

Chatterji et al. describe a novel interplay between LIN28B and IMP1 in intestinal epithelial cells. Their data provide new evidence for the opposing effects of the LIN28B–IMP1 axis on post-transcriptional regulation of canonical WNT signaling, with implications in intestinal homeostasis, regeneration, and tumorigenesis.
Keywords: LIN28B, IMP1, intestinal tumorigenesis, post-transcriptional regulation, ribosome profiling
Abstract
RNA-binding proteins (RBPs) are expressed broadly during both development and malignant transformation, yet their mechanistic roles in epithelial homeostasis or as drivers of tumor initiation and progression are incompletely understood. Here we describe a novel interplay between RBPs LIN28B and IMP1 in intestinal epithelial cells. Ribosome profiling and RNA sequencing identified IMP1 as a principle node for gene expression regulation downstream from LIN28B. In vitro and in vivo data demonstrate that epithelial IMP1 loss increases expression of WNT target genes and enhances LIN28B-mediated intestinal tumorigenesis, which was reversed when we overexpressed IMP1 independently in vivo. Furthermore, IMP1 loss in wild-type or LIN28B-overexpressing mice enhances the regenerative response to irradiation. Together, our data provide new evidence for the opposing effects of the LIN28B–IMP1 axis on post-transcriptional regulation of canonical WNT signaling, with implications in intestinal homeostasis, regeneration and tumorigenesis.
Post-transcriptional regulation of key growth and oncogenic pathways by RNA-binding proteins (RBPs) is an emerging hallmark of cancer (Chatterji and Rustgi 2018). Understanding the relative contribution and regulation of gene transcription and translation to functional and phenotypic outcomes is essential for generating effective therapeutics. Given the large number of putative binding targets and effector pathways downstream from RBPs, their evaluation in vivo is critical to understanding the prevailing effects of specific RBP:RNA target interactions in variable biological contexts. Intestinal epithelial cells exhibit remarkable plasticity in their ability to rapidly proliferate, differentiate, and undergo repair—processes that are aberrantly regulated during tumorigenesis. While the critical cellular pathways for epithelial injury repair and tumor development are well studied, the relative contribution of RBPs as key functional regulators of these pathways are just beginning to be understood. A primary role of RBPs is to bind to and regulate functionally interrelated target transcripts, forming functional units referred to as “regulons” (Keene 2007; Morris et al. 2010; Chatterji and Rustgi 2018). The integration of transcriptional and translational data has revealed that a significant number of genes are highly regulated at the translational level, underscoring the potential significant contribution of RBPs to numerous basic biological functions (Schafer et al. 2015). Pathways critical to epithelial homeostasis, such as WNT signaling, are aberrantly activated in and can contribute to colorectal cancer (CRC) pathogenesis, resulting in challenges for direct therapeutic targeting.
As a master regulator of the let-7 family of microRNAs (miRs), LIN28B exerts its effects via suppression of let-7 processing and thus up-regulation of let-7 targets, including HMGA2 and IGF2BP1 (IMP1) (Boyerinas et al. 2008; Piskounova et al. 2011). In addition, LIN28B acts as a pluripotency factor and promotes tumorigenesis in multiple tissues (Viswanathan et al. 2009; Tu et al. 2015; Rahkonen et al. 2016; Zhang et al. 2016). We demonstrated previously that LIN28B overexpression promotes migration and invasion in vitro and that CRC cells overexpressing LIN28B exhibit smaller yet more metastatic tumors in subcutaneous xenograft mouse models. (King et al. 2011a,b). In addition, we and others have established LIN28B is a bone fide oncogene in the intestinal epithelium (Madison et al. 2013; Tu et al. 2015). Furthermore, tumorigenesis was accelerated in mice with specific deletion of let-7c2/let-7b as revealed by us, suggesting that the oncogenic effects of LIN28B are predominantly via its regulation of let-7. In support of this notion, deletion of a single allele of the let-7 target Hmga2 repressed Lin28b-driven tumorigenesis (Madison et al. 2015). Other key let-7 targets up-regulated in VillinLin28b mice include Igf2bp1, Igf2bp2, E2f5, cyclinD1, and Acvr1c. Recognizing that LIN28B functions as an oncogene in CRC and blocks processing of pre/pri -let-7, we reasoned that let-7 mRNA targets may be effectors of LIN28B-mediated oncogenic transformation. Specifically, we hypothesized that IMP1 may be a critical effector of LIN28B's actions.
IMP1 is a RBP with oncofetal functions that is expressed minimally in adult tissues under homeostatic conditions. We demonstrated previously in subcutaneous xenografts that IMP1 overexpression in SW480 CRC cells exhibit increased tumor volume and elevated levels of its mRNA-binding target, CD44 (Hamilton et al. 2013). Conversely, loss of both Imp1 alleles in stromal cells led to enhanced tumor load in colitis-associated colon tumorigenesis (Hamilton et al. 2015), indicating a tumor-suppressive role for IMP1 in this nonepithelial compartment. Others have reported diverse findings in other cancers; for example, overexpression of the IMP1 mouse homolog CRD-BP in mammary epithelial cells resulted in mammary tumor formation, with a subset of mice exhibiting metastasis (Tessier et al. 2004). Conversely, studies in orthotopic breast fat pad xenografts demonstrated that IMP1 overexpression suppressed the growth of MDA231 cell-derived tumors and subsequent lung metastasis as well as an associated decrease in IMP1 mRNA-binding targets PTGS2, GDF15, and IGF2 (Wang et al. 2016). Finally, ectopic Imp1 expression in the MMTV-PyMT breast cancer mouse model significantly reduced metastatic tumors (Nwokafor et al. 2016). Based on these functional studies, we conclude that IMP1 may exhibit oncogenic or tumor-suppressive functions in a context-dependent manner that may vary from tissue to tissue and even within a tissue (epithelial vs. mesenchymal). Furthermore, these studies demonstrate that IMP1 may enhance or suppress its mRNA-binding targets.
In the present study, we used the intestinal epithelium as a prototypical model for understanding the tissue-specific roles of two key RBPs—LIN28B and IMP1—in the context of fundamental homeostasis and tumorigenesis. We performed novel and unbiased transcriptome and translatome studies as a function of LIN28B expression and found that IMP1 is the top regulator of differentially expressed transcripts with LIN28B overexpression. We next evaluated the in vivo consequences of LIN28B expression with and without IMP1 expression, elucidating their functional relevance in intestinal homeostasis and tumorigenesis with the finding that WNT signaling is pivotal. Taken together, our studies reveal a LIN28B–IMP1 axis governing a post-transcriptional regulon capable of modulating intestinal epithelial cell growth and tumorigenesis via global control of the WNT pathway.
Results
IMP1 loss is a significant regulator of translation in the context of LIN28B overexpression
To understand how LIN28B may modulate translation of the transcriptome and how this is impacted by IMP1, we used ribosome profiling to obtain a genome-wide snapshot of translation in the context of LIN28B overexpression. First, we evaluated SW480 CRC cells overexpressing LIN28B (Supplemental Fig. 1A; King et al. 2011a,b). We used translatome (Tebaldi et al. 2014) to identify putative regulatory factors upstream of differentially translated genes in LIN28B-overexpressing versus control cells. This method computes a Fisher test P-value indicating whether binding sites for each regulator are significantly enriched in differentially expressed genes lists. This analysis allows the user to identify possible regulatory factors responsible for the translational regulation of differentially expressed genes in the experiment under consideration. The list of genes regulated by each post-transcriptional element was obtained from the Atlas of UTR Regulatory Activity (AURA) database (Dassi et al. 2014). Through this analysis, we identified IMP1 as a significant post-transcriptional regulator in LIN28B-overexpressing cells (Fig. 1A).
Figure 1.

IMP1 is a significant translational regulator downstream from LIN28B. (A) Radar map demonstrating the top post-transcriptional regulators of differentially expressed genes with LIN28B overexpression in SW480 cells as compared with control cells. A single point on the radar map indicates −10 log P-value of enrichment of any post-transcriptional regulator. Differentially expressed genes at each level were identified using the DEGseq, and the list of post-transcriptional regulators was obtained from the AURA database (see the Materials and Methods). (B) Scatter plot of differential expression between LIN28B-overexpressing lines with and without IMP1 deletion.The log2 fold changes between ribosome-bound RNAs (ribosome-protected fragments [RPFs]) and total mRNA are plotted. The plot indicates that IMP1 regulates both mRNA abundance and translation. (C) Scatter plot of genes with significant (in blue) differential translational efficiencies between LIN28B-overexpressing cells with and without IMP1 deletion. Translation efficiencies (TEs) of the transcripts were calculated as the ratio of reads of ribosome-protected fragments to the reads in total mRNA abundance. (D) Pathway analysis using DAVID software of the differentially expressed genes from C to see what signaling/effector pathways are enriched with IMP1 deletion (note the Wnt signaling pathway).
Next, we analyzed differential gene expression between LIN28B-overexpressing lines with and without CRISPR/Cas9-mediated IMP1 deletion (Supplemental Fig. 1A) and calculated log fold changes between ribosome-bound RNAs (ribosome-protected fragments [RPFs]) and total mRNA. A total of 1284 genes was regulated by IMP1 exclusively at the translational level, whereas 264 genes were regulated only at the transcriptional level. Additionally, 458 genes were regulated via translational antagonism (where genes exhibited increased mRNA levels but lower translational efficiencies or vice versa) or translational reinforcement (where translational changes amplified the changes at the mRNA level) (Fig. 1B; Supplemental Table 11). These data demonstrate that IMP1 is a major translational regulator of target genes. Translational regulation can occur through differential translation efficiencies (TEs) of transcripts, which were calculated as the ratio of RPF reads to total mRNA abundance. We observed several transcripts with significantly different TE values between cells overexpressing LIN28B with or without IMP1 deletion (Fig. 1C; Supplemental Table 9). Pathway enrichment analysis for TE genes differentially expressed in LIN28B cells with or without IMP1 deletion revealed a significant representation of pathways linked to cancer, including the WNT signaling pathway (Fig. 1D; Supplemental Tables 8, 10). Finally, gene set enrichment analysis (GSEA) of RNA sequencing (RNA-seq) data from the same cells suggest that IMP1 deletion up-regulates WNT signaling at the mRNA level as well (Supplemental Table 1), demonstrating IMP1 modulation of WNT at both transcript abundance/stability and TE levels in this context. We further validated this in the SW480 cells overexpressing LIN28B with and without IMP1 deletion (Supplemental Fig. 1C,D). We saw a significant up-regulation of Wnt targets with IMP1 deletion via quantiative PCR (qPCR) and up-regulation of β-catenin protein levels via Western blot in these cells. Finally, to validate up-regulation of Wnt targets identified via RNA-seq, we used siRNA to knock down IMP1 in Caco2 cells (Supplementary Fig. 4A,B) that endogenously express LIN28B (Mizuno et al. 2018). We observed significant up-regulation of multiple WNT targets with IMP1 knockdown, corroborating our findings in a second, independent cell line (Supplemental Fig. 4A,B).
In an attempt to elucidate whether direct binding may correlate with TEs, we compared binding intensities of IMP1 targets identified via previously published enhanced UV cross-linking and immunoprecipitation (eCLIP) in human pluripotent stem cells (hPSCs) (Conway et al. 2016) with differential TEs of these targets from the present study (in human CRC cells). However, we found no correlation between binding intensities and TEs (Supplemental Fig. 1B). In addition, a large number of targets identified in eCLIP studies did not change significantly in our TE data set and vice versa (Supplemental Table 2). While we cannot exclude the possibility that differences between mRNA-binding efficiencies and TEs may be due in part to comparing hPSCs with CRC cells, these data nevertheless reinforce the concept that eCLIP-identified IMP1-bound targets may not necessarily correlate with gene expression changes (Conway et al. 2016).
IMP1 loss enhances LIN28B-mediated tumorigenesis in vivo
Gene expression and ribosome profiling data suggest that IMP1 loss causes dysregulation of several key signaling pathways that are involved in cellular proliferation and maintenance. To understand the phenotypic consequence of these changes, we used LIN28B-overexpressing CRC cells with and without IMP1 loss in subcutaneous xenograft experiments (Supplemental Fig. 2A). Consistent with our prior published studies, LIN28B overexpression caused a significant decrease in tumor volume as compared with controls, irrespective of IMP1 status (Supplemental Fig. 2B; King et al. 2011a). LIN28B-overexpressing tumors exhibited a more differentiated phenotype as compared with controls, which had poorly differentiated tumors. This differentiation phenotype was reversed with IMP1 loss in LIN28B-overexpressing cells (Supplemental Fig. 2C), suggesting that IMP1 loss may promote an increase in stemness markers and/or down-regulate differentiation pathways in CRC cells. We also observed an up-regulation of nuclear β-catenin staining with IMP1 loss and a significant increase in Wnt signature in these tumors (Supplemental Fig. 2C,D).
To confirm that LIN28B expression in transformed cells can promote differentiation, we evaluated LIN28B expression and differentiation status in human colon tumors in colon cancer tissue microarrays (King et al. 2011a). We found that the majority of LIN28B overexpressing tumors was either moderately or well differentiated (Supplemental Fig. 2E). The mechanistic explanation for this is not known; however, recent studies have demonstrated that, in some CRC cell lines, overexpression of pluripotency factors (OCT3/4, SOX2, and KLF4) can lead to tumors with increased differentiation (Oshima et al. 2014). This is in line with our finding, since LIN28B is also a pluripotency factor (Rahkonen et al. 2016; Zhang et al. 2016; Tsanov et al. 2017), although this does not mean that pluripotency and differentiation are necessarily linked in this context.
We next evaluated transgenic mice with intestinal epithelial overexpression of Lin28b (Villin-Lin28b) and intestinal epithelial-specific deletion of Imp1 (VillinCre;Imp1fl/fl) (Supplemental Fig. 2F; Hamilton et al. 2013; Madison et al. 2013). Our prior work demonstrated de novo intestinal tumor growth in Villin-Lin28b mice, confirming that Lin28b can act as an oncogene in the intestine (Madison et al. 2013). Imp1 expression is enhanced significantly in Villin-Lin28b mice and completely eliminated in the intestinal epithelium of Villin-Lin28b;VillinCre;Imp1fl/fl mice (Fig. 2A). We found that, at 12 mo of age, 18 out of 25 Villin-Lin28b mice (72%) had at least one tumor, while 10 out of 12 (83.33%) Villin-Lin28b;VillinCre;Imp1fl/fl mice had tumors, the majority of which had multiple lesions. Aged mice overexpressing LIN28B exhibit a significant increase in the number of proliferating cells (Fig. 2B,C) in normal tissue. Villin-Lin28b;VillinCre;Imp1fl/fl mice exhibit a significant increase in intestinal tumor number per mouse compared with Villin-Lin28b mice (Fig. 2D). We did not observe hyperplasia or tumor growth with Imp1 loss alone, evaluated out to 12 mo of age. Histological grading revealed a higher percentage of adenocarcinoma lesions in Villin-Lin28b;VillinCre;Imp1fl/fl mice compared with Villin-Lin28b mice (Fig. 2E,F), demonstrating that Imp1 loss promotes higher tumor number and grade in the context of Lin28b overexpression.
Figure 2.

IMP1 loss enhances LIN28B-mediated tumorigenesis in vivo. Imp1fl/fl (wild type), Villin-Cre;Imp1fl/fl, Villin-Lin28b, and Villin-Cre;Villin-Lin28b;Imp1fl/fl mice were aged up to 1 yr and then sacrificed to evaluate tumor growth. (A) Igf2bp1/Imp1 expression in the epithelia isolated from the jejuna of 12-mo-old mice. n > 4 mice per genotype. (B) Representative immunofluorescence staining for EdU incorporation in mouse intestinal epithelium. Bars, 500 µm. (C) Quantification of EdU+ve cells using flow cytometry. n = 3 mice per genotype. (D) The number of intestinal tumors in mice with or without Imp1 in the context of Lin28b overexpression. n > 9 mice per genotype. (E) The percentage of tumors classified as adenocarcinomas by histological scoring. (F) Representative hematoxylin and eosin (H&E) staining of intestinal epithelia in aged mice. Mice lacking Lin28b overexpression exhibited normal intestinal morphology at 12 mo of age. Mice with Lin28b overexpression exhibited tumor development that worsened with Imp1 loss. Bars, 500 µm. (G) Relative expression of different let-7 targets in the intestinal epithelium via qPCR. n > 5 mice per genotype. (H) Relative expression of stem cell markers in the intestinal epithelium via qPCR expressed as fold change with respect to Imp1fl/fl mice. n > 5 mice per genotype. (I) Relative expression of Wnt target genes in the intestinal epithelium via qPCR expressed as fold change with respect to Imp1fl/fl mice. n > 5 mice per genotype. (J) Representative immunohistochemical staining for β-catenin in mouse intestinal epithelium (magnified in the inset). (K) Representative β-catenin protein levels in mouse intestinal crypts from the four genotypes. All data are expressed as mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P<0.0001 by ordinary one-way ANOVA test followed by Tukey's multiple comparison test. The significance is shown compared with control/wild-type samples unless indicated otherwise.
Our prior studies demonstrated that Lin28b overexpression results in increased expression of other let-7 targets, including Hmga2, Arid3a, Igf2bp2, and CycD1, as well as enhanced expression of stem cell-related genes (Madison et al. 2015). We evaluated isolated epithelia from aged mice and found no significant change in Arid3a, Igf2bp2, Hmga2, and CycD1 expression in Villin-Lin28b;VillinCre;Imp1fl/fl mice compared with Villin-Lin28b mice (Fig. 2G). We found a robust increase in stem cell and broader crypt cell markers, including Prom1, Hopx, Bmi1, Lgr5, and Olfm4 (van der Flier et al. 2009; Zhu et al. 2009; Takeda et al. 2011; Tian et al. 2011; de Sousa e Melo et al. 2017), suggesting an expansion of the stem cell compartment in Villin-Lin28b;VillinCre;Imp1fl/fl mice compared with Villin-Lin28b mice (Fig. 2H). Consistent with an expansion of the crypt base stem cell compartment, we observed an increase in WNT target gene expression with Imp1 loss at both the mRNA and protein levels (Fig. 2I–K; Supplemental Fig. 3D). This effect was also observed ex vivo in enteroids generated from isolated crypts, where Villin-Lin28b;VillinCre;Imp1fl/fl enteroids exhibit increased AXIN2 expression compared with wild-type or Villin-Lin28b mouse enteroids (Supplemental Fig. 2G). Together, these data support the hypothesis that IMP1 may modulate stem cell signature and canonical WNT target gene expression in the intestine and that these effects persist in isolated crypt enteroid cultures.
To understand these findings in the context of patients with CRC, we evaluated The Cancer Genome Atlas (TCGA) (Cline et al. 2013) for LIN28B and IMP1 expression levels in colon and rectal adenocarcinoma (COADREAD) patients. We found a positive correlation between the two transcripts in patient tumors (Pearson's coefficient = 0.266, P < 0.0001) (Supplemental Fig. 3A). However, tumor stage-wise evaluation revealed higher IMP1 expression in all tumor stages, whereas LIN28B expression increased in stage IV, supporting the concept that IMP1 expression is regulated via additional mechanisms beyond LIN28B (Supplemental Fig. 3B).
IMP1 contributes to intestinal epithelial regeneration
Building on our observation that Imp1 loss up-regulates stem cell and WNT target genes in the context of LIN28B overexpression, we sought to understand broader roles for IMP1 in the context of intestinal homeostasis and repair from injury. Radiation-mediated injury has been increasingly used to study stem cell function and regeneration of the intestinal epithelium (Potten 2004; Zhou et al. 2013; Gregorieff et al. 2015). Imp1 expression is low in the adult intestinal crypt but is increased in the regenerating epithelium following whole-body irradiation at both the RNA and protein levels (Fig. 3A). We observed IMP1 protein expression specifically in regenerative foci 4 d following irradiation, a time point at which surviving stem cells are undergoing rapid proliferation to replenish the epithelium and restore barrier function (Fig. 3B). We observed that Villin-Lin28b mice lose significantly more weight at day 4 after irradiation as compared with wild-type controls. This phenotype is reversed in Villin-Lin28b;VillinCre;Imp1fl/fl mice, where Imp1 deletion results in significantly less weight loss as compared with Villin-Lin28b mice, returning to wild-type levels (Fig. 3C). Furthermore, the number of regenerative crypts at day 4 following radiation is significantly higher in Villin-Lin28b;VillinCre;Imp1fl/fl mice compared with Villin-Lin28b mice (Fig. 3D). Villin-Lin28b;VillinCre;Imp1fl/fl mice also display decreased H2AX foci (Zhou et al. 2015) as compared with Villin-Lin28b mice, thus indicating less DNA damage at day 4 after radiation (Supplemental Fig. 3C). Thus, the beneficial effect may be due to both enhanced WNT signaling and WNT-independent protective effects.
Figure 3.
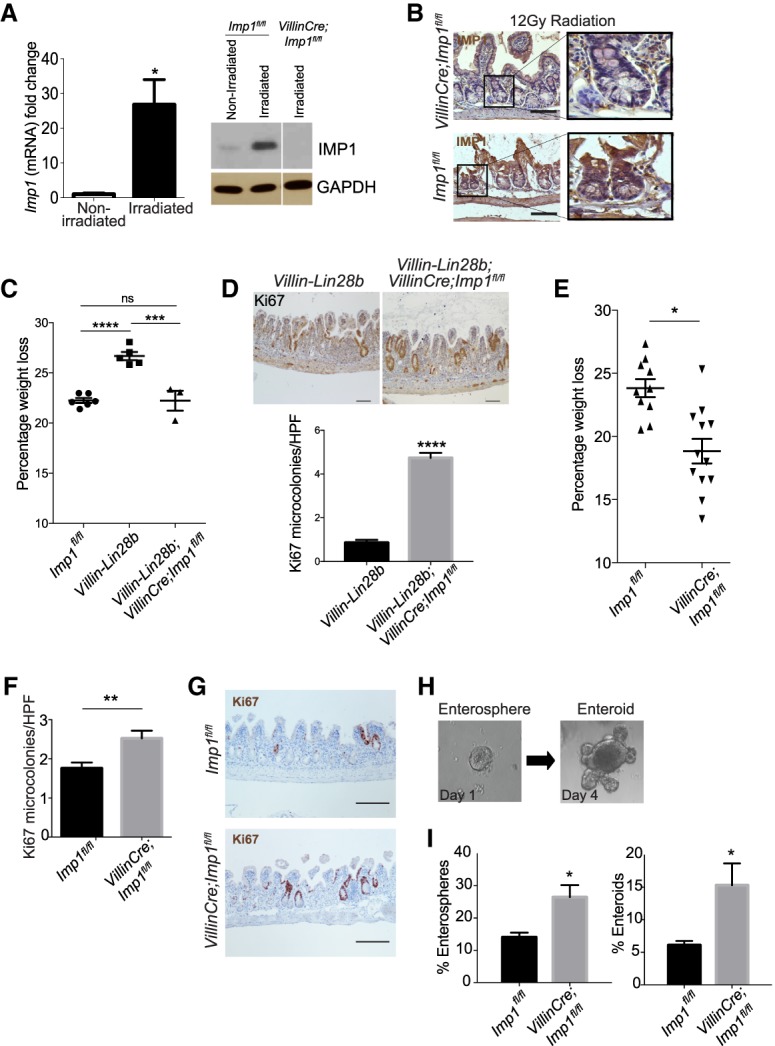
IMP1 regulates intestinal epithelial regeneration following irradiation. Mice were evaluated for Imp1 expression in nonirradiated conditions and 4 d after 12 Gy of whole-body irradiation. (A) qRT–PCR and representative Western blot for Imp1 in isolated crypts from these mice. (*) P < 0.05 versus nonirradiated. n = 3–4 mice per genotype. (B) Representative immunohistochemical staining showing Imp1 increase in wild-type (Imp1fl/fl) mice following radiation. VillinCre;Imp1fl/fl mice were used as controls. Bars, 500 µm. (C) Villin-Lin28b;Villin-Cre;Imp1fl/fl mice lost significantly less weight at sacrifice following irradiation than Villin-Lin28b mice (22.23% ± 0.9905% mean weight loss in Villin-Cre;Villin-Lin28b;Imp1fl/fl mice [n = 3] vs. 26.68% ± 0.4076% in Villin-Lin28b [n = 5]). The weight loss in Villin-Lin28b mice was significantly higher than in controls (22.24% ± 0.2556% mean weight loss). (D) Analysis of Ki67+ cells revealed a significant increase in Ki67+ regenerative crypt foci in VillinCre Villin-Lin28b;Imp1fl/fl mice at 4 d following irradiation. n = 3–4 mice per genotype; n = 20–30 high-power fields (HPFs) per mouse. Representative immunohistochemical staining for Ki67+ foci in the mouse intestinal epithelium is shown. Bars, 500 µm. (E) VillinCre;Imp1fl/fl mice lost significantly less weight at sacrifice following irradiation than controls (18.83% ± 0.98% in VillinCre;Imp1fl/fl mice vs. 23.34% ± 0.56% mean weight loss in controls). n = 14 Imp1fl/fl mice; n = 12 VillinCre;Imp1fl/fl mice. (F) Analysis of Ki67+ cells revealed a robust increase in Ki67+ regenerative crypt foci in VillinCre;Imp1fl/fl mice at 4 d following irradiation. n = 4 mice per genotype; n = 20–30 HPFs per mouse. (G) Representative immunohistochemical staining for Ki67+ foci in the mouse intestinal epithelium. Bars, 500 µm. (H) Representative pictures of the enterosphere and enteroid with buds. Enhanced growth of post-irradiation enteroids from VillinCre;Imp1fl/fl mice. Regenerative crypt units were plated in enteroid culture on the day of sacrifice to evaluate ex vivo survival and growth. (I) Evaluation of enterosphere growth 1 d following plating and enteroid growth 4 d after plating revealed a significant increase in VillinCre;Imp1fl/fl compared with Imp1fl/fl. All data are expressed as mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001 by ordinary one-way ANOVA test followed by Tukey's multiple comparison test. The significance is shown compared with control/wild-type samples unless indicated otherwise.
Evaluating IMP1 separately from its function downstream from LIN28B, we observed that VillinCre;Imp1fl/fl mice exhibit less weight loss following irradiation compared with control Imp1fl/fl mice (Fig. 3E) and that the number of regenerative crypt foci is increased in VillinCre;Imp1fl/fl mice (Fig. 3F,G). Crypt regeneration in VillinCre;Imp1fl/fl mice was evaluated further by plating post-irradiated crypts in three-dimensional (3D) enteroid culture (Fig. 3H; Fuller et al. 2012). Crypt enteroids from untreated Imp1fl/fl and VillinCre;Imp1fl/fl mice establish and grow at similar rates under homeostasis (Supplemental Fig. 4C); however, post-irradiated crypts from VillinCre;Imp1fl/fl mice exhibit a significant increase in enterosphere formation as well as growth into enteroids 4 d after plating (Fig. 3I).
Our data demonstrating that Imp1 deletion enhances Lin28b-mediated tumorigenesis and regeneration in both wild-type and Villin-Lin28b mice may be counterintuitive to current views of IMP1 function. We therefore generated mice in which Imp1 is overexpressed in the intestinal epithelium (VillinCre Imp1OE) (Supplemental Fig. 4D,E). We analyzed aged mice (10–12 mo) and found no tumors (Fig. 4A) or any significant difference in intestinal morphology in VillinCre Imp1OE or Villin-Lin28b; VillinCre Imp1OE mice compared with wild-type control mice (Fig. 4B). We did not observe a significant difference in Paneth, goblet, or enteroendocrine cell numbers (Supplemental Fig. 5A). Intriguingly, irradiated VillinCre;Imp1OE mice did not exhibit significant differences in weight loss compared with control mice at day 4, although there was a trend for increased weight loss in VillinCre Imp1OE mice (Supplemental Fig. 5B). Taken together, these data confirm in an independent manner that IMP1 overexpression by itself is insufficient to initiate tumors.
Figure 4.
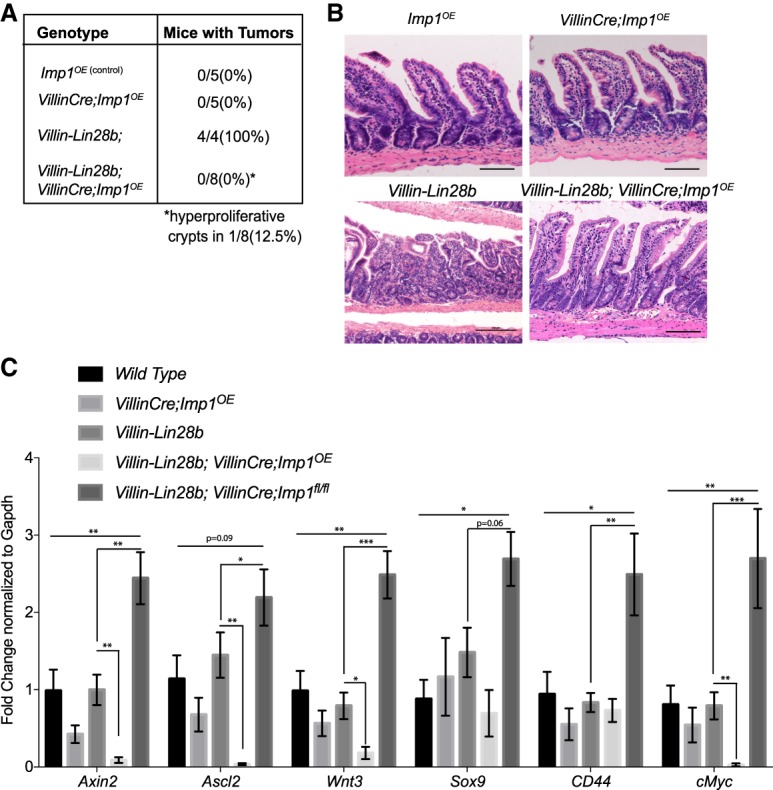
IMP1 overexpression does not initiate tumors in vivo. (A) The VillinCre;Imp1OE mice did not exhibit tumor formation between 10 and 12 mo of age. (B) Representative H&E and Ki67 staining of intestinal epithelia in aged (10–12 mo) mice. Mice overexpressing IMP1 exhibited normal intestinal morphology and did not show a tumor phenotype. Bars, 500 µm. (C) Relative expression of Wnt target genes in the intestinal epithelium via qPCR expressed as fold change with respect to wild-type control mice at 10–12 mo of age. n = >4 mice per genotype. All data are expressed as mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001 by ordinary one-way ANOVA test followed by Tukey's multiple comparison test.
We observed up-regulation of Wnt targets in Lin28b;VillinCre;Imp1fl/fl mice. To evaluate whether VillinCre;Imp1OE mice exhibit opposing effects, we analyzed gene expression in Villin-Lin28b;VillinCre;Imp1OE mice. Analysis of 10- to 12-mo-old mice revealed a decrease in Wnt target gene expression in Villin-Lin28b;VillinCre;Imp1OE mice compared with Villin-Lin28b mice. This supports the hypothesis that Imp1 loss may serve as a “switch” for canonical Wnt target gene regulation in the context of Lin28b overexpression (Fig. 4C). Finally, analysis of aged Villin-Lin28b;VillinCre;Imp1OE mice revealed a suppression of tumor incidence compared with Villin-Lin28b mice (Fig. 4A). In summary, the functional effects observed in mice with Imp1 loss or overexpression provide evidence that Imp1 acts as a suppressor of the epithelium's proliferative response under nontransformed conditions, due in part to regulation of Wnt signaling. In this regard, high IMP1 in CRC patients is likely an “effect” rather than a “cause” of tumor initiation.
IMP1 plays a functional role in reserve intestinal stem cells
We performed RNA-seq followed by GSEA on isolated crypts from Imp1fl/fl and VillinCre;Imp1fl/fl mice to evaluate baseline changes in gene expression with Imp1 loss. Similar to Villin-Lin28b;VillinCre;Imp1fl/fl mice, crypt gene expression analysis in VillinCre;Imp1fl/fl mice revealed an increase in intestinal stem cell and Wnt target gene expression (Fig. 5A; Supplemental Fig. 6A). GSEA revealed enrichment of gene targets involved in proliferation, regeneration, and Wnt signaling in VillinCre;Imp1fl/fl mice compared with control mice (P-value < 0.001; false discovery rate [FDR] < 0.001) (Supplemental Table 6). We validated up-regulation of Wnt targets identified via RNA-seq using siRNA to knock down IMP1 in Caco2 cells (Supplemental Fig. 4A,B), which endogenously express LIN28B (Mizuno et al. 2018). We observed significant up-regulation of multiple WNT targets with IMP1 knockdown in these cells.
Figure 5.

IMP1 plays a functional role in reserve intestinal stem cells. (A) qRT–PCR for stem cell markers and Wnt target genes in isolated crypts from VillinCre;Imp1fl/fl mice compared with Impfl/fl mice. n = 3–4 mice per genotype. (B) qPCR in Sox9-EGFP reporter mice, where epithelial cell populations are subclassified viafluorescent-activated cell sorting (FACS) into Sox9-EGFP0-negative, Sox9-EGFP0 sublow, Sox9-EGFP0 low, and Sox9-EGFP0 high cells. Imp1 is expressed at low levels in all represented cell types except Sox9-EGFP-negative cells (white bars). Imp1 expression is significantly higher in Sox9-EGFP low and Sox9-EGFP high cells, which encompass intestinal stem cells. n = 5 animals per group. (C) HopxCreERT2;Imp1fl/fl mice lost significantly less weight at sacrifice following irradiation than controls (18.8% ± 1.951% mean weight loss in HopxCreERT2;Imp1fl/fl mice vs. 23.92% ± 1.015% in controls). n = 8 HopxCreERT2;Imp1fl/fl; n = 12 control mice. (D) Analysis of Ki67+ cells revealed there was a robust increase in Ki67+ regenerative crypt foci at 4 d following irradiation in HopxCreERT2;Imp1fl/fl mice compared with control mice. n = 4 mice per genotype; n = 20–30 HPFs per animal. Bars, 500 µm. (E) Representative immunohistochemical staining for Ki67+ foci in the mouse intestinal epithelium quantified in D. (F) qPCR for Imp1 expression in Lgr5+ and Paneth (CD24/cKit/SSC high) cells sorted from Lgr5-eGFP-IRES-CreERT2 mouse crypts. All data are expressed as mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001 by unpaired t-test.
VillinCre;Imp1fl/fl mice lack Imp1 in all intestinal epithelial cells, including stem, progenitor, and differentiated cells. Based on the up-regulation of stem cell-related and Wnt target genes in VillinCre;Imp1fl/fl mice, we reasoned that Imp1 may exhibit a functional role within the intestinal crypt compartment. To confirm Imp1 gene expression in small intestine crypt cells, we used Sox9-EGFP reporter mice in which subpopulations of intestinal epithelial cells are fractionated by their relative EGFP expression using fluorescent-activated cell sorting (FACS). Sox9-EGFP-negative, Sox9-EGFP sublow, Sox9-EGFP low, and Sox9-EGFP high segregate into populations of differentiated cells, progenitor/transit-amplifying cells, active crypt base stem cells, and a mixed population of reserve stem cells/secretory progenitors/enteroendocrine cells, respectively (Formeister et al. 2009; Gracz et al. 2010; Van Landeghem et al. 2012; Roche et al. 2015). Subpopulations were validated previously with gene expression and functional assays. In Sox9-EGFP mice, Imp1 expression is significantly higher in Sox9-EGFP low and Sox9-EGFP high cells as compared with differentiated lineages (Sox9-EGFP-negative cells) (Fig. 5B). Furthermore, Imp1 expression is up-regulated in all cell populations following whole-body irradiation, with a significant increase in Sox9-EGFP high cells relative to other cells types (Supplemental Fig. 6B).
To determine whether the protective effect of Imp1 loss persists in a more restricted epithelial population, we crossed Imp1fl/fl mice to mice harboring the HopxCreERT2 knock-in allele (Li et al. 2016), which is expressed specifically in WntNegative quiescent reserve stem cells (Takeda et al. 2011; Li et al. 2014; Yousefi et al. 2016). Similar to VillinCre;Imp1fl/fl mice, we found that HopxCreERT2;Imp1fl/fl mice exhibit less weight loss following irradiation compared with controls (Fig. 5C) and an increase in regenerative crypt foci, consistent with enhanced recovery following injury (Fig. 5D,E). To verify that Imp1 is enriched in the crypts using a second model, we used Lgr5-eGFP-IRES-CreERT2 mice. We performed gene expression analysis for Imp1 in Lgr5+ and Paneth (CD24/cKit/SSC high) cells sorted from Lgr5-eGFP-IRES-CreERT2 mouse crypts. We observed that Imp1 is enriched in both Lgr5+ and Paneth cells compared with unsorted crypts (Fig. 5F). Taken together, these data support a role for IMP1 induction to negatively regulate WNT signaling during homeostasis (putatively in crypt cells with an inactive canonical Wnt signaling pathway during homeostasis).
In aggregate, we propose a model in which LIN28B overexpression promotes increased proliferation and tumorigenesis and enhances IMP1 expression, which may then feed back and suppress these pathways during homeostasis (Fig. 6). This mechanism is reinforced by data in Villin-Lin28b;VillinCre;Imp1fl/fl mice (where Imp1 loss enhances Lin28b-mediaated tumorigenesis and up-regulates Wnt signaling) and Villin-Lin28b;VillinCre;Imp1OE mice (where high Imp1 expression reduces Lin28b-mediated tumorigenesis).
Figure 6.

IMP1 is the principal node for post-transcriptional regulation downstream from LIN28B. We propose a model in which IMP1 plays an important regulatory role downstream from LIN28B. Both LIN28B overexpression and whole-body irradiation enhance IMP1 expression in the intestinal epithelium. Deletion of IMP1 causes a significant increase in LIN28B-mediated tumorigenesis, likely due in part to observed increases in Wnt signaling and, potentially, stem cell signature. Furthermore, IMP1 loss (specifically in Hopx+ stem cells) causes increased regeneration following radiation injury. Taken together, these data suggest IMP1 as a regulator of intestinal epithelial homeostasis downstream from both LIN28B and radiation.
Discussion
In the present study, we used ribosome profiling to demonstrate that IMP1 acts as a pivotal regulator downstream from LIN28B and that IMP1 deletion promotes changes to both mRNA levels and TEs. Globally, there is partial overlap in genes that are regulated at the mRNA and translation levels, suggesting that IMP1 may regulate both homeostatic transcript abundance and TE for a subset of target transcripts. In addition, we found no correlation between the binding efficiencies of targets from eCLIP studies (Conway et al. 2016) and the changes in their TEs through ribosome profiling. This notion is supported by recent studies comparing eCLIP-identified IMP1-bound targets with gene expression changes in IMP1-depleted cells, where there was no significant correlation between binding and trends in gene expression changes (Conway et al. 2016). Additionally, there are targets that are not revealed in the eCLIP studies but are found to be differentially regulated by ribosome profiling, highlighting the tissue-specific as well as potentially direct/indirect roles of IMP1 on gene expression. Our data support the significance of evaluating both the transcriptome and translatome simultaneously to understand RBP function and the notion that evaluating translational reinforcement, antagonism, and buffering may uncover additional roles for RBPs to refine the functional outcome of a gene or pathway.
LIN28B is a critical pluripotency factor and is engaged in diverse cancer types, including CRC (Rahkonen et al. 2016; Zhang et al. 2016; Tsanov et al. 2017). LIN28B acts to a major extent through the post-transcriptional down-regulation of the let-7 miR family. This enables the induction of let-7 mRNA targets, which are normally suppressed. In most contexts, this should lead to enhanced proliferation, growth, and, presumably, a pathway toward transformation. While the let-7 mRNA targets are many (e.g., CyclinD1, HMGA2, IMP1, and IMP2), it remains elusive how precisely LIN28B exerts its actions through the let-7 mRNA targets. Here, we found that IMP1 regulates the TEs of genes downstream from LIN28B, with one node through Wnt signaling pathway components. Pathway analyses highlight several pathways involved in cancer (affecting proliferation, migration, differentiation, and apoptosis), including WNT signaling, that are significantly enriched with Imp1 deletion and concurrent Lin28b expression. These genes are regulated by changes in translational efficiencies by differential binding of ribosomes on the transcripts—a finding that expands substantially our knowledge about IMP1 and WNT signaling, building significantly on a previous study showing that β-catenin induces IMP1 transcription and that IMP1 stabilizes β-catenin mRNA (Gu et al. 2008).
We demonstrate that Imp1 loss in Lin28b transgenic mice promotes a robust increase in high-grade tumors exhibiting phenotypically less differentiation as well as a more invasive phenotype, up-regulation of stem/progenitor cell genes, and enhanced nuclear β-catenin, suggesting that Imp1 loss may promote expansion of the intestinal stem cell compartment. This is in contrast to data in neural stem cells, where Imp1 deletion promoted differentiation (Nishino et al. 2013). Similarly, the finding that Imp1 loss promotes tumorigenesis in the intestinal epithelium is in contrast to prior studies suggesting that IMP1 may act as an oncogene in different tissues (Tessier et al. 2004; Kobel et al. 2007; Mayr and Bartel 2009; Hamilton et al. 2013; Gutschner et al. 2014). However, more recent studies have identified tumor-suppressive roles of IMP1 as well (Hamilton et al. 2015; Wang et al. 2016). These divergences likely depend on numerous factors, including age, the compartment in which Imp1 is expressed (for example, epithelial vs. mesenchymal), the specific tissue type (intestinal, breast, brain, etc.), and whether Imp1 loss is acting alone or in concert with known oncogenes. IMP1 stabilizes β-catenin mRNA in breast cancer cells (Gu et al. 2008) and is activated by it in turn in a feedback mechanism (Gu et al. 2009). In other studies, IMP1 was shown to bind and stabilize β-TRCP1, a β-catenin antagonist (Elcheva et al. 2009) . Hence, we used unbiased approaches (RNA-seq and ribosome profiling/sequencing) to evaluate global changes in pathways with IMP1 loss and then used in vivo mouse models to understand the net effects on WNT pathway signaling. However, our data do not necessarily exclude a role for IMP1 in tumor progression, where it may exhibit different roles based on target transcript abundance.
Although LIN28B has been reported to range from 30% to 75% of CRC tumors (Piskounova et al. 2011; Tu et al. 2015), IMP1 is overexpressed in up to 80% of CRCs (King et al. 2011a; Mongroo et al. 2011). This suggests that the LIN28B–Let7–IMP1 axis may function under certain scenarios, but other LIN28B-independent factors yet to be identified in CRC may still regulate IMP1 expression. This is supported by our analysis using TCGA, where LIN28B and IMP1 expression do not positively correlate at all stages of colon cancer. Importantly, our finding that VillinCre;Imp1OE mice do not exhibit spontaneous tumor initiation suggests that IMP1 overexpression is itself insufficient to drive tumor initiation. To that end, it is possible that IMP1 overexpression may serve as a “brake” or “checkpoint” to the oncogenic effects of LIN28B. The “checkpoint” regulation may be achieved through a variety of mechanisms ascribed to IMP1, such as sequestration of transcripts in IMP1-containing ribonucleoprotein (RNP) granules (to avoid mRNA degradation and/or release the transcripts for translation at critical time points) (Jonson et al. 2007; Weidensdorfer et al. 2009), reduction of miR-mediated silencing of mRNA transcripts (Elcheva et al. 2009; Nishino et al. 2013; Busch et al. 2016; Degrauwe et al. 2016; for review, see van Kouwenhove et al. 2011), or displacement of miR-containing RISCs (Elcheva et al. 2009). IMP1 may also enhance the expression of LIN28B by shielding LIN28B mRNA from regulation by let-7 in vitro (Busch et al. 2016). Additionally, Lin28b and Drosophila Imp1 (dImp1) cooperate in mitotic activity and oncogenesis (Narbonne-Reveau et al. 2016). Furthermore, LIN28B and IMP1 may cooperate to augment cellular bioenergetics and, in the case of LIN28B, cell metabolism (Tu et al. 2015). Nevertheless, the LIN28B–IMP1 post-transcriptional regulon may be viewed potentially as a node in CRCs that feature APC loss/β-catenin activation/Wnt signaling activation.
Targeting key pathways in CRC remains challenging, as so many of these pathways are important for normal epithelial homeostasis. The targeting of RBPs can be refined to interrupt specific RBP:RNA transcript interactions. LIN28B and IMP1 exhibit low expression in most adult tissues, suggesting that targeting them may have few deleterious effects. Inhibitors for LIN28B would prevent aberrant blockade of tumor suppressor let-7, whereas a specific IMP1 inhibitor would require targeting of tumor-promoting interactions while sparing tumor suppressor functions. For example, a novel IMP1 inhibitor that targets c-Myc specifically has demonstrable effects on melanoma and ovarian cell proliferation; however, studies have been limited to in vitro assays thus far (Mahapatra et al. 2014, 2017). In summary, our findings highlight the LIN28B/IMP1 signaling axis as a key regulon for normal homeostasis, radiation-mediated injury, and tumorigenesis in the intestinal epithelium, providing mechanistic insights into the basic biology of RBPs as well as underscoring the importance of understanding post-transcriptional regulation of principle oncogenic pathways in vivo.
Materials and methods
Cell lines and cell culture
CRC cell lines were obtained from American Type Culture Collection and authenticated by short tandem repeat (STR). Stable LIN28B expression in SW480 and LoVo cells was achieved using MSCV-PIG-LIN28B and empty vector control plasmids (gifts from Dr. Joshua Mendell) using the protocol described previously (King et al. 2011b). IMP1 was knocked out by cotransfecting cells with IMP-1 CRISPR/Cas9 knockout plasmid (h) (Santa Cruz Biotechnology, sc-401703) and IMP-1 HDR plasmid (h) (Santa Cruz Biotechnology, sc-401703 HDR) followed by sorting and clonal expansion of RFP+ve cells. The knockout was verified by Western blotting for IMP1. IMP1 was knocked down in CaCo2 cells by siRNA transfection using Lipofectamine RNAiMax as per the manufacturer's guidelines. Silencer select negative control #1 siRNA from Thermo Fisher was used as a negative control. qRT–PCR was used to validate knockdown. Cell lines were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS (GE Healthcare Life Sciences) and 1% penicillin–streptomycin (Thermo Fisher Scientific) in a 37°C incubator with 5% CO2 under puromycin selection. Cells are tested for mycoplasma at least every 2 mo in the laboratory.
Animal models
VillinCre;Vil-Lin28b;Imp1fl/fl mice were obtained by mating Vil-Lin28bMed and VillinCre;Imp1fl/fl mice that have been described previously (Hamilton et al. 2013; Madison et al. 2013) and were maintained via backcrosses to C57BL/6J. Imp1fl/fl mice were considered wild type and expressed LIN28B and IMP1 insignificantly differently from wild-type mice. Lgr5-EGFP-IRES-creERT2 mice were purchased from Jackson Laboratories. The HopxCreERT2;Imp1fl/fl mice were generated by crossing Imp1fl/fl mice with tamoxifen-inducible Hopx-CreERT2 (JAX strain 017606) mice that were generated at the University of Pennsylvania in the laboratory of J. Epstein (Philadelphia, PA) and have been characterized previously (Yousefi et al. 2016). Sox9-EGFP mice were maintained as described previously (Van Landeghem et al. 2012). Cyagen was used to generate Imp1OE mice. Briefly, the knock-in construct was generated by amplification of mouse genomic fragments from a BAC clone using high-fidelity DNA polymerase. The targeting vector was assembled to include Flag-tagged Imp1 (Igf2bp1) cDNA targeted to the Rosa locus downstream from the EF1α promoter and a polyA sequence flanked by loxP sites (Supplemental Fig. 4B). In addition, we included an IRES;tdTomato;SV40EpolyA following the Imp1 cDNA. We crossed these mice with Villin-Cre mice in order to specifically overexpress Imp1 in intestinal epithelial cells. All experimental analyses were performed on three or more individual mice (male or female mice at 12-mo of age for tumor studies and 8- to 12-wk-old mice for irradiation studies). Controls and experimental groups were either sex-matched littermates or age-matched sex-matched nonlittermates. To ablate Imp1 in HopxCreERT2;Imp1fl/fl mice, tamoxifen (Sigma-Aldrich) was dissolved in peanut oil at 10 mg/mL, and 200 µg of tamoxifen per gram of body weight was injected intraperitoneally for each dose. Two doses were administered 24 h apart before the irradiation experiments. Whole-body γ-irradiation (12 Gy) was administered to at least three mice in each group. For irradiation experiments using VillinCre;Impfl/fl mice, animals were given a whole-body single dose of 12 Gy using the Gammacell 40 cesium 137 irradiation unit. Irradiation of Sox9-EGFP mice was performed as described previously (Van Landeghem et al. 2012). Body weights were recorded daily, and mice were euthanized before losing a maximum of 25% total body weight at day 4 after irradiation. All mouse protocols were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania under protocol 804607.
Ribosome profiling
Ribosome profiling libraries from three pooled cell culture plates were prepared using a standard protocol (McGlincy and Ingolia 2017) with minor modifications. Separate 5′ and 3′ linkers were ligated to the RNA fragment instead of 3′ linker followed by circularization (Subtelny et al. 2014). 5′ linkers contained four random nucleotide unique molecular identifiers (UMIs), similar to a 5-nucleotide (nt) UMI in 3′ linkers. During size selection, we restricted the footprint lengths to 18–34 nt. Matched RNA-seq libraries were prepared using RNA that was randomly fragmentated by incubating for 15 min at 95°C in 1 mM EDTA, 6 mM Na2CO3, and 44 mM NaHCO3 (pH 9.3). RNA-seq fragments were restricted to 18–50 nt. Ribosomal RNA (rRNA) were removed from pooled RNA-seq and footprinting samples using RiboZero (Epicenter, MRZH116). cDNA for the pooled library were PCR-amplified for 15 cycles.
Ribosome profiling data processing and analysis
RNA-seq and footprinting reads were mapped to the human transcriptome using the riboviz pipeline (Carja et al. 2017). Sequencing adapters were trimmed from reads using Cutadapt 1.10 (Martin 2011) using parameters --trim-n -O 1 --minimum-length 5. The reads from different samples were separated based on the barcodes in their 3′ linkers using fastx_barcode_splitter (FASTX toolkit, Hannon laboratory) with one mismatch allowed at most. UMIs and barcodes were removed from reads in each sample using Cutadapt (--trim-n -m 10 -u 4 -u -10). Trimmed reads that aligned to human/mouse rRNA were removed using Bowtie version 1.1.2 (Langmead et al. 2009). Remaining reads were mapped to a set of 19,192 principal transcripts for each gene in the APPRIS database (Rodriguez et al. 2013) using HISAT2 version 2.1.0 (Kim et al. 2015). Only reads that mapped uniquely were used for all downstream analyses. For genes with multiple principal transcripts, the first one in the list was chosen. Codes for selecting these transcripts were obtained from the riboviz package (https://github.com/shahpr/riboviz) (Carja and Plotkin 2017). For the radar map, the list of post-transcriptional regulators was obtained from the AURA database (Dassi et al. 2014), and the plot was generated using modified functions of the translatome (Tebaldi et al. 2014) package in R. The translatome package was modified to restrict the list of post-transcriptional regulators to the regulators of the 19,192 genes used for mapping reads. All analyses of ribosome profiling data sets were performed in R. We restricted analyses to genes with at least one data set (RNA-seq or footprinting across conditions) with 64 mapped reads, and genes with 0 read counts in any data set were ignored unless the mean read counts across all eight data sets were >64. Differentially expressed genes were identified using the DEGseq package (Wang et al. 2010) using a false discovery rate (FDR) cutoff of 0.001. Gene ontology (GO) enrichment analyses and identification of post-transcriptional regulators of differentially expressed genes at the transcriptome and translatome levels were performed using the translatome package.
RNA-seq
RNA was isolated from freshly isolated mouse intestine crypts and cell lines using the GeneJET RNA purification kit according to the manufacturer's instructions, with DNase treatment. The cDNA libraries were then generated using the Illumina TruSeq stranded mRNA library preparation kit with Ribo Zero treatment (RS-122-2201). The cDNA was sequenced using HiSeq 50 cycle single-read sequencing version 4 by the High-Throughput Genomics Core at the Huntsman Cancer Institute, University of Utah, for a fee. All sequencing reads were first trimmed to remove 3′ sequencing adapters and aligned to the hg19 human genome with STAR using the default parameters (Dobin et al. 2013). Read counts for each gene were measured using HTSeq run in “intersection-strict” mode (Anders et al. 2015), and differential expression analysis was performed using DESeq2 (Love et al. 2014). Taking all differentially expressed genes with an FDR of <0.05, GO analysis was performed using DAVID (Huang da et al. 2009a,b). Genotypes were run in triplicates, and DESeq2 analysis was performed on them. GO analysis was performed on the differentially expressed genes. GSEA was performed according to previously established guidelines (Subramanian et al. 2005).
TCGA analyses
Publicly available gene expression data from TCGA were downloaded from the Genomic Data Commons (GDC) data portal, and graphs were generated using Graphpad Prism. Tumor type and stage data were acquired from the University of California at Santa Cruz (UCSC) Xena (http://xena.ucsc.edu). Pooled COADREAD data contained 433 samples in different tumor stages. For staging, high expression and low expression groups were determined by cutoff at the 75th percentile.
Correlation of expression was determined via χ2 analysis, and 95% confidence interval was calculated for confirmation of statistical significance in Graphpad Prism.
Tumor tissue microarray analysis
A tumor tissue microarray comprised of a uniform cohort of 228 (133 male and 95 female) patients with colon carcinoma (88 in stage 2 and 140 in stage 3) diagnosed between November 1993 and October 2006 was used. Rectal tumors were excluded (King et al. 2011a). LIN28B staining intensity was scored by a pathologist (A.J.K.-S.); 1 was used to signify low Lin28b intensity, 2 was used to signify intermediate intensity, and 3 was used to signify high intensity. We used the high-expressing samples and analyzed the differentiation status by the three-tier tumor grading system recommended by the American Joint Commission (Edge and Compton 2010).
Enteroid culture and analyses
The proximal jejunum (6 cm) was flushed with cold PBS and cut lengthwise to expose the luminal surface. Tissue was placed in calcium–magnesium-free Hank's buffered salt solution (CMF-HBSS) with 1 mM N-acetyl cysteine, vortexed briefly, and placed on ice. The tissue was then transferred to CMF-HBSS containing 10 mM EDTA and 1 mM NAC and incubated for 45 min at 4°C with end-to-end rotation. Epithelial cells were then mechanically dissociated by vortexing for 30 sec following a 30-sec rest on ice; this was repeated a total of four times. The villi were removed by filtering over a 70-µm filter. Crypts were pelleted at 100g for 4 min and resuspended in Basal+ medium (advanced DMEM/F12 containing 2 mM, 10 mM HEPES, 1× penicillin/streptomycin, 5 µM CHIR99021, 1 mM NAC, 1× N2 supplement, 1× B27 supplement for quantification). Crypts were defined as small U-shaped structures (or partial U-shaped structures for irradiated mice). Crypts were plated in four wells of a 48-well plate per mouse at a density of 150 crypts per well in an 80/20 mixture of Matrigel matrix and Basal+ medium also containing 50 ng/mL mouse epidermal growth factor and 2.5% Noggin/R-spondin conditioned medium (Sato et al. 2009). After Matrigel solidified, crypt-containing patties were overlaid with ENR medium. The number of surviving crypt spheres (enteroids) was counted on days 1–4 following plating to determine enteroid plating efficiency. On day 4 after plating, the number of surviving enteroids was scored. All counting was performed at 10× magnification using the Olympus IX81 inverted microscope. A crypt sphere was defined as a 3D sphere or oval shape surrounding a lumen (i.e., enterosphere). A “budded” enteroid was defined as a 3D structure with one or more de novo projections of any length representing crypts emerging from the spheroid body.
qRT–PCR
Small intestine crypt RNA was isolated using the GeneJet RNA purification kit. Equal amounts of total RNA were reverse-transcribed using the TaqMan RT reagent kit, and the resulting cDNA was used with Power SYBR Green PCR master mix and validated primer sets listed in Supplemental Table 3. Non-reverse-transcribed samples were used as negative controls, and gene expression was calculated using the R = 2(−ΔΔCt) method, where changes in Ct values for the genes of interest were normalized to housekeeping genes Tbp, Rsp6, or Gapdh. Gene expression data are expressed as fold change versus mean values for wild-type or untreated controls. All experiments were replicated in at least three independent experiments with technical replicates (duplicates) in each experiment. For analyses of Sox9-EGFP mice, FACS isolation of Sox9-EGFP cell populations was performed as described previously (Van Landeghem et al. 2012). Total RNA from all Sox9-EGFP populations and nonsorted epithelial cells was extracted and reverse-transcribed. qRT–PCR was performed using platinum qPCR SuperMix-UDG and the following TaqMan probes: Mm00501602_m1 (Igf2bp1) and Mm02342456_g1 (Rsp6). Samples were run in duplicate, and expression was determined by a standard curve of pooled nonsorted intestinal epithelial cells and normalized to the invariant control gene Rps6.
Histology, immunohistochemistry, and tissue analyses
Tissues were fixed in zinc formalin fixative overnight at 4°C, washed in PBS, and moved to 70% ethanol before paraffin embedding and sectioning. Hematoxylin and eosin (H&E) staining was performed according to standard procedures in the Molecular Pathology and Imaging Core of the Penn Center for Molecular Studies in Digestive and Liver Diseases. For immunostaining, antigen retrieval was performed by heating slides in 10 mM citric acid buffer (2.1 g of citric acid monohydrate in 1 L of di-H2O at pH 6.0) in a pressure cooker. The images were taken using a Nikon Eclipse E600 microscope and analyzed with the NIS-Elements basic research software version 4.51. Imaging was performed at room temperature. The following primary antibodies were used for immunostaining: anti-Ki67 (1:200), anti-lysozyme (1:1000), anti-chromagranin A (1:1000), anti-H2AX (1:1000), anti-IMP1 (1:1000), anti-AXIN2 (1:4000), and anti-β-catenin (1:1000). Cy2- or Cy3-conjugated secondary antibodies were obtained from Jackson Immunoresearch Laboratories, Inc. Biotinylated secondary antibodies and the DAB substrate kit for immunohistochemistry were purchased from Vector Laboratories. All quantifications were done across at least 30 high-powered fields per animal that were randomly selected areas throughout the small intestine of at least four mice in each genotype. For quantifying regeneration after irradiation, regenerative microcolonies/foci were defined as a cluster of five or more Ki67-positive cells from a single clone (colony or hyperproliferative crypt).
Flow cytometry and sorting
For analysis of EdU-positive cells, the mice were injected with 20 mg/kg EdU intraperotoneally. The mice were sacrificed 2 h after the injection. The single-cell suspensions of crypt-enriched intestinal epithelium from the small intestine (jejunum) of the mice were then stained for EdU following the manufacture's instructions. For sorting, mouse intestinal epithelial cells were isolated into single cells. CD24-PE (Biolegend) and cKit PE-Cy7 (eBioscience) staining was done for sorting as per the manufacturer's instructions. Flow cytometry was performed using a FACS LSR (BD Biosciences) with a 100-µm nozzle. Sorting was done using a FACS Aria (BD Biosciences) sorter. Gating and compensation were performed using negative and single-color controls. Three biological replicates (and three technical replicates) for each genotype were used.
Quantification and statistical analysis
Statistical analysis was performed using Graphpad Prism software version 7.0. One-way ANOVA with Tukey's post-hoc test was performed to compare the differences among groups in all studies. Unpaired t-test (two-tailed) was used when two groups were compared. For all experiments with error bars, the standard error of the mean (SEM) was calculated, and the data are presented as mean ± SEM. The sample size for each experiment is included in the figure legends.
Data and software availability
See “Ribosome profiling data processing and analysis” for software information. The accession numbers for all new RNA-seq data and ribosome-profiling data reported here have been deposited in Gene Expression Omnibus (accession no. GSE115647).
Supplementary Material
Acknowledgments
We thank members of the Rustgi laboratory for helpful discussions, and Dr. Dan Dixon for critical reading of this manuscript. We thank Dr. Kay Lund for valuable input. Financial support was provided by National Institutes of Health (NIH) R01 DK056645 (to P.C., R.M., S.F.A., K.E.H., and A.K.R.), NIH P30DK050306 and its pilot grant program (to K.E.H. and A.K.R.), NIH K01 DK100485 (to K.E.H.), a Crohn's and Colitis Foundation Career Development Award (to K.E.H.), NIH F32DK107052 (to S.F.A.), NIH T32CA1152999 (to S.F.A.), the American Cancer Society (to A.K.R.), the Lustgarten Family Colon Cancer Fund (to A.K.R.), NIH R35 GM124976 (to S.L., H.R.S.W., and P.S.), National Institute of General Medical Sciences T32GM008216-29 (to S.W.F.), start-up funds from the Human Genetics Institute of New Jersey and Rutgers University (to S.L., H.R.S.W., and P.S.), NIH R03DK114463 (to K.E.H.), and startup funds from the Children's Hospital of Philadelphia Research Institute (to K.E.H.).
Author contributions: P.C., K.E.H., and A.K.R. conceived the study. P.C., K.E.H., B.B.M., and A.K.R. performed the methodology. S.W.F., B.D.G., S.L., H.R.S.W., P.S., and J.R.P. ran the software and performed the formal analysis. P.C., K.E.H., S.F.A., S.L., H.R.S.W., R.M., L.A.S., P.D.H., A.J.K.-S., A.T.M., L.V.L., and P.S. performed the investigation. P.C., K.E.H., P.S., and A.K.R. wrote the original draft of the manuscript. P.C., K.E.H., S.F.A., C.J.L., P.S., and A.K.R. reviewed and edited the manuscript. K.E.H. and A.K.R. acquired the funding.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.314369.118.
References
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Shomron N, Hedegaard MM, Vinther J, Andersen JS, Feig C, Xu J, Burge CB, Peter ME. 2008. Identification of let-7-regulated oncofetal genes. Cancer Res 68: 2587–2591. [DOI] [PubMed] [Google Scholar]
- Busch B, Bley N, Muller S, Glass M, Misiak D, Lederer M, Vetter M, Strauss HG, Thomssen C, Huttelmaier S. 2016. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res 44: 3845–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carja O, Plotkin JB. 2017. Evolutionary rescue through partly heritable phenotypic variability. bioRxiv 10.1101/092718. [DOI] [PMC free article] [PubMed]
- Carja O, Xing T, Plotkin JB, Shah P. 2017. riboviz: analysis and visualization of ribosome profiling datasets. bioRxiv 10.1101/100032. [DOI] [PMC free article] [PubMed]
- Chatterji P, Rustgi AK. 2018. RNA binding proteins in intestinal epithelial biology and colorectal cancer. Trends Mol Med 24: 490–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J. 2013. Exploring TCGA pan-cancer data at the UCSC cancer genomics browser. Sci Rep 3: 2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway AE, Van Nostrand EL, Pratt GA, Aigner S, Wilbert ML, Sundararaman B, Freese P, Lambert NJ, Sathe S, Liang TY, et al. 2016. Enhanced CLIP uncovers IMP protein–RNA targets in human pluripotent stem cells important for cell adhesion and survival. Cell Rep 15: 666–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassi E, Re A, Leo S, Tebaldi T, Pasini L, Peroni D, Quattrone A. 2014. AURA 2: Empowering discovery of post-transcriptional networks. Translation 2: e27738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degrauwe N, Schlumpf TB, Janiszewska M, Martin P, Cauderay A, Provero P, Riggi N, Suva ML, Paro R, Stamenkovic I. 2016. The RNA binding protein IMP2 preserves glioblastoma stem cells by preventing let-7 target gene silencing. Cell Rep 15: 1634–1647. [DOI] [PubMed] [Google Scholar]
- de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, Anderson JE, Storm EE, Modrusan Z, Koeppen H, et al. 2017. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543: 676–680. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge SB, Compton CC. 2010. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 17: 1471–1474. [DOI] [PubMed] [Google Scholar]
- Elcheva I, Goswami S, Noubissi FK, Spiegelman VS. 2009. CRD-BP protects the coding region of βTrCP1 mRNA from miR-183-mediated degradation. Mol Cell 35: 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formeister EJ, Sionas AL, Lorance DK, Barkley CL, Lee GH, Magness ST. 2009. Distinct SOX9 levels differentially mark stem/progenitor populations and enteroendocrine cells of the small intestine epithelium. Am J Physiol Gastrointest Liver Physiol 296: G1108–G1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MK, Faulk DM, Sundaram N, Shroyer NF, Henning SJ, Helmrath MA. 2012. Intestinal crypts reproducibly expand in culture. J Surg Res 178: 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracz AD, Ramalingam S, Magness ST. 2010. Sox9 expression marks a subset of CD24-expressing small intestine epithelial stem cells that form organoids in vitro. Am J Physiol Gastrointest Liver Physiol 298: G590–G600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. 2015. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526: 715–718. [DOI] [PubMed] [Google Scholar]
- Gu W, Wells AL, Pan F, Singer RH. 2008. Feedback regulation between zipcode binding protein 1 and β-catenin mRNAs in breast cancer cells. Mol Cell Biol 28: 4963–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Pan F, Singer RH. 2009. Blocking β-catenin binding to the ZBP1 promoter represses ZBP1 expression, leading to increased proliferation and migration of metastatic breast-cancer cells. J Cell Sci 122: 1895–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschner T, Hammerle M, Pazaitis N, Bley N, Fiskin E, Uckelmann H, Heim A, Grobeta M, Hofmann N, Geffers R, et al. 2014. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology 59: 1900–1911. [DOI] [PubMed] [Google Scholar]
- Hamilton KE, Noubissi FK, Katti PS, Hahn CM, Davey SR, Lundsmith ET, Klein-Szanto AJ, Rhim AD, Spiegelman VS, Rustgi AK. 2013. IMP1 promotes tumor growth, dissemination and a tumor-initiating cell phenotype in colorectal cancer cell xenografts. Carcinogenesis 34: 2647–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton KE, Chatterji P, Lundsmith ET, Andres SF, Giroux V, Hicks PD, Noubissi FK, Spiegelman VS, Rustgi AK. 2015. Loss of stromal IMP1 promotes a tumorigenic microenvironment in the colon. Mol Cancer Res 13: 1478–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Jonson L, Vikesaa J, Krogh A, Nielsen LK, Hansen T, Borup R, Johnsen AH, Christiansen J, Nielsen FC. 2007. Molecular composition of IMP1 ribonucleoprotein granules. Mol Cell Proteomics 6: 798–811. [DOI] [PubMed] [Google Scholar]
- Keene JD. 2007. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8: 533–543. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CE, Cuatrecasas M, Castells A, Sepulveda AR, Lee JS, Rustgi AK. 2011a. LIN28B promotes colon cancer progression and metastasis. Cancer Res 71: 4260–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CE, Wang L, Winograd R, Madison BB, Mongroo PS, Johnstone CN, Rustgi AK. 2011b. LIN28B fosters colon cancer migration, invasion and transformation through let-7-dependent and -independent mechanisms. Oncogene 30: 4185–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobel M, Weidensdorfer D, Reinke C, Lederer M, Schmitt WD, Zeng K, Thomssen C, Hauptmann S, Huttelmaier S. 2007. Expression of the RNA-binding protein IMP1 correlates with poor prognosis in ovarian carcinoma. Oncogene 26: 7584–7589. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Yousefi M, Nakauka-Ddamba A, Jain R, Tobias J, Epstein JA, Jensen ST, Lengner CJ. 2014. Single-cell analysis of proxy reporter allele-marked epithelial cells establishes intestinal stem cell hierarchy. Stem Cell Reports 3: 876–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Yousefi M, Nakauka-Ddamba A, Tobias JW, Jensen ST, Morrisey EE, Lengner CJ. 2016. Heterogeneity in readouts of canonical wnt pathway activity within intestinal crypts. Dev Dyn 245: 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Liu Q, Zhong X, Hahn CM, Lin N, Emmett MJ, Stanger BZ, Lee JS, Rustgi AK. 2013. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes Dev 27: 2233–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Jeganathan AN, Mizuno R, Winslow MM, Castells A, Cuatrecasas M, Rustgi AK. 2015. Let-7 represses carcinogenesis and a stem cell phenotype in the intestine via regulation of Hmga2. PLoS Genet 11: e1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra L, Mao C, Andruska N, Zhang C, Shapiro DJ. 2014. High-throughput fluorescence anisotropy screen for inhibitors of the oncogenic mRNA binding protein, IMP-1. J Biomol Screen 19: 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. 2017. A novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol 10: 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10–12. [Google Scholar]
- Mayr C, Bartel DP. 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, Ingolia NT. 2017. Transcriptome-wide measurement of translation by ribosome profiling. Methods 126: 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno R, Chatterji P, Andres SF, Hamilton KE, Simon L, Foley SW, Jeganathan AN, Gregory BD, Madison BB, Rustgi AK. 2018. Differential regulation of LET-7 by LIN28B isoform-specific functions. Mol Cancer Res 16: 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongroo PS, Noubissi FK, Cuatrecasas M, Kalabis J, King CE, Johnstone CN, Bowser MJ, Castells A, Spiegelman VS, Rustgi AK. 2011. IMP-1 displays cross-talk with K-Ras and modulates colon cancer cell survival through the novel proapoptotic protein CYFIP2. Cancer Res 71: 2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AR, Mukherjee N, Keene JD. 2010. Systematic analysis of posttranscriptional gene expression. Wiley Interdiscip Rev Syst Biol Med 2: 162–180. [DOI] [PubMed] [Google Scholar]
- Narbonne-Reveau K, Lanet E, Dillard C, Foppolo S, Chen CH, Parrinello H, Rialle S, Sokol NS, Maurange C. 2016. Neural stem cell-encoded temporal patterning delineates an early window of malignant susceptibility in Drosophila. Elife 5: e13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J, Kim S, Zhu Y, Zhu H, Morrison SJ. 2013. A network of heterochronic genes including Imp1 regulates temporal changes in stem cell properties. Elife 2: e00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwokafor CU, Sellers RS, Singer RH. 2016. IMP1, an mRNA binding protein that reduces the metastatic potential of breast cancer in a mouse model. Oncotarget 7: 72662–72671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima N, Yamada Y, Nagayama S, Kawada K, Hasegawa S, Okabe H, Sakai Y, Aoi T. 2014. Induction of cancer stem cell properties in colon cancer cells by defined factors. PLoS One 9: e101735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Polytarchou C, Thornton JE, LaPierre RJ, Pothoulakis C, Hagan JP, Iliopoulos D, Gregory RI. 2011. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 147: 1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potten CS. 2004. Radiation, the ideal cytotoxic agent for studying the cell biology of tissues such as the small intestine. Radiat Res 161: 123–136. [DOI] [PubMed] [Google Scholar]
- Rahkonen N, Stubb A, Malonzo M, Edelman S, Emani MR, Narva E, Lahdesmaki H, Ruohola-Baker H, Lahesmaa R, Lund R. 2016. Mature Let-7 miRNAs fine tune expression of LIN28B in pluripotent human embryonic stem cells. Stem Cell Res 17: 498–503. [DOI] [PubMed] [Google Scholar]
- Roche KC, Gracz AD, Liu XF, Newton V, Akiyama H, Magness ST. 2015. SOX9 maintains reserve stem cells and preserves radioresistance in mouse small intestine. Gastroenterology 149: 1553–1563.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JM, Maietta P, Ezkurdia I, Pietrelli A, Wesselink JJ, Lopez G, Valencia A, Tress ML. 2013. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res 41: D110–D117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265. [DOI] [PubMed] [Google Scholar]
- Schafer S, Adami E, Heinig M, Rodrigues KE, Kreuchwig F, Silhavy J, van Heesch S, Simaite D, Rajewsky N, Cuppen E, et al. 2015. Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat Commun 6: 7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP. 2014. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. 2011. Interconversion between intestinal stem cell populations in distinct niches. Science 334: 1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebaldi T, Dassi E, Kostoska G, Viero G, Quattrone A. 2014. tRanslatome: an R/Bioconductor package to portray translational control. Bioinformatics 30: 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier CR, Doyle GA, Clark BA, Pitot HC, Ross J. 2004. Mammary tumor induction in transgenic mice expressing an RNA-binding protein. Cancer Res 64: 209–214. [DOI] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. 2011. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478: 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov KM, Pearson DS, Wu Z, Han A, Triboulet R, Seligson MT, Powers JT, Osborne JK, Kane S, Gygi SP, et al. 2017. LIN28 phosphorylation by MAPK/ERK couples signalling to the post-transcriptional control of pluripotency. Nat Cell Biol 19: 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu HC, Schwitalla S, Qian Z, LaPier GS, Yermalovich A, Ku YC, Chen SC, Viswanathan SR, Zhu H, Nishihara R, et al. 2015. LIN28 cooperates with WNT signaling to drive invasive intestinal and colorectal adenocarcinoma in mice and humans. Genes Dev 29: 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, Haegebarth A, Stange DE, van de Wetering M, Clevers H. 2009. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology 137: 15–17. [DOI] [PubMed] [Google Scholar]
- van Kouwenhove M, Kedde M, Agami R. 2011. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer 11: 644–656. [DOI] [PubMed] [Google Scholar]
- Van Landeghem L, Santoro MA, Krebs AE, Mah AT, Dehmer JJ, Gracz AD, Scull BP, McNaughton K, Magness ST, Lund PK. 2012. Activation of two distinct Sox9-EGFP-expressing intestinal stem cell populations during crypt regeneration after irradiation. Am J Physiol Gastrointest Liver Physiol 302: G1111–G1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O'Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. 2009. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 41: 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, Zhang X. 2010. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138. [DOI] [PubMed] [Google Scholar]
- Wang G, Huang Z, Liu X, Huang W, Chen S, Zhou Y, Li D, Singer RH, Gu W. 2016. IMP1 suppresses breast tumor growth and metastasis through the regulation of its target mRNAs. Oncotarget 7: 15690–15702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidensdorfer D, Stohr N, Baude A, Lederer M, Kohn M, Schierhorn A, Buchmeier S, Wahle E, Huttelmaier S. 2009. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA 15: 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi M, Li N, Nakauka-Ddamba A, Wang S, Davidow K, Schoenberger J, Yu Z, Jensen ST, Kharas MG, Lengner CJ. 2016. Msi RNA-binding proteins control reserve intestinal stem cell quiescence. J Cell Biol 215: 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ratanasirintrawoot S, Chandrasekaran S, Wu Z, Ficarro SB, Yu C, Ross CA, Cacchiarelli D, Xia Q, Seligson M, et al. 2016. LIN28 regulates stem cell metabolism and conversion to primed pluripotency. Cell Stem Cell 19: 66–80. [DOI] [PubMed] [Google Scholar]
- Zhou WJ, Geng ZH, Spence JR, Geng JG. 2013. Induction of intestinal stem cells by R-spondin 1 and Slit2 augments chemoradioprotection. Nature 501: 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Ma X, Wang Z, Sun C, Wang Y, He Y, Zhang H. 2015. Radiation-induced hyperproliferation of intestinal crypts results in elevated genome instability with inactive p53-related genomic surveillance. Life Sci 143: 80–88. [DOI] [PubMed] [Google Scholar]
- Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. 2009. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 457: 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.