

Abstract
Spinal muscular atrophy (SMA) is a progressive neurodegenerative disease that is the leading genetic cause of infantile death. It is caused by a severe deficiency of the ubiquitously expressed Survival Motor Neuron (SMN) protein. SMA is characterized by α-lower motor neuron loss and muscle atrophy, however, there is a growing list of tissues impacted by a SMN deficiency beyond motor neurons. The non-neuronal defects are observed in the most severe Type I SMA patients and most of the widely used SMA mouse models, however, as effective therapeutics are developed, it is unclear whether additional symptoms will be uncovered in longer lived patients. Recently, the immune system and inflammation has been identified as a contributor to neurodegenerative diseases such as ALS. To determine whether the immune system is comprised in SMA, we analyzed the spleen and immunological components in SMA mice. In this report, we identify: a significant reduction in spleen size in multiple SMA mouse models and a pathological reduction in red pulp and extramedullary hematopoiesis. Additionally, red pulp macrophages, a discrete subset of yolk sac-derived macrophages, were found to be altered in SMA spleens even in pre-symptomatic post-natal day 2 animals. These cells, which are involved in iron metabolism and the phagocytosis of erythrocytes and blood-borne pathogens are significantly reduced prior to the development of the neurodegenerative hallmarks of SMA, implying a differential role of SMN in myeloid cell ontogeny. Collectively, these results demonstrate that SMN deficiency impacts spleen development and suggests a potential role for immunological development in SMA.
Introduction
Spinal muscular atrophy (SMA) is a leading genetic cause of infantile death. SMA is an autosomal recessive neurodegenerative disorder caused by a severe reduction in the levels of survival motor neuron (SMN) protein resulting from homozygous loss or mutation of SMN1 gene (1). SMN is encoded by two nearly identical genes: SMN1 and SMN2. SMN1 primarily produces the full length SMN protein, however, SMN2 produces a low amount of full-length SMN due to an alternative splicing event common to ∼80–90% of its transcripts. This unstable and truncated isoform is referred to as SMNΔ7 (2). While the genetic basis of the disease is clear, the specific SMN-associated function that leads to SMA development is still not thoroughly understood (3,4). SMN is ubiquitously expressed and essential for viability in all tissues, however, SMA develops mainly due to loss of α-lower motor neurons (LMNs) in the spinal cord and subsequent weakness of the proximal muscles (5,6), thus its categorization as a progressive neurodegenerative and neuromuscular disease.
The basis for the selective susceptibility of α-LMNs remains poorly understood, however, recent studies have demonstrated important roles for non-neuronal tissues within the central nervous system such as astrocytes (7) and Schwann cells (8,9) in the development of the SMA phenotype. In addition, a growing number of studies focused largely upon SMA mouse models and to a lesser degree in SMA patients, have reported defects in peripheral organs including heart and vasculature (10,11), pancreas (12,13), intestines (14), liver (15), and testis (16). Consistent with this notion, there is growing appreciation for the role of the immune system in multiple neurodegenerative diseases including amyotrophic lateral sclerosis (17,18), Alzheimer’s Disease (19,20) and Huntington disease (21). In SMA, the development of the thymus and spleen has recently been examined in a severe mouse model and shown to be defective (22).
The spleen is a multifunctional organ, positioned in the abdomen underneath the diaphragm, and is the largest secondary source of lymphoid tissue. The spleen is comprised primarily of two functionally distinct zones: the red pulp and the white pulp while the boundary between the red and white pulp is referred to as the marginal zone. The red pulp is densely packed connective tissue, comprised of a variety of blood cell populations as well as splenic sinuses. In rodents, the splenic red pulp also serves as a major source of extramedullary hematopoiesis. In contrast, the white pulp is the lymphoid component of the spleen and is composed of a T-cell zone arranged around a central arteriole, termed the periarteriolar lymphoid sheath (23), as well as the surrounding follicles or B-cell zone. The red pulp is the body’s blood filtration system; it digests erythrocytes and recycles iron by the red pulp macrophage population. The white pulp is the center for generating an adaptive immune response, whereas the marginal zone is populated with macrophages, B lymphocytes, and dendritic cell populations and is involved in innate and adaptive immune responses. These functions highlight the importance of the spleen in immune system homeostasis, as well as serving as an efficient site for inflammatory immune responses.
In this study, we report selective hypoplasia of splenic tissue in three important SMA mouse models, ranging from severe to intermediate. In the severe “SMNΔ7” mouse, red pulp was selectively reduced in SMA spleens with a reduction in extramedullary hematopoiesis. A greater frequency of lymphocytes and CD11b+ macrophages was identified, yet, conversely red pulp macrophages that represent the yolk sac-derived population were reduced in SMA spleens. Finally, utilizing a potent SMN-inducing antisense oligonucleotide (ASO), treatment with the ASO rescued spleen pathology in SMA model mice. These findings illustrate the role SMN serves in the development and maturation of immune tissue, revealing for the first time a selective role of SMN in myeloid cells while also potentially further expanding the scope of peripheral tissues that contribute to the complex SMA pathology.
Results
Selective and progressive hypoplasia of SMA spleens
To assess the role of SMN within the spleen, we examined spleen growth and development in three widely used SMN-deficient SMA mouse models: “SMNΔ7”, “Taiwanese” SMA mice, and Smn2B/- mice. In the SMNΔ7 mouse, which is a severe model, spleen size was initially examined at post-natal day (PND) 12, which would be considered a symptomatic stage in disease development. At PND 12, the SMNΔ7 SMA spleen was significantly smaller (Fig. 1A) and weighed significantly less than the wild type (WT) spleen. WT spleen average weight was 53 mg (±2.3, n = 11) while the SMA spleen average weight was 9.7 mg (±1.4, n = 11) (Fig. 1B). In a time-course analysis, spleen weights were determined from PND 5 (pre-symptomatic) through PND 13 (symptomatic) in the SMNΔ7 mice, demonstrating that the weight of SMA spleens was significantly less at PND 5 compared to WT (SMA spleen: 16.6 mg ± 1.7; WT spleen: 27.6 mg ± 2.7; n = 4) and continued to decrease over time (Fig. 1B). In addition to the weight, spleen size was selectively and progressively decreased in SMNΔ7 mice, as evidenced by the continued reduction in mass compared to other organs including the brain (Fig. 1C) or as a percentage of body weight (Fig. 1D). Similar results were observed when comparing the size of the Smn2B/- organs and in the “Taiwanese” SMA model at PND 9, confirming the spleen defects in the three most commonly used SMA mouse models (Fig. 1A) (Supplementary Material, Fig. S1A and B). The selective hypoplasia of the spleen was even more pronounced in the intermediate model of SMA, Smn2B/-, as evidenced by the continued decrease in relative size from 10% down to 1% from PND 7 to 21, respectively (Fig. 1A, Supplementary Material, Fig. S1C and D). Collectively, these results demonstrate that SMN deficiency selectively impacts the development and size of the spleen in severe and intermediate mouse models of SMA.
Figure 1.

SMA spleens exhibit severe and progressive size reduction (A) Organs harvested from WT (right) and SMA (left) of late symptomatic SMNΔ7 (left panel), late symptomatic Smn2B/- mice (middle) at PND 21, and late symptomatic “Taiwanese” SMA mice at PND 9 (right panel). (B) Spleen weights from pre- (PND 5), to late (PND 13) symptomatic time points of SMNΔ7 SMA and WT littermates control mice. Spleens are significantly smaller than control spleens at all time points. (C) Mass ratio of SMA to WT for body weight (BW), brain, and spleen (SP). SMA spleens are disproportionately smaller than WT spleens. (D) Ratio of spleen mass to body weight. The WT spleens are growing proportionally with the body, SMA spleens start to regress after PND 7. (n = 4 to 6 for each genotype at each time point) (*P < 0.005, **P < 0.01, ****P < 0.001).
Red pulp is selectively reduced in SMNΔ7 SMA spleens
To determine whether splenic structure was disrupted in SMA mice, the relative composition of red and white pulp was determined at PND 12 in SMNΔ7 mice. Hematoxylin and eosin (H&E) staining of SMA and WT spleen sections revealed normal structure, distribution and abundance of white pulp. In contrast, red pulp distribution and composition was reduced in SMA spleens with a decrease in extramedullary splenic hematopoiesis compared to WT spleens (Fig. 2A). We performed immunohistochemistry on spleen sections using anti-B220 and anti-CD3, pan markers of B- and T-lymphocytes, respectively. Analysis of PND 12 spleen samples demonstrates that B- and T-cell zones were well preserved in the SMA spleen. While a higher B-cell density is observed, this is mainly due to the reduction in the red pulp area which indirectly concentrates the B-cell regions (Fig. 2B, Supplementary Material, Fig. S2). No differences were detected between WT and SMA periarteriolar lymphoid sheaths (Fig. 2C, Supplementary Material, Fig. S2). The reduction in red pulp and extramedullary hematopoiesis was evident, at PND 10, but not PND 5 (Supplementary Material, Fig. S3).
Figure 2.
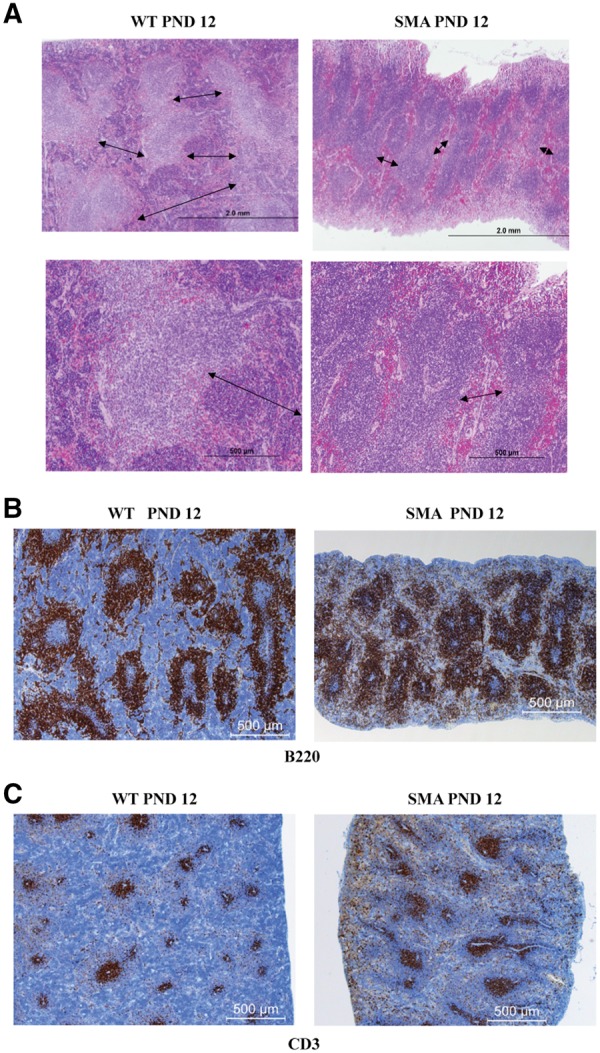
Red pulp is reduced in SMNΔ7 SMA spleens. (A) H&E staining of 4 µm sections from WT (left) and SMA (right) spleens of SMNΔ7 mice at PND 12. The red pulp (margins depicted by arrows) is smaller in SMA mice with marked decrease in extramedullary hematopoiesis. (B, C) Representative immunohistochemistry for the expression of B220 (B) and CD3 (C) of fixed paraffin embedded spleen sections from PND 12 SMNΔ7 SMA mice (right) and their littermate WT controls (left). SMA spleen has a greater concentration of B220+, with a reduction of red pulp area, while the white comprises most of the spleen space. Sections are visualized with immunoperoxidase (brown) for expression of CD3 on T-cells (A); and B220 on B cells (B). Sections are counterstained with hematoxylin (blue).
SMA spleen has increased lymphocyte concentrations
As one of the primary functions of the spleen involves the maintenance of immune cells and the generation of immune responses, we next wanted to determine whether the relative composition of immune cells was impacted by SMN deficiency. To assess the immune cell composition in SMA spleens relative to WT, total splenocytes from individual mice were stained with macrophage and lymphocyte surface markers and analyzed by flow cytometry. Our results demonstrated that at PND 5, the relative composition of CD4+ and CD8+ T lymphocytes was similar to WT (Fig. 3A and B); however, at the symptomatic time point, PND 12, SMNΔ7 spleens had a higher percentage of CD4+ and CD8+ T-cells (Fig. 3C and D). CD4+ T-cells comprised approximately 11.2% (±1) of the SMA spleen cell population compared to 5.6% (± 0.3) in the WT spleens (n = 7, P<0.001) (Fig. 3C and D). CD8+ T-cells were detected at a higher frequency in SMA spleen, resulting in an average of 4.3 (±0.5) compared to 2.1 (±0.2) in the WT spleens (n = 7, P = 0.003) (Fig. 3C and D). Similar to the T-cell populations, the frequency of B lymphocytes was increased to 52.6% (±2.9), significantly higher than the 30.3% (±2.7) detected in WT spleens (n = 10, P<0.001) (Fig. 3E and F). A primary function of B-cells is to present antigens on their cell surface bound to MHC class II surface proteins. To determine whether B-cells within the spleen exhibited defects in antigen presentation machinery, B-cell MHC class II expression was determined by staining with anti-I-A/I-E, and a pan B-cell marker B220. FACS results demonstrated that MHC class II expression on B cells was similar between WT and SMA splenocytes (Fig. 3G), suggesting that while B-cell populations were increased in SMA spleens, MHC class II expression was largely unchanged. Collectively, these results demonstrate that SMN deficiency impacts splenic development as well as the relative abundance of mediators of the immune response.
Figure 3.

Higher frequency of lymphocytes in SMNΔ7 SMA spleens. (A) Total splenocytes from individual WT (left) and SMA (right) spleens of SMNΔ7 mice at PND 5 were surface labeled. Frequency of (CD4+, CD8-) and (CD4-, CD8+) T cells are indicated relative to the total live splenocytes in representative FACS plots. (B) Bar graph showing the mean percentages of CD4+ and CD8+ T lymphocytes ± SEM for three mice per group. (C,D) are the same as in (A) and (B) respectively for splenocytes from mice at PND 12 (n = 7 for each group). (E) Total splenocytes from individual WT (left) and SMA (right) spleens of SMNΔ7 mice at PND12 were surface labeled. Frequencies of B lymphocytes (CD3-, B220+) are indicated relative to the total live splenocytes in representative FACS plots. (F) Bar graph showing the mean percentages ± SEM for 10 mice per group. (G) To study the level of expression of MHC class II on B cells, total splenocytes from individual WT (left) and SMA (right) spleens were stained with anti-I-A/I-E (MHC II) and analyzed by flow. Frequencies of (B220+ I-A/I-E+) subsets are indicated relative to (CD3-, B220+) splenocytes in representative FACS plots.
Vasculature and proliferation activity are not altered in SMA spleen
To determine possible causes of the structural defects and differences in immune cell populations in the SMA spleen, splenic vasculature and splenocyte proliferation was examined. Previously SMN was shown to have a role in the proper vasculature development (24). Since the spleen, and in particular the red pulp, is rich in blood and sinuses, we postulated that vasculature defects may be the underlying basis for the reduction in the red pulp. To examine the vasculature, immunostaining was performed using CD31, a marker for endothelial cells. Surprisingly, CD31+ staining between SMA and WT spleen sections did not identify a significant difference in the vascular structure (Fig. 4A, Supplementary Material, Fig. S4). Similarly, immunohistochemistry was performed using anti-Ki67 antibody, a nuclear protein used to monitor cellular proliferation rates. Immunohistochemistry at PND 12 identified no difference in the relative abundance of Ki67-positive staining, demonstrating that the proliferation rates did not account for the differences in lymphocytic abundance in the SMA spleens (Fig. 4B, Supplementary Material, Fig. S4).
Figure 4.
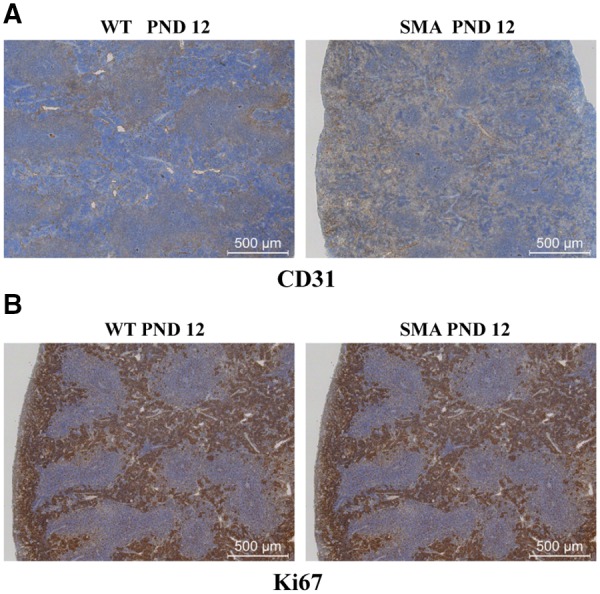
SMA spleens exhibit no difference in proliferation activity or vasculature density. (A) Representative immunohistochemistry images for CD31 (A) and Ki673 (B) of fixed paraffin embedded spleen sections from PND 12 SMNΔ7 SMA mice (right) and their littermate WT controls (left). SMA spleen showed no obvious difference of CD31 expression or Ki67 staining. Sections are stained by immunoperoxidase (brown) for the expression of CD31 or Ki67 and are counterstained with hematoxylin (blue).
SMN deficiency alters macrophage populations in SMA spleen
To examine the relative abundance of macrophages in the SMA spleen, total splenocytes from PND 12 SMA and WT littermate mice were stained with anti-CD11b and analyzed by flow cytometry. Compared to WT splenocytes, a significant increase in CD11b+ macrophages was observed in SMA spleens at PND 12 (Fig. 5A and B). Macrophages are diverse in phenotype, location, and function. In splenic macrophages, two major populations exist: 1) white pulp macrophages which originate from the bone marrow through conventional hematopoiesis and are renewed from hematopoietic stem cells (HSCs); 2) red pulp macrophages (RPM) which are derived from embryonic precursors in the yolk sac (YS). F4/80 and CD11b surface markers can efficiently distinguish these two macrophage populations since the bone marrow-derived population is CD45+ F4/80low CD11bhigh and yolk sac-derived macrophages are CD45+ F4/80high and CD11blow. Flow cytometry analysis on CD45-gated cells revealed that the HSC-derived macrophages (F4/80low CD11bhigh) were present at a higher frequency in SMA spleens whereas the RP yolk sac-derived macrophages (F4/80high CD11blow) were reduced in the SMA spleen. Further staining with anti-CX3CR1, a signature chemokine of the YS-derived macrophages, confirmed the origin (YS) and identity (resident RPM) of this population, (Fig. 5C and D). To determine whether the observed yolk sac-derived macrophage reduction occurred at an early developmental stage, PND 2 a pre-symptomatic time point was analyzed similarly. FACS analysis revealed a reduction in yolk sac-derived macrophage population from SMA mice compared to WT littermates even at this early stage (Fig. 5E and F).
Figure 5.

Higher frequency of CD11b+ macrophages and reduction of the yolk sac-derived population in SMA spleens. Total splenocytes from individual WT (left) and SMA (right) spleens of SMNΔ7 mice at PND 12 were surface labeled. (A) Frequency of CD11b macrophages (CD3-, B220-, CD11b+) are indicated relative to live CD3-, B220- splenocytes in representative FACS plots. (B) Bar graph showing the mean percentages ± SEM for six mice per group. (C) Total splenocytes from individual WT (upper) and SMA (lower) spleens were stained with anti-CD11b and anti-F4/80 antibodies and the different subsets of macrophages were identified by flow cytometry. Frequency of (F4/80low CD11bhigh) and (F4/80high CD11blow) subsets are indicated relative to live CD45+ splenocytes in representative FACS plots. Right panels depict the percentage of cells expressing CX3CR1 in each of the two macrophage populations. (D) Bar graph showing the mean percentages ± SEM for six mice per group. (E,F) Same as (C) and (D) for spleens harvested at PND 2.
SMN restoration corrects spleen pathology
Previously, we have developed a highly effective SMN2 exon 7 specific splice-switching antisense oligonucleotide (ASO) that increases levels of full length SMN, significantly extends survival, and decreases the motor neuron pathology in SMA mice (25). To determine whether ASO treatment can rescue the SMN-deficiencies in the SMA spleen, SMA mice were treated at PND 1 with the ASO via an intracerebroventricular injection. In spleen sections from ASO-treated SMA mice, the red and white pulp structure and accumulation was similar to WT, demonstrating that ASO treatment rescues the spleen pathology (Fig. 6A). Additionally, the B- and T-cell populations were restored to WT levels in ASO-treated samples at PND 12 as determined by B220 and CD3 immunohistochemistry (Fig. 6B and C). Collectively, this work identifies structural and functional defects in SMA spleens in SMA mice, demonstrating a role for SMN in normal spleen development, but also suggesting an immunological component to the complex SMA pathology observed in SMA model mice.
Figure 6.

Restoration of SMN via ASO treatment corrects SMA spleen pathology. Representative images of spleen sections from PND 12 ASO-treated SMA mice (right), sham-treated SMA control (center), and WT control (left). (A) H&E staining, treated SMA spleen displays architecture and red pulp similar to WT. (B) Immunostaining for B lymphocytes using anti-B220. (C) Immunostaining for T lymphocytes using anti-CD3.
Discussion
Motor neuron degeneration is a hallmark of SMA. However, SMN is a ubiquitously expressed protein and the possibility that it has important functional roles related to SMA pathology in non-neuronal cells and tissues has been an ongoing question in the field. Investigating SMN function and understanding whether SMN activities relate to SMA is important from a clinical stand point but also from an animal modelling perspective. Here we demonstrate that SMN deficiency significantly impacts spleen development and the relative composition of splenic macrophages. In two different SMA mouse models, the spleen displays significant reduction in size and weight at post-symptomatic stage. Further, a third model demonstrated these characteristics at a pre-symptomatic stage. SMA spleens are not only significantly smaller than their WT control littermates but also proportionally more reduced than the overall reduction in the body weight and size. Even though nearly all organs are reduced in SMA mice, this disproportional reduction in size was specific to spleen in the three SMA mouse models, whereas the non-lymphoid organs had milder size alteration. At an early stage in development (PND 9) in a SOD1 ALS mouse model of disease, spleen size was indistinguishable from wildtype (Supplementary Material, Fig. S5), suggesting a degree of SMA-centric specificity for early spleen hypoplasia. In SMNΔ7, a well-established SMA mouse model, we found that SMN deficiency alters the architecture of the spleen micro-anatomy; red pulp space is markedly reduced while the white pulp structure is widely preserved. Extramedullary hematopoiesis, a common finding in rodent red pulp spleens was markedly decreased in spleens from SMA mice. Looking into the vasculatures, immunohistochemistry did not show reduced density in SMA spleens, hence though compromised vasculatures was previously reported in SMA muscle and GI tract (14,24), vasculature is not contributing to spleen pathology and could not provide for the moment an explanation for the red pulp reduction. Our flow and immunohistochemistry data revealed a higher density of T- and B-lymphocytes and CD11b macrophages. Ki67 staining showed similar proliferation activity, hence SMN deficiency is not altering lymphocyte or macrophage proliferation in neonatal SMA spleens.
The spleen is a unique lymphoid organ and an important site for innate and adaptive immune response generation and has been previously associated with neurodegeneration (26). Reduction in spleen size has been reported in end-stage SOD1 mouse models of ALS with major disruptions within the red and white pulp zones (27). Beyond structural differences, splenectomy reduced neurodegeneration in a brain injury model and a transient size reduction in the spleen has been recorded following stroke induction in the middle cerebral artery occlusion mouse model (28). In SMA, spleen and thymus atrophy has been reported (22). Additionally, a recent study in the Taiwanese SMA mouse model reported severe atrophy in the SMA spleen with major disruptions of the red and white zones at disease end stage.
Recently, F4/80 and CD11b markers have been used efficiently to identify developmentally and functionally distinct macrophage populations in the spleen and other tissues (30,31). We investigated these myeloid subsets and found that CD45+ F4/80low CD11bhigh macrophages were present at a greater frequency in SMA spleens whereas CD45+ F4/80high CD11blow macrophages were largely depleted in the SMA spleen. The spleen contains highly adapted macrophage subpopulations each possessing distinct functions and residing in micro-anatomical locations (32). Furthermore, fate-mapping studies showed that macrophages have different ontogenies. The F4/80low CD11bhigh macrophages originate from the bone marrow and conventional hematopoiesis. These cells are renewed from haematopoietic stem cell precursors and circulating blood monocytes. In the spleen, this population resides mainly in the white pulp and the marginal zone, performing crucial roles in the immune system homeostasis and in mounting adaptive and innate responses during infection and inflammation. The F4/80high and CD11blow population has an earlier origin and is derived from embryonic precursors in the yolk sac (30,33–35). In mice, this population circulates and colonizes the embryo between E9.5 and E10.5 (30). Though it is present in most neonatal tissues, in adulthood the yolk sac derived macrophages disappear in some tissue like intestines and persist throughout adulthood in most organs where it constitutes the resident macrophages (30,31). This subset is long lived, difficult to divide, still it is self-renewing and does not depend on HSC renewal. In spleens, it constitutes the red pulp macrophages (RPM) and is responsible for erythrocyte phagocytosis, iron recycling, and contributes to blood borne pathogen and debris clearing.
Our results showed that F4/80low CD11bhigh (bone marrow derived) and F4/80high CD11blow (yolk sac derived) subsets are differently altered in the SMNΔ7 SMA spleens. These two populations differ not only in their precursors and spatiotemporal regulation, but are fundamentally distinct as they are derived from different transcription factor cascades (32,36,37). The CD11bhigh HSC derived monocytes and macrophages depend on the transcription factor Myb for their development. By contrast, Myb is dispensable for the development of the embryonic yolk sac-derived F4/80high macrophages which require the PU.1 related transcription factor Spi-C (30,32). Whether SMN deficiency is affecting the Spi-C pathway and F4/80high development, or whether RPM deficiency is a result of the red pulp destruction in SMA spleen, it is not currently clear.
Recent work that focused upon the “Taiwanese” SMA mouse model (Jackson Laboratory Stock 5058) has shown a number of similarities to the work in this report, although some differences were also identified (29). At post-natal time points, spleen weight (raw or as a percent of total body weight) was significantly reduced in SMA mice and red pulp morphology was significantly altered. Additionally, vascular density was not affected in the SMA spleen. However, several differences are clearly noted between the various animal models, including that cellular proliferation and cell death were altered in the 5058 spleen based upon Ki67 and TUNEL staining, in contrast to the current study in which cell proliferation was not impacted. Additionally, developing B-cell follicles are absent in the 5058 spleen. Interestingly, splenic defects were also reported in routine autopsy results from SMA patients, although the nature and degree of pathology varied (29). Collectively, these results reveal the impact of SMN-deficiency upon the developing spleen as well as a routine splenic function in SMA models as well as patients, further highlighting the importance of peripheral tissues in SMA development.
Materials and Methods
Animals
All mice were housed and handled in accordance with the Animal Care and Use Committees of the University of Missouri. SMNΔ7 mice were purchased from Jackson Laboratory (stock numbers #5025). Smn2B/2B mice, which have a hybrid background of C57BL/6 and CD1, were a kind gift from Dr. R. Kothary, Ottawa, Canada. FVB Smn+/- mice were purchased from the Jackson Laboratory. Mice were housed under a 12 h light/dark cycle and the colonies were maintained as heterozygote breeding pairs under specific pathogen-free conditions. SMNΔ7 mice investigated in this study had a homozygous deletion of the murine Smn gene (mSmn) and carried homozygous transgenes for human SMN2 (SMN2) and an SMN cDNA lacking exon 7 (38). The heterozygous mice for the mSmn (SMN2+/+;SMNΔ7+/+;mSmn+/−) were interbred to generate the mSmn knock-out mice (KO) (SMN2+/+;SMNΔ7+/+;mSmn−/−) used as the SMA mouse, and the homozygous wild-type (WT) for the mSmn (SMN2+/+;SMNΔ7+/+;mSmn+/+) used as unaffected positive controls. SMNΔ7 mice live to an average of 14 days of age and show signs of progressive muscle paralysis starting day 7. Smn2B/2B mice which contain a targeted mutation in exon 7 of the mSmn gene (39) were bred to the FVB Smn+/- which have a heterozygous deletion of mSmn. The Smn2B/+ offspring have normal phenotype and lifespan and were used as unaffected positive controls. The Smn2B/- mice show signs of SMA around day 10, and have an average lifespan of approximately 30 days (40). Mice were genotyped using tail biopsies and standard PCR protocol as previously described (41). For all experiments, equal numbers of KO and WT pups were investigated from a litter.
Tissue collection
SMA mice and their WT littermates control were sacrificed at PND 5, 10, 12 (pre- and post-symptomatic) and weighed. Various organs were harvested and weighed. Spleens were removed and fixed overnight in 4% paraformaldehyde (PFA) in 0.1 M PBS, washed in PBS and embedded in paraffin blocks. 4 μm sections were prepared and stained with hematoxylin and eosin following standard procedures.
Immunohistochemistry
IHC was performed on 4 μm sections from the paraffin block. The sections were deparaffinized and rehydrated through graded alcohols to water. The endogenous peroxidase activity was inhibited using 3% hydrogen peroxide for 5 min. Sections were blocked with 5% BSA then incubated for 1 h at room temperature with the primary antibody (rabbit polyclonal anti-CD31, Abcam, Cambridge; or rabbit polyclonal anti-Ki67, Thermo-Scientific, or rabbit anti-CD3 clone G7, DAKO, or rat anti-F4/80 clone C1:A3-1, AbD Serotec, or rat anti-CD45R (B220) clone Ra3-6b2, Invitrogen). After washing, the secondary Ab was applied for 30 min at room temperature, staining was visualized by addition of hydrogen peroxide substrate and diaminobenzidine chromogen (DAB). Finally, the slides were counterstained with Mayer’s hematoxylin, dehydrated, and mounted. Sections were imaged using 5x objectives on the Leica-DM5500-Widefield microscope. All images were taken using the same imaging parameters and quantified using Fiji ImageJ software (NIH).
Flow cytometry
Fluorophore-conjugated murine antibodies directed against the following epitopes were purchased from BioLegend or BD Biosciences: CD3ε (145-2C11); CD4 (RM4-5); CD8a (53-6.7); CD45R/B220 (30-F11); CD11b (M1/70); F4/80 (BM8 from BioLegend); CX3CR1 (SA011F11); I-A/I-E (M5/114.15.2). F4/80 (Cl:A3-1 from Serotec). 7-amino-actinomycian D (7AAD) was used to gate out dead cells. Single cell suspensions from total individual splenocytes were obtained by grinding and filtering through 70μm mesh filters. Following erythrocytes lysing, total cells were counted using dead cell exclusion Trypan blue stain. (1–3) x 106 of a total splenocyte suspension were incubated with various antibodies for 30 min at 4 °C. Cells were acquired on a Beckman Coulter CyAn (Brea, CA) and data were analyzed using FlowJo software.
ASO delivery
Intracerebroventricular (ICV) injections were used to deliver a bolus of 2 μl of 4 nmol Morpholino-modified ASO (MOE1v11) or sham within the first 24 h after birth as previously described (42). Briefly, ICV injections were performed using sterilized glass micropipettes. The needles were inserted perpendicular to the skull at the injection site approximately 0.25 mm lateral to the sagittal suture and 0.5 mm rostral to the coronary suture.
Statistical analyses
Comparisons between two groups were performed by two-tailed Student’s t-tests. Graph generation and statistical analysis were performed using GraphPad Prism software. The following notations were used to indicate statistical significance: ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We would like to thank the Lorson lab for helpful discussion and analysis as well as Taylor Paskoff, Madeline Bolding, Madeline Simon, Zachary Lorson and Elinor Stanley for technical assistance.
Conflict of Interest statement. None declared.
Funding
This work was supported by the following grants: C.L.L. (FightSMA; Gwendolyn Strong Foundation); C.L.F. (NIH U42 OD010918); E.J.A (IU FORCES: Funding Opportunities for Research Commercialization and Economic Success); S.K.C. (CureSMA Young Investigator Award); and M.T.K. (Department of Veterinary Pathobiology).
References
- 1. Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80, 155–165. [DOI] [PubMed] [Google Scholar]
- 2. Lorson C.L., Hahnen E., Androphy E.J., Wirth B. (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA, 96, 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Donlin-Asp P.G., Bassell G.J., Rossoll W. (2016) A role for the survival of motor neuron protein in mRNP assembly and transport. Curr. Opin. Neurobiol., 39, 53–61. [DOI] [PubMed] [Google Scholar]
- 4. Li D.K., Tisdale S., Lotti F., Pellizzoni L. (2014) SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin. Cell. Dev. Biol., 32, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wirth B., Barkats M., Martinat C., Sendtner M., Gillingwater T.H. (2015) Moving towards treatments for spinal muscular atrophy: hopes and limits. Expert. Opin. Emerg. Drugs, 20, 353–356. [DOI] [PubMed] [Google Scholar]
- 6. Kolb S.J., Kissel J.T. (2015) Spinal Muscular Atrophy. Neurol. Clin., 33, 831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rindt H., Feng Z., Mazzasette C., Glascock J.J., Valdivia D., Pyles N., Crawford T.O., Swoboda K.J., Patitucci T.N., Ebert A.D., et al. (2015) Astrocytes influence the severity of spinal muscular atrophy. Hum. Mol. Genet., 24, 4094–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hunter G., Aghamaleky Sarvestany A., Roche S.L., Symes R.C., Gillingwater T.H. (2014) SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet., 23, 2235–2250. [DOI] [PubMed] [Google Scholar]
- 9. Hunter G., Powis R.A., Jones R.A., Groen E.J., Shorrock H.K., Lane F.M., Zheng Y., Sherman D.L., Brophy P.J., Gillingwater T.H. (2016) Restoration of SMN in Schwann cells reverses myelination defects and improves neuromuscular function in spinal muscular atrophy. Hum. Mol. Genet., 25, 2853–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bevan A.K., Hutchinson K.R., Foust K.D., Braun L., McGovern V.L., Schmelzer L., Ward J.G., Petruska J.C., Lucchesi P.A., Burghes A.H., et al. (2010) Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet., 19, 3895–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shababi M., Habibi J., Yang H.T., Vale S.M., Sewell W.A., Lorson C.L. (2010) Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet., 19, 4059–4071. [DOI] [PubMed] [Google Scholar]
- 12. Bowerman M., Michalski J.P., Beauvais A., Murray L.M., DeRepentigny Y., Kothary R. (2014) Defects in pancreatic development and glucose metabolism in SMN-depleted mice independent of canonical spinal muscular atrophy neuromuscular pathology. Hum. Mol. Genet., 23, 3432–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bowerman M., Swoboda K.J., Michalski J.P., Wang G.S., Reeks C., Beauvais A., Murphy K., Woulfe J., Screaton R.A., Scott F.W., et al. (2012) Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol., 72, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sintusek P., Catapano F., Angkathunkayul N., Marrosu E., Parson S.H., Morgan J.E., Muntoni F., Zhou H. (2016) Histopathological Defects in Intestine in Severe Spinal Muscular Atrophy Mice Are Improved by Systemic Antisense Oligonucleotide Treatment. PLoS One, 11, e0155032.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szunyogova E., Zhou H., Maxwell G.K., Powis R.A., Francesco M., Gillingwater T.H., Parson S.H. (2016) Survival Motor Neuron (SMN) protein is required for normal mouse liver development. Sci. Rep., 6, 34635.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ottesen E.W., Howell M.D., Singh N.N., Seo J., Whitley E.M., Singh R.N. (2016) Severe impairment of male reproductive organ development in a low SMN expressing mouse model of spinal muscular atrophy. Sci. Rep., 6, 20193.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boillee S., Vande Velde C., Cleveland D.W. (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron, 52, 39–59. [DOI] [PubMed] [Google Scholar]
- 18. Lobsiger C.S., Cleveland D.W. (2007) Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci., 10, 1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bettcher B.M., Kramer J.H. (2013) Inflammation and clinical presentation in neurodegenerative disease: a volatile relationship. Neurocase, 19, 182–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scali C., Prosperi C., Bracco L., Piccini C., Baronti R., Ginestroni A., Sorbi S., Pepeu G., Casamenti F. (2002) Neutrophils CD11b and fibroblasts PGE(2) are elevated in Alzheimer's disease. Neurobiol. Aging, 23, 523–530. [DOI] [PubMed] [Google Scholar]
- 21. Valekova I., Jarkovska K., Kotrcova E., Bucci J., Ellederova Z., Juhas S., Motlik J., Gadher S.J., Kovarova H. (2016) Revelation of the IFNalpha, IL-10, IL-8 and IL-1beta as promising biomarkers reflecting immuno-pathological mechanisms in porcine Huntington's disease model. J. Neuroimmunol., 293, 71–81. [DOI] [PubMed] [Google Scholar]
- 22. Dachs E., Hereu M., Piedrafita L., Casanovas A., Caldero J., Esquerda J.E. (2011) Defective neuromuscular junction organization and postnatal myogenesis in mice with severe spinal muscular atrophy. J. Neuropathol. Exp. Neurol., 70, 444–461. [DOI] [PubMed] [Google Scholar]
- 23. Gurumoorthy A., Gopalsamy V., K R., Piplani P., Malik R. (2011) N-[4-(2-Morpholino-eth-oxy)phen-yl]acetamide monohydrate. Acta. Crystallogr. Sect. E. Struct. Rep. Online, 67, o262.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Somers E., Stencel Z., Wishart T.M., Gillingwater T.H., Parson S.H. (2012) Density, calibre and ramification of muscle capillaries are altered in a mouse model of severe spinal muscular atrophy. Neuromuscul. Disord., 22, 435–442. [DOI] [PubMed] [Google Scholar]
- 25. Osman E.Y., Washington C.W. 3rd, Kaifer K.A., Mazzasette C., Patitucci T.N., Florea K.M., Simon M.E., Ko C.P., Ebert A.D., Lorson C.L. (2016) Optimization of Morpholino Antisense Oligonucleotides Targeting the Intronic Repressor Element1 in Spinal Muscular Atrophy. Mol. Ther., 24, 1592–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ajmo C.T. Jr., Vernon D.O., Collier L., Hall A.A., Garbuzova-Davis S., Willing A., Pennypacker K.R. (2008) The spleen contributes to stroke-induced neurodegeneration. J. Neurosci. Res., 86, 2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Banerjee R., Mosley R.L., Reynolds A.D., Dhar A., Jackson-Lewis V., Gordon P.H., Przedborski S., Gendelman H.E. (2008) Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS One, 3, e2740.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seifert H.A., Hall A.A., Chapman C.B., Collier L.A., Willing A.E., Pennypacker K.R. (2012) A Transient Decrease in Spleen Size Following Stroke Corresponds to Splenocyte Release into Systemic Circulation. J. Neuroimmune Pharma, 7, 1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thomson A.K., Somers E., Powis R.A., Shorrock H.K., Murphy K., Swoboda K.J., Gillingwater T.H., Parson S.H. (2016) Survival of motor neurone protein is required for normal postnatal development of the spleen. J. Anat., doi: 10.1111/joa.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schulz C., Gomez Perdiguero E., Chorro L., Szabo-Rogers H., Cagnard N., Kierdorf K., Prinz M., Wu B., Jacobsen S.E., Pollard J.W., et al. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science, 336, 86–90. [DOI] [PubMed] [Google Scholar]
- 31. Bain C.C., Bravo-Blas A., Scott C.L., Gomez Perdiguero E., Geissmann F., Henri S., Malissen B., Osborne L.C., Artis D., Mowat A.M. (2014) Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol., 15, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kohyama M., Ise W., Edelson B.T., Wilker P.R., Hildner K., Mejia C., Frazier W.A., Murphy T.L., Murphy K.M. (2009) Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature, 457, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davies L.C., Jenkins S.J., Allen J.E., Taylor P.R. (2013) Tissue-resident macrophages. Nat. Immunol., 14, 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davies L.C., Taylor P.R. (2015) Tissue-resident macrophages: then and now. Immunology, 144, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mebius R.E., Kraal G. (2005) Structure and function of the spleen. Nat. Rev. Immunol., 5, 606–616. [DOI] [PubMed] [Google Scholar]
- 36. DeKoter R.P., Walsh J.C., Singh H. (1998) PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. Embo J., 17, 4456–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sumner R., Crawford A., Mucenski M., Frampton J. (2000) Initiation of adult myelopoiesis can occur in the absence of c-Myb whereas subsequent development is strictly dependent on the transcription factor. Oncogene, 19, 3335–3342. [DOI] [PubMed] [Google Scholar]
- 38. Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. (2005) SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet., 14, 845–857. [DOI] [PubMed] [Google Scholar]
- 39. DiDonato C.J., Lorson C.L., De Repentigny Y., Simard L., Chartrand C., Androphy E.J., Kothary R. (2001) Regulation of murine survival motor neuron (Smn) protein levels by modifying Smn exon 7 splicing. Hum. Mol. Genet., 10, 2727–2736. [DOI] [PubMed] [Google Scholar]
- 40. Bowerman M., Murray L.M., Beauvais A., Pinheiro B., Kothary R. (2012) A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord., 22, 263–276. [DOI] [PubMed] [Google Scholar]
- 41. Coady T.H., Lorson C.L. (2010) Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J. Neurosci., 30, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Osman E.Y., Miller M.R., Robbins K.L., Lombardi A.M., Atkinson A.K., Brehm A.J., Lorson C.L. (2014) Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum. Mol. Genet., 23, 4832–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.