

Abstract
The gonadoblastoma gene, testis-specific protein Y-encoded (TSPY), on the Y chromosome and its X-homologue, TSPX, are cell cycle regulators and function as a proto-oncogene and a tumor suppressor respectively in human oncogenesis. TSPY and TSPX competitively bind to the androgen receptor (AR) and AR variants, such as AR-V7, at their conserved SET/NAP domain, and exacerbate and repress the transactivation of the AR/AR-V7 target genes in ligand dependent and independent manners respectively. The inhibitory domain has been mapped to the carboxyl acidic domain of TSPX, truncation of which renders TSPX to be stimulatory while its transposition to the C-terminus of TSPY results in an inhibitory hybrid protein. TSPY and TSPX co-localize with the endogenous AR, in the presence of ligand, on the promoters and differentially regulate the expression of the endogenous AR target genes in the androgen-responsive LNCaP prostate cancer cells. Transcriptome analysis shows that TSPY and TSPX expressions differentially affect significant numbers of canonical pathways, upstream regulators and cellular functions. Significantly, among the common ones, TSPY activates and TSPX inhibits numerous growth-related and oncogenic canonical pathways and cellular functions in the respective cell populations. Hence, TSPY and TSPX exert opposing effects on the transactivation functions of AR and AR-Vs important for various physiological and disease processes sensitive to male sex hormone actions, thereby not only affecting the pathogenesis of male-specific prostate cancer but also likely contributing to sex differences in the health and diseases of man.
Introduction
The male sex hormone androgen and its receptor, androgen receptor (AR), play key roles in various developmental pathways, physiology and disease processes, such as prostate differentiation and oncogenesis (1,2), and sexually dimorphic physiology and diseases, such as cardiovascular functions/diseases (3) and brain development and neural diseases (4,5). At present, the contributions of genes on the sex chromosomes, i.e. X and Y chromosome, in sex-specific and sexually dimorphic human cancers and diseases have not been fully investigated. In the case of cancers, abnormal activation of a Y-located proto-oncogene could have a positive effect(s) on oncogenesis in the affected cells in males while inactivation of a X-located tumor suppressor could predispose males to oncogenesis. Indeed, the testis-specific protein Y-encoded (TSPY) gene on the Y chromosome and its X-homologue, TSPX (6), represent such a pair of homologues on the sex chromosomes that are potentially at the two extremes of the human oncogenic spectrum.
TSPY is a small gene, tandemly repeated 30–60 times at the critical region harboring the gonadoblastoma locus (GBY) (7), the only oncogenic locus on the Y chromosome (8). It is normally expressed and likely serves normal functions in prespermatogonia of fetal testis (9), and spermatogonia and spermatocytes of adult testis (10). Significantly, TSPY is also abundantly expressed in gonadoblastoma and various testicular germ cell tumors (11–13), as well as somatic cancers, such as prostate cancer and hepatocellular carcinoma (14,15). Ectopic expression of TSPY in incompatible cells, such as female/dysfunctional germ cells and somatic cells incapable of entering male germ cell lineage, promotes cell proliferation and tumorigenesis (16). It accelerates G2/M transition by stimulating the mitotic cyclin B-cyclin dependent kinase 1 (CDK1) activities (17), and likely affects the G2/M checkpoints (11). Aberrant expression of TSPY in transgenic mice results in gonadoblastoma-like structures in the ovaries (18). Hence, TSPY is a male-specific proto-oncogene for the GBY locus on the Y chromosome, and likely contributes to various human cancers.
TSPX, also known as TSPYL2, CDA1, CINAP and DENTT, is a single-copy homologue of TSPY on the X chromosome (6). TSPY and TSPX originated from the same ancestral gene with similar exon–intron organization at their conserved SET/NAP domain, initially identified in the SET oncoprotein and the nucleosome assemble protein (NAP), but differ at their flanking sequences, as results of the evolutionary divergence of the sex chromosomes. In particular, TSPX harbors a large acidic domain at its carboxyl terminus, which is absent in TSPY. Importantly, it possesses contrasting properties in cell cycle regulation, i.e. retardation of cell proliferation (19) and repression of cyclin B-CDK1 activities (17), to those of TSPY, and has been considered as a tumor suppressor on the X chromosome for various human cancers (15,19,20).
In this report, we show that TSPY and TSPX competitively bind to AR, but stimulate and repress AR transactivation of responsive genes, respectively. We have identified the respective binding domains and mapped the TSPX repressor function to its carboxyl acidic domain, absent in TSPY. Importantly, such interactions and modulations could be extended to constitutively active AR variants, lacking the carboxyl ligand binding domain, and endogenous androgen-responsive genes in the androgen-responsive prostate cancer LNCaP cells. Transcriptome analysis suggests that this pair of homologues differentially affect various pathways and cellular functions in this prostate cancer cell line. Hence, TSPY and TSPX serve as co-activator and co-repressor of AR and AR variants, and could function as an oncogene and a tumor suppressor respectively in prostate cancer and other sexually dimorphic tumors and diseases affected by the actions of androgen and its receptor and receptor variants.
Results
Interactions of TSPY and TSPX with the androgen receptor
After the initial identification of AR as a TSPY-binding protein in a yeast two-hybrid study (21), also see Materials and Methods, co-immunoprecipitation was used to demonstrate the interactions between TSPY and AR, in transfection assays with FLAG-tagged TSPY and HA-tagged AR constructs in HEK293 cells (17,21). Our results showed that AR was specifically precipitated together with TSPY in the presence or absence of the synthetic ligand, R1881 (Fig. 1A). Similar study showed that TSPX and AR could also be co-immunoprecipitated as protein complexes (Fig. 1B), suggesting that both TSPY and TSPX are capable of interacting with AR in similar manners.
Figure 1.

(A–B) Co-immunoprecipitation of FLAG-tagged TSPY (A) and TSPX (B) with full length AR in the presence and absence of the ligand R1881; and (C-D) with AR variants, AR-V7 (C) and AR-V4 (D) in HEK293 cells. Co-immunoprecipitation was performed with FLAG-specific antibody (against TSPY or TSPX), and analyzed with antibodies against FLAG (TSPY/TSPX) and HA (AR and AR-Vs). Input, total cell lysates before; Co-IP:FLAG, after co-immunoprecipitation. IP, immunoprecipitation; WB, western blot; HA, anti-HA antibody; FLAG, anti-FLAG antibody.
To confirm their interactions, GST-TSPY and GST-TSPX fusion proteins were used in pull-down assays with in vitro35S-labeled AR (17,21). Our results showed that GST-TSPY and GST-TSPX were capable of binding to AR (Fig. 2A, left), which were confirmed by affinity assays with HA-tagged AR and 35S-lableled TSPY or TSPX (Fig. 2A, right). To determine the domain(s) in AR responsible for interacting with TSPY, the N-terminal domain (NTD), DNA-binding domain (DBD) and the ligand-binding domain (LBD) (Fig. 3F) were in vitro labeled, and analyzed similarly with GST-pull down assays. The results showed that both NTD and DBD were being retained by GST-TSPY fusion protein (Fig. 2B), suggesting that they are the interactive domains in AR.
Figure 2.

Interacting domain mapping and functional analysis of TSPY isoforms with androgen receptor. (A) GST-TSPY or GST-TSPX pulldown of in vitro synthesized AR (left) and HA-AR affinity pulldown of in vitro synthesized TSPY or TSPX (right). (B) GST-TSPY pulldown of in vitro synthesized N-terminal domain (NTD), DNA-binding domain (DBD) and ligand-binding domain (LBD) of AR. NTD and DBD were retained by GST-TSPY fusion protein, albeit minimal background binding of NTD to the GST alone. (C) Co-immunoprecipitation of TSPY isoforms (as illustrated in E) with co-transfected FLAG-TSPY isoform and HA-AR expression vectors in HEK293 cells. (D) Effects of TSPY isoforms in AR transactivation of ARR2PB-luciferase reporter in HEK293 cells. All isoforms of TSPY are capable of co-activating the probasin promoter-directed luciferase reporter in the presence of the R1881 ligand. (E) Schematic illustrations of the structures of TSPY and TSPY isoforms encoded by alternatively spliced transcripts (22) and H25 is a new variant TSPY transcript, recently identified in our laboratory.
Figure 3.

TSPY stimulates and TSPX inhibits the transactivation of a probasin promoter-luciferase reporter by AR and AR variants. (A–B) AR up regulates the probasin promoter-luciferase in the presence of synthetic ligand, R1881, and is stimulated by TSPY but repressed by TSPX in dosage and ligand (R1881) dependent manners. TSPX competitively counters the TSPY stimulation of AR in a dosage dependent manner (B, right). (C) Identification of the carboxyl acidic domain as the inhibitory domain in TSPX, truncation of which (TSPXΔC) renders it be stimulatory as TSPY while its transposition to the carboxyl terminus of TSPY (TSPY-TSPX) results in an inhibitory protein as the full-length TSPX (illustrated in E). (D) TSPY exacerbation and TSPX repression of AR variant, AR-V7 (left) and AR-V4 (right), transactivation of probasin promoter-luciferase in ligand-independent manners. (E) Schematic representation of TSPY, TSPX and modular derivatives used in the probasin-reporter assays in C. (F) AR variants, AR-V7 and AR-V4, domain structures as compared to full length AR, AR-FL.
Previous studies showed that TSPY transcriptional units can produce alternatively spliced transcripts, coding for different isoforms with various in-frame deletions/truncations of the full-length protein (Fig. 2E) (22). To identify the likely domain(s) responsible for TSPY interaction with AR, co-immunoprecipitation experiments were performed with FLAG-TSPY isoforms and HA-AR. Our results showed that AR was co-immunoprecipitated with all TSPY isoforms (Fig. 2C). Comparison with the encoded sequences of the various TSPY isoforms showed that the N-terminal portion of the SET/NAP domain, encoded by exon 2 and 3, was responsible for interacting with AR (Fig. 2E). Alignment of the SET, TSPY and TSPX protein sequences showed that this region harbors the most conserved stretch of amino acids among members of this protein family (Supplementary Material, Fig. S1A) and encompasses the α4, α5, and β1-3 structures of the SET/NAP domain in the corresponding crystal structure of the SET protein (23) (Supplementary Material, Fig. S1B).
TSPY stimulates and TSPX represses AR transactivation of a target gene
To explore the effects of TSPY and TSPX interactions with AR on its transcriptional activities, a luciferase reporter directed by the androgen-responsive ARR2PB rat probasin promoter (24) was used in transfection assays with various combinations of AR, TSPY and TSPX expression vectors in the presence or absence of the synthetic R1881 ligand. Our results showed that AR up regulated the ARR2PB-luciferase reporter only in the presence of the R1881 ligand (Fig. 3A and B). TSPY exacerbated and TSPX repressed such ligand-dependent AR transactivation in dosage dependent manners (Fig. 3A). Significantly, the TSPY exacerbation of AR transactivation could be competitively inhibited by TSPX co-expression in a dosage dependent manner (Fig. 3B). Interestingly, as demonstrated in co-immunoprecipitation analysis (Fig. 2C), the TSPY isoforms could interact and co-activate with AR in its transactivation of the ARR2PB-luciferase reporter in similar ligand-dependent manner (Fig. 2D).
As discussed above, the major difference between TSPY and TSPX is the acidic domain at the carboxyl terminus of TSPX, absent in TSPY (Fig. 3E). To explore the potential function of this acidic domain, an abbreviated TSPX gene (TSPXΔC) coding for a TSPX protein with a truncated acidic domain was used in similar reporter assays. Our results showed that TSPXΔC stimulated the ligand-dependent AR transactivation. Conversely, transposition of this acidic domain to the C-terminus of TSPY (TSPY-TSPX) rendered this fusion protein to be inhibitory, similar to the full-length TSPX protein (Fig. 3C). These results mapped the inhibitory function of TSPX to the acidic domain, the absence of which converted it to be stimulatory as TSPY, in AR transactivation.
TSPY and TSPX interact with AR variants and modulate their transactivation activities in ligand-independent manners
The identification of the NTD and DBD as the interactive domains for AR with TSPY and TSPX suggest that such protein-protein interactions could occur with AR splice variants, lacking the ligand-binding domain (LBD) (Fig. 3F). These AR variants can bind to the androgen-responsive elements (ARE) of target genes and regulate their expression in ligand-independent manners (25,26). They have been postulated to play key roles in development of drug resistance to androgen deprivation therapy (ADT) and advances to metastatic and castration resistant prostate cancer (CRPC) (27). Using the AR-V7 (also known as AR3) and AR-V4 (aka AR5) variants (26) as examples (Fig. 3F), we had conducted similar studies in co-immunoprecipitation and reporter assays. Our results showed that both AR variants were co-immunoprecipitated with TSPY or TSPX (Fig. 1C and D). Significantly, such TSPY and TSPX interactions led to exacerbation and repression respectively on the AR variant-mediated and ligand-independent transactivation of the probasin-luciferase reporter (Fig. 3D). Our findings suggest that TSPY and TSPX are co-regulators for not only the full-length AR but also the constitutively active AR variants lacking the LBD in ligand dependent and independent manners, respectively.
TSPY and TSPX differentially co-regulate AR targets in the prostate cancer LNCaP cell line
To determine the TSPY and TSPX effects on androgen-responsive cells, a doxycycline inducible lentiviral expression system (28) was used to express the TSPY or TSPX gene and a control EGFP-containing vector in the androgen-responsive prostate cancer LNCaP cell line. Under this system, the transgene can be activated with doxycycline in the culture media. Cell proliferation analysis showed that TSPY stimulated and TSPX repressed the growth of LNCaP cells, as compared to those with EGFP vector under doxycycline induction (Fig. 4A). To determine the TSPY and TSPX effects on AR transcriptional functions in LNCaP cells, specific primers were synthesized for five androgen-responsive genes, i.e. KLK3 (PSA), TMPRSS2, FKBP5, NKX3.1, and KLK2, and a non-responsive gene, HPRT (Supplementary Material, Table S1) (29) and used in qRT-PCR analysis of RNAs isolated from LNCaP cells transduced with TSPY, TSPX or EGFP, in the presence of doxycycline (activation of transgene) and the synthetic ligand R1881. Our results showed that 3 of 5 AR target genes, i.e. KLK3 (PSA), TMPRSS2 and KLK2, were considerably and one (FKBF5) was slightly up regulated in TSPY-expressing LNCaP, as compared to those expressing EGFP only (Fig. 4B). The NKX3.1 gene is usually down regulated in prostate cancer (2), and its expression was lower in TSPY-expressing LNCaP cells than that for the control cells. Minimal effects on HPRT were observed in LNCaP cells expressing any of these transgenes. Our results suggest that TSPY could co-activate the AR transcriptional regulation of selected endogenous target genes in the androgen-responsive LNCaP cells. On the other hand, TSPX repressed the AR transcriptional regulation of all five targets (Fig. 4B). To confirm TSPY and TSPX interactions with AR in regulation of these target genes, chromatin immunoprecipitation analysis was performed with LNCaP cells expressing either TSPY or TSPX transgene using specific antibodies against AR and the epitopes for TSPY, TSPX (10,17,20,21,30). Our results showed that TSPY and TSPX co-localized with AR in the promoter sequences of the respective target genes (Fig. 4C and D), supporting the notion that TSPY and TSPX, and perhaps other co-factors, form complexes with AR, which bind to the promoters of the endogenous androgen-responsive genes and co-activate and co-repress respectively their expression in the presence of the R1881 ligand.
Figure 4.

Effects of TSPY and TSPX expression in prostate cancer LNCaP cells in the presence of the synthetic ligand R1881. (A) TSPY promotes and TSPX inhibits cell proliferation, as compared to EGFP alone, in LNCaP cells. (B) qRT-PCR analysis of five endogenous androgen-responsive genes (KLK3, TMPRSS2, FKBP5, NKX3.1, KLK2) and HPRT control in the above transduced LNCaP cells. (C–D) Chromatin immunoprecipitation assays confirm TSPY (C) and TSPX (D) are co-localized with AR in bindings to the promoters of the five androgen-responsive genes in LNCaP cells in the presence of R1881.
TSPY and TSPX differentially affect a variety of signaling pathways, upstream regulators and cellular functions in LNCaP cells
To explore the likely global effects of TSPY and TSPX in LNCaP cells under androgen stimulation, the LNCaP cells ectopically expressing TSPY, TSPX and EGFP vector alone were analyzed with RNA-Seq strategies (31–33). The gene expression patterns were compared between LNCaP cells expressing either TSPY or TSPX versus those expressing the EGFP vector alone, in the presence of the synthetic ligand R1881. The differential expression levels of the genes were calculated and tabulated as log2 fold changes (33). Preliminary analysis confirmed the up and down regulation of the five target genes examined previously (Fig. 4) in the respective transcriptomes of TSPY and TSPX expressing cells.
With a false discover rate (FDR) < 0.05 and differential gene expression level cutoff of 0.8 at log2 scale (i.e. with differential expression >1.71 or < 0.57 fold changes), there were 2682 and 1423 genes differentially expressed between TSPY- or TSPX-expressing and EGFP-expressing cells, respectively (Supplementary Material, Table S2). These gene sets were uploaded to the knowledge base Ingenuity Pathway Analysis (IPA) system (http://www.ingenuity.com), and analyzed with the Core Analysis suite, which provides statistical analyses on the canonical pathways, upstream regulators and diseases or functions likely to be affected by the differentially expressed genes. IPA identified 132 canonical pathways with P value < 0.05 affected by TSPY over-expression in the LNCaP cells (Supplementary Material, Table S3A). Forty-two common pathways were identified among the canonical pathways with z scores in TSPX expressing LNCaP cells (Fig. 5A). Most common pathways, except the PTEN signaling, were activated (z score > +2.0) among TSPY-expressing cells while most of the same pathways were either inhibited or changed minimally (z scores ± < 2) in TSPX-expressing cells (Supplementary Material, Table S3B). TSPY inhibition of the PTEN (phosphatase and tensin homologue deleted on chromosome 10) signaling pathway in the prostate cancer LNCaP cells is an interesting observation. PTEN is a phospholipase that opposes PI3K (phosphoinositol-3-kinase) activity, important for cell proliferation and oncogenesis (34,35). Inactivation/loss of PTEN is frequently observed in prostate and other cancers (2,35). Hence, TSPY inhibition of PTEN signaling pathway and activation of other oncogenic pathways in the prostate cancer LNCaP cells under androgen stimulation could be significant observations, supporting the postulation of TSPY involvement in the prostatic oncogenic process(es).
Figure 5.

TSPY and TSPX differentially affect numerous canonical pathways, upstream regulators and diseases or functions in prostate cancer LNCaP cells in the presence of R1881 ligand (Supplementary Material, Tables S3–S5, respectively). Among the common ones, TSPY differentially activates most canonical pathways (Y-zScore > +2.0) associated with cell growth and proliferation and oncogenesis (A), which are either inhibited or minimally affected by TSPX. Such differences are less obvious among common upstream regulators (B) and diseases or functions (C), postulated to contribute in TSPY- or TSPX-mediated differential gene expression patterns in LNCaP cells. Notably, invasion of cells, movement and migration of tumor cells are activated, and morbidity or mortality, organismal death and apoptosis are inhibited among the cellular functions/diseases in TSPY-expressing cells; while the same functions were minimally affected in TSPX-expressing cells. X-zScore = activation z scores for TSPX-expressing cells; Y-zScore = activation z scores for TSPY-expressing cells. Ingenuity Pathway Analysis (http://www.Ingenuity.com/) was performed on July 1, 2016.
Ingenuity Pathway Analysis identified 471 and 222 upstream regulators with P value <0.05 to be likely associated with the differential gene expression patterns in TSPY and TSPX expressing cells respectively (Supplementary Material, Table S4A and B). Among them, 40 common upstream regulators showed z scores of ± >2, likely to be activated or inhibited in respective cell populations. In addition to differentially activated or inhibited upstream regulators, e.g. UCP1, PGR, HSF1, PRL, TP73, TGM2, TCR and BNIP3L, some shared similar activation or inhibition states, e.g. IFNG, MAPK1, IFNL1, IL1RN, SREBF1/2, INSIG1, IL1B, ATP7B, EBI3 and GAPDH, between TSPY and TSPX expressing cells (Fig. 5B). Although the proportions of upstream regulators with similar z scores were relatively small, these observations suggest that TSPY and TSPX could exert similar effects on gene expression on selected common upstream regulators in LNCaP cells. Interestingly, only 16 diseases or functions were commonly affected by TSPY and TSPX expression in LNCaP cells, with P < 0.05 and z scores of ± >2 (Supplementary Material, Table S5). Among these common cellular functions, morbidity or mortality, organismal death and apoptosis were inhibited while invasion of cells, cell movement and migration of epithelial cells and tumor cell lines were activated in TSPY expressing cells (Fig. 5C). The same functions were minimally affected in TSPX expressing cells.
The canonical androgen signaling pathway was moderately activated (z score = 1.89) in TSPY expressing cells, but not in those expressing TSPX (Fig. 5A, last row). Similarly as an upstream regulator, AR was slightly activated (z score = 0.622) and inhibited (z score = −0.682) in TSPY and TSPX expressing cells, respectively (Fig. 5B, last row). Since AR plays critical roles in androgen signaling pathway, its expression was also slightly up regulated in TSPY expressing cells (log2 fold change, + 0.355), but was significantly repressed in TSPX expressing cells (log2 fold change, −1.093), respectively. The TSPX inhibition on AR expression in LNCaP cells could be confirmed by qRT-PCR analysis (Supplementary Material, Fig. S2 and Table S1). Hence, these observations suggest that TSPY and TSPX could directly or indirectly affect AR expression, thereby potentially exerting their stimulatory and inhibitory effects on AR functions not only through their protein–protein interactions with AR, but also likely through differential modulations of AR transcription in LNCaP cells.
Discussion
TSPY and TSPX act as a proto-oncogene and a tumor suppressor respectively in androgen-sensitive cancers
Prostate and prostate cancers are greatly dependent on the male sex hormone and its receptors in development, growth, oncogenic initiation and progression (1,2,36). Studies showed that many men could harbor indolent cancer in their prostates, but only some will develop symptoms of clinical prostate cancer (2,36). The mechanism(s) responsible for such selective transition is still uncertain, although androgen and AR are presumed to play key parts in the process(es). Once developed, prostate cancer can be effectively treated and controlled with androgen deprivation therapy (ADT), especially adjunct to radiotherapy or prostatectomy (37). ADT targets AR, via antagonist such as enzalutamide and androgen synthetic pathway inhibitors, such as abiraterone acetate (38). Most patients respond positively, but would eventually develop drug resistance and progress into the lethal and/or metastatic castration resistant prostate cancer (CRPC) (38). Currently, the molecular mechanisms contributing to drug resistance and advance to CRPC are uncertain. Various studies suggest that gene amplification and alternative splicing with deleted LBD of AR could restore AR signaling in a constitutive manner(s), thereby sustaining AR signaling without the need of the ligand in the oncogenic processes (39–41). Among the various AR variants, AR-V7 is closely linked to drug resistance in patients and prostate cancer cell lines (41–44). Over-expression of AR-V7 promotes epithelial-to-mesenchymal transition (EMT), autocrine/paracrine actions and tumor stem cell development, thereby advancing aggressive tumorigenesis (45). AR-V7 mRNA has been successfully detected in circulating tumor cells and/or whole blood of CRPC patients, and is an effective biomarker in prognosis for drug resistance and treatment assessment in prostate cancer (42,46,47). Despite these advances, AR-V7 is also detectable in non-tumor prostate, early prostate cancer and other human tissues (48,49), suggesting that additional oncogenic factors or events might be required to fully realize its oncogenic potential(s) (50). At present, the synergistic factors and associated molecular mechanisms for AR/AR-V7 oncogenic actions in prostate cancer development and aggressive progression are uncertain (51). As a male-specific proto-oncogene on the Y chromosome, TSPY stimulation of both full-length AR and constitutively active AR-V7 suggests that it could serve as a promoter and greatly amplify the androgen and AR/AR-V oncogenic actions. Indeed, we have demonstrated TSPY expression in indolent (latent) cancers of patients without any clinical symptoms (14), and in clinical prostate cancer at all Gleason grades (14,22). Hence, TSPY could be a key factor synergizing and exacerbating the oncogenic actions of AR and AR variants, important for initiation of prostatic oncogenesis/transition from indolent to clinical cancer and for aggressive advance to metastatic CRPC.
As a postulated tumor suppressor, TSPX suppresses lung cancer cell proliferation (19) and promotes proteosomal degradation of the HBx oncoprotein of the hepatitis B virus (HBV) (20), an etiological agent for liver cancer. TSPX is an essential component of the REST/NRSF transcriptional complex important for TGFβ signaling activation (52), which in turn up-regulates the cell cycle inhibitor, p21CDKN1A and p53 actions (53). The present study clearly demonstrated that TSPX is a co-repressor for AR and its constitutively active variants, thereby minimizing their corresponding oncogenic actions. Hence, TSPX is likely an X-linked tumor suppressor for prostate cancer; any mutational/epigenetic inactivation could predispose males to oncogenesis, not only in prostate but also other cancers, such as liver and bladder cancer (54,55). We postulate that TSPY and TSPX could be important biomarkers for prostate cancer; the presence or absence of each could signify a likely exacerbation or repression respectively of AR and/or AR-V-mediated oncogenic evolution of such AR-addictive cancer (2,26,27).
Mechanisms of TSPY and TSPX actions in oncogenesis
In addition to their opposing effects on AR and AR-V functions in transactivation of AR/AR-V target genes (Fig. 6), TSPY exacerbates and TSPX represses the cyclin B-CDK-1 activities (17), resulting in an expedited and retarded G2/M transition in the cell cycle, respectively (16). TSPY co-localizes with cyclin B at the mitotic spindles and its stimulation of cyclin B-CDK1 activities could compromise the spindle assembly checkpoint (SAC), thereby increasing the potential for chromosome nondisjunction and genomic instability (11). TSPX, on the other hand, could also co-localize with cyclin B at the mitotic spindle, but by modulating the cyclin B-CDK1 activities, it could insure an orderly G2/M transition of the cell cycle, and maintain the SAC integrity (Fig. 6). Significantly, its C-terminal acidic domain has been identified to be responsible for its inhibition on cyclin B-CDK1 activities (17), similar to its repression on AR/AR-V transactivation (Fig. 3C). Again, truncation of this domain renders TSPX to be stimulatory as TSPY, and its transposition to the C-terminus of TSPY results in an inhibitory hybrid protein on the cyclin B-CDK1 activities (17). Hence, the loss of the acidic domain in TSPY during the evolution of the sex chromosomes (6) could be key in its specialized functions in male germ cell biology and differentiation, distinct from the ancestral gene-like TSPX. Thus, TSPY and TSPX could contribute to oncogenesis as opposing cell cycle modulators and co-regulators of AR and AR-V oncogenic functions in human cancers.
Figure 6.
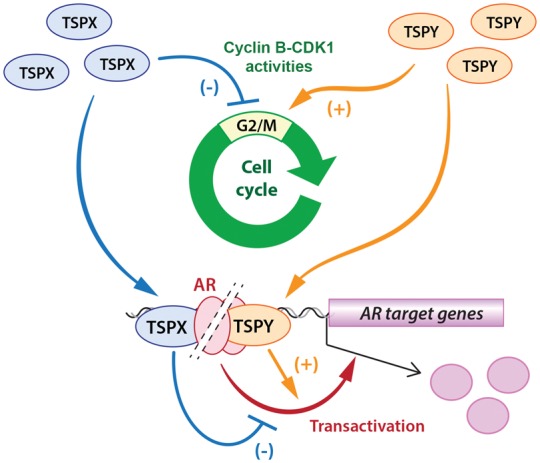
Diagrammatic illustration of TSPY and TSPX effects in cell cycle regulation via their actions on cyclinB-CDK1 activities at G2/M stage (upper); and differential actions in androgen receptor transactivation of responsive genes (lower). Both opposing functions of TSPY and TSPX could likely be attributed to their evolutionary divergence, which resulted in the carboxyl terminal acidic domain in the protein encoded by their ancestral gene being retained in TSPX but lost in TSPY.
Importantly, truncation of the carboxyl terminal domain of TSPX renders the abbreviated protein to possess similar oncogenic properties as those for TSPY in cyclin B-CDK1 stimulation/cell cycle regulation (17) and AR transactivation (present study). Therefore, any genetic mutations or alternative splicing events, deleting off the carboxyl domain from the TSPX protein, could potentially convert such a tumor suppressor to a TSPY-like oncogenic molecule theoretically capable of promoting cell proliferation, genomic instability and/or exacerbating male sex hormone actions in oncogenesis (11,16,17). Indeed, a prostate cancer susceptibility locus has been identified with GWAS at Xp11.22, in the proximity of TSPX on the X chromosome (56); and epigenetic abnormalities and mutations on TSPX gene have been reported in uterine leiomyomas (57) and serous endometrial tumors (58). Although the exact nature of the prostate cancer susceptibility locus at Xp11.22 and those for genetic/epigenetic changes in TSPX in uterine and endometrial tumors are uncertain, our results raise the possibility of TSPX involvement in the pathogenesis of these and other human cancers.
TSPY and TSPX in sexual dimorphisms in health and diseases of man
Sex differences are prevalent in normal development and physiology, such as brain structures, muscle development, cardiovascular physiology and nervous functions (3–5), as well as pathogeneses of sexual dimorphic diseases, such as heart diseases, hypertension, neurodegenerative diseases, schizophrenia, autism, depression, diabetes and metabolic syndrome (3–5,59,60). The sex hormones and their receptors could play crucial roles in these developmental, physiological and pathogenic processes. As a gene on the man-only Y chromosome, the TSPY stimulatory actions on AR transactivation will greatly amplify the male-specific effects, thereby contributing to the overall male biases in various diseases affected by the male sex hormone.
In addition to its cell cycle regulation and TGFβ activation (17,19,52), TSPX also serves a variety of functions associated with quantitative trait diseases, such as diabetes, atherosclerosis and cognitive disorders (52,61–66). It regulates the expression of the GluN2A and GluN2B subunits of the N-methyl-d-aspartate (NMDA) receptor in the hippocampus, and could play an important role(s) in synaptogenesis and synaptic functions (63). It interacts with the calcium/calmodulin-dependent serine protein kinase (CASK) and regulates the expression of various genes important for neurodevelopment (65). Loss-of-function mutation of Tspx (Tspyl2) results in neurodevelopmental and behavioral abnormalities in mice (61). Mutations and micro-deletions involving TSPX have been detected in patients with attention deficit hyperactivity disorder (ADHD), schizophrenia and mild intellectual disability (62,64). Since men have only one X chromosome, mutations/inactivation of TSPX could preferentially predispose males to such diseases. Significantly, we have previously demonstrated that an intact human TSPY transgene and a TSPY promoter-directed reporter transgene could be activated and express in various parts, including the hippocampus, of the brain in transgenic mice, in similar spatiotemporal manner as those for the endogenous Tspx (10,18). Hence, abnormal activation of TSPY could exert a male-specific effect(s) on the biological and disease process(es) and competitively interfere with the TSPX functions in the brain and other tissues, resulting in male-biased physiological phenotypes and/or diseases. Our studies and those of others on TSPY and TSPX have identified the opposing multi-functional nature of these sex chromosome homologues, which could play critical roles in various quantitative traits in the health and diseases of man.
Materials and Methods
Plasmids and antibodies
The rat probasin promoter ARR2PB (67) was inserted immediately upstream of the coding sequence for luciferase in pGL3-basic vector (Promega Corp., Madison, WI). Full length, truncated and retrofitted cDNAs for TSPX and TSPY isoforms were inserted into p3XFLAG-CMV-7 expression vector (Sigma, St. Louis) for the mammalian expression, as previously described (17,21). The coding sequences for TSPY and TSPX were inserted into either pGEX4T3 or pET41b vector (Promega Corp., Madison, WI) and used for GST fusion proteins synthesis. The pET28b vector was used for in vitro transcription and translation synthesis of various labeled proteins. The full-length human androgen receptor cDNA clone, pS6-HA-AR and pS6 vector was provided by Dr. Keith Yamamoto, UCSF. The cDNAs for AR variants, AR3 (AR-V7) and AR5 (AR-V4) were previously cloned in the laboratory of Dr Yun Qiu, University of Maryland (25). The doxycycline inducible lentiviral vectors, FUW-tetO and FUW-M2rTA (28), and the lentiviral packaging plasmids, pMD2G and pPAX2 were obtained from the Addgene Repository.
TSPY monoclonal antibodies were originally generated in our laboratory, as previously reported (10). All antibodies against respective epitopes were purchased from various vendors and used as previously described (17,20,21). The rabbit polyclonal anti-AR antibody (sc-816) was purchased from Santa Cruz Biotechnology.
Yeast two-hybrid screen and analysis
Initially, a bait harboring the SET/NAP domain (residues 151–308) of TSPY was used in a yeast two-hybrid screen of an expression cDNA library of E11.5 mouse embryonic gonads (21,68), resulting in identification of NONO, a component of an AR repressor complex (69), as one of several TSPY interacting proteins (21). Preliminary studies with NONO and AR showed that TSPY and TSPX could bind directly with AR and affect its transactivation functions, independent of their bindings to NONO. Accordingly, our subsequent studies were focused on characterization of the TSPY/TSPX interactions with AR and their effects on AR transcriptional functions.
GST pull down assay
GST-TSPY and GST-TSPX recombinant proteins were synthesized in E. coli strain BL21(DE3) bacteria and purified with affinity chromatograph and glutathione-Sepharose beads. The full-length AR and subdomains NTD, DBD and LBD were synthesized and labeled using the TnT T7 Quick Coupled Transcription/Translation System (Promega Corp., Madison) in the presence of 35S-methionine (Amersham Biosciences, Piscataway). GST pull down assays were performed with respective GST fusion proteins and labeled proteins, as previously described (17,21). The bound proteins were eluted and analyzed in 10% SDS-PAGE gels and detected by autoradiography.
Co-immunoprecipitation
Human HEK293 cells were transfected with various combinations of p3xFLAG-TSPY, p3xFLAG-TSPX, pS6-HA-AR, pS6-HA-AR3, pS6-HA-AR4 plasmids using the X-tremeGene 9 reagents (Roche, Indianapolis), as previously described (17,21). Transfected cells were harvested at 48 hours after transfection. Co-immunoprecipitation was performed with EZview Red ANTI-FLAG M2 Affinity Gel. The immunocomplexes were separated by SDS-PAGE electrophoresis and analyzed by Western blot and anti-FLAG, anti-HA or anti-AR polyclonal antibodies, as before (17,20,21).
Promoter assay and functional domain identification
The ARR2PB-luciferase was used as a reporter to analyze the effect of TSPY or TSPX on AR transactivation in HEK293 cells. Briefly, HEK293 cell were cultured in DMEM containing 10% charcoal treated medium overnight, and transfected with ARR2PB-luciferase reporter and TSPY or TSPX expression vectors, as above (17,20). The β-galactosidase reporter, pCMV-LacZ, was included in the transfection as an internal control. Forty-eight hours after transfection, the cells were harvested and analyzed with the luciferase assay system, and β-galactosidase enzyme assay system (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions. The relative luciferase activities were calculated with respect to the β-galactosidase activities in the same cell populations. To identify which domain of TSPX responsible for the inhibiting activity on AR transactivation, truncated TSPX and retrofitted TSPY-TSPX expression vectors were used in the promoter assays, as described before (17).
Lentiviral transduction, cell proliferation and expression analysis
Lentiviral particles were generated by transfection of HEK293T cells with combinations of FUW-tetO-TSPY, FUW-tetO-TSPX, FUW-tetO-EGFP or FUW-M2rTA and envelope plasmids pMD2G and packaging plasmid pPAX2, collected from the culture media at 48 and 72 h after transfection, filtered through a 0.45 mm filter, and centrifuged at 13,000 rpm for 10 min. The supernatants contained the respective lentiviral particles.
LNCaP cells were seeded at a density of 1x105/well on a 6-well plate and cultured overnight with RPMI-1640 containing 10% FBS (tetracycline-free). They were transduced with lentiviral particles containing FUW-tetO-TSPY, FUW-tetO-TSPX and FUW-tetO-EGFP transgenes and polybrene (final concentration 8 μg/ml). Doxycycline at final concentration of 1 μg/ml was added to the respective transduced cell populations (28). The synthetic ligand R1881 at final concentration of 10nM was also added to the culture media to activate the endogenous AR. The transduced cells were harvested at 48 h after ligand stimulation; total RNAs were extracted using TRIzol reagents (Life Technologies, Inc., San Carlos, CA). One μg of total RNAs were used to synthesize cDNAs by Transcriptor First Strand cDNA Synthesis Kit (Roche). Expression levels of selected AR target genes were analyzed by quantitative RT-PCR (15) with gene-specific PCR primer pairs (Supplementary Material, Table S1) (29).
Cell proliferation analysis was performed with the respectively transduced cells, as above, in the presence of 1 μg/ml of doxycycline and in biological triplicates using the WST-1 Cell Proliferation Assay Kit from the Clontech Laboratories according to the manufacturer’s instruction.
Detection of the bindings of TSPY or TSPX and AR on endogenous target genes
LNCaP cells transduced with TSPY, TSPX and EGFP transgenes were cultured in 10cm dish in the presence of 1 μg/ml doxycycline and 10 nM R1881 for 48 h. The cells were harvested and cross-linked with 1% formaldehyde for 10 min at room temperature. Chromatin immunoprecipitation was performed with normal mouse IgG, anti-FLAG and anti-AR antibodies using Magna ChIP™ A/G Chromatin Immunoprecipitation Kit (EMD Millipore) (30). ChIPed DNAs were detected by quantitative PCR with primer pairs of at the promoters of respective AR target genes (Supplementary Material, Table S1). Fold enrichments were calculated with reference to EGFP transduced cells.
Transcriptome analysis
Two micrograms of total RNA extracted from LNCaP cells expressing TSPY, TSPX and EGFP vector alone, as described above, was used in sequencing library preparations with the KAPA Stranded RNA-Seq Library Preparation kit for Illumina Platform (KR0934, KAPA Biosystems). The NEBNext Multiplex Oligos primer sets (New England Bio Laboratory) were used to bar code the respective libraries, and sequenced with the High-Output Kit (75 cycles, FC404-1005, Illumina) using a NextSeq 500 Sequencer (Illumina, Inc.). The sequencing was performed in technical duplicates, which generated ∼10 × 106 of 75-nucleotide raw reads per sample. Sequence analyses were performed with the BaseSpace software suite and the Galaxy Portal (70). The raw reads were first checked for quality by FastQC program. Adaptor contaminations and/or low quality reads were trimmed by FastQ tool kit. They were then aligned to the Human Reference Genome, GRCH37/hg19, and assigned to specific genes by FeatureCounts (31,32). Differential gene expression were identified by DESeq2 (33), which generated the relative fold changes between two RNA-Seq samples and calculated the P-values and adjusted P values (as false discovery rates, FDR).
The final differential gene expression patterns between TSPY and TSPX expressing cells versus EGFP expressing cells were analyzed with the Excel program. The lists of differentially expressed genes were selected with two criteria for Ingenuity Pathway Analysis. First, differentially expressed genes with adjusted P-values (FDR) <0.05 were selected as statistically significant in their fold change values. Second, a cutoff of their differential gene expression level was implemented to further refine the respective gene selections. Initial cutoff values at 0.5- and 1.0-fold changes at log2 were used to generate differentially expressed gene lists for the Ingenuity Pathway Analysis. Based on the preliminary results, a final optimal cutoff value of ±0.8 at log2 (i.e. with differential expression > 1.71- or < 0.57-fold changes) was adopted, which resulted in lists of 2682 and 1423 differentially expressed genes in TSPY and TSPX cells respectively. The Core analysis module was used for subsequent analyses, which identified the canonical pathways, likely upstream regulators, and disease or functions, associated with the differential gene expression patterns, with statistical significance (P values from Fisher’s Exact Test) and predicted activation (±z score corresponding to activation or inhibition) of specific pathways, upstream regulators and diseases or functions. An initial cutoff at P < 0.05 was applied to the IPA results, which were then sorted by the respective z scores. Those with z score of > +2 or < −2 were considered to be statistically significant in activation and inhibition respectively in their predicted states among the differentially expressed genes in the TSPY or TSPX versus EGFP expressing LNCaP cells. The common pathways, upstream regulators and diseases or functions, which have z scores of > +2 or < −2 on the activation prediction analysis of either TSPY or TSPX expressing cells, were compared with Excel. The results were sorted in ascending order based on the p values for TSPY expressing cells and presented in a 3-color graphic scheme with red, yellow and green as the highest, numerical 0 and lowest z scores, respectively.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank Drs Keith Yamamoto and Robert Matusik for providing the androgen receptor plasmids and ARR2PB promoter construct, respectively.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (grant 1R21 CA152589); Merit-reviewed grant 1I01BX000865 from the Department of Veterans Affairs to Y.-F.C.L. Y.-F.C.L. is a Research Career Scientist of the Department of Veterans Affairs.
References
- 1. Cunha G.R., Ricke W., Thomson A., Marker P.C., Risbridger G., Hayward S.W., Wang Y.Z., Donjacour A.A., Kurita T. (2004) Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J. Steroid Biochem. Mol. Biol., 92, 221–236. [DOI] [PubMed] [Google Scholar]
- 2. Shen M.M., Abate-Shen C. (2010) Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev., 24, 1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang C.K., Lee S.O., Chang E., Pang H., Chang C. (2016) Androgen receptor (AR) in cardiovascular diseases. J. Endocrinol., 229, R1–R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grgurevic N., Majdic G. (2016) Sex differences in the brain-an interplay of sex steroid hormones and sex chromosomes. Clin. Sci. (Lond.), 130, 1481–1497. [DOI] [PubMed] [Google Scholar]
- 5. Bayless D.W., Shah N.M. (2016) Genetic dissection of neural circuits underlying sexually dimorphic social behaviours. Phil. Trans. R. Soc. London B Biol. Sci., 371, 20150109.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lau Y.F.C., Kido T., Li Y. (2007) The TSPY Gene Family In The Y Chromosome and Male Germ Cell Biology in Health and Diseases. Lau Y.-F.C., Chan W.-Y. (eds.), World Scientific Publishers, pp. 73–90. [Google Scholar]
- 7. Skaletsky H., Kuroda-Kawaguchi T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., et al. (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature, 423, 825–837. [DOI] [PubMed] [Google Scholar]
- 8. Page D.C. (1987) Hypothesis: a Y-chromosomal gene causes gonadoblastoma in dysgenetic gonads. Development, 101 Suppl, 151–155. [DOI] [PubMed] [Google Scholar]
- 9. Honecker F., Stoop H., de Krijger R.R., Chris Lau Y.F., Bokemeyer C., Looijenga L.H. (2004) Pathobiological implications of the expression of markers of testicular carcinoma in situ by fetal germ cells. J. Pathol., 203, 849–857. [DOI] [PubMed] [Google Scholar]
- 10. Kido T., Lau Y.F. (2005) A Cre gene directed by a human TSPY promoter is specific for germ cells and neurons. Genesis, 42, 263–275. [DOI] [PubMed] [Google Scholar]
- 11. Lau Y.F., Li Y., Kido T. (2009) Gonadoblastoma locus and the TSPY gene on the human Y chromosome. Birth Defects Res. C. Embryo Today, 87, 114–122. [DOI] [PubMed] [Google Scholar]
- 12. Li Y., Tabatabai Z.L., Lee T.L., Hatakeyama S., Ohyama C., Chan W.Y., Looijenga L.H., Lau Y.F. (2007) The Y-encoded TSPY protein: a significant marker potentially plays a role in the pathogenesis of testicular germ cell tumors. Hum. Pathol., 38, 1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y., Vilain E., Conte F., Rajpert-De Meyts E., Lau Y.F. (2007) Testis-specific protein Y-encoded gene is expressed in early and late stages of gonadoblastoma and testicular carcinoma in situ. Urol. Oncol., 25, 141–146. [DOI] [PubMed] [Google Scholar]
- 14. Kido T., Hatakeyama S., Ohyama C., Lau Y.F. (2010) Expression of the Y-encoded TSPY is associated with progression of prostate cancer. Genes (Basel), 1, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kido T., Lo R.C., Li Y., Lee J., Tabatabai Z.L., Ng I.O., Lau Y.F. (2014) The potential contributions of a Y-located protooncogene and its X homologue in sexual dimorphisms in hepatocellular carcinoma. Hum. Pathol., 45, 1847–1858. [DOI] [PubMed] [Google Scholar]
- 16. Oram S.W., Liu X.X., Lee T.L., Chan W.Y., Lau Y.F. (2006) TSPY potentiates cell proliferation and tumorigenesis by promoting cell cycle progression in HeLa and NIH3T3 cells. BMC Cancer, 6, 154.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Lau Y.F. (2008) TSPY and its X-encoded homologue interact with cyclin B but exert contrasting functions on cyclin-dependent kinase 1 activities. Oncogene, 27, 6141–6150. [DOI] [PubMed] [Google Scholar]
- 18. Kido T., Schubert S., Schmidtke J., Lau Y.F. (2011) Expression of the human TSPY gene in the brains of transgenic mice suggests a potential role of this Y chromosome gene in neural functions. J. Genet. Genomics, 38, 181–192. [DOI] [PubMed] [Google Scholar]
- 19. Kandalaft L.E., Zudaire E., Portal-Nunez S., Cuttitta F., Jakowlew S.B. (2008) Differentially expressed nucleolar transforming growth factor-beta1 target (DENTT) exhibits an inhibitory role on tumorigenesis. Carcinogenesis, 29, 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kido T., Ou J.H., Lau Y.F. (2011) The X-linked tumor suppressor TSPX interacts and promotes degradation of the hepatitis B viral protein HBx via the proteasome pathway. PLoS One, 6, e22979.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kido T., Lau Y.F. (2008) The human Y-encoded testis-specific protein interacts functionally with eukaryotic translation elongation factor eEF1A, a putative oncoprotein. Int. J. Cancer, 123, 1573–1585. [DOI] [PubMed] [Google Scholar]
- 22. Lau Y.F., Lau H.W., Komuves L.G. (2003) Expression pattern of a gonadoblastoma candidate gene suggests a role of the Y chromosome in prostate cancer. Cytogenet. Genome Res., 101, 250–260. [DOI] [PubMed] [Google Scholar]
- 23. Muto S., Senda M., Akai Y., Sato L., Suzuki T., Nagai R., Senda T., Horikoshi M. (2007) Relationship between the structure of SET/TAF-Ibeta/INHAT and its histone chaperone activity. Proc. Natl. Acad. Sci. USA, 104, 4285–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu X., Wu J., Huang J., Powell W.C., Zhang J., Matusik R.J., Sangiorgi F.O., Maxson R.E., Sucov H.M., Roy-Burman P. (2001) Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech. Dev., 101, 61–69. [DOI] [PubMed] [Google Scholar]
- 25. Guo Z., Yang X., Sun F., Jiang R., Linn D.E., Chen H., Chen H., Kong X., Melamed J., Tepper C.G., et al. (2009) A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res., 69, 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu J., Van der Steen T., Tindall D.J. (2015) Are androgen receptor variants a substitute for the full-length receptor? Nat. Rev. Urol., 12, 137–144. [DOI] [PubMed] [Google Scholar]
- 27. Antonarakis E.S., Armstrong A.J., Dehm S.M., Luo J. (2016) Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis., 19, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hockemeyer D., Soldner F., Cook E.G., Gao Q., Mitalipova M., Jaenisch R. (2008) A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell, 3, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin C., Yang L., Xie M., Lin C., Merkurjev D., Yang J.C., Tanasa B., Oh S., Zhang J., Ohgi K.A., et al. (2014) Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc. Natl. Acad. Sci. USA, 111, 9235–9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y., Zheng M., Lau Y.F. (2014) The sex-determining factors SRY and SOX9 regulate similar target genes and promote testis cord formation during testicular differentiation. Cell Rep., 8, 723–733. [DOI] [PubMed] [Google Scholar]
- 31. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liao Y., Smyth G.K., Shi W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 33. Love M.I., Huber W., Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bertrand F.E., McCubrey J.A., Angus C.W., Nutter J.M., Sigounas G. (2014) NOTCH and PTEN in prostate cancer. Adv. Biol. Regul., 56, 51–65. [DOI] [PubMed] [Google Scholar]
- 35. Milella M., Falcone I., Conciatori F., Cesta Incani U., Del Curatolo A., Inzerilli N., Nuzzo C.M., Vaccaro V., Vari S., Cognetti F., et al. (2015) PTEN: multiple functions in human malignant tumors. Front. Oncol., 5, 24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Packer J.R., Maitland N.J. (2016) The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta, 1863, 1238–1260. [DOI] [PubMed] [Google Scholar]
- 37. Heidenreich A., Bastian P.J., Bellmunt J., Bolla M., Joniau S., van der Kwast T., Mason M., Matveev V., Wiegel T., Zattoni F., et al. (2014) EAU guidelines on prostate cancer. part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur. Urol., 65, 124–137. [DOI] [PubMed] [Google Scholar]
- 38. Watson P.A., Arora V.K., Sawyers C.L. (2015) Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer, 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dehm S.M., Tindall D.J. (2011) Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer, 18, R183–R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo Z., Qiu Y. (2011) A new trick of an old molecule: androgen receptor splice variants taking the stage?! Int. J. Biol. Sci., 7, 815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakazawa M., Antonarakis E.S., Luo J. (2014) Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm. Cancer, 5, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Antonarakis E.S., Lu C., Wang H., Luber B., Nakazawa M., Roeser J.C., Chen Y., Mohammad T.A., Chen Y., Fedor H.L., et al. (2014) AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med., 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu L.L., Xie N., Sun S., Plymate S., Mostaghel E., Dong X. (2014) Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene, 33, 3140–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu C., Luo J. (2013) Decoding the androgen receptor splice variants. Transl. Androl. Urol., 2, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun F., Chen H.G., Li W., Yang X., Wang X., Jiang R., Guo Z., Chen H., Huang J., Borowsky A.D., et al. (2014) Androgen receptor splice variant AR3 promotes prostate cancer via modulating expression of autocrine/paracrine factors. J. Biol. Chem., 289, 1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Antonarakis E.S. (2015) Predicting treatment response in castration-resistant prostate cancer: could androgen receptor variant-7 hold the key? Expert Rev. Anticancer Ther., 15, 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sprenger C., Uo T., Plymate S. (2015) Androgen receptor splice variant V7 (AR-V7) in circulating tumor cells: coming of age of AR splice variants? Ann. Oncol., 26, 1805–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takeuchi T., Okuno Y., Hattori-Kato M., Zaitsu M., Mikami K. (2016) Detection of AR-V7 mRNA in whole blood may not predict the effectiveness of novel endocrine drugs for castration-resistant prostate cancer. Res. Rep. Urol., 8, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu D.G., Hickey T.E., Irvine C., Wijayakumara D.D., Lu L., Tilley W.D., Selth L.A., Mackenzie P.I. (2014) Identification of androgen receptor splice variant transcripts in breast cancer cell lines and human tissues. Horm. Cancer, 5, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foley C., Mitsiades N. (2016) Moving beyond the androgen receptor (AR): targeting AR-interacting proteins to treat prostate cancer. Horm. Cancer, 7, 84–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Culig Z. (2016) Androgen receptor coactivators in regulation of growth and differentiation in prostate cancer. J. Cell. Physiol., 231, 270–274. [DOI] [PubMed] [Google Scholar]
- 52. Epping M.T., Lunardi A., Nachmani D., Castillo-Martin M., Thin T.H., Cordon-Cardo C., Pandolfi P.P. (2015) TSPYL2 is an essential component of the REST/NRSF transcriptional complex for TGFbeta signaling activation. Cell Death Differ., 22, 1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Toh B.H., Tu Y., Cao Z., Cooper M.E., Chai Z. (2010) Role of cell division autoantigen 1 (CDA1) in cell proliferation and fibrosis. Genes (Basel), 1, 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McBeth L., Grabnar M., Selman S., Hinds T.D. Jr. (2015) Involvement of the androgen and glucocorticoid receptors in bladder cancer. Int. J. Endorcinol., 2015, 384860.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ma W.L., Lai H.C., Yeh S., Cai X., Chang C. (2014) Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocr. Relat. Cancer, 21, R165–R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gudmundsson J., Sulem P., Rafnar T., Bergthorsson J.T., Manolescu A., Gudbjartsson D., Agnarsson B.A., Sigurdsson A., Benediktsdottir K.R., Blondal T., et al. (2008) Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat. Genet., 40, 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sato S., Maekawa R., Yamagata Y., Asada H., Tamura I., Lee L., Okada M., Tamura H., Sugino N. (2014) Potential mechanisms of aberrant DNA hypomethylation on the x chromosome in uterine leiomyomas. J. Reprod. Dev., 60, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Le Gallo M., O'Hara A.J., Rudd M.L., Urick M.E., Hansen N.F., O'Neil N.J., Price J.C., Zhang S., England B.M., Godwin A.K., et al. (2012) Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet., 44, 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lai M.C., Lombardo M.V., Baron-Cohen S. (2014) Autism. Lancet, 383, 896–910. [DOI] [PubMed] [Google Scholar]
- 60. Abel K.M., Drake R., Goldstein J.M. (2010) Sex differences in schizophrenia. Int. Rev. Psychiatry, 22, 417–428. [DOI] [PubMed] [Google Scholar]
- 61. Li Q., Chan S.Y., Wong K.K., Wei R., Leung Y.O., Ding A.Y., Hui T.C., Cheung C., Chua S.E., Sham P.C., et al. (2016) Tspyl2 loss-of-function causes neurodevelopmental brain and behavior abnormalities in mice. Behav. Genet., 46, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moey C., Hinze S.J., Brueton L., Morton J., McMullan D.J., Kamien B., Barnett C.P., Brunetti-Pierri N., Nicholl J., Gecz J., et al. (2016) Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. Eur. J. Hum. Genet., 24, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tsang K.H., Lai S.K., Li Q., Yung W.H., Liu H., Mak P.H., Ng C.C., McAlonan G., Chan Y.S., Chan S.Y. (2014) The nucleosome assembly protein TSPYL2 regulates the expression of NMDA receptor subunits GluN2A and GluN2B. Sci. Rep., 4, 3654.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vasli N., Ahmed I., Mittal K., Ohadi M., Mikhailov A., Rafiq M.A., Bhatti A., Carter M.T., Andrade D.M., Ayub M., et al. (2016) Identification of a homozygous missense mutation in LRP2 and a hemizygous missense mutation in TSPYL2 in a family with mild intellectual disability. Psychiatr. Genet., 26, 66–73. [DOI] [PubMed] [Google Scholar]
- 65. Wang G.S., Hong C.J., Yen T.Y., Huang H.Y., Ou Y., Huang T.N., Jung W.G., Kuo T.Y., Sheng M., Wang T.F., et al. (2004) Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron, 42, 113–128. [DOI] [PubMed] [Google Scholar]
- 66. Chai Z., Dai A., Tu Y., Li J., Wu T., Wang Y., Hale L.J., Koentgen F., Thomas M.C., Cooper M.E. (2013) Genetic deletion of cell division autoantigen 1 retards diabetes-associated renal injury. J. Am. Soc. Nephrol., 24, 1782–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Y., Kasper S., Yuan J., Jin R.J., Zhang J., Ishii K., Wills M.L., Hayward S.W., Matusik R.J. (2006) Androgen-dependent prostate epithelial cell selection by targeting ARR(2)PBneo to the LPB-Tag model of prostate cancer. Lab. Invest., 86, 1074–1088. [DOI] [PubMed] [Google Scholar]
- 68. Oh H.J., Li Y., Lau Y.F. (2005) Sry associates with the heterochromatin protein 1 complex by interacting with a KRAB domain protein. Biol. Reprod., 72, 407–415. [DOI] [PubMed] [Google Scholar]
- 69. Dong X., Sweet J., Challis J.R., Brown T., Lye S.J. (2007) Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Cell. Biol., 27, 4863–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Giardine B., Riemer C., Hardison R.C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J., et al. (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome Res., 15, 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.