

Abstract
Sialic acids are monosaccharides found in terminal sugar chains of cell surfaces and proteins; they have various biological functions and have been implicated in health and disease. Genetic defects of the GNE gene which encodes a critical bifunctional enzyme for sialic acid biosynthesis, lead to GNE myopathy, a disease manifesting with progressive muscle atrophy and weakness. The likely mechanism of disease is a lack of sialic acids. There remains, however, an unexplained link between hyposialylation and the muscle atrophy and weakness. In this study, we found that muscle proteins were highly modified by S-nitrosylation, and that oxidative stress-responsive genes were significantly upregulated, in hyposialylated muscles from human GNE myopathy patients and model mice. In both in vitro and in vivo models, the production of reactive oxygen species (ROS) was elevated with cellular hyposialylation, and increasing overall sialylation by extrinsic sialic acid intake reduced ROS and protein S-nitrosylation. More importantly, the antioxidant, oral N-acetylcysteine led to amelioration of the muscle atrophy and weakness in Gne mutant mice. Our data provide evidence of additional important function of sialic acids as a ROS scavenger in skeletal muscles, expanding our understanding on how sialic acid deficiency contributes to disease pathology, and identify oxidative stress as a therapeutic target in GNE myopathy.
Introduction
Sialic acids are N- and O-acyl forms of neuraminic acids and widely distributed in many organisms. They are present in the peripheral end of the sugar chains in glycoproteins and glycolipids and are involved in various biological events by acting as interface structures on cell-cell recognition and receptor-ligand interaction, and determining the fate and turnover of modified proteins/lipids (1–3). In mammals, sialic acid is known to be produced by a single biosynthetic pathway. The first enzyme, UDP-GlcNAc 2-epimerase/ManNAc kinase is important and critical for determining sialic acid production, and encoded by the GNE gene (4). So far, two diseases caused by the genetic mutations in GNE gene have been known. The dominant mutation in the allosteric feedback inhibition site of GNE causing excessive production of sialic acids (5), leads to sialuria (OMIM #269921), a rare congenital disorder presenting with hepatomegaly, coarse facies and varying degrees of developmental delay. On the other hand, bi-allelic recessive mutations cause GNE myopathy (OMIM #605820), a progressive muscle disorder, which has been posited to be due to the reduction in sialic acid biosynthesis (6,7).
GNE myopathy, which is also known as distal myopathy with rimmed vacuoles (8) or hereditary inclusion body myopathy (9), is an autosomal recessive myopathy characterized by skeletal muscle atrophy and weakness that preferentially involve the distal muscles. The symptoms of muscle weakness start from the second or third decade, and most of the affected patients become wheelchair-bound within 12 years in average (10,11). Muscle biopsy findings include myofiber atrophy, intracellular amyloid deposits, and presence of rimmed vacuoles (10,12). Additionally, reversal of the muscle symptoms in a mouse model (Gne-/-hGNED207V-Tg) by sialylation-increasing therapies further strengthened the role of hyposialylation in the mechanism of disease (13–15). The preclinical studies in mice paved the way for phase 1 (NCT01359319, sialic acid; NCT01634750, ManNAc), phase 2 (NCT01517880, sialic acid; NCT02346461, ManNAc), and phase 3 (NCT02377921, sialic acid) clinical trials for human patients. In spite of extensive experimental works during the last decade, there remains a question on hyposialylation leads to muscle disease, and whether sialic acids or sialylation have a role in skeletal muscle biology and maintenance. Another question that remains is on how efficient the salvage pathway in sialic acid supply is utilized in GNE myopathy, allowing the use of dietary sources and lysosomal production of free sialic acid to patients as a natural supplement of sialic acids. The results of the clinical trials will likely provide the efficacy of the extrinsic sialic acid supplementation and some answers to this question.
Using GNE myopathy mouse model, we have demonstrated that atrophy precedes the intracellular histological changes in the muscle (16). Muscle atrophy occurs when the protein degradation rate is higher than the synthesis rate, and this phenomenon is seen in several situations like disuse, aging, starvation, and several disease states (17,18). We hypothesize that oxidative stress may be generated by sialic acid deficiency and it may contribute to the muscle atrophy in GNE myopathy, as muscle-derived oxidants have been linked to skeletal muscle weakness and atrophy via muscle protein modifications and enhanced proteolysis (19–21). In this study, we investigated the relationship between hyposialylation and ROS production/oxidative stress in GNE myopathy models, and then the contribution of oxidative stress to muscle weakness and atrophy by the treatment of GNE myopathy mice with an antioxidant compound. We provided several pieces of evidence to demonstrate that sialic acids work on the scavenging of ROS in skeletal muscles, and the oxidative stress induced by sialic acid deficiency might be the upstream pathomechanism of GNE myopathy, which can be a new therapeutic target.
Results
Muscle proteins were highly S-nitrosylated in GNE myopathy
Analysis of oxidation-related protein/lipid modifications in skeletal muscles from GNE myopathy patients revealed elevation of overall protein S-nitrosylation (Fig. 1A and C),which is produced from reactive nitrogen oxide species under oxidized conditions (22,23), although there is no difference in the patterns of S-nitrosyl proteins on two-dimensional PAGE between samples from patient and control (Fig. 1E). We also observed the elevation of protein S-nitrosylation in muscles of GNE myopathy mice, although the findings did not reach statistical significance (Fig. 1B and D). We then performed a shotgun proteomic analyses to identify which proteins were S-nitrosylated proteins in muscles from GNE myopathy patients and mice, and among the >200 proteins identified, the most abundant 30 proteins are listed in Table 1 and Supplementary Material, Table S1. Proteins in the contractile system, energy production, and calcium homeostasis in sarcoplasmic reticulum were abundant in both human patient and mouse muscles. Analysis of protein carbonylation and tyrosine nitrosylation, did not show any difference in the oxidative levels between GNE myopathy and control muscles.
Figure 1.

Elevated S-nitrosylation of skeletal muscle proteins in GNE myopathy muscles. (A) Detection of protein S-nitrosylation in human skeletal muscles. Control, diseased control; GM, GNE myopathy patients (left, Patient 1; center, Patient 2; right, Patient 3). (B) Detection of S-nitrosylated proteins from mouse skeletal muscles. Control, littermates; GM, GNE myopathy mice. (C) Total protein S-nitrosylation rates in human GNE myopathy muscles (n = 4). (D) Total S-nitrosylation rates in mouse muscle proteins (n = 7). Total intensities were normalized by total protein amounts. Data represent mean ± SEM of each group. *P < 0.05. (E)S-nitrosylated protein profiles on 2-Dimensional PAGE. GM, GNE myopathy patient 5.
Table 1.
List of the major S-nitrosylated proteins in skeletal muscles of GNE myopathy patients
| Rank | Protein | Molecular Mass (kDa) | Function | No. of Identified fragments |
|---|---|---|---|---|
| 1 | Titin | 3816 | Contraction | 150 |
| 2 | Filamin-C | 291 | Cytoplasm | 90 |
| 3 | Myomesin-2 | 165 | Contraction | 67 |
| 4 | Actin, alpha skeletal muscle | 42 | Contraction | 60 |
| 5 | Glycogen phosphorylase, muscle | 97 | Energy production | 55 |
| 6 | Alpha-actinin-2 | 104 | Contraction | 53 |
| 7 | Beta-enolase | 47 | Energy production | 47 |
| 8 | Myosin-7 | 223 | Contraction | 45 |
| 9 | Myosin-2 | 223 | Contraction | 45 |
| 10 | Creatine kinase, muscle | 43 | Energy production | 38 |
| 11 | Myosin-1 | 223 | Contraction | 31 |
| 12 | Fructose-bisphosphate aldolase A | 39 | Energy production | 31 |
| 13 | Carbonic anhydrase 3 | 30 | Antioxidation | 30 |
| 14 | Myosin-binding protein C, slow-type | 128 | Contraction | 27 |
| 15 | Myomesin-1 | 188 | Contraction | 25 |
| 16 | Four and a half LIM domains protein 1 | 36 | Contraction | 25 |
| 17 | Triosephosphate isomerase | 31 | Energy production | 22 |
| 18 | Glyceraldehyde-3-phosphate dehydrogenase | 36 | Energy production | 21 |
| 19 | Aconitate hydratase, mitochondrial | 85 | Energy production | 20 |
| 20 | Sarcoplasmic reticulum calcium ATPase 1 | 110 | SR/ER | 20 |
Atrogenes and oxidative stress related genes are upregulated in the GNE myopathy muscles
Two muscle-specific ubiquitin ligases, atrogin-1/Fbxo32 and MuRF1/Trim63, are upregulated in various models of muscle atrophy (24,25). Quantitative RT-PCR revealed these two skeletal muscle atrophy markers are highly expressed with a 1.8-fold increase in atrogin-1 expression and a 2.0-fold increase in MuRF1 in GNE myopathy mice muscles as compared to littermate muscles (Fig. 2A), consistent with the atrophy we observe in our model mice (16) and suggested that common proteolytic systems of muscle atrophy are involved in the pathomechanism of GNE myopathy.
Figure 2.

Upregulation of atrogenes and oxidative stress-responsive genes in GNE myopathy muscles. (A) Expression of Fbxo32, Trim63, Srxn1, Mts, and Map1lc3b in untreated (white bars, n = 17), high dose (HD, 1.0%) or low dose (LD, 0.1%) N-acetylcysteine (NAC) treated (gray bars, n = 6 per group), and littermate controls (black bars, n = 6) as fold changes of littermate controls. Data represent mean ± SEM of each group. *P < 0.05. (B-E) Microarray analysis followed by gene ontology profiling: (B) muscle atrophy-related genes, (C) oxidative stress and redox homeostasis-related genes, (D) autophagy-related genes, (E) collagen-related genes. Each dot represents average expression values for the same gene from GNE myopathy (vertical axis, n = 9) and littermates (horizontal axis, n = 3) muscles. Inverted triangles show recovered expression values with N-acetylcysteine treatment (vertical axis, n = 6) for significantly upregulated genes.
In order to determine the pathologic pathways related to loss of GNE function, we investigated the expression of genes expected to be dysregulated in GNE myopathy by microarray analysis (Fig. 2B–E, Supplementary Material, Table S2A–D) using mouse tissues. Through genetic ontology profiling, we found several genes related to muscle atrophy, redox homeostasis, autophagy, and collagen organization that were highly expressed in GNE myopathy (muscles when compared to littermate muscles). Among these genes, we verified by quantitative RT-PCR that oxidative stress responsive genes, Mt1, Mt2, and Mt3 genes and Srxn1 gene were upregulated. The expression of Mt1, Mt2, and Mt3 were increased by 3.8-, 3.4-, and 4.4-fold, respectively, and Srxn1 expression was increased by 1.5-fold in murine GNE myopathy muscles (Fig. 2A). We found MT1X and MT2A were also upregulated in human GNE myopathy muscles (Supplementary Materials, Fig. S1).
Reactive oxygen species (ROS) is increased in GNE myopathy muscles
To directly establish that oxidative stress is associated with the pathomechanism of GNE myopathy, we analysed the levels of ROS in skeletal muscles in vivo (Fig. 3A and B). Hydroxyl radicals in microdialysate, representing ROS, from resting muscles during baseline were similar in both GNE myopathy mice (2.3 ± 0.8 pM) and littermates (2.4 ± 1.2 pM), but were considerably increased during and after muscle contraction by electrical stimulation of gastrocnemius muscles. Notably, the degree of ROS increments was significantly greater in GNE myopathy mice (14.5 ± 8.8 pM) than those in littermates (6.3 ± 2.8 pM) (Fig. 3C and D), implying oxidative stress is indeed increased in the affected muscles.
Figure 3.

Measurement of ROS production in the GNE myopathy muscle in vivo using microdialysis. (A-B) Experimental set up and representative HPLC chromatograms. Perfusion medium containing 5 mM salicylate (Sal) was pumped through a microdialysis probe and DHBA in dialysate was detected by HPLC–electrochemical detection system: (A) baseline, (B) post-stimulus. (C) Electrical stimulus (ES) consisted of 50V, 40 Hz, 3 ms pulses for 300 trains. HPLC profils of DHBA during the experiments. (D) DHBA increments after contraction in GNE myopathy mice (n = 9) and littermates (n = 5). Pre-ES, before ES. ES1, 1st 20 min after ES. ES2, 2nd 20 min after ES. Data represent mean ± SEM. Each circle represents a DHBA level from an individual mouse. *P < 0.05.
Anti-oxidant capacity is impaired in hyposialylated myotubes of GNE myopathy
To further explore oxidative stress and sialic acid deficiency, we measured intracellular ROS production in cultured GNE myopathy myotubes. Intracellular ROS level, quantified by dichloro-fluorescein (DCF) diacetate labeling, was increased in GNE myopathy myotubes compared to controls (Fig. 4A and B). Mean DCF fluorescence measured in GNE myopathy myotubes was 1113 ± 30 AFU, which was ten times higher than fluorescence measured in littermate controls (115 ± 15 AFU). By giving NeuAc in the medium, the increased DCF fluorescence levels in GNE myopathy myotubes were significantly reduced to near-normal levels (349 ± 19 AFU), suggesting that sialylation is important for maintenance of the redox homeostasis in skeletal muscle cells.
Figure 4.

Impaired antioxidant capacity in hyposialylated GNE myopathy myotubes. (A) Intracellular ROS generation with green DCF staining. (B) Measurement of DCF fluorescence by a plate reader. Data represent mean ± SEM of each group (n = 14). (C-D) ROS levels and cells viability were analysed with the addition of increasing concentration of H2O2 (0.5, 1.0, 2.0, and 4.0 mM) or menadione (5, 10, 20, and 40 μM) to culture media. Control myotubes of each group were cultured in the same condition without adding H2O2, menadione: (C) increased ROS levels with the addition of increasing concentration of H2O2, (D) menadione. (E-F) Relative viability of cells: (E) with the addition of increasing concentration of H2O2, (F) menadione. N-acetylcysteine (NAC). Each point is mean ± SEM of four determinations. *P < 0.05; **P < 0.01.
We then investigated the susceptibility of GNE myopathy myotubes to oxidative stress when exposed to increasing concentration of pro-oxidants (Fig. 4C–F). Exposure of cells to H2O2 (Fig. 4C) or menadione (Fig. 4D) led to an increase in intracellular ΔDCF fluorescence (fluorescence at each concentration - fluorescence at zero concentration) in a dose dependent manner. Hyposialylated GNE myopathy myotubes showed more accelerated increase in ΔDCF fluorescence levels compared to littermate cells. When GNE myopathy myotubes were incubated with NeuAc before and during exposure to pro-oxidants, the ΔDCF fluorescence increments were significantly decreased in both H2O2 and menadione. Importantly, with pro-oxidants treatment, cell viability decreased sharply in GNE myopathy myotubes, whereas NeuAc supplementation rescued GNE myopathy myotubes to be more resistant to higher concentrations of H2O2 or menadione (Fig. 4E and F).
Sialic acid administration suppresses muscle protein S-nitrosylation in GNE myopathy mice
We have previously shown the prophylactic or therapeutic effects of sialic acid administration on the myopathic phenotypes of GNE myopathy model mice (14,15). We analysed protein S-nitrosyl modification of gastrocnemius muscles from the GNE myopathy mice whose muscle phenotypes were well improved after oral administration of NeuAc at 20 mg/kg body wt/d for 10 mo (Fig. 5A). S-nitrosylation was significantly reduced by NeuAc administration (Fig. 5B), suggesting the relationship between hyposialylation, oxidative stress and the onset of myopathy. We thus hypothesized that oxidative stress can be a critical mediator of hyposialylation and muscle weakness, and scavenging ROS with an antioxidant can ameliorate the muscle phenotypes in GNE myopathy.
Figure 5.

Suppression of protein S-nitrosylation in GNE myopathy mice muscles by sialic acid administration. (A,B) Protein S-nitrosyl modification in gastrocnemius muscles from the GNE myopathy mice with/without oral administration of NeuAc at 20 mg/kg body wt/d for 10 mo. Control, untreated controls (n = 3); GM, untreated GNE myopathy mice (n = 3); NeuAc NeuAc-treated GNE myopathy mice (n = 3). Data presented with mean ± SEM. *P < 0.05.
N-acetylcysteine treatment restores muscle weakness and atrophy in GNE myopathy model mice
To determine whether pharmacological antioxidants are protective against oxidative stress in GNE myopathy myotubes, we incubated cells with N-acetylcysteine and found remarkable decrease in DCF fluorescence level (191 ± 10 AFU, P < 0.01) (Fig. 4A and B). N-acetylcysteine also worked effectively in the high oxidative stress condition and the protective effect appeared to be more evident for menadione (Fig. 4C–F). N-acetylcysteine treated GNE myopathy myotubes showed comparable ΔDCF fluorescence and even better cell viability compared to those of the littermate myotubes under menadione exposure. Our data suggest that impaired antioxidant capacity possibly due to a decreased ROS scavenging activity in hyposialylated GNE myopathy myotubes can be recovered by normalization of sialylation levels or administration of exogenous antioxidants.
To clarify the implication of impaired antioxidant capacity in the myopathic phenotype of GNE myopathy, we gave N-acetylcysteine to the model mouse. We treated GNE myopathy mice continuously from 20-35 wk to 55–57 wk of age with two doses of N-acetylcysteine (0.15 g/kg and 1.5 g/kg per day) as a prophylactic and/or therapeutic application to early phase of the disease course and analysed motor performance, muscle force generation, and changes in muscle histology with emphasis on myofiber atrophy.
In the high dose treatment group, both treadmill performance and endurance tests were significantly improved and the low dose group also showed a better motor function compared to the untreated group (Fig. 6A and B). Consistently, gastrocnemius contractile properties were remarkably improved with N-acetylcysteine treatment. The peak isometric twitch force and maximum tetanic force were improved both in the high dose and low dose group, compared with those in the untreated group. Interestingly, however, after normalization of absolute forces with cross section area (CSA), specific isometric force and specific tetanic force were also increased as well in the treated groups compared to those in the untreated GNE myopathy mice. Some littermate mice also presented increased absolute contractile forces with N-acetylcysteine supplement (Fig. 6C–F), without statistical significance. No adverse side effects were found in long term N-acetylcysteine treatment. Serum aspartate aminotransferase, alkaline phosphatase, and blood urea nitrogen showed no difference between treated and untreated mice (data not shown).
Figure 6.

Oral N-acetylcysteine administration improved muscle force generation and motor performance in GNE myopathy mice. GNE myopathy mice were treated with low dose (LD, n = 13) or high dose (HD, n = 13) N-acetylcysteine (NAC) and compared to untreated group (n = 17). Littermate controls were treated in the same conditions (LD, n = 7; HD, n = 7; untreated, n = 6). (A) Treadmil performance test evaluating the distance mice could run. (B) Treadmill endurace test evaluating the number of beam breaks during a constant loading. (C-F) Contractile forces of gastrocnemius muscles: (C) in peak isometric twitch force, (D) maximum tetanic force, (E) specific isometric force normalized by CSA, (F) specific tetanic force normalized by CSA. Values from each mouse are shown with mean ± SEM. *P < 0.05; **P < 0.01.
To determine the effects of N-acetylcysteine on muscle atrophy, we analysed myofiber diameters in gastrocnemius from the treated mice (Fig. 7A and B). Mean fiber diameters of treated mice were significantly increased in both high dose (35.2 ± 1.2 μm, P < 0.05) and low dose (35.7 ± 0.8 μm, P < 0.01) groups compared to the untreated group (31.7 ± 0.8 μm). From the analysis of muscle transverse images (Fig. 7A) and histograms of myofiber diameters (Fig. 7C), we noted that the increase of average fiber diameters in treated mice was attributed to the decreased number of atrophic fibers.
Figure 7.

Skeletal muscle atrophy in GNE myopathy mice was ameliorated by N-acetylcysteine treatment. (A) Representative sarcolemmal staining images with caveolin-3 antibody from gastrocnemius muscles. NAC, N-acetylcysteine treated mice. Bar = 20 μm. (B) Muscle fiber diameters in low dose (LD, n = 13) or high dose (HD, n = 13) N-acetylcysteine treated GNE myopathy mice (NACLD or NACHD) were compared to those in untreated controls (n = 17). Data presented with mean ± SEM. *P < 0.05; **P < 0.01. (C) Fiber diameter histogram from a mouse in each group was compared.
Taken together, our data revealed the ability of N-acetylcysteine to recover muscle strength and myofiber size in GNE myopathy mice, similar to sialic acid supplementation (14,15). To understand the effects of N-acetylcysteine at molecular level, we then performed gene expression analysis with the high dose and low dose treated muscles. The upregulation of atrogenes (Fbxo32 and Trim63) and oxidative stress responsive genes (Mt1 and Mt2) in the GNE myopathy muscle was downregulated with treatment (Fig. 2A), as expected. Map1lc3b, a marker used for the presence of autophagy, was lowered as well (Fig. 2 A and D) and β-amyloid expression by ELISA in muscle homogenates was decreased after treatment (Fig. S2). Similarly, we observed the effective reduction of S-nitrosyl modification in muscle proteins after N-acetylcysteine treatment (Fig. 8A). S-nitrosylation of skeletal muscle actin, creatine kinase, aldolase A was significantly decreased (Fig. 8B). We also observed the reduction in the modification of other proteins, myosin heavy chain fast-type, carbonic anhydrase 3, triosephosphate isomerase 1 in average, without statistical significance (Supplementary Material, Fig. S3).
Figure 8.

Reduction of S-nitrosyl modification in skeletal muscle proteins by N-acetylcysteine treatment. (A) Profiles of S-nitrosylation of mouse skeletal muscle proteins. (B) Total S-nitrosylation rates nomalized by protein amounts. (C–E)S-nitrosylation rates of skeletal muscle: (C) actin (Acta1), (D) muscle creatine kinase (Ckm), (E) fructose-bisphosphate aldolase A (Aldoa). Control, untreated controls (n = 8); GM, untreated GNE myopathy mice (n = 7); LD/NACLD, GM with low dose of N-acetylcysteine (n = 8); HD/NACHD, GM with high dose of N-acetylcystine (n = 8). Data presented with mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In this paper, we define an important role of sialic acid in normal muscle physiology and disease. Figure 9 represents a schematic model for an additional role of sialic acid in healthy skeletal muscles and the proposed pathomechanism in GNE myopathy from hyposialylation to muscle atrophy and weakness. In healthy muscles, sialic acids normally produced by the biosynthetic pathway are involved with cell-cell interaction, glycoprotein stability, and signal transduction. They also play a role on suppressing ROS, which is generated during muscle contraction. In GNE myopathy muscles, sialic acids are deficient due to GNE mutations. The dysregulated ROS production is a consequence of reduced antioxidant capacity in the muscles brought about by decreased overall sialylation, causing upregulation of atrogenes and leading to myofiber atrophy and enhancing S-nitrosyl modification of the contractile and metabolic proteins. In our study, N-acetylcysteine and NeuAc supplementation showed similar efficacy with regards to reducing oxidative stress in myotubes; this observation likely supports the antioxidant property of sialic acids. As elevated levels of S-nitrosylation due to oxidative stress have been detected in various muscle disorders (22), studies investigating the benefit of NeuAc supplementation in these disorders will likely define the antioxidant role of sialic acid.
Figure 9.
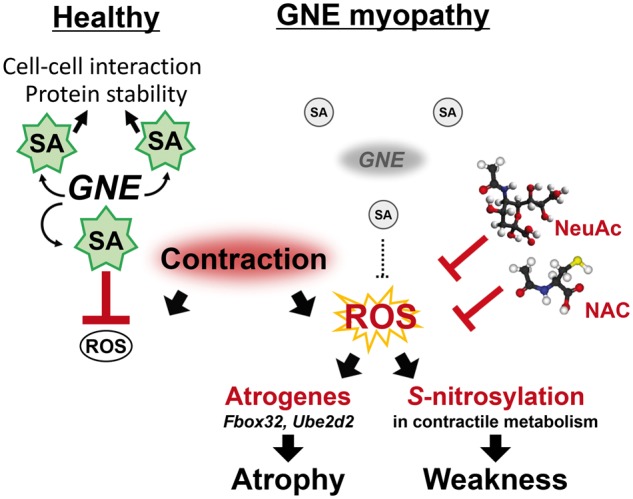
Role of sialic acids on antioxidant defense system. Left, sialic acids (SA) have a role to scavenge ROS generated on muscle contraction in healthy skeletal muscles. Right, proposed pathomechanism in GNE myopathy. Muscle atrophy and weakness are promoted by enhanced oxidative stress in reduced SA in skeletal muscles due to GNE mutations. Extrinsic NeuAc or antioxidant (NAC, N-acetylcysteine) supplementation has an effect against myopathic phenotypes in GNE myopathy model mice.
The direct relationship between sialic acid and oxidative stress has not been completely clarified, few studies infer to the roles of sialic acid in redox balance as an ROS scavenger or by reciprocal action with glycoproteins. It has been shown that sialic acids have a biological function as a direct scavenger of radicals such as H2O2 (26,27). Moreover, it has been found that non-reducing sialic acid residues can be a target of ROS and the sialic acid content of some glycoproteins are apparently linked with oxidative stress (28,29). One of those glycoproteins being associated with diseases of free radical etiology is transferrin, a metal binding monomeric protein, of which the degree of sialylation has been suggested to play a role in the pathophysiology of Parkinson’s disease and Alzheimer’s disease (30,31). Our study supports an important biological function of sialic acid, i.e. its antioxidative activity, especially in a GNE myopathy, a disease with a primary defect of hyposialylation.
While the biomedical literature abounds with claims that oxidative stress is important in various human disorders, it is still challenging to show that ROS is involved in the pathomechanism of a specific disease (32). In this study, we demonstrated the increase in oxidation-related protein modification in human and mouse GNE myopathy muscles, and elevated intracellular ROS in both in vitro and in vivo models. Interestingly, we found remarkable increments in contraction-induced production of ROS in the GNE myopathy muscles, suggesting that the ROS increase may either be a consequent of muscle contraction or caused by decreased overall antioxidant capacity in sialic acid-deficient muscles. Analysing kinetics of hydroxyl radical after muscle contraction may give an answer that we could not evaluate in this study due to the limited sensitivity of current methods of measurement. In cell culture experiments, however, we noted a logarithmic acceleration in ROS level and apoptosis of cells by the treatment with the pro-ROS drugs, H2O2 and menadione, indicating a reduction of ROS scavenging capacity in hyposialylated myotubes from GNE myopathy mice. Similarly, the upregulation of ROS-responsive genes such as Mts and Srxn1 in GNE myopathy muscles suggests that the affected muscles are highly exposed to oxidative stress. Metallothioneins belong to the group of intracellular cysteine-rich proteins and the synthesis of them was known to be increased by several-fold under oxidative stress (33,34). Sulfiredoxin1 is an endogenous antioxidant protein that was initially identified by its H2O2-induced upregulation (35) and a recent study showed that Sulfiredoxin1 is critical to maintaining redox balance in cells, especially under exposure to low steady-state levels of H2O2 (36).
Based on our data, chronic dysregulation of redox homeostasis in hyposialylated muscles directs cells into a catabolic state and ultimately leads to cell death in vitro and muscle atrophy in vivo in GNE myopathy. The role of oxidative stress in disease has been described in other muscle diseases. In muscular dystrophy, dysregulated ROS production might be caused by constitutive defects or tissue responses to the primary pathology (37–39). In contrast, enhanced ROS production in GNE myopathy is not a consequence of tissue injury, but rather an upstream phenomenon to muscle atrophy. We and others have shown that skeletal muscles in GNE myopathy are hyposialylated (7,40). This lack of sialylation, and the consequently impaired ROS scavenging activity, could lead to increased production of oxidative stress and contribute significantly to muscle atrophy, even at an earlier stage in the course of the disease. Muscle atrophy then, is followed by abnormalities in the contractile proteins in the muscle, which subsequently lead to muscle degeneration. Regardless of the primary etiology and mechanism of disease, however, increased radical injury can trigger a vicious cycle that can accelerate disease progression and thus remains as a common and rational therapeutic target in diseases of skeletal muscle.
The improvement of muscle atrophy and force generation by antioxidant treatment supports our initial hypothesis that ROS is associated with muscle atrophy and weakness in GNE myopathy. The muscle force generation in treated GNE myopathy mice was very similar to the littermates, which supports the previous suggestion that muscle force generation is proportional to and influenced by myofiber diameter (14). On the other hand, contractile forces of GNE myopathy mice normalized with CSA were improved (Fig. 6E and F), suggesting the existence of other factors contributing to muscle weakness in GNE myopathy mice. In this paper, we clearly demonstrated that S-nitrosylated modification of skeletal muscle proteins was significantly increased in GNE myopathy muscles and they were reduced by sialic acid or N-acetylcysteine administration in GNE myopathy model. Many proteins which work on muscle contraction, metabolic pathway, and calcium regulation in sarcoplasmic reticulum, were identified in the fraction containing S-nitrosylated proteins. These results indicate that the elevation of oxidation-related modification may cause the functional defects of these proteins, leading to the skeletal muscle weakness due to impaired contractile system and energy production as suggested previously (41). Furthermore, we previously showed that structural changes including intracellular protein deposits and rimmed vacuole formation start to affect muscle contraction after 40 wk of age in GNE myopathy mice (16). Our data thus imply that the abnormalities in muscle pathology, which appeared in advanced stages of GNE myopathy, are also associated with oxidative stress as well and can be prevented by antioxidant treatment. Decrease of β-amyloid and LC3b expression by N-acetylcysteine treatment supports additional evidences that infer to relate redox imbalance to autophagy dysregulation (42,43), which is known to be important in the pathomechanism of various rimmed vacuolar myopathies (44,45).
Our results show that N-acetylcysteine can ameliorate myopathic phenotypes of GNE myopathy model mice. As N-acetylcysteine has free radical scavenging properties and is able to increase the pool of glutathione in the body, it has been regarded as a powerful antioxidant (46–49). In the skeletal muscle, it has been reported that N-acetylcysteine can protect an isolated muscle preparation from contraction-induced oxidative stress (50,51) and can inhibit muscle fatigue in humans (52). In the current study, the decrease in both of the muscle protein modification and expression of oxidative stress responsive genes in treated mice suggests that N-acetylcysteine can counteract the oxidative status and oxidative stress in hyposialylated GNE myopathy muscles, which seems to be the upstream event.
So far, how hyposialylation causes muscle weakness and degeneration has been unclear. Our data provided the novel functions of sialic acids in skeletal muscles and a novel insight into the pathomechanism of GNE myopathy by revealing an important involvement of oxidative stress in this disease. Increased oxidative stress in hyposialylated muscles invariably leads to skeletal muscle atrophy and weakness in GNE myopathy mice. The success of N-acetylcysteine treatment in normalizing this phenomenon suggests that a similar approach may benefit human GNE myopathy patients.
Materials and Methods
Human specimens
Skeletal muscles from four GNE myopathy patients who harbor the mutations in GNE gene (Patient 1, D207V/V362A; Patient 2, c.832 + 4A > G/V603L; Patient 3, M60R/V603L; Patient 4, D207V/W544X; Patient 5, c.1474 + 5G > A/V603L) were used. The ethics committee of the National Center of Neurology and Psychiatry approved this study and the use of human subjects for this study (A2011-081). Biopsied muscles from GNE patients and diseased and control patient, were obtained with informed consent.
Mice
Generation of Gne-knockout mice that express the human GNE mutation D207V (Gne-/-hGNED207V-Tg) was described previously (13). The same line of GNE myopathy mice (Gne-/-hGNED207V-Tg) and littermate mice (Gne+/-hGNED207V-Tg) as controls were used throughout the study. All mouse experiments conducted in this study were approved (Approval No. 2011003 and 2014004) by the Ethical Review Committee on the Care and Use of Rodents in the National Institute for Neuroscience, National Center of Neurology and Psychiatry.
Identification of S-nitrosylated proteins
Protein S-nitrosylation was analysed by S-Nitrosylated Protein Detection Assay Kit (Cayman Chemical). Muscle proteins containing biotin-labeled cysteines from S-nitrosocysteines (53) were prepared from tibialis anterior muscles from human GNE myopathy patients and biceps brancii muscle from diseased control individuals, and gastrocnemius muscles from GNE myopathy mouse. Then prepared proteins were separated on single and two-dimensional PAGE. For detection and quantification of the band intensities, ImageQuant LAS 4000 and ImageQuant TL (GE healthcare) were used. For two-dimensional PAGE experiment, Immobiline Drystrip (pH3-10, GE Healthcare) was used for the first dimension separation. For quantification of S-nitrosylated proteins, biotin labeled proteins were purified using Neutravidin beads (Thermo Scientific) after addition of biotinylated-ribonuclease A as an internal standard for correction of the recovery of the proteins. LC-tandem mass shotgun analyses of the purified fractions from tibialis anterior muscles from 5 human GNE myopathy patients and gastrocnemius muscle from GNE myopathy mouse, were performed with Mascot Server by Aproscience. The detected proteins were listed in abundance as predicted by the number of assigned fragments. On western blotting, anti-skeletal muscle actin (Sigma), anti-muscle creatine kinase (Genetex), anti-aldolase A (Genetex) antibodies, anti-fast myosin heavy chain (Novocastra), anti-carbonic anhydrase 3 (Abcam) and anti-triosephosphate isomerase (Abcam) antibodies and corresponding alkaline phosphatase-labeled secondary antibodies (Cell Signaling Technology) were used. Anti-nitrotyrosine antibody (Sigma) and Oxidized Protein Western Blot Detection Kit (Abcam) were also used for detection of tyrosine-nitrosylation and protein carbonylation, respectively.
Quantitative RT-PCR and microarray analysis
Total RNA was extracted from triceps brachii muscles using TRIzol reagent and subsequently treated with DNase I (Invitrogen). cDNA was synthesized using SuperScript VILO cDNA Synthesis Kit (Invitrogen).Microarray experiments were carried out using a CodeLink Mouse Whole Genome Bioarray (Applied Microarrays Inc.) at Filgen Inc. The arrays were scanned using a GenePix 4000A Array Scanner (Molecular Devices Inc.). The data were analysed by using Microarray Data Analysis Tool version 3.2 (Filgen Inc.). For quantitative PCR, TaqMan probes were used in combination with the TaqMan Gene Expression Master Mix (Applied Biosystems) in a total reaction of 20 μl. StepOnePlus Real-Time PCR System (Applied Biosystems) was used to quantify mRNA expression. mRNA relative expression was normalized to internal control (GAPDH) and determined as fold change in the average expression value of littermate muscles. The TaqMan probes used were as follows: F-box protein 32 (Fbxo32), Mm00499523_m1; tripartite motif-containing 63 (Trim63), Mm01185221_m1; Sulfiredoxin1 homolog (Srxn1), Mm00769566_m1; metallothionein 1 (Mt1), Mm00496660_g1; metallothionein 2 (Mt2), Mm00809556_s1; metallothionein 3 (Mt3), Mm00496661_g1; microtubule-associated protein 1 light chain 3 (Map1lc3b), Mm00782868_sH.
In vivo hydroxyl radical measurement
Hydroxyl radicals in living muscles from mice were measured by salicylate trapping method combined with microdialysis (54,55). 2,5-Dihydroxybenzoic acid (DHBA) generated from the salicylate in the microdialysis fluids were measured as an index of reaction with hydroxyl radicals (56). Anesthesia was induced in mice with intraperitoneal sodium pentobarbital (50 mg per kg body wt) and was maintained with supplemental doses. Microdialysis probes OP-100-075 (Eicom) were placed into the gastrocnemius muscle of the left limb and perfused with 5 mM salicylate in Ringer’s solution (8.6 g NaCl, 0.25 g CaCl2 and 0.3 g KCl in 1 liter of ultrapure water) at a flow rate of 1 μl/min. All the liquid flow lines were shielded from light exposure to avoid oxidation. Microdialysates were collected every 20 min resulting in a total of 20 μl of dialysate per collection. Following 80 to 100 min of baseline microdialysis collections, the left gastrocnemius muscles were subjected to contract by electrical stimulation of surface electrodes. Muscles were stimulated to contract at 40 Hz with 3 ms pulses for 300 trains at 50 V. Following the contractions, at least 5 further 20-min microdialysate collections were taken. DHBA in microdialysates was detected by HPLC-electrochemical detection system (Eicom) and the chromatograms were analysed using PowerChrom software (eDAQ).
Cell cultures and myotubes analysis
Myocytes from Gne-/-hGNED207V-Tg mice and littermate controls were grown in DMEM containing 20% FBS and 1% chicken embryo extracts in a 5% CO2 and differentiated in a serum free medium, OptiPROTM SFM (Gibco), to make the cells hyposialylated. Where indicated, myotubes were treated with 5 mM NeuAc (Japan Food and Liquor Alliance) or 5 mM N-acetylcysteine (Sigma) for 72 h. For the analysis of intracellular ROS, a the fluorescence-based probe, 2',7'-dichlorodihydrofluorescein diacetate (57,58) was used. Cells were loaded with 5 μM dichlorodihydrofluorescein diacetate (Invitrogen) in PBS for an hour and recovered with phenol red-free DMEM (Gibco) for 30 min at 37 °C in a 5% CO2 incubator. The cells were then placed in a CytoFluor Series 4000 multi-well fluorescence plate reader (PerSeptive Biosystems) with temperature maintained at 37 °C. The excitation filter was set at 485 nm and the emission filter was set at 530 nm. The fluorescence from each well was captured and recorded.
To evaluate the ROS generation under pro-oxidants, myotubes cultured in each condition were loaded with 5 μM dichlorodihydrofluorescein diacetate for 1 h and recovered in the different concentrations of H2O2 or menadione-containing medium. After 30 min incubation in dark at 37 °C, the microplate containing cells with pro-oxidants were placed in a plate reader and fluorescence was captured at every 5 min until it reached a plateau. Maximum fluorescence of each well was used for analysis.
For the cell viability assay, myotubes were exposed to H2O2 for 24 h and to menadione for 6 h before staining. Nuclear staining with Hoechst 33342 (Thermo Scientific) and propidium iodide (Dojindo) was used for morphological assessment of apoptosis by fluorescence microscopy (59). Minimum 200 nuclei were counted and cell viability was calculated by the exclusion of propidium iodide-stained nuclei from Hoechst 33342-stained nuclei.
N-acetylcysteine treatment protocols
Gne-/-hGNED207V-Tg mice were randomly divided into the high dose (n = 13), low dose (n = 13) N-acetylcysteine treated, or untreated control (n = 17) groups. For treatment groups, mice were provided ad libitum access to drinking water containing high dose (1.0% w/v) or low dose (0.1% w/v) N-acetylcysteine (Sigma), which gives an average dose of 1.5 g/kg for high-dose group and of 0.15 g/kg for low-dose group per day. There was no difference in water consumption between mice receiving N-acetylcysteine supplemented water or regular drinking water. We initiated treatment from the average age of 26 wk until the mice reached at least 55 wk. Three littermate groups (high dose; n = 7, low dose; n = 7, or untreated control; n = 6) were treated in the same way.
Motor performance analysis
The motor performance was evaluated using treadmill exercise as previously reported (9,10) with minor modification. Briefly, after 7 days of acclimation on the treadmill, two exercise tests were performed on separate days. The performance test started with a speed of 10 m/min for 5 min and the speed was gradually increased by 10 m/min every min until the mouse was exhausted and could no longer run. The time of exhaustion was used to calculate the total distance of the mice ran during the exercise. The endurance exercise consisted of a 60-min treadmill run at 20 m/min with a 7° incline, during which the number of beam breaks or rests were recorded. Both tests were done twice with 2 d rest in between.
Muscle contractile properties analysis
We measured the contractile properties of the gastrocnemius and tibialis anterior muscles according to the previous protocol (14,15).
Fiber diameter analysis
Muscle tissues were processed according to the previous protocols (13–15). For the morphometric analysis, we stained frozen transverse sections (6 μm) of gastrocnemius muscles with rabbit polyclonal antibody to caveolin 3 (BD Transduction Laboratories), followed by Alexa fluor-conjugated goat IgG antibody to rabbit (Invitrogen). Five randomly selected images per mouse were used to measure fiber diameters using ImageJ software (NIH). Minimal inner diameters of 1,000 myofibers from each mouse were measured.
Statistics
Statistics were calculated using GraphPad Prism 5 software (GraphPad). Quantitative RT-PCR data were analysed using Mann-Whitney test. Between-group comparison for in vivo N-acetylcysteine treatment was performed using one-way analysis of variance (ANOVA) with Dunnett’s post-test. All values are expressed as means ± SEM. We performed two-sided tests with a P < 0.05 level of significance.
Microarray data
GEO accession number is GSE95151.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
M.C.V.M. was supported by the NHGRI Intramural research program of the National Institutes of Health, USA and the NIH Common Fund from the Office of the Director. I. Nishino had financial support from Ultragenex Inc. and grants from Astellas Pharma Inc., grants and personal fees from Genzyme Japan, outside the submitted work. S.N. has grants from Novartis Pharmaceuticals Japan.
Conflict of Interest statement. A.C., M.M., I.Nonaka, report no financial disclosures deemed relevant to the submitted manuscript outside the submitted work. M.C.V.M., I.Nishino and S.N. have a patent WO 2010/131712A1 issued.
Funding
Intramural Research Grant for Neurological and Psychiatric Disorders of National Center of Neurology and Psychiatry [grant numbers 29-4, 28-6]; Research and Development Grants for Comprehensive Research for Persons with Disabilities from Japan Agency for Medical Research and Development, AMED [grant number 16dk0310072h0001]; Japan Society for the Promotion of Science (JSPS) KAKENHI [JP15H04846]; and by the NHGRI Intramural research program of the National Institutes of Health, USA.
References
- 1. Pillai S., Netravali I.A., Cariappa A., Mattoo H. (2012) Siglecs and immune regulation. Annu. Rev. Immunol., 30, 357–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jaeken J. (2013) Congenital disorders of glycosylation. Handb. Clin. Neurol., 113, 1737–1743. [DOI] [PubMed] [Google Scholar]
- 3. Schnaar R.L., Gerardy-Schahn R., Hildebrandt H. (2014) Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol. Rev., 94, 461–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keppler O.T., Hinderlich S., Langner J., Schwartz-Albiez R., Reutter W., Pawlita M. (1999) UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science, 284, 1372–1376. [DOI] [PubMed] [Google Scholar]
- 5. Seppala R., Lehto V.P., Gahl W.A. (1999) Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am. J. Hum. Genet., 64, 1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eisenberg I., Avidan N., Potikha T., Hochner H., Chen M., Olender T., Barash M., Shemesh M., Sadeh M., Grabov-Nardini G.. et al. (2001) The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat. Genet., 29, 83–87. [DOI] [PubMed] [Google Scholar]
- 7. Noguchi S., Keira Y., Murayama K., Ogawa M., Fujita M., Kawahara G., Oya Y., Imazawa M., Goto Y., Hayashi Y.K.. et al. (2004) Reduced sialylation and abnormal glycosylation in distal myopathy with rimmed vacuoles. J. Biol. Chem., 279, 11402–11407. [DOI] [PubMed] [Google Scholar]
- 8. Nonaka I., Sunohara N., Ishiura S., Satoyoshi E. (1981) Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J. Neurol. Sci., 51, 141–155. [DOI] [PubMed] [Google Scholar]
- 9. Askanas V., Engel W.K. (1995) New advances in the understanding of sporadic inclusion-body myositis and hereditary inclusion-body myopathies. Curr. Opin. Rheumatol., 7, 486–496. [DOI] [PubMed] [Google Scholar]
- 10. Nonaka I., Noguchi S., Nishino I. (2005) Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr. Neurol. Neurosci. Rep., 5, 61–65. [DOI] [PubMed] [Google Scholar]
- 11. Mori-Yoshimura M., Monma K., Suzuki N., Aoki M., Kumamoto T., Tanaka K., Tomimitsu H., Nakano S., Sonoo M., Shimizu J.. et al. (2012) Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. J. Neurol. Sci., 318, 100–105. [DOI] [PubMed] [Google Scholar]
- 12. Nishino I., Malicdan M.C., Murayama K., Nonaka I., Hayashi Y.K., Noguchi S. (2005) Molecular pathomechanism of distal myopathy with rimmed vacuoles. Acta Myol., 24, 80–83. [PubMed] [Google Scholar]
- 13. Malicdan M.C., Noguchi S., Nonaka I., Hayashi Y.K., Nishino I. (2007) A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum. Mol. Genet., 16, 2669–2682. [DOI] [PubMed] [Google Scholar]
- 14. Malicdan M.C., Noguchi S., Hayashi Y.K., Nonaka I., Nishino I. (2009) Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat. Med., 15, 690–695. [DOI] [PubMed] [Google Scholar]
- 15. Yonekawa T., Malicdan M.C., Cho A., Hayashi Y.K., Nonaka I., Mine T., Yamamoto T., Nishino I., Noguchi S. (2014) Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain, 137, 2670–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malicdan M.C., Noguchi S., Hayashi Y.K., Nishino I. (2008) Muscle weakness correlates with muscle atrophy and precedes the development of inclusion body or rimmed vacuoles in the mouse model of DMRV/hIBM. Physiol. Genomics, 35, 106–115. [DOI] [PubMed] [Google Scholar]
- 17. Kandarian S.C., Jackman R.W. (2006) Intracellular signaling during skeletal muscle atrophy. Muscle Nerve., 33, 155–165. [DOI] [PubMed] [Google Scholar]
- 18. Tisdale M.J. (2005) The ubiquitin-proteasome pathway as a therapeutic target for muscle wasting. J. Support Oncol., 3, 209–217. [PubMed] [Google Scholar]
- 19. Moylan J.S., Reid M.B. (2007) Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve, 35, 411–429. [DOI] [PubMed] [Google Scholar]
- 20. Powers S.K., Smuder A.J., Criswell D.S. (2011) Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid. Redox Signal, 15, 2519–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doria E., Buonocore D., Focarelli A., Marzatico F. (2012) Relationship between human aging muscle and oxidative system pathway. Oxid. Med. Cell. Longev., 2012, 830257.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Foster M.W., Hess D.T., Stamler J.S. (2009) Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med., 15, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fioramonti X., Deak A., Deshpande S., Carneiro L., Zhou C., Sayed N., Orban B., Berlin J.R., Pénicaud L., Leloup C.. et al. (2013) Hypothalamic S-nitrosylation contributes to the counter-regulatory response impairment following recurrent hypoglycemia. PLoS One, 8, e68709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K.. et al. (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science, 294, 1704–1708. [DOI] [PubMed] [Google Scholar]
- 25. Gomes M.D., Lecker S.H., Jagoe R.T., Navon A., Goldberg A.L. (2001) Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U. S. A, 98, 14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iijima R., Takahashi H., Namme R., Ikegami S., Yamazaki M. (2004) Novel biological function of sialic acid (N-acetylneuraminic acid) as a hydrogen peroxide scavenger. FEBS Lett., 561, 163–166. [DOI] [PubMed] [Google Scholar]
- 27. Ogasawara Y., Namai T., Yoshino F., Lee M.C., Ishii K. (2007) Sialic acid is an essential moiety of mucin as a hydroxyl radical scavenger. FEBS Lett., 581, 2473–2477. [DOI] [PubMed] [Google Scholar]
- 28. Goswami K., Nandakumar D.N., Koner B.C., Bobby Z., Sen S.K. (2003) Oxidative changes and desialylation of serum proteins in hyperthyroidism. Clin. Chim. Acta, 337, 163–168. [DOI] [PubMed] [Google Scholar]
- 29. Rajendiran S., Lakshamanappa H.S., Zachariah B., Nambiar S. (2008) Desialylation of plasma proteins in severe dengue infection: possible role of oxidative stress. Am. J. Trop. Med. Hyg., 79, 372–377. [PubMed] [Google Scholar]
- 30. van Kamp G.J., Mulder K., Kuiper M., Wolters E.C. (1995) Changed transferrin sialylation in Parkinson's disease. Clin. Chim. Acta, 235, 159–167. [DOI] [PubMed] [Google Scholar]
- 31. van Rensburg S.J., Berman P., Potocnik F., MacGregor P., Hon D., de Villiers N. (2004) 5- and 6-glycosylation of transferrin in patients with Alzheimer's disease. Metab. Brain Dis., 19, 89–96. [DOI] [PubMed] [Google Scholar]
- 32. Halliwell B., Gutteridge J.M.C. (2007) Free Radicals in Biology and Medicine 4th Ed.Oxford University Press, Oxford, New York: 888 pp. [Google Scholar]
- 33. Ruttkay-Nedecky B., Nejdl L., Gumulec J., Zitka O., Masarik M., Eckschlager T., Stiborova M., Adam V., Kizek R. (2013) The role of metallothionein in oxidative stress. Int. J. Mol. Sci., 14, 6044–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato M., Bremner I. (1993) Oxygen free radicals and metallothionein. Free Radic. Biol. Med., 14, 325–337. [DOI] [PubMed] [Google Scholar]
- 35. Biteau B., Labarre J., Toledano M.B. (2003) ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature, 425, 980–984. [DOI] [PubMed] [Google Scholar]
- 36. Baek J.Y., Han S.H., Sung S.H., Lee H.E., Kim Y.M., Noh Y.H., Bae S.H., Rhee S.G., Chang T.S. (2012) Sulfiredoxin protein is critical for redox balance and survival of cells exposed to low steady-state levels of H2O2. J. Biol. Chem., 287, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tidball J.G., Wehling-Henricks M. (2007) The role of free radicals in the pathophysiology of muscular dystrophy. J. Appl. Physiol., 102, 1677–1686. [DOI] [PubMed] [Google Scholar]
- 38. Menazza S., Blaauw B., Tiepolo T., Toniolo L., Braghetta P., Spolaore B., Reggiani C., Di Lisa F., Bonaldo P., Canton M. (2010) Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet., 19, 4207–4215. [DOI] [PubMed] [Google Scholar]
- 39. Lawler J.M. (2011) Exacerbation of pathology by oxidative stress in respiratory and locomotor muscles with Duchenne muscular dystrophy. J. Physiol., 589, 2161–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chan Y.M., Lee P., Jungles S., Morris G., Cadaoas J., Skrinar A., Vellard M., Kakkis E. (2017) Substantial deficiency of free sialic acid in muscles of patients with GNE myopathy and in a mouse model. PLoS One, 12, e0173261.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barreiro E., de la Puente B., Busquets S., López-Soriano F.J., Gea J., Argilés J.M. (2005) Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett., 579, 1646–1652. [DOI] [PubMed] [Google Scholar]
- 42. Kiffin R., Bandyopadhyay U., Cuervo A.M. (2006) Oxidative stress and autophagy. Antioxid. Redox Signal, 8, 152–162. [DOI] [PubMed] [Google Scholar]
- 43. Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J.. et al. (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab., 6, 458–471. [DOI] [PubMed] [Google Scholar]
- 44. Malicdan M.C., Noguchi S., Nishino I. (2007) Autophagy in a mouse model of distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Autophagy, 3, 396–398. [DOI] [PubMed] [Google Scholar]
- 45. Tresse E., Salomons F.A., Vesa J., Bott L.C., Kimonis V., Yao T.P., Dantuma N.P., Taylor J.P. (2010) VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy, 6, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aruoma O.I., Halliwell B., Hoey B.M., Butler J. (1989) The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med., 6, 593–597. [DOI] [PubMed] [Google Scholar]
- 47. Cotgreave I.A. (1997) N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv. Pharmacol., 38, 205–227. [PubMed] [Google Scholar]
- 48. Zafarullah M., Li W.Q., Sylvester J., Ahmad M. (2003) Molecular mechanisms of N-acetylcysteine actions. Cell Mol. Life Sci., 60, 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parasassi T., Brunelli R., Costa G., De Spirito M., Krasnowska E., Lundeberg T., Pittaluga E., Ursini F. (2010) Thiol redox transitions in cell signaling: a lesson from N-acetylcysteine. Scientific World Journal, 10, 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Terrill J.R., Radley-Crabb H.G., Grounds M.D., Arthur P.G. (2012) N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord., 22, 427–434. [DOI] [PubMed] [Google Scholar]
- 51. Sandström M.E., Zhang S.J., Bruton J., Silva J.P., Reid M.B., Westerblad H., Katz A. (2006) Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J. Physiol., 575, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reid M.B., Stokic D.S., Koch S.M., Khawli F.A., Leis A.A. (1994) N-acetylcysteine inhibits muscle fatigue in humans. J. Clin. Invest., 94, 2468–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ckless K., Reynaert N.L., Taatjes D.J., Lounsbury K.M., van der Vliet A., Janssen-Heininger Y. (2004) In situ detection and visualization of S-nitrosylated proteins following chemical derivatization: Identification of Ran GTPase as a target for S-nitrosylation. Nitric Oxide, 11, 216–227. [DOI] [PubMed] [Google Scholar]
- 54. Close G.L., Ashton T., McArdle A., Jackson M.J. (2005) Microdialysis studies of extracellular reactive oxygen species in skeletal muscle: factors influencing the reduction of cytochrome c and hydroxylation of salicylate. Free Radic. Biol. Med., 39, 1460–1467. [DOI] [PubMed] [Google Scholar]
- 55. Pattwell D., McArdle A., Griffiths R.D., Jackson M.J. (2001) Measurement of free radical production by in vivo microdialysis during ischemia/reperfusion injury to skeletal muscle. Free Radic. Biol. Med., 30, 979–985. [DOI] [PubMed] [Google Scholar]
- 56. Richmond R., Halliwell B., Chauhan J., Darbre A. (1981) Superoxide-dependent formation of hydroxyl radicals: detection of hydroxyl radicals by the hydroxylation of aromatic compounds. Anal. Biochem., 118, 328–335. [DOI] [PubMed] [Google Scholar]
- 57. Wang H., Joseph J.A. (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med., 27, 612–616. [DOI] [PubMed] [Google Scholar]
- 58. Halliwell B., Whiteman M. (2004) Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean?. Br. J. Pharmacol., 142, 231–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee Y.J., Shacter E. (1999) Oxidative stress inhibits apoptosis in human lymphoma cells. J. Biol. Chem., 274, 19792–19798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.