

Abstract
Motivation
Comprehensive catalogue of genes that drive tumor initiation and progression in cancer is key to advancing diagnostics, therapeutics and treatment. Given the complexity of cancer, the catalogue is far from complete yet. Increasing evidence shows that driver genes exhibit consistent aberration patterns across multiple-omics in tumors. In this study, we aim to leverage complementary information encoded in each of the omics data to identify novel driver genes through an integrative framework. Specifically, we integrated mutations, gene expression, DNA copy numbers, DNA methylation and protein abundance, all available in The Cancer Genome Atlas (TCGA) and developed iDriver, a non-parametric Bayesian framework based on multivariate statistical modeling to identify driver genes in an unsupervised fashion. iDriver captures the inherent clusters of gene aberrations and constructs the background distribution that is used to assess and calibrate the confidence of driver genes identified through multi-dimensional genomic data.
Results
We applied the method to 4 cancer types in TCGA and identified candidate driver genes that are highly enriched with known drivers. (e.g.: P < 3.40 × 10−36 for breast cancer). We are particularly interested in novel genes and observed multiple lines of supporting evidence. Using systematic evaluation from multiple independent aspects, we identified 45 candidate driver genes that were not previously known across these 4 cancer types. The finding has important implications that integrating additional genomic data with multivariate statistics can help identify cancer drivers and guide the next stage of cancer genomics research.
Availability and Implementation
The C ++ source code is freely available at https://medschool.vanderbilt.edu/cgg/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Cancer is a disease of the genome and responsible for one in eight deaths worldwide (Stratton et al., 2009). With the development of next-generation sequencing technologies, recent cancer genomic profiling projects such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC), present new challenges but also unprecedented opportunities for unraveling the complexity of cancer genome landscapes. The mutations found in a cancer cell genome have accumulated over the lifetime of the cancer patient and may cause a series of DNA sequence variations, including point mutations, somatic copy-number alterations (SCNAs) and genomic rearrangements (Macconaill and Garraway, 2010; Stratton et al., 2009). However, most of the aberrations are passengers and one of the fundamental challenges in cancer genomics is to distinguish drivers from passengers (Garraway and Lander, 2013).
Tremendous work has been done to identify driver genes based on point mutations (Davies et al., 2005; Kandoth et al., 2013; Larson et al., 2012; Lawrence et al., 2014); however, most mutations are at intermediate (2–20%) or even lower frequencies (Lawrence et al., 2014), leaving infrequently mutated driver genes difficult to find. Other forms of genomic aberrations, such as SCNAs and epigenetic changes, can also drive tumor initiation and progression in different mechanisms. SCNAs, which affect a larger fraction of the genome, play an important role in activating oncogenes and inactivating tumor suppressors (Akavia et al., 2010; Beroukhim et al., 2007, 2010), and studies of SCNAs dramatically expand the set of cancer therapeutic targets (Mo et al., 2013). Given that SCNAs often affect a large chunk of the genome, it is more challenging to pinpoint the specific genes or DNA elements driving cancer progression from the passengers (Beroukhim et al., 2007; Krasnitz et al., 2013). It is estimated that over 70% of 140 recurrently altered regions did not contain any known oncogenes or tumor suppressors (Zack et al., 2013). Epigenetic changes of the genome can also drive tumor development by altering chromatin structure and gene expression (Stratton et al., 2009). More recently, DNA methylation abnormalities, gene expression and other additional genomic data like protein expression, have been utilized for the discovery of ‘Epi-driver genes’ (Vogelstein et al., 2013). Epi-drivers are usually not frequently mutated and play their roles through altering gene regulation, providing a more complete characterization of the molecular architecture of cancers coupled with DNA mutations and structural aberrations (Cancer Genome Atlas, 2012; Wang et al., 2014).
Tumor development is a far more complex process than appreciated, revealed recently by extensive intratumor heterogeneity (ITH) in multiple cancers (Michor and Polyak, 2010). Besides a few well-known mutated genes, most mutations are subclonal, occupying only a small fraction of cancer cells (Michor and Polyak, 2010). Moreover, multiple subclones usually exist in a tumor, and subclones may cooperate in a complex manner to promote tumor development (Diaz-Cano, 2012). It is reasonable to assume that individual driver genes in subclones have weaker effects on their own, making it challenging to identify these driver genes, for both mutated genes and Epi-drivers. Traditional studies usually focused on one dimensional data, primarily mutations, to identify driver genes, and therefore have lower power to detect weaker drivers that are not frequently observed in cancer patients. It is crucial to expand the driver catalog by identifying novel driver genes to have a more complete view of genomic processes governing tumor development.
In this study, we aim to achieve the goal through a novel framework to integrate multiple genomic data using a multivariate approach. We hypothesize that driver genes often show a constellation of aberrations in multiple genomic aspects, and jointly modeling multi-omics data has the advantage of aggregating both genomic and epigenomics signals to increase power to identify drivers not detectable by individual omics data. The multi-omics data for each gene are modeled as a multivariate Gaussian distribution, and the omics data for all genes in the genome are modeled as a mixture of Gaussians with an unknown number of components that represent different groups of genes with distinct genomics aberration profiles. The underlying rationale for this modeling is that the vast majority of passengers would exhibit random albeit similar aberrations to form the background, while driver genes often show distinct profiles with one or more omics data showing deviations from the background. For each gene, each dimension of the multivariate data is a statistic representing the strength that this gene is a driver gene based on the corresponding omics data; such a framework is flexible in that it can take advantage of improved estimates of the driver signals by new development of numerous methods based on signal omics data. One particular example is MutSigCV, which has been constantly updated to fine tune the driver signals based on mutational data. One key challenge is to specify the number of mixture components, which may vary across cancer types, and may potentially depend on the scale and dimension of the data as finer mixture modeling requires larger scale of data. To achieve robust clustering, we adopted a non-parametric Bayesian approach to automatically determine the optimal number of components, while at the same time imposing our prior belief via the Bayesian modeling. The strength of a gene being a driver is assessed by the magnitude of the shift of the centers of background clusters with and without the candidate gene, based on the rationale that a driver gene with strong strength in multi-omics data is able to disturb the background model to a greater extent. Intuitively, the shift reflects the distance from the center of the adapted model to the center of the background model, with true driver genes farther away from the background.
We applied our method to the TCGA data and analyzed copy number, methylation, gene expression, pathway, protein abundance and somatic point mutations in 2944 patients across 4 cancer types: breast adenocarcinoma (BRCA), glioblastoma multiforme (GBM), colon and rectal carcinoma (CRC) and ovarian serous carcinoma (OV). We obtained robust clustering for all cancer types, and the identified candidate driver genes are significantly enriched for known driver genes. In particular, multiple line of evidence independently support that novel candidates are likely to be genuine driver genes. We showed that the more cancer genomic data are pumped in, the better the accuracy of the predicted driver genes is, reflecting that simultaneously modeling multi-omics data has the advantage of incorporating independent and complementary supporting evidence.
2 Methods
iDriver is composed of three components: iDriver-Clust, iDriver-Adapt and iDriver-Score. iDriver-Clust is a non-parametric Bayesian framework to cluster multi-omics gene profiles through a Dirichlet Process (DP). We use X to denote the n-dimensional vector for each gene in which each dimension represents the evidence for a gene being a driver gene from one of the omics platforms. iDriver-Clust is to identify clusters of genes with distinct genome profiles encoded in X. We assume that X follows an infinite mixture of Gaussian distribution, a priori, and infer the clustering of genes based on the genomic profiles of all genes with DP as the prior. Briefly, somatic mutations and Epigenetic genome change profiles for each gene were represented in X via a feature extraction process (see SI Materials and Methods) to summarize the driver-evidence across all patients (Fig. 1A). Each dimension of X was normalized to range from 0 to 1 for all genes (Fig. 1B). Next we apply a DP clustering to obtain the background distribution of all gene’s feature vectors. We used a variational Bayesian inference algorithm (Blei and Jordan, 2006) to increase the computational efficiency. The number of model mixture components is automatically determined based on feature vectors during the model inference procedure (Fig. 1C). Upon building the background model, we applied iDriver-Adapt algorithm to each gene to assess the gene’s impact on the overall background model. Finally we applied the iDriver-Score algorithm to calculate the distance between each gene and their background (Fig. 1D). Of particular note is that iDriver models the multivariate Gaussian distribution with a full covariance matrix to allow for the inter-dependency among omics features. Given the flexibility of the framework, it can readily incorporate additional features, regardless of the dependency with the existing ones, to further boost the performance.
Fig. 1.
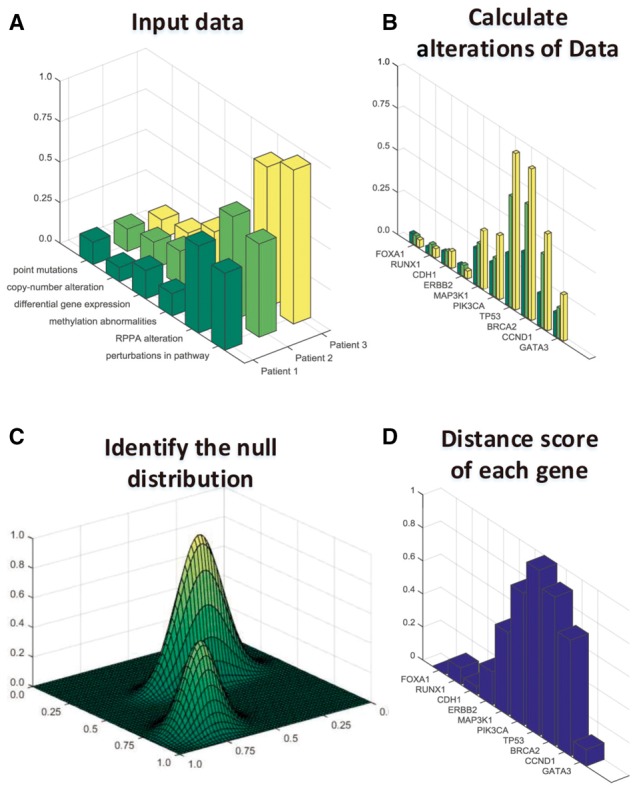
Overview of the iDriver method. (A) Features extracted from different patients to compute the vector represent of each gene. (B) Combine each gene's all kinds of features from different data sources. (C) iDriver-Clust identifying the background distribution of point mutations, copy-number alteration, differential gene expression in RNA-seq data, DNA methylation abnormalities, reverse-phase protein array (RPPA) alteration, gene perturbations in pathway. (D) iDriver-Adapt algorithm and DPM-Dscore algorithm is used to identify driver genes from passengers
2.1 iDriver-Clust using Dirichlet Process Mixtures Model
Dirichlet Process Mixtures is a non-parametric Bayesian mixture model based on Dirichlet process (Antoniak, 1974):
| (1) |
where G is a random sample distribution drawn from a Dirichlet process, G0 is a base distribution and α is a positive scaling parameter. Parameters for different mixture components are generated by drawing N times from G. is the observed variable. The construction of Dirichlet Process is based on the stick-breaking, provided by the Kolmogorov consistency theorem (Ferguson, 1973). The representation of G by stick-breaking is as follows:
| (2) |
where πk is the kth mixing weight constructed in the stick-breaking manner, is one successive piece of breaking a unit length ‘stick’ and represents the mixture components. With the introduction of latent variable zn as the mixture component associated with observable data xn, the data can be described as arising from the following process:
Draw
Draw
Draw
- For the nth observable data:
- a) Draw
- b) Draw
We place a Gamma prior on the scaling parameter α that can help unbiased detection of the number of mixture components. Most typically, we drew the data point from a set of Gaussian distributions. Even then, there is no direct method to compute the posterior distribution of variables in DPM. Two main kinds of approximate inference methods can be used: Markov chain Monte Carlo (MCMC) sampling (Liu, 2008) and Variational Bayesian (VB) inference (Attias, 2000). In this paper, we use VB inference for DPM due to its capability to handle large scale applications without incurring high computational cost and to provide a deterministic methodology for approximating likelihoods and posteriors. The idea for VB approximation was developed from theoretical physics where it is called mean field theory (He et al., 1998). We denoted the observed variables as and the set of all latent variables and parameters as , and the log marginal probability p(X) can be decomposed as:
| (3) |
where q(Z) is the lower bound of the log-likelihood that needs to be maximized and is the Kullback-Leibler distance. According to this mean field theory of VB inference, q(Z) is partitioned into M partitions and the q distribution factorizes with respect to these partitions, so that:
| (4) |
Finally, we need to get the optimal solution of each :
| (5) |
where denotes the expectation of q distributions over all variables except zm. The approximate posterior distribution q(Z) is then iteratively updated until convergence, since they depend on the statistics of each other (see Supplementary Materials for details).
2.2 iDriver-Adapt algorithm
We developed iDrive-Adapt based on the following three important model assumptions. (i) Driver genes tend to be far away from the mean of the background distribution; (ii) Passenger genes are likely to be close to the center of background distribution; (iii) the more a gene’s data vector is far away from the mean of null distribution the more likely it is a driver. Accordingly, iDriver-Adapt algorithm is proposed to project the feature vector of each gene to an individual distribution based on background distribution. Unlike the standard approach of VB inference of a model for each gene independently, the idea in the adapt approach is to derive the model by only updating the mean parameter of well-trained background model based on the mean field theory. Note that not all parameters in the background need to be updated because the training set for the background model contains almost 20 000 gene vectors and represents a distribution over a large space while the projection algorithm has only a single gene vector whose influence on the overall weights and variances is negligible. In the implementation of iDriver-Adapt the weights and variances of individual distribution are shared and only mean parameters are updated with new estimates. In detail, first, the posterior rk of the kth mixture component can be calculated:
| (6) |
where x is each gene’s feature, denotes the expectation of the distributions. Then, based on mean field theory, we update the kth mixture’s mean parameter mk:
| (7) |
where mk0 is the background model’s mean parameter. Finally, the individual distribution of each gene is generated with new estimates of means and the original weights and variances.
2.3 iDriver-Score algorithm
iDriver-Adapt not only allows for an effective estimation of each gene’s individual distribution but also helps producing a fast-scoring algorithm for driver gene discovery. Since only the mean parameter among each gene and background is different, based on model assumption 3), iDriver-Score algorithm is designed to rank genes of all 20 000 protein-coding genes based on the Euclidean distance. The distance score for a test gene is computed as the sum of all the mixture’s Euclidean distance between the mean parameters of individual gene model and background model:
| (8) |
The higher the score of a gene, the more likely it is a cancer driver.
3 Results
3.1 Analysis of the clustering of the genes
We applied iDriver to 4 cancer types in TCGA data, including 1098 breast cancer tumors, 631 CRC tumors, 613 GBM tumors and 602 OV tumors, all with multiple types of omics data. Clusters are assigned by selecting the component that maximizes the posterior probability. We found that genes were automatically grouped into 2 categories across all 4 cancer types, with a major cluster containing ∼94% genes and a minor cluster with ∼6% of genes. To examine the distribution of known driver genes in these two clusters, we collected a total of 86 curated driver genes from Cancer Gene Census (CGC) (Futreal et al., 2004) and found that known driver genes were significantly enriched in the minor cluster for all 4 cancer types (pbrca < 3.00 × 10 − 16, pcrc < 1.72 × 10 − 14, pgbm < 2.14 × 10 − 5, pov < 1.49 × 10 − 3, Supplemental Table 1). For example, in breast cancer the ‘driver’ cluster containing 7% of the genes harbors 81% of known driver genes, demonstrating the effectiveness of iDriver to capture omics profiles characteristic of driver genes. We accordingly termed the two clusters as the ‘passenger’ and ‘driver’ clusters, respectively.
To have a global view of the genomics aberrations on the genome-level, we used Principal Component Analysis (PCA) to reduce the original omics features into three dimensions and carried out the clustering based on the reduced data. The transformed data along their cluster memberships were visualized in a 3-D plot for different tumor types (Fig. 2). Most genes do not have genomic alterations in the passenger group and a small set of genes has one or more types of genomic alterations in the driver group. The clustering results are similar to the original results (Supplementary Tables 1 and 2).
Fig. 2.
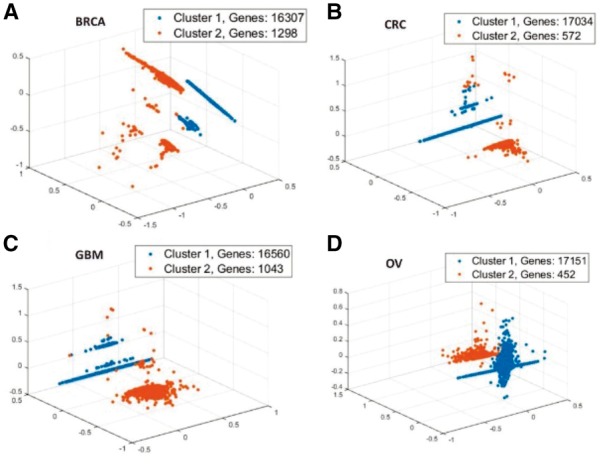
Cluster visualization with iDriver-Clust on different tumor types. In order to do the visualization, firstly, linear dimensionality reduction use Principal component analysis keeping only the most significant vectors to project the data to a 3-dimensional space. Then, get clusters of all genes by choosing the component that has max posterior probability on TCGA datasets of four tumor types: (A) BRCA, (B) CRC, (C) GBM and (D) OV
In addition to the advantage of DP for the automatic determination of the number of clusters, we further explored whether the prior distribution imposed in the non-parametric Bayesian framework is advantageous in clustering. As a comparison, we carried out traditional clustering using the k-means algorithm, with pre-specified cluster number as 2 for all tumor types. Although the minor clusters identified by k-means are also enriched for known driver genes, (pbrca < 1.84 × 10 − 13, pcrc < 2.52 × 10 − 9, pgbm < 6.65 × 10 − 3, pov < 1.49 × 10 − 3), as a comparison, iDriver achieved superior performance over the k-means, indicating that the prior distribution embedded in the DP fits the genomic data well and is critical for effective grouping of omics profiles.
3.2 Scoring and identifying driver genes
Note that not all genes in the driver cluster are driver genes, and conversely that the passenger cluster also contains driver genes. We applied iDriver-Adapt and iDriver-Score to obtain an integrated driver-score for each gene (Methods), representing the calibrated strength for a specific gene being a driver. The performance of our approach was compared against two other state-of-the-art model-based algorithms, MutSig2CV (Lawrence et al., 2013) and GISTIC2 (Mermel et al., 2011), which are based on point mutations and SCNAs respectively. For all 4 tumor types, the top-scoring genes are significantly enriched for known drivers, with the enrichment far greater than the two algorithms that are based on single genomics data types (Table 1). In BRCA, the 22 top genes are significantly enriched for known drivers (15/22, p < 3.40 × 10 − 36), a large improvement over the genes reported by MutSig2CV and GISTIC2 (14/89, p < 1.39 × 10 − 24 and 7/264, p < 5.25 × 10 − 7). In particular, all of the five top-scoring genes are well-known breast cancer genes (TP53, PIK3CA, CDH1, GATA3, MAP3K1). In CRC, the top 49 genes are also significantly enriched for known CRC drivers (12/49, p < 2.85 × 10 − 22), outperforming both MutSig2CV (20/1450, p < 1.30 × 10 − 10) and GISTIC2 (2/75, p < 7.99 × 10 − 3). Among the top 10 scoring genes, 8 are known CRC genes (KRAS, TP53, APC, SMAD4, FBXW7, PIK3CA, BRAF, TCF7L2). In GBM, 5 of the top 10 genes (PIK3R1, IDH1, PIK3CA, STAG2, PDGFRA), and in OV 3 of the top 18 genes (BRCA1, BRCA2, CDK12) are known driver genes, respectively, both achieving greater enrichment of driver genes than Mutsig2CV and GISTIC2. We also compared the performance using the same number of candidate genes and observed the same pattern (Supplementary Table 6). All of these suggest the effectiveness of our integrative approach over the state-of-the-art methods that are based on single genomic data types for identifying driver genes.
Table 1.
A comparison of enrichment for CGC genes sets of cancer drivers between iDriver and two state of the art background modeling methods on TCGA datasets of 4 tumor types: BRCA, CRC, GBM, OV
| Cancer | Method | Genes | In CGC | Coverage | Enrichment |
|---|---|---|---|---|---|
| BRCA | iDriver | 22 | 15 | 48% | 3.40 × 10−36 |
| MutSig2CV | 89 | 14 | 45% | 1.39 × 10−24 | |
| Gistic2 | 264 | 7 | 22% | 5.25 × 10−7 | |
| CRC | iDriver | 49 | 12 | 35.29% | 2.85 × 10−22 |
| Mutsig2CV | 1450 | 20 | 58.8% | 1.30 × 10−10 | |
| Gistic2 | 75 | 2 | 5.88% | 7.99 × 10−3 | |
| GBM | iDriver | 10 | 5 | 48% | 4.11 × 10−14 |
| Mutsig2CV | 20 | 5 | 41.6% | 3.55 × 10−12 | |
| Gistic2 | 111 | 1 | 8.00% | 6.94 × 10−2 | |
| OV | iDriver | 18 | 3 | 15.7% | 8.03 × 10−7 |
| Mutsig2CV | 10 | 2 | 10.5% | 4.69 × 10−5 | |
| Gistic2 | 107 | 1 | 5.20% | 1.01 × 10−1 |
3.3 Evaluation of novel candidate driver genes
After the enrichment analysis of known cancer drivers, we were primarily interested in assessing whether the novel candidate cancer genes are truly drivers. Specifically, we selected the top 20 candidates for each cancer type, and excluded known driver genes from the list, and focused only on novel genes for the evaluation. We carried out a systematic evaluation from multiple aspects to assess the enrichment of genuine novel driver genes in our top-scoring candidate genes. To achieve unbiased evaluation, all lines of the evidence are not used in the scoring of candidate genes.
First, for the novel candidates in individual tumor types, with the ‘guilt-by-association’ principle (Altshuler et al., 2000; Oliver, 2000) that genes physically or functionally close to each other tend to be involved in the same biological pathways and have similar effects on phenotypes, we evaluated whether the novel gene set is significantly closer to the known driver genes than a randomly selected set in gene networks. For this evaluation we used the HumanNet (Lee et al., 2011) protein-protein interaction network. We used the Dijkstra’s algorithm (Misa and Frana, 2010) to calculate the shortest path between each gene in the candidate and each gene in the known gene set. We defined the distance between the novel gene set and known gene set in the PPI network as the median of the shortest paths among all pairs of genes. Shown in Table 2 are the distances between novel and known driver genes. It is evident that for all the 4 tumor types novel genes are significantly closer to the known drivers, indicating that our top novel candidates harbor genuine driver genes. Note that, specifically for this analysis, we obtained the top candidates by removing the pathway-derived feature in the input vector so that the evidence revealed in the PPI network is unbiased.
Table 2.
iDriver novel driver genes validation results
| Analysis | BRCA | CRC | GBM | OV | |
|---|---|---|---|---|---|
| Gene sets distances | Distance | 5 | 4 | 5 | 5 |
| p value | 0.002 | 0.001 | 0.001 | 0.009 | |
| Connectivity analysis | Degree | 5.5 | 5.5 | 5 | 3 |
| p value | 0.004 | 0.003 | 0.003 | 0.023 | |
| Purifying selection analysis | Count | 6 | 4 | 4 | 4 |
| p value | 0.003 | 0.015 | 0.015 | 0.015 | |
| Pathway analysis | Cancer pathwaysa | 1 | 2 | 5 | 3 |
| Expression analysis | p value | 1.1 × 10−5 | 0.020 | 0.046 | 7.3 × 10−5 |
| ShRNA screen analysis | Count | 6 | 3 | 3 | 3 |
| p value | 9.0 × 10−5 | 0.042 | 0.042 | 0.042 |
Number of enriched KEGG cancer related pathways. Detail is in Supplementary Table 8.
Second, for each of the novel gene sets, we tested the hypothesis whether the connectivity of novel genes in the HumanNet network is larger than random-select gene sets. Cancer genes have been shown to have greater interaction partners compared with non-cancer genes (Jonsson and Bates, 2006). We defined the connectivity of a gene set as the median of the degrees of all the genes in the HumanNet network, and compared the gene set connectivity with randomly selected gene sets to obtain empirical P values. The results in Table 2 show that the connectivity of novel genes is significantly larger than random for all four cancer types (P = 0.004, 0.003, 0.003 and 0.023), further supporting the enrichment of genuine driver genes in our top lists.
Third, we also evaluated whether the novel genes are under strong purifying selection, as known driver genes are often evolutionarily conserved (Cheng et al., 2014; Gonzalez-Angulo et al., 2010; Samocha et al., 2014; Sweet-Cordero et al., 2005). We collected a set of genes that are under strong evolution constraints (Samocha et al., 2014) and observed strong enrichment of our novel candidate genes with the constraint genes for all four cancer type, with corresponding P values of 0.00032, 0.015, 0.015 and 0.015 (Table 2).
Fourth, we also evaluated whether the novel genes are highly expressed genes, as known cancer-associated genes tend towards higher expression (Lawrence et al., 2013). We used t-test to compare the expression of novel candidate genes with the rest of the coding genes. In this evaluation, we dropped the expression level feature in iDriver to obtain candidate driver genes. As a result (Table 2), each cancer type’s 20 novel genes are significantly highly-expressed than the others (P = 1.1 × 10−5, 0.020, 0.046 and 7.3 × 10−5 for the four cancer types, respectively).
Fifth, also by the ‘guilt-by-association’ assumption, we tried to evaluate whether the novel genes are enriched in cancer pathways. We used WebGestalt (Wang et al., 2013; Zhang et al., 2005) for the enrichment analysis and corrected for the multiple testing using false discovery rate (Benjamini and Hochberg, 1995). We observed that novel candidates are highly enriched in cancer-related pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) such as the MAPK signaling pathway, cell cycle and p53 signaling pathway (Table 2 and Supplementary Table 8). To deeply construct an integrated view of genomic alterations in the cancer pathways, we selected 11 known driver genes and 6 candidate driver genes of BRCA, CRC, GBM, OV and mapped them onto 2 major pathways in human cancer: PI3K/RAS pathway and TGF-β/SMAD4 pathway (Fig. 3). Some of the genes in these two pathways encode receptors for the growth factors themselves. We observed that some novel drivers are in the upstream of known drivers in the two pathways, and their inactivation or activation may result in the activation of growth-promoting signal downstream of the pathway and enhance cancer cell’s growth and survival. For examples: inactivation of PTEN results in activation of the AKT kinase; inactivation of NF1 results in constitutive activity of oncogenes such as KRAS (Brems et al., 2009).
Fig. 3.
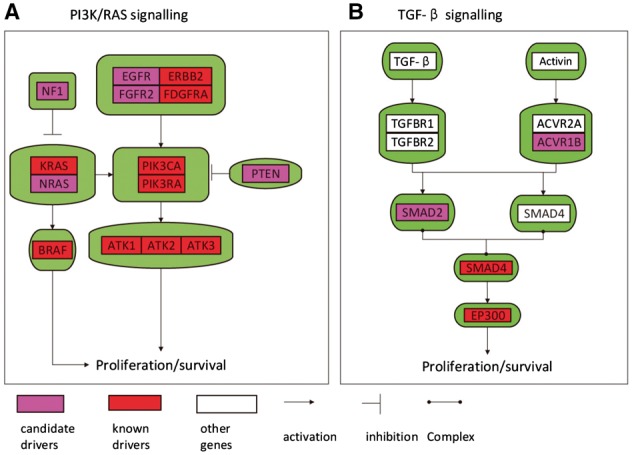
Two signal transduction pathways affected by mutations and epigenetic alterations in human cancer. (A) PI3K/RAS pathways illustrated by curated analysis, (B) TGF-β pathways illustrated by curated analysis. Known driver genes are red coded, novel driver genes are purple coded, protein components encoded by other genes are write coded
Finally, we are interested in investigating whether the novel candidate can significantly affect cancer cell survival/proliferation in cancer cell line experiments. Large-scale short hairpin RNA (shRNA) knock-out screening is able to identify those genes that affect cell survival and facilitate driver gene discovery (Cheung et al., 2011; Luo et al., 2008). We used the Achilles v2.4.3 dataset of 216 cell lines using 54k shRNA library (Cowley et al., 2014) to determine whether the novel driver candidates significantly affect cancer cell survival. We generated a gene list including 793 genes with the median ATARiS gene level q-values <0.05, representing the genes implicated in cancer cell survival assessed via shRNA. We compared our candidates with this gene list and observed significant enrichment for all four tumor types (Table 2).
In total, we reported 45 high confidence driver candidate genes, and the full list of these genes for all cancer types are in Supplemental Tables 3 4. Some of the genes in the full list, (e.g. CASZ1 and TPX2) already have supportive evidence in literature (Liu et al., 2011; Wei et al., 2013).
3.4 Analysis of feature combination
iDriver integrates somatic mutations and epigenetic changes of genome data in an aim to achieve improved sensitivity and specificity. Here we gave some examples of genes that showed aberrations in multi-omics platforms to illustrate the complementary roles of various genomics features. We also visualized the top 10 genes’ features of each tumor type with PCA in 3D to show how these genes stand out from the mean of the features (Supplementary Fig. S2). In BRCA, all top10 candidate drivers have 4 or more feature aberrations (total feature dimension is 7), demonstrating the importance of integrating multi-omics data. The top candidate gene list includes BRCA2, a well-known tumor suppressor gene that was difficult to identify by other computational approaches (Beroukhim et al., 2010). Although BRCA2 was mutated at a low frequency (6%) and was not in the deleted regions, it underwent significant differential expression and perturbations in pathways. It is a combination of 4 feature-aberrations in our framework that was able to identify BRCA2 as a driver gene. In CRC, 8 of the top10 candidate genes have 4 or more feature-aberrations. The top candidate gene list includes some known drivers with low mutation frequencies, like PIK3R1, MSH6 and CTNNB1. PIK3R1 has five feature aberrations, especially the extreme DNA methylation abnormality and over expression of the protein. MSH6 and CTNNB1 also have large RPPA abnormalities that suggested them to be cancer drivers. Constitutive activity of CTNNB1 in CRC is mainly due to the suppressor gene APC's inactivation, resulting in the activation of some growth-promoting signals downstream of the Wnt/β-catenin signaling pathway (He et al., 1998; Morin et al., 1997). In GBM, all of the genes in the list of top 10 genes have 4 or more feature-aberrations, and in particular, PIK3R1, a known GBM driver, reached the first place of the gene list with 5 feature-aberrations. In OV, 90% of top10 genes have 4 or more feature-aberrations. Although the list of known driver genes included 19 genes, they are very difficult to pinpoint because almost all of them have low frequency mutations. iDriver pinpointed 3 known genes with top 18 candidate genes reported in OV: BRCA1, BRCA2 and CDK12. CDK12, a cancer genes involved in RNA splicing regulation in OV (Chen et al., 2006), is challenging to find due to its low mutation frequency and showed up in our top list since it has 4 feature aberrations including slight SCNA amplification and gene underexpression.
To further investigate the relative contribution of various omics types, we organized all of the seven features into three groups: Feature I includes a two-dimensional feature of sequence mutations; Feature II includes the features of SCNAs; Feature III includes a combination of the other 4 features. We carried out the same integrative analyses using different combinations of the three groups of features, and generated the precision-recall curves (PRC) across the focused four types of tumor datasets as shown in Fig. 4. The area under the precision-recall curve (AUC) is used to measure the performance. In all cases, Feature I achieved much better performance than other features (Feature II and Feature III) (Fig. 4), consistent with the notion that mutations that directly disrupt the gene structure are more important than epigenoimcs features that impact the expression of the target genes and often varied with types of cell, developmental stage and patient age (Pelizzola and Ecker, 2011). Also, SCNAs’ amplification and deletion regions typically cover a large number of genes, making it hard to tease the cancer drivers apart from the passengers. However, integration of other features with mutation data is useful and usually can significantly improve the performance of iDriver (Supplemental Table 7). As a result, the integrative model with all features achieved improvements in four tumor types, compared with only using the mutation data, e.g. the percentage of improvement is 16.2% in BRCA, 18.3% in CRC, 45.7% in GBM and 1.57% in OV. Most top genes have more than half aberrations of all features, indicating that Feature I, Feature II, Feature III groups provide complementary evidence and supporting our hypothesis that driver genes do not only harbor driver mutations but at the same time also harbor a constellation of epigenetic alterations. It is also clear that the more features were pumped in, the better the accuracy of the predicted driver genes is (Fig. 4), supporting the importance of integrating complementary omics data for driver gene identification.
Fig. 4.
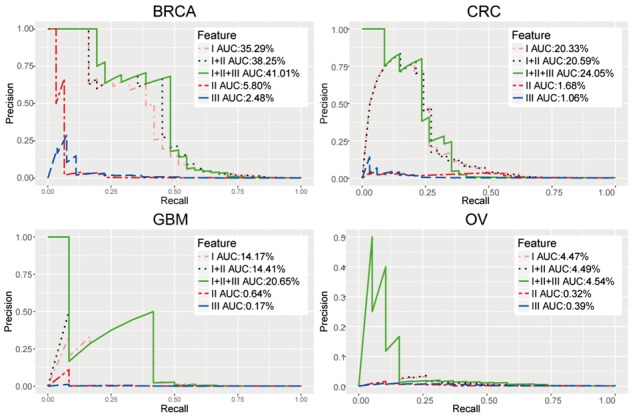
A comparison of precision-recall curves for almost 20,000 protein-coding genes in four tumor types to show the contribution of three categories of feature types across four cancer types: (A) BRCA, (B) CRC, (C) GBM and (D) OV. We grouped the original features into three categories: I-mutation (MutSig2CV, mutation frequency), II-SCNA and III-epigenomics feature (expression, methylation, pathway aberrations, rppa)
4 Discussion
One particular challenge facing the field of cancer genomics is to identify cancer driver genes that contribute to oncogenesis and cancer progression. Although defining drivers as genes conferring a selective growth advantage in physiologic terms is easy, it is more difficult to identify drivers from a sea of passengers using genomics data (Vogelstein et al., 2013). In this study, we developed iDriver, a model-based framework that integrates multi-omics data from TCGA to prioritize novel cancer driver genes across multiple tumor types. A key aspect of our approach is the integrative clustering of genes to discover different patterns of driver and passenger genes with a comprehensive data view. We reasoned that driver genes and passenger genes exhibit different patterns; however, the exact patterns are unknown and it is not unreasonable to assume that the patterns can be complex given the widespread genomic aberrations across multiple omics data. To cope with the complexity, we took an unsupervised approach in a Bayesian framework to automatically determine the number of clusters and to reveal the intrinsic characteristics of genomics aberrations of drivers and passengers. Intriguingly, our framework revealed two major clusters, named as ‘driver’ and ‘passenger’ clusters, for all cancer types we investigated, probably reflecting a general pattern of cancer genomics, the investigation of which is beyond the scope of the current study.
We adopted a non-parametric Bayesian framework for its advantages beyond its ability of automatically determining the optimal cluster number. Dirichlet Process Mixtures is a highly effective multivariate statistical model to describe the background distribution of aberrations of all types of useful omic data. Unlike most computational approaches which assume that the input data are independent, iDriver models the input vectors as multivariate Gaussian random variables with a full covariance matrix to take into account the dependence among omics data.
In addition, the prior distribution imposed in the DP reflects our prior belief that the background cluster is dominantly larger than others. Such a prior fits the data well as the resulting clustering of iDriver is superior to the traditional K-means algorithm. Finally, iDriver-Adapt and iDriver-Score algorithms are developed to score each gene based on its impact (or ‘perturbation’) to the global distribution of the background model to get reliable assessment. These advantages of our framework exceled in this particular study and facilitated the identification and prioritization of candidate driver genes in the four cancer types we investigated.
One of the major rationales in our modeling strategy is that driver genes often show constellation of genomics aberrations across multiple omics data. Our results are consistent with this rationale and supported our approach to modeling multi-omics data in an effort to identify driver genes with weak signals in individual omics data. We identified several known cancer drivers, e.g. BRCA2, which are difficult to identify by computational approaches based on single omics data. In addition, we also identified novel candidate drivers with such patterns, e.g. CASZ1 and TPX2, which also have supportive evidence in literature. CASZ1 has been reported as a candidate tumor-suppressor gene which suppresses neuroblastoma tumor growth through reprogramming gene expression (Liu et al., 2011); TPX2 has been detected as a novel prognostic marker for the growth and metastasis of colon cancer and its expression in colon metastatic lesions is significantly higher than that in primary cancerous tissue and normal colon mucosa (Wei et al., 2013). iDriver identified 6 novel driver gene candidates in 4 cancer types in 2 major cancer pathways and these genes are often in the upstream of known drivers in the pathways, expanding the candidates for understanding the tumorigenesis mechanisms as well as therapeutics development.
In summary, we have shown that iDriver can extract useful insights from integrated omic data to fully exploit the different patterns of driver genes. Ever increasing genomics data are being generated spanning a wide range of cancer types. Given the flexibility of our framework, other cancer types in TCGA or any other cancer genomics projects can be analyzed comprehensively in future studies to help identify and prioritize candidate driver genes.
Funding
This work has been partially supported by the NIH grant R01HG00680 and internal funding from Vanderbilt University.
Conflict of Interest: none declared.
Supplementary Material
References
- Akavia U.D. et al. (2010) An integrated approach to uncover drivers of cancer. Cell, 143, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D. et al. (2000) Guilt by association. Nat. Genet., 26, 135–137. [DOI] [PubMed] [Google Scholar]
- Antoniak C.E. (1974) Mixtures of Dirichlet processes with applications to Bayesian nonparametric problems. Ann. Stat., 1152–1174. [Google Scholar]
- Attias H. (2000) A variational Bayesian framework for graphical models. Adv. Neural Inf. Process. Syst., 12, 209–215. [Google Scholar]
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological), 289–300. [Google Scholar]
- Beroukhim R. et al. (2007) Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl. Acad. Sci. U. S. A., 104, 20007–20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R. et al. (2010) The landscape of somatic copy-number alteration across human cancers. Nature, 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blei D.M., Jordan M.I. (2006) Variational inference for Dirichlet process mixtures. Bayesian Anal., 1, 121–143. [Google Scholar]
- Brems H. et al. (2009) Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol., 10, 508–515. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas,N. (2012) Comprehensive molecular portraits of human breast tumours. Nature, 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F. et al. (2014) Studying tumorigenesis through network evolution and somatic mutational perturbations in the cancer interactome. Mol. Biol. Evol., 31, 2156–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.H. et al. (2006) Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol. Cell Biol., 26, 2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung H.W. et al. (2011) Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. U. S. A., 108, 12372–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley G.S. et al. (2014) Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data, 1, 140035.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. et al. (2005) Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res., 65, 7591–7595. [DOI] [PubMed] [Google Scholar]
- Diaz-Cano S.J. (2012) Tumor heterogeneity: mechanisms and bases for a reliable application of molecular marker design. Int. J. Mol. Sci., 13, 1951–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson T.S. (1973) A Bayesian analysis of some nonparametric problems. Ann. Stat., 209–230. [Google Scholar]
- Futreal P.A. et al. (2004) A census of human cancer genes. Nat. Rev. Cancer, 4, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway L.A., Lander E.S. (2013) Lessons from the cancer genome. Cell, 153, 17–37. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Angulo A.M. et al. (2010) Future of personalized medicine in oncology: a systems biology approach. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol., 28, 2777–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T.C. et al. (1998) Identification of c-MYC as a target of the APC pathway. Science, 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Jonsson P.F., Bates P.A. (2006) Global topological features of cancer proteins in the human interactome. Bioinformatics, 22, 2291–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C. et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature, 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnitz A. et al. (2013) Target inference from collections of genomic intervals. Proc. Natl. Acad. Sci. U. S. A., 110, E2271–E2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson D.E. et al. (2012) SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics, 28, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S. et al. (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature, 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S. et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature, 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. et al. (2011) Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res., 21, 1109–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.S. (2008) Monte Carlo Strategies in Scientific Computing. Springer Science & Business Media, New York. [Google Scholar]
- Liu Z. et al. (2011) CASZ1, a candidate tumor-suppressor gene, suppresses neuroblastoma tumor growth through reprogramming gene expression. Cell Death Diff., 18, 1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B. et al. (2008) Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. U. S. A., 105, 20380–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macconaill L.E., Garraway L.A. (2010) Clinical implications of the cancer genome. J. Clin. Oncol., 28, 5219–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel C.H. et al. (2011) GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol., 12, R41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michor F., Polyak K. (2010) The origins and implications of intratumor heterogeneity. Cancer Prev. Res., 3, 1361–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misa T.J., Frana P.L. (2010) An interview with Edsger w. Dijkstra. Commun. ACM, 53, 41–47. [Google Scholar]
- Mo Q. et al. (2013) Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. U. S. A., 110, 4245–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin P.J. et al. (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science, 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- Oliver S. (2000) Guilt-by-association goes global. Nature, 403, 601–603. [DOI] [PubMed] [Google Scholar]
- Pelizzola M., Ecker J.R. (2011) The DNA methylome. FEBS Lett., 585, 1994–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha K.E. et al. (2014) A framework for the interpretation of de novo mutation in human disease. Nat. Genet., 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M.R. et al. (2009) The cancer genome. Nature, 458, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet-Cordero A. et al. (2005) An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat. Genet., 37, 48–55. [DOI] [PubMed] [Google Scholar]
- Vogelstein B. et al. (2013) Cancer genome landscapes. Science, 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. (2013) WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res., 41, W77–W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. et al. (2014) Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet., 46, 573–582. [DOI] [PubMed] [Google Scholar]
- Wei P. et al. (2013) TPX2 is a novel prognostic marker for the growth and metastasis of colon cancer. J. Trans. Med., 11, 313.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack T.I. et al. (2013) Pan-cancer patterns of somatic copy number alteration. Nat. Genet., 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. et al. (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res., 33, W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.