DNA fragments corresponding to the ORFs of GFP and full-length Mdm2 and Mdm4 generated by PCR using these plasmids as templates were cloned downstream from the GUSB promoter of vectors harboring AAV2 inverted terminal repeats (ITRs) for the production of self-complementary AAV9. All constructs were verified by DNA sequencing.
AAV9 production
DNA for the production of AAV9 vectors was purified using endotoxin-free Mega preparaton kit (Qiagen) according to the manufacturer's instructions. The recombinant plasmids were each packaged into AAV9 capsid by triple-plasmid transfection of human HEK293 cells, and virions were purified by CsCl gradient centrifugation as described previously (Mueller et al. 2012). The resulting vectors were concentrated to final titers of ∼2 × 1013 genome copies per milliliter using Amicon Ultracel centrifugal filter devices with a 30,000 nominal molecular weight limit (Millipore).
Cell lines and treatments
NIH3T3 cell lines used in this study were described previously (Lotti et al. 2012; Ruggiu et al. 2012) and were treated with 100 ng/mL Dox for 5 d (SmnRNAi and SMN/SmnRNAi) or 60 h (SmBRNAi) prior to analysis.
SMA mice and experimental procedures
All mouse work was performed in accordance with the National Institutes of Health Guide on the Care and Use of Animals and approved by the Institutional Animal Care and Use Committee of Columbia University. Equal proportions of mice of both sexes were used, and aggregated data are presented because gender-specific differences were not found. The SMNΔ7 mouse line (Smn+/−/ SMN2+/+/SMNΔ7+/+) used in this study to generate SMA mice was on a pure FVB background and was obtained from Jackson Mice (Jax stock no. 005025). Genotyping of the Smn knockout allele was performed using tail DNA PCR and the primers listed in Supplemental Table S1 as described previously (Fletcher et al. 2017). All ICV injections were carried out in P0 mice anesthetized by isoflurane inhalation by a single injection in the right lateral ventricle of the brain. After 30 min of recovery, pups were placed in their breeder cage and monitored daily for weight and righting time. For AAV9 gene delivery, we delivered 1 × 1011 genome copies of the indicated AAV9 vectors in a PBS solution containing a vital dye (Fast Green, Sigma). For MO injections, varying doses (25–400 µg) of the described oligonucleotides in a PBS solution containing a vital dye (Fast Green, Sigma) were delivered. MOs were obtained from GeneTools, and the sequences are listed in Supplemental Table S1. Mice were sacrificed, and tissue collection was performed in a dissection chamber under continuous oxygenation (95%O2/5%CO2) in the presence of cold (∼12°C) artificial cerebrospinal fluid (aCSF) containing 128.35 mM NaCl, 4 mM KCl, 0.58 mM NaH2PO4, 21 mM NaHCO3, 30 mM D-glucose, 1.5 mM CaCl2, and 1 mM MgSO4.
Motor neuron isolation by LCM
LCM of motor neurons was carried out essentially as described previously (Lotti et al. 2012). Briefly, to retrogradely label motor neurons, the IL and QL muscles of P2 wild-type and SMA mice were exposed, and ∼1 µL of CTb conjugated to Alexa 488 was delivered by intramuscular injection using a finely pulled glass microelectrode. At P6, the spinal cord was dissected, and the L1–L3 segments were embedded in OCT and flash-frozen. Cryosections of 14 µm were mounted on PEN-membrane slides 2.0 (Zeiss) and fixed in 100% ethanol for 15 sec prior to LCM. Following air drying for 30 sec, individual motor neurons were microdissected using a DM6000B microscope equipped with a LMD6000 laser capture unit (Leica). Approximately 200 motor neurons were collected bilaterally from L1–L3 spinal segments of one or more mice for each sample group and biological replicate.
RNA and protein analysis
For RNA analysis, purification of total RNA from mouse spinal cords and NIH3T3 cells was carried out using TRIzol reagent (Invitrogen) as per the manufacturer's instructions followed by treatment with RNase-free DNaseI (Ambion). cDNA was generated using RevertAid RT reverse transcription kit (Thermo Fisher) with random hexamer and oligo dT primers. RT–PCRs were first performed at increasing cycle numbers to ensure that analysis was performed within the linear range of amplification using AmpliTaq Gold DNA polymerase (Thermo Fisher). The identities of all of the RT–PCR products were confirmed by DNA sequencing. RT-qPCR analysis was done using SYBR Green (Applied Biosystems) in technical triplicates. Total RNA was purified from LCM motor neurons using the Absolutely RNA Nanoprep kit (Agilent). Amplified cDNA was prepared from total RNA using the Ovation PicoSL WTA system V2 kit (Nugen) and purified with the MinElute reaction cleanup kit (Qiagen). RNA quality and quantity were assessed using the 2100 Bioanalyzer (Agilent). The primers used for RT–PCR and RT-qPCR experiments are listed in Supplemental Table S1.
For Western blot analysis, proteins from spinal cord tissue or NIH3T3 cells were homogenized in SDS-PAGE sample buffer and quantified using RC DC protein assay (Bio-Rad). Protein extracts were run on 12% polyacrylamide gels and transferred to nitrocellulose membranes for probing. Blocking was done in 5% milk in PBS/0.1% Tween, and primary and secondary antibodies were diluted in PBS/0.1% Tween. The antibodies used for these experiments are listed in Supplemental Table S2.
Immunohistochemistry and immunofluorescence analysis
For spinal cord analysis, dissected tissue was fixed in 4% PFA overnight, and then specific lumbar segments were identified by the ventral roots. Segments were embedded in agar and sectioned at 75 µm with a VT1000 S vibratome (Leica). Sections were blocked in 10% donkey serum in 0.01 M PBS/0.4% Triton-X for 1.5 h and then incubated with primary antibodies overnight at room temperature. Six washing steps of 10 min each were done prior to incubation with secondary antibodies (Jackson ImmunoResearch) in PBS for 3 h at room temperature. Another six washing steps were performed before sections were mounted in 30% glycerol/PBS.
For NMJ analysis of skeletal (QL) muscle, freshly dissected tissue was fixed in 4% PFA for 1 h, then cryoprotected in 30% sucrose overnight, embedded in OCT, and frozen. Thirty-micrometer cryosections were cut and then blocked with 5% donkey serum in TBS/0.2% Triton-X for 1 h at room temperature prior to staining. Sections were incubated with primary antibodies in blocking buffer overnight at 4°C. Following incubation, sections were washed three times for 10 min in TBS/0.2% Triton-X and then incubated with secondary antibodies for 1 h at room temperature followed by three washing steps. Slides were mounted with Fluoromount-G (SouthernBiotech).
For immunofluorescence analysis, NIH3T3 fibroblasts plated on coverslips were fixed in 4% PFA for 15 min and then permeabilized with 0.5% Triton X-100 in PBS for 10 min at room temperature. Samples were blocked in 3% BSA/0.05% sodium azide in PBS for 1 h and incubated with primary antibodies in blocking buffer for 2 h. Following three 5-min washes, coverslips were incubated with secondary antibodies (Jackson ImmunoResearch) and DAPI diluted in blocking buffer for 1 h, washed three times, and mounted using ProLong Gold anti-fade mountant (Thermo Fisher).
The antibodies used for immunohistochemistry and immunofluorescence experiments are listed in Supplemental Table S2.
Confocal microscopy and quantification
All images were acquired using a Leica SP5 confocal microscope and analyzed offline using the Leica LASAX software from z-stack images as described previously (Mentis et al. 2011; Fletcher et al. 2017). Images for motor neuron number quantification were acquired using a 20× objective at 3-µm steps in the Z-axis. Images for VGluT1+ inputs were acquired using a 40× objective at 0.3-µm steps in the Z-axis. Images for NMJ innervation quantification were obtained using a 20× objective at 2-µm steps in the Z-axis. Images for SmB intensity analysis were acquired using a 40× objective at identical settings for wild-type and SMA samples at 3-µm steps, and analysis was performed using LASAX software by measuring the peak intensity per unit area of the nuclear region of motor neurons.
Electrophysiology
Electrophysiological experiments were carried out as described previously (Simon et al. 2017). For recordings of the monosynaptic reflex, the intact ex vivo spinal cord preparation was perfused continuously with oxygenated (95% O2/5% CO2) aCSF at ∼10 mL/min. The dorsal root and ventral root of the L2 segment were placed into suction electrodes for stimulation or recording, respectively. The extracellular recorded potentials were recorded (DC, 3 kHz, Cyberamp, Molecular Devices) in response to a brief (0.2-msec) stimulation (A365, current stimulus isolator, World Precision Instruments) of the L2 dorsal root. The stimulus threshold was defined as the current at which the minimal evoked response was recorded in three out of five trials. Recordings were fed to an A/D interface (Digidata 1440A, Molecular Devices) and acquired with Clampex (version 10.2, Molecular Devices) at a sampling rate of 10 kHz. Data were analyzed offline using Clampfit (version 10.2, Molecular Devices). Measurements were taken from averaged traces of five trials elicited at 0.1 Hz. The temperature of the physiological solution ranged between 21°C and 25°C.
The function of the NMJs of the QL muscle at P11 was assessed ex vivo as described previously (Fletcher et al. 2017). Briefly, after the removal of the spinal cord, the remaining vertebral column with the ventral root L1–L3 in continuity to the QL was transferred to the recording chamber into aCSF. The ventral root L2 was placed into a suction electrode to stimulate the motor neuron axons. Visual twitching of the QL after stimulation of the ventral root L2 ensured proper stimulation of the muscle. Subsequently, to measure the CMAP, a concentric bipolar electrode was inserted in the QL between the insertion points of ventral roots L1 and L2. The stimulus threshold was defined as the current at which the minimal evoked response was recorded in three out of five trials. The nerve was stimulated at 1×, 2×, 5×, and 10× thresholds to ensure a supramaximal stimulation of the muscle. The maximum CMAP amplitude (baseline to peak) was determined as the average from five measurements.
Statistics
Results are expressed as mean ± standard error of the mean (SEM) from at least three independent experiments and biological replicates unless indicated otherwise. For statistical analysis, differences between two groups were analyzed by two-tailed unpaired Student's t-test, and differences among three or more groups were analyzed by one-way or two-way ANOVA followed by Tukey's post-hoc test as appropriate. GraphPad Prism 5 was used for all statistical analyses, and P-values are indicated as follows: P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Supplementary Material
Acknowledgments
We thank James Manley and Neil Shneider for critical comments on this manuscript. This work was supported by grants R01NS069601 (to L.P.), R01NS102451 (to L.P.), and R01NS078375 (to G.Z.M.) from the National Institute of Neurological Disorders and Stroke, and grant GR.10235006 (to G.Z.M.) from the U.S. Department of Defense.
Author contributions: M.V.A. and L.P. designed the study. M.V.A. performed all of the experiments except for the electrophysiology studies and the isolation of motor neurons by LCM, which were carried out by C.M.S. S.P.S. and L.S.S. produced the AAV9 vectors. M.V.A., C.M.S., G.Z.M., and L.P. analyzed the data. M.V.A. and L.P. wrote the paper with input from all authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.316059.118.
References
- Bardot B, Bouarich-Bourimi R, Leemput J, Lejour V, Hamon A, Plancke L, Jochemsen AG, Simeonova I, Fang M, Toledo F. 2015. Mice engineered for an obligatory Mdm4 exon skipping express higher levels of the Mdm4-S isoform but exhibit increased p53 activity. Oncogene 34: 2943–2948. [DOI] [PubMed] [Google Scholar]
- Bäumer D, Lee S, Nicholson G, Davies JL, Parkinson NJ, Murray LM, Gillingwater TH, Ansorge O, Davies KE, Talbot K. 2009. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet 5: e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebee TW, Dominguez CE, Samadzadeh-Tarighat S, Akehurst KL, Chandler DS. 2012. Hypoxia is a modifier of SMN2 splicing and disease severity in a severe SMA mouse model. Hum Mol Genet 21: 4301–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, Guccione E. 2013. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 27: 1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AHM, Beattie CE. 2009. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10: 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Manley JL. 2017. RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev 31: 1509–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. 2009. RNA and disease. Cell 136: 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP. 2005. p53 in neuronal apoptosis. Biochem Biophys Res Commun 331: 761–777. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Sohn YK, Wands JR. 1997. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci 152: 73–83. [DOI] [PubMed] [Google Scholar]
- Dewaele M, Tabaglio T, Willekens K, Bezzi M, Teo SX, Low DHP, Koh CM, Rambow F, Fiers M, Rogiers A, et al. 2016. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest 126: 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin-Asp PG, Bassell GJ, Rossoll W. 2016. A role for the survival of motor neuron protein in mRNP assembly and transport. Curr Opin Neurobiol 39: 53–61. [DOI] [PubMed] [Google Scholar]
- Donlin-Asp PG, Fallini C, Campos J, Chou C-C, Merritt ME, Phan HC, Bassell GJ, Rossoll W. 2017. The survival of motor neuron protein acts as a molecular chaperone for mRNP assembly. Cell Rep 18: 1660–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu Q-S, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. 2002. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol 52: 597–606. [DOI] [PubMed] [Google Scholar]
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, et al. 2017. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 377: 1723–1732. [DOI] [PubMed] [Google Scholar]
- Fletcher EV, Simon CM, Pagiazitis JG, Chalif JI, Vukojicic A, Drobac E, Wang X, Mentis GZ. 2017. Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat Neurosci 20: 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen WJ, Paushkin S, Wyce A, Massenet S, Pesiridis GS, Van Duyne G, Rappsilber J, Mann M, Dreyfuss G. 2001. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol 21: 8289–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Mancini F, Pellegrino M, Di Conza G, Puxeddu E, Sacchi A, Pontecorvi A, Moretti F. 2010. Regulation of MDM4 (MDMX) function by p76(MDM2): a new facet in the control of p53 activity. Oncogene 29: 5935–5945. [DOI] [PubMed] [Google Scholar]
- Gogliotti RG, Quinlan KA, Barlow CB, Heier CR, Heckman CJ, Didonato CJ. 2012. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci 32: 3818–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen EJN, Talbot K, Gillingwater TH. 2018. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol 14: 214–224. [DOI] [PubMed] [Google Scholar]
- Hamilton G, Gillingwater TH. 2013. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 19: 40–50. [DOI] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. 2011. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478: 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, Fleet C, Cullen P, Gupta SV, Mekhoubad S, Chiao E, Allaire N, Bennett CF, Rigo F, Krainer AR, et al. 2017. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc Natl Acad Sci 114: E2347–E2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodelka FM, Ebert AD, Duelli DM, Hastings ML. 2010. A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum Mol Genet 19: 4906–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T. 1997. Changes of p53 in the brains of patients with Alzheimer's disease. Biochem Biophys Res Commun 232: 418–421. [DOI] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach MER, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AHM. 2005. SMNΔ7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet 14: 845–857. [DOI] [PubMed] [Google Scholar]
- Li DK, Tisdale S, Lotti F, Pellizzoni L. 2014. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin Cell Dev Biol 32: 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling KKY, Gibbs RM, Feng Z, Ko C-P. 2012. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet 21: 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F, Imlach WL, Saieva L, Beck ES, Hao LT, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, et al. 2012. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151: 440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine J-C, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. 2006. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ 13: 927–934. [DOI] [PubMed] [Google Scholar]
- Martinez TL, Kong L, Wang X, Osborne MA, Crowder ME, Van Meerbeke JP, Xu X, Davis C, Wooley J, Goldhamer DJ, et al. 2012. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J Neurosci 32: 8703–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern VL, Iyer CC, Arnold WD, Gombash SE, Zaworski PG, Blatnik AJ, Foust KD, Burghes AHM. 2015. SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNΔ7 mouse model of SMA. Hum Mol Genet 24: 5524–5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Bühler D, Pillai R, Lottspeich F, Fischer U. 2001. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol 3: 945–949. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, et al. 2017. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 377: 1713–1722. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ. 2011. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69: 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Kondo T, Mizuno Y, Nagatsu T. 2007. p53 protein, interferon-γ, and NF-κB levels are elevated in the parkinsonian brain. Neurosci Lett 414: 94–97. [DOI] [PubMed] [Google Scholar]
- Mueller C, Ratner D, Zhong L, Esteves-Sena M, Gao G. 2012. Production and discovery of novel recombinant adeno-associated viral vectors. Curr Protoc Microbiol 26: 14D.1.1–14D.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LM, Beauvais A, Gibeault S, Courtney NL, Kothary R. 2015. Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/−) mouse model of spinal muscular atrophy. Acta Neuropathol Commun 3: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskoui M, Darras BT, De Vivo DC. 2017. Spinal muscular atrophy: 125 years later and on the verge of a cure. In Spinal muscular atrophy: disease mechanisms and therapy (ed. Sumner CJ, et al. ), pp. 3–19. Academic Press, San Diego. [Google Scholar]
- Pellizzoni L, Yong J, Dreyfuss G. 2002. Essential role for the SMN complex in the specificity of snRNP assembly. Science 298: 1775–1779. [DOI] [PubMed] [Google Scholar]
- Perry ME, Mendrysa SM, Saucedo LJ, Tannous P, Holubar M. 2000. p76(MDM2) inhibits the ability of p90(MDM2) to destabilize p53. J Biol Chem 275: 5733–5738. [DOI] [PubMed] [Google Scholar]
- Pillai RS, Grimmler M, Meister G, Will CL, Lührmann R, Fischer U, Schümperli D. 2003. Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev 17: 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Davis B, Chiang Y-H, Filichia E, Barnett A, Greig NH, Hoffer B, Luo Y. 2016. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson's disease model. J Neurochem 138: 746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiu M, McGovern VL, Lotti F, Saieva L, Li DK, Kariya S, Monani UR, Burghes AHM, Pellizzoni L. 2012. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol 32: 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo LJ, Myers CD, Perry ME. 1999. Multiple murine double minute gene 2 (MDM2) proteins are induced by ultraviolet light. J Biol Chem 274: 8161–8168. [DOI] [PubMed] [Google Scholar]
- Shadfan M, Lopez-Pajares V, Yuan Z-M. 2012. MDM2 and MDMX: alone and together in regulation of p53. Transl Cancer Res 1: 88–89. [PMC free article] [PubMed] [Google Scholar]
- Simic G, Seso-Simic D, Lucassen PJ, Islam A, Krsnik Z, Cviko A, Jelasic D, Barisic N, Winblad B, Kostovic I, et al. 2000. Ultrastructural analysis and TUNEL demonstrate motor neuron apoptosis in Werdnig-Hoffmann disease. J Neuropathol Exp Neurol 59: 398–407. [DOI] [PubMed] [Google Scholar]
- Simon CM, Dai Y, Van Alstyne M, Koutsioumpa C, Pagiazitis JG, Chalif JI, Wang X, Rabinowitz JE, Henderson CE, Pellizzoni L, et al. 2017. Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep 21: 3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers E, Lees RD, Hoban K, Sleigh JN, Zhou H, Muntoni F, Talbot K, Gillingwater TH, Parson SH. 2016. Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann Neurol 79: 217–230. [DOI] [PubMed] [Google Scholar]
- Tisdale S, Pellizzoni L. 2015. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci 35: 8691–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale S, Lotti F, Saieva L, Van Meerbeke JP, Crawford TO, Sumner CJ, Mentis GZ, Pellizzoni L. 2013. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell Rep 5: 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valianatos G, Valcikova B, Growkova K, Verlande A, Mlcochova J, Radova L, Stetkova M, Vyhnakova M, Slaby O, Uldrijan S. 2017. A small molecule drug promoting miRNA processing induces alternative splicing of MdmX transcript and rescues p53 activity in human cancer cells overexpressing MdmX protein. PLoS One 12: e0185801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Alstyne M, Pellizzoni L. 2016. Advances in modeling and treating spinal muscular atrophy. Curr Opin Neurol 29: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Wade M, Li Y-C, Wahl GM. 2013. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L-Y, Greig NH, Huang Y-N, Hsieh T-H, Tweedie D, Yu Q-S, Hoffer BJ, Luo Y, Kao Y-C, Wang J-Y. 2016. Post-traumatic administration of the p53 inactivator pifithrin-α oxygen analogue reduces hippocampal neuronal loss and improves cognitive deficits after experimental traumatic brain injury. Neurobiol Dis 96: 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. 2008. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.