

Abstract
Peptidyl-tRNA hydrolase 2 (PTRH2) regulates integrin-mediated pro-survival and apoptotic signaling. PTRH2 is critical in muscle development and regulates myogenic differentiation. In humans a biallelic mutation in the PTRH2 gene causes infantile-onset multisystem disease with progressive muscle weakness. We report here that the Ptrh2 knockout mouse model recapitulates the progressive congenital muscle pathology observed in patients. Ptrh2 null mice demonstrate multiple degenerating and regenerating muscle fibers, increased central nuclei, elevated creatine kinase activity and endomysial fibrosis. This progressive muscle pathology resembles the muscular dystrophy phenotype in humans and mice lacking the α7 integrin. We demonstrate that in normal muscle Ptrh2 associates in a complex with the α7β1 integrin at the sarcolemma and Ptrh2 expression is decreased in α7 integrin null muscle. Furthermore, Ptrh2 expression is altered in skeletal muscle of classical congenital muscular dystrophy mouse models. Ptrh2 levels were up-regulated in dystrophin deficient mdx muscle, which correlates with the elevated levels of the α7β1 integrin observed in mdx muscle and Duchenne muscular dystrophy patients. Similar to the α7 integrin, Ptrh2 expression was decreased in laminin-α2 dyW null gastrocnemius muscle. Our data establishes a PTRH2 mutation as a novel driver of congenital muscle degeneration and identifies a potential novel target to treat muscle myopathies.
Introduction
Integrins are αβ heterodimeric cell surface receptors that link the cytoskeleton to the underlying extracellular matrix (ECM) and activate downstream signaling pathways necessary for normal development and skeletal muscle function. In muscle, integrins play a critical role in the pathogenesis of muscular dystrophies (1). Muscular dystrophies are a group of genetic diseases that result in progressive muscle degeneration, functional disability and premature patient death (1). For example, loss of or mutations within the laminin binding α7 integrin gene cause a congenital muscular dystrophy with delayed motor milestones in humans and mice (2,3). Moreover, a skeletal muscle-specific knockout mouse model of the downstream α7 integrin effector serine/threonine integrin-linked kinase (ILK) results in a phenotype similar to the α7 integrin null mice (4) suggesting that integrin effector proteins play key roles in the pathogenesis of muscle myopathies. Not all genetic mutations that cause muscle myopathies have been identified.
PTRH2 (also called BIT1; Bit-1) functions on an integrin-mediated signal transduction pathway to regulate cell survival and apoptosis (5–7). In cells attached to the ECM, PTRH2 associates in a complex with focal adhesion kinase (FAK) and activates a pro-survival PI3K-AKT-NFκB-bcl-2 pathway (6). Loss of integrin-mediated attachment, on the other hand, promotes the association of PTRH2 with a pro-apoptotic Groucho-TLE complex and induces apoptosis (5).
Using whole exome sequencing, we recently identified a homozygous nonsense mutation in the PTRH2 gene in two siblings of a consanguineous Turkish family (8). Both patients developed a progressive congenital myopathy with delayed motor milestones and distal muscle weakness and wasting. Patients were unable to roll from prone to supine until 8 (male II.1) or 12 (female II.4) months of age and did not walk or sit without assistance until 18 (II.1) or 36 (II.4) months compared to healthy aged-matched controls at 5, 5 and 10 months of age respectively. Recently, at 15 years-of-age, the older and more severely affected female patient was no longer mobile and became wheelchair dependent due to muscle weakness and ataxia (8). These findings point to a PTRH2 mutation as a novel driver in progressive human congenital muscle degeneration with delayed motor milestones.
Similar to the patients, Ptrh2 null mice appear healthy at birth, however, they develop a runting syndrome and die within the first two weeks of life. At this early age, the primary phenotype is muscle weakness and epaxial muscle fiber diameters are smaller in the Ptrh2 null mice (9), suggesting impaired or delayed muscle development. Indeed, Ptrh2 null mice exhibit a myopathy with hypotrophic myofibers (10).
We report here that the Ptrh2 knockout mice recapitulate the muscle degeneration identified in patients with a PTRH2 gene mutation. The mice develop a postnatal progressive muscle pathology with multiple degenerating and regenerating muscle fibers, increased central nuclei, elevated creatine kinase activity and endomysial fibrosis. Moreover, PTRH2 expression is differentially regulated in skeletal muscle of mouse models for congenital muscular dystrophy.
Results
Ptrh2 null mice develop congenital progressive muscle pathology
Similar to the human phenotype (8), Ptrh2 null mice did not exhibit any muscle weakness at birth, but became progressively weaker within the first week. By postnatal day 7 (P7; Fig. 1A) these mice were unable to right themselves, displayed severe joint contractures, could no longer walk and all the mice died by P10 (Fig. 1B;Supplementary Video S1).
Figure 1.

Ptrh2 null mice demonstrate progressive skeletal muscle pathology at P7. (A) Ptrh2 KO mice exhibit severe joint contractures at P7 compared to wild-type (WT) littermates. (B) The viability of ptrh2 KO mice (n = 25) is severely reduced compared with wild-type (n = 25). All Ptrh2 KO died at 7-10 days. (C) Quantitation of centrally-located nuclei in triceps, tibialis anterior and gastrocnemius skeletal muscle at P7 (n = 3 per genotype and muscle type; 1,000 myofibers/animal; *P < 0.01; **P < 0.001). (D) Quantitation of creatine kinase levels in WT and Ptrh2 KO gastrocnemius muscle at P7 (n = 4 per genotype, **P < 0.001). Creatine kinase detects severe muscle damage. (E) Myofibers ≤ 10 μm diameter were increased in Ptrh2 KO gastrocnemius muscle compared to WT (n = 8 per genotype; 1000 myofibers/animal, ***P < 0.0001) indicating a significant increase in small regenerating fibers. (F) Increased endomysial fibrosis in gastrocnemius muscle (arrows) as determined by Trichrome staining in Ptrh2 KO mice compared to WT (longitudinal sections; bar 50 μm.) (G) Quantitation of Trichrome staining demonstrated a significant increase of fibrosis in the Ptrh2 KO mice compared to aged matched littermate controls at P7. (n = 4 per genotype, *P < 0.01). (H,I)Ptrh2 KO mice had reduced sarcolemmal integrity as determined by the uptake of Evans blue dye (red stain; arrows) in gastrocnemius muscle compared to WT (n = 4 per genotype; 1,500 myofibers/animal; *P < 0.01). Myofibers were counterstained with Oregon Green-488-conjugated WGA (green; bar 50 μm). (I) Increased eMyHC expression in Ptrh2 KO mouse gastrocnemius muscle compared to age-matched WT littermates at P7 (n = 4 per genotype; ***P < 0.0001).
Ptrh2 null mice present with muscle damage
To determine the pathology underlying Ptrh2 loss in skeletal muscle, we examined H&E-stained cryosections of gastrocnemius muscle from Ptrh2 null mice and age-matched littermate controls at P7. We detected severe myopathic changes typical of muscular dystrophies, including centrally nucleated fibers (Fig. 1C) in triceps, tibialis anterior and gastrocnemius skeletal muscle, elevated creatine kinase activity (Fig. 1D), increased fiber size variation (Fig. 1E) and endomysial fibrosis (Fig. 1F and G) in the null mice. Evans blue dye (EBD) uptake (11,12), a measure of sarcolemmal defects, revealed multiple damaged myofibers in the gastrocnemius muscle of Ptrh2 null mice, while no myofibers were EBD positive in control littermates (Fig. 1H and I). Active regeneration was observed in Ptrh2 null skeletal muscle by the presence of muscle fiber clusters expressing embryonic myosin heavy chain (eMyHC), which is a marker of regeneration (13). There was a significant increase in myofibers expressing eMyHC in Ptrh2 null muscle compared to age-matched littermate controls (Fig. 1J). Muscle regeneration in the null mice was further supported by a significant increase of centrally located nuclei (approximately 15%), an indication of newly formed muscle fibers (4) and by a 10-fold increase in myofibers with diameters below 10 μm (Fig. 1C and E). Taken together, these data demonstrate the presence of small regenerating fibers by P7 in the Ptrh2 nulls. Furthermore, creatine kinase activity, which is a marker of muscle damage, was significantly increased in the null mice at P7 (Fig. 1D) supporting the occurrence of muscle damage at this early age.
Ptrh2 associates with α7β1 integrin in skeletal muscle
Because Ptrh2 functions on an integrin regulated pathway and the Ptrh2 null mouse phenotype described here is similar to that of α7 integrin null mice that develop congenital muscular dystrophy with delayed motor milestones, (3,14,15) we next examined whether Ptrh2 was in a complex with the α7β1 integrin. Indeed, Ptrh2 localized to the sarcolemma of myofibrils and co-localized with the muscle-specific α7B integrin in WT gastrocnemius muscle (Fig. 2A). To further test that Ptrh2 and α7B integrin are in a complex, we immunoprecipitated Ptrh2 from the gastrocnemius muscle lysate and examined for co-precipitation of the α7B integrin by immunoblotting. The α7B integrin was detected in Ptrh2 immunoprecipitates, and in reciprocal co-immunoprecipitations Ptrh2 was detected in α7B integrin immunoprecipitates (Fig. 2B and C). Ptrh2 did not co-immunoprecipitate with either the alpha 5 or beta 1 integrin (Fig. 2D). Taken together, Ptrh2 and the muscle-specific α7B integrin associate in a complex in skeletal muscle.
Figure 2.
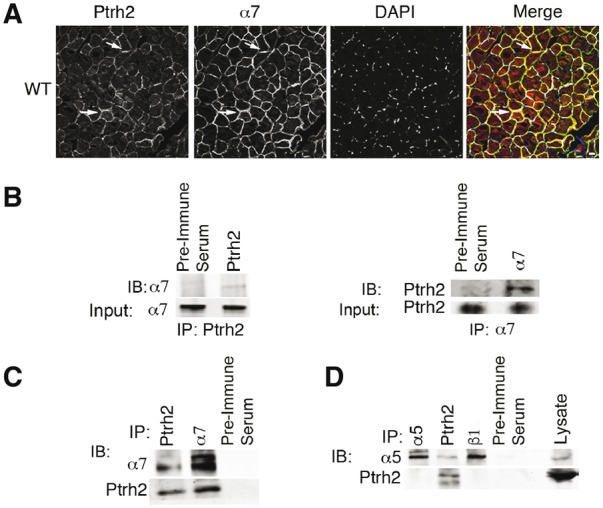
Ptrh2 associates in a complex with the α7B integrin in skeletal muscle. (A) Ptrh2 co-localizes with the muscle-specific α7B integrin at the sarcolemma of myofibrils in gastrocnemius muscle of WT mice as examined by immunofluorescence microscopy. Arrows indicate co-localization of Ptrh2 and α7 integrin that is displayed in yellow in the merged image. Nuclei were stained with DAPI (blue). Immunofluorescence magnification 63x. (B) Ptrh2 and α7B integrin co-immunoprecipitate in WT gastrocnemius muscle. Gastrocnemius muscle was collected, lysed and immunoprecipitated (IP) with anti-Ptrh2, anti-α7B integrin antibodies or pre-immune serum as a control. Control and immunoprecipitates were analyzed by immunoblotting (IB) with either anti-α7B integrin (left) or anti-Ptrh2 antibody (right). Lower images are of 2% input lysate based on overall protein content immunoblotted with anti-α7B (left) or anti-Ptrh2 (right) antibodies. (C) Efficiency of antibodies used for IPs in gastrocnemius muscle that was collected, lysed and immunoprecipitated (IP) with anti-Ptrh2, anti-α7B integrin antibodies or pre-immune serum as a control. Control and immunoprecipitates were analyzed by immunoblotting (IB) with either anti-α7B integrin or anti-Ptrh2 antibody. Lysate of 2% input based on overall protein content was immunoblotted with anti-α7B or anti-Ptrh2 antibodies. (D) Ptrh2 does not co-immunoprecipitate with α5 or β1 integrin. Gastrocnemius muscle was collected, lysed and immunoprecipitated (IP) with anti-Ptrh2, anti-α5, anti-β1 integrin antibodies or pre-immune serum as a control. Control and immunoprecipitates were analyzed by immunoblotting (IB) with either anti-α5 or anti-Ptrh2 antibodies. Lysate of 2% input based on overall protein content was immunoblotted with anti-α5 integrin or anti-Ptrh2 antibodies. Data shown in A–D are representative of three independent experiments.
We next examined whether loss of Ptrh2 altered α7 integrin expression and localization in skeletal muscle. Immunofluorescence and Western blot analysis demonstrated a significant decrease in α7 integrin protein expression in Ptrh2 null gastrocnemius muscle (Fig. 3A and B), suggesting that Ptrh2 positively regulates α7 integrin expression. In contrast, laminin α2 (a ligand for the α7 integrin) expression was similar in Ptrh2 null and WT skeletal muscle (Supplementary Material, Fig. S1), indicating Ptrh2 is not involved in laminin matrix deposition in the basal lamina of muscle at P7. Moreover, at P7 Ptrh2 null muscle showed no increase in inflammatory cell infiltrate as measured by CD4 and CD11 immunofluorescence (Supplementary Material, Fig. S2).
Figure 3.

α7 integrin expression is decreased in Ptrh2 null skeletal muscle. (A) Immunofluorescence and Western blot (B) of the α7 integrin showed reduced protein levels in gastrocnemius muscle in Pthr2 null (Ptrh2-/-) mice at P7 compared to WT littermate controls (n = 3 per genotype for immunoblots and immunofluorescence; bar 10 μm). (C) Western blot analysis of the gastrocnemius muscle lysate indicated that Ptrh2 and pAKT/tAKT are decreased in Ptrh2-/- and α7-/-mice compared to WT (n = 3 per genotype for immunoblots and immunofluorescence; **P<0.001). Data shown in A–C are representative of three independent experiments.
Integrin-mediated signaling is decreased in Ptrh2 null mice
We previously reported that Ptrh2 provides a protective function by activating a FAK-PI3K-AKT pro-survival pathway (6). Indeed, the α7β1 integrin relays a similar pro-survival effect by activating AKT (16). We analyzed gastrocnemius muscle from Ptrh2 and integrin α7 null mice and found that pAKT/AKT levels were decreased in both null mouse models (Fig. 3C). Taken together, these findings suggest that Ptrh2 may function, at least in part, as a cytoplasmic effector of α7β1 integrin pro-survival signaling in muscle.
Ptrh2 expression is altered in mouse models of muscular dystrophy
Because we observed that mutant PTRH2 causes distal muscle pathology in patients and mice, Ptrh2 and the α7 integrin associate in a complex and loss of the α7 integrin promotes muscular dystrophy in humans and mice (3,4), we next examined the Ptrh2 expression in classical mouse models of muscular dystrophy including α7 integrin deficient, laminin-α2 dyW null and dystrophin deficient mdx mice. Ptrh2 and pAKT/AKT expression was decreased in both α7 deficient and laminin-α2 dyW nulls (Fig. 4A and B) but increased in mdx mice (Fig. 4B), suggesting that Ptrh2 may play an important role in these muscle pathologies.
Figure 4.

Ptrh2 is differentially expressed in mouse models of congenital muscular dystrophy. (A) Ptrh2 immunofluorescence was lower in α7 integrin and laminin-α2 dyW null gastrocnemius muscle compared to WT. The absence of Ptrh2 expression was confirmed in Ptrh2-/- mice (n = 3 per genotype; bar 10 μm). (B) Western blot analysis demonstrated that Ptrh2 and pAKT/tAKT levels were increased in mdx (right panel) and decreased in laminin-α2 dyW null (left panel) gastrocnemius muscle compared to WT (n = 3 per genotype; **P<0.001, ***P<0.0001). Tubulin was used as a loading control. Data shown in A and B are representative of three independent experiments.
Discussion
Muscle disease occurs when the ECM-integrin-cytoskeleton complex is disrupted, thereby altering downstream pro-survival signals. Our data demonstrate that Ptrh2 associates in a complex with the α7β1 integrin at the sarcolemma, as determined by co-localization studies and co-immunoprecipitation experiments. Thus, in skeletal muscle, Ptrh2 is localized at sites of integrin signaling. PTRH2 is a regulator of myogenic differentiation and loss of PTRH2 expression promotes early muscle differentiation (10). In humans, mutations in the PTRH2 gene cause progressive congenital muscle weakness resulting in the most affected patients being wheelchair bound by age 15 (8). Ptrh2 is localized, in part, at the plasma membrane and associates in a complex with FAK to activate a PI3K-AKT-pro-survival signaling pathway in cancer and endothelial cells (6). Indeed, patient fibroblasts with a homozygous deletion in the PTRH2 gene are sensitive to stress-induced apoptosis suggesting that PTRH2 expression is important in promoting cell survival during tissue development (8).
The α7β1 integrin is involved in muscle cell survival and activates a pro-survival AKT pathway when re-expressed in dystrophic muscle (16). Similar to earlier in vitro findings that PTRH2 blocks staurosporine-induced apoptosis (6), α7 integrin expression prevents staurosporine-induced apoptosis in myoblasts in vitro (17). We demonstrate here that pAKT signaling is reduced in skeletal muscle of Ptrh2 and α7 integrin null mice. We further show that loss of Ptrh2 expression decreases α7 integrin expression but not laminin. Taken together, these findings suggest Ptrh2 may function as an α7 cytoplasmic effector in skeletal muscle.
Deletion of ILK, a known cytoplasmic effector of the α7 integrin, induces a mild muscular dystrophy similar to the α7 integrin null phenotype (4), establishing α7 integrin-mediated signals as essential for normal muscle development and function. Our data points to a critical role for Ptrh2-mediated signaling in muscle cell development and function. The skeletal muscle phenotype of the Ptrh2 null mouse is more severe than that of the α7 integrin null mice and resembles, in severity, the dystrophin and α7 integrin double knockout mice (mdx/α7-/-;) (18). These mice develop early-onset muscular dystrophy with fibrosis and die at 2–4 weeks of age (18) whereas the Ptrh2 null mice develop severe muscle pathology and die by day 10. The lack of Ptrh2-mediated signaling induces a more severe phenotype compared to α7 integrin nulls as detected by the presence of muscle regeneration, central nuclei, elevated creatine kinase activity and endomysial fibrosis at P7. Skeletal muscle from the α7 nulls present with approximately 2% central nuclei. Ptrh2 nulls present with approximately 15% central nuclei as examined in triceps, tibialis anterior and gastrocnemius skeletal muscle. This observed increase in central nuclei is similar to the mdx/α7 nulls, which demonstrate approximately 15% central nuclei in the tibialis anterior muscle (18). Moreover, we observe a higher number of eMyHC positive fibers from day 7 Ptrh2 null skeletal muscle compared with eMyHC positive fibers from 3 week old alpha 7 null skeletal muscle. eMyHC is expressed during normal muscle development in the embryo and transiently during muscle repair after birth. The observed increase in eMyHC may be due, in part, to the finding that loss of PTRH2 expression promotes early skeletal muscle differentiation and that Ptrh2 null myoblasts prematurely express muscle-specific proteins (10). Nevertheless, the eMyHC expression in postnatal skeletal muscle is a gauge for active muscle regeneration and we observe increased eMyHC positive fibers in the Ptrh2 null skeletal muscle. To address this further, we also evaluated creatine kinase activity, which is a marker of muscle damage. Creatine kinase activity was significantly increased in the Ptrh2 null mice at P7 further supporting the occurrence of muscle damage at this early age.
This data indicates loss of Ptrh2 expression in muscle alters the normal myofiber regenerative process. Interestingly, we found skeletal muscle from dystrophin deficient mdx mice up-regulate Ptrh2 levels, which correlate with elevated levels of the α7β1 integrin observed in mdx muscle and Duchenne muscular dystrophy (DMD) patients (19). Similar to the α7 integrin, Ptrh2 expression was decreased in laminin-α2 dyW null gastrocnemius muscle. These findings point to a key role for PTRH2 in DMD and merosin deficient congenital muscular dystrophy (MDC1A) disease progression.
In humans, a mutation in the PTRH2 gene promotes a similar phenotype as mutations in the α7 integrin gene both of which result in progressive congenital muscle degeneration with delayed motor milestones. Our data points to a new complex whereby Ptrh2 associates with the α7 integrin and functions to regulate pro-survival signaling pathways in skeletal muscle. Further studies are required to delineate the precise role PTRH2 plays in muscle disease. Identification of PTRH2 as a novel gene in human muscle pathologies may provide a potential new therapeutic target for these fatal muscle diseases.
Materials and Methods
PTRH2 null mice
All animals received care in compliance with the principles of laboratory animal care and use formulated by the Institutional Animal Care & Use Committee (USA). The generation of PTRH2 null mice inbred in a black 6 pure strain genetic background that die by P10 have been described previously (9).
Hematoxylin and eosin staining
For histological studies paraformaldehyde fixed, paraffin embedded sections were stained with Hematoxylin and Eosin (H&E) as described (18). Briefly, gastrocnemius muscles were cyrosectioned and 10-μm sections were placed on Surgipath microscope slides. Tissue sections were fixed in ice-cold 95% ethanol followed by 70% ethanol and re-hydrated in water. Tissue sections were then stained with Gill’s hematoxylin (Fisher Scientific, Fair Lawn, NJ), rinsed in water and stained with eosin (Sigma-Aldrich, St Louis, MO), rinsed and mounted with DePeX medium (electron Microscopy Sciences, Washington, PA).
Centrally located nuclei from gastrocnemius skeletal muscle were counted at 63X magnification by bright-field microscopy. The number of central nuclei per muscle fiber was determined by counting 1000 muscle fibers each for triceps, tibialis anterior and gastrocnemius muscle per animal. The percentage of central nuclei was expressed as the number of central nuclei/total number of fibers counted. At least three animals from each genotype were analyzed (wild type, Ptrh2-/-).
Immunofluorescence
Gastrocnemius muscles were embedded in Tissue-TEK Optimal Cutting temperature compound (Sakura Finetek USA Inc., Torrance, CA) as described (18). Ten-μm sections were cut using a Leica MC1850 cryostat and placed onto Surgipath microscope slides (Surgipath Medical Industries, Richmond, IL). Integrin α7 was detected with anti-CA5.5 rat monoclonal antibody (Sierra Biosource, Morgan Hill, CA) followed by FITC-anti-rat secondary (see the Supplementary Appendix). PTRH2 was detected by rabbit polyclonal anti-Ptrh2 antibody (developed at Washington Biotechnology Columbia, MD using the PTRH2 amino acid sequence GPADLIDKVTAGHLKL;(6). Laminin-α2 chain was detected with a rabbit anti-α2G polyclonal antibody. Slides were mounted using Vectashield with DAPI (Vector Laboratories) and visualized using a Zeiss Axioskop 2 Plus fluorescent microscope and images were captured using a Zeiss AxioCam HRc digital camera with Axiovision 4.1 software.
Central nuclei from tibialis anterior and triceps muscle were determined from embedded tissue that was sectioned into 10 micron thick sections using a cryosectioner. Sections were incubated in 1X PBS for 5 mins to reconstitute tissue and then for 10 mins in 1:100 wheat germ agglutinin – alexa 647 conjugated (to delineate muscle fibers), washed 2 min three times with 1X PBS, mounted in Vectashield containing DAPI stain (stains nuclei) and imaged using confocal microscope. Central nuclei were counted as nuclei located in the center of muscle fibers (more than one nuclei per fiber is only counted as one CLN fiber) and averaged to the total fiber number. The number of central nuclei per muscle fiber was determined by counting 1000 muscle fibers each muscle type per animal. The percentage of central nuclei was expressed as the number of central nuclei/total number of fibers counted. At least three animals from each genotype were analyzed (wild type, Ptrh2-/-).
Embryonic myosin heavy chain
Embryonic myosin heavy chain (eMyHC) was used in immunofluorescence experiments to measure muscle regeneration. Immunofluorescence was performed on 10-μm sections from the gastrocnemius muscle from day 7 PTRH2 null mice and wild type littermate controls with an anti-eMyHC antibody (FL.652, Developmental Studies Hybridoma Bank, University of Iowa IA). The primary antibody was detected with a FITC-conjugated mouse secondary antibody. Myofibers were incubated with rhodamine labeled WGA to outline the myofibers. Multiple adjacent sections were analyzed with twenty random, non-overlapping microscopic fields that were counted per animal at 630X magnification using a Zeiss Axioskop 2 Plus fluorescent microscope and images were captured using a Zeiss AxioCam HRc digital camera with Axiovision 4.1 software. Data were reported as percent of eMYHC-positive fibers.
Creatine kinase activity assay
Gastrocnemius muscle from WT and Ptrh2-/- null mice was isolated, washed with PBS and weighed (1 mg of muscle was lysed in 5ul 50mM potassium phosphate lysis buffer and sonicated). Creatine kinase activity was quantitated as per the kit protocol (MAK 116; Sigma Aldrich, St Louis, MO).
Western blotting
The gastrocnemius muscle from day 7 after birth (P7) was ground in liquid nitrogen. Protein was extracted in 200 mM octyl-β-D-glucopyranoside (Sigma Aldrich, St Louis, MO), 50 mM Tris-HCL pH 7.4, 150 mM NaCl, 1mM CaCl2, ImM MgCl2, 2mM PMSF and a 1:200 dilution of Protease Inhibitor Cocktail Set III (Calbiochem, EMD Biosciences, San Diego, CA). For the detection the α7 integrin: protein was quantified by Bradford assay and 80 μg of total protein were separated on 8%% SDS-PAGE gels under non-reduced conditions, and transferred to nitrocellulose membranes. Membranes were blocked in Odyssey Blocking buffer (LiCor Biosciences, Lincoln, NE) that was diluted 1:1 in phosphate-buffered saline (PBS). The α7 integrin was detected with a rabbit anti-α7A (a2 345) polyclonal antibody and an Alexa Fluor 680-coupled goat anti-rabbit IgG (Molecular Probes, Eugene OR) to detect primary antibodies.
For the detection of PTRH2, pAKT, tAKT, tubulin: 30 μg total protein was separated on a 4–12% SDS-PAGE gel under reducing conditions and transferred to nitrocellulose membranes. Membranes were blocked in the Odyssey Blocking buffer and incubated with either anti-PTRH2 (developed at Washington Biotech), anti-AKT or anti-phospho-AKT (Cell Signaling) or anti-microtubulin (BioLegend) for 2 h. Membranes were washed and incubated with Li-Cor secondary antibodies (ant-rabbit IR 800 CW and anti-mouse IR 680 CW; Li-Cor) for 1 h. Immunoblots were analyzed via Odyssey software, provided with the Li-Cor system.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 6 software. Averaged data are reported as the mean ± the standard error of the mean. Individually reported data points were reported as mean ± standard deviation. Comparison of two groups was performed using a Student's t-test and between multiple groups using one-way analysis of variance with a Bonferroni post-test with P < 0.05 considered statistically significant.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank the family members who participated in this study and Anna Leychenko for immunoblotting and mouse husbandry expertise.
Conflict of Interest statement. None declared.
Funding
This work was supported by: National Institutes of Health (NCRR P20-RR016453 to M.L.M; RO1-AR064338 to D.J.B.), the Ingeborg Foundation (M.L.M), the German Research Foundation (SFB665, DFG, A.M.K.), the Berlin Institute of Health (BIH, A.M.K.), the Sonnenfeld Stiftung (A.M.K), the Deutsche Gesellschaft für Muskelkranke DGM (A.M.K.), the Max-Planck Society (H.H.), and the EU FP 7 project GENCODYS, #241995 (H.H.).
References
- 1. Carmignac V., Durbeej M. (2012) Cell-matrix interactions in muscle disease. J. Pathol., 226, 200–218. [DOI] [PubMed] [Google Scholar]
- 2. Hayashi Y.K., Chou F.L., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B.L., Kramer R.H., Kaufman S.J., et al. (1998) Mutations in the integrin alpha7 gene cause congenital myopathy. Nat. Genet., 19, 94–97. [DOI] [PubMed] [Google Scholar]
- 3. Mayer U., Saher G., Fässler R., Bornemann A., Echtermeyer F., von der Mark H., Miosge N., Pöschl E., von der Mark K. (1997) Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat. Genet., 17, 318–323. [DOI] [PubMed] [Google Scholar]
- 4. Gheyara A.L., Vallejo-Illarramendi A., Zang K., Mei L., Rene St.-Arnaud, Dedhar S., Reichardt L.F. (2007) Deletion of integrin-linked kinase from skeletal muscles of mice resembles muscular dystrophy due to alpha 7 beta 1-integrin deficiency. Am. J. Pathol., 171, 1966–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jan Y., Matter M., Pai J.T., Chen Y.L., Pilch J., Komatsu M., Ong E., Fukuda M., Ruoslahti E. (2004) A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors 1. Cell, 116, 751–762. [DOI] [PubMed] [Google Scholar]
- 6. Griffiths G.S., Grundl M., Leychenko A., Reiter S., Young-Robbins S.S., Sulzmaier F.J., Caliva M.J., Ramos J.W., Matter M.L. (2011) Bit-1 Mediates Integrin-dependent Cell Survival through Activation of the NF{kappa}B Pathway. J. Biol. Chem., 286, 14713–14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matter M.L., Ginsberg M.H., Ramos J.W. (2001) Identification of cell signaling molecules by expression cloning. Sci.STKE, 2001, L9.. [DOI] [PubMed] [Google Scholar]
- 8. Hu H., Matter M.L., Issa-Jahns L., Jijiwa M., Kraemer N., Musante L., de la Vega M., Ninnemann O., Schindler D., Damatova N., et al. (2014) Mutations in PTRH2 cause novel infantile-onset multisystem disease with intellectual disability, microcephaly, progressive ataxia, and muscle weakness. Ann. Clin. Transl.Neurol., 1, 1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kairouz-Wahbe R., Biliran H., Luo X., Khor I., Wankell M., Besch-Williford C., Oshima J., Pascual R., Ruoslahti E. (2008) Anoikis effector Bit1 negatively regulates Erk activity 8. Proc. Natl. Acad. Sci. U.S.A, 105, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Griffiths G.S., Doe J., Jijiwa M., Van Ry, Cruz V., de la Vega M., Ramos J.W., Burkin D.J., Matter M.L. (2015) Bit-1 is an essential regulator of myogenic differentiation. J. Cell Sci., 128, 1707–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grady R.M., Teng H., Nichol M.C., Cunningham J.C., Wilkinson R.S., Sanes J.R. (1997) Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell, 90, 729–738. [DOI] [PubMed] [Google Scholar]
- 12. McGeachie J.K., Grounds M.D., Partridge T.A., Morgan J.E. (1993) Age-related changes in replication of myogenic cells in mdx mice: quantitative autoradiographic studies. J. Neurol. Sci., 119, 169–179. [DOI] [PubMed] [Google Scholar]
- 13. Webster C., Silberstein L., Hays A.P., Blau H.M. (1988) Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell, 52, 503–513. [DOI] [PubMed] [Google Scholar]
- 14. Burkin D.J., Kaufman S.J. (1999) The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res., 296, 183–190. [DOI] [PubMed] [Google Scholar]
- 15. Lopez M.A., Mayer U., Hwang W., Taylor T., Hashmi M.A., Jannapureddy S.R., Boriek A.M. (2005) Force transmission, compliance, and viscoelasticity are altered in the alpha7-integrin-null mouse diaphragm. Am. J. Physiol. Cell Physiol., 288, C282–C289. [DOI] [PubMed] [Google Scholar]
- 16. Boppart M.D., Burkin D.J., Kaufman S.J. (2011) Activation of AKT signaling promotes cell growth and survival in alpha7beta1 integrin-mediated alleviation of muscular dystrophy. Biochim. Biophys. Acta, 1812, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J., Burkin D.J., Kaufman S.J. (2008) Increasing alpha 7 beta 1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am. J. Physiol. Cell Physiol., 294, C627–C640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rooney J.E., Welser J.V., Dechert M.A., Flintoff-Dye N.L., Kaufman S.J., Burkin D.J. (2006) Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J. Cell Sci., 119, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 19. Hodges B.L., Hayashi Y.K., Nonaka I., Wang W., Arahata K., Kaufman S.J. (1997) Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J. Cell Sci., 110 (Pt 22), 2873–2881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.