

Abstract
Niemann-Pick disease, type C1 (NPC1) is a heritable lysosomal storage disease characterized by a progressive neurological degeneration that causes disability and premature death. A murine model of NPC1 disease (Npc1-/-) displays a rapidly progressing form of NPC1 disease which is characterized by weight loss, ataxia, increased cholesterol storage, loss of cerebellar Purkinje neurons and early lethality. To test the potential efficacy of gene therapy for NPC1, we constructed adeno-associated virus serotype 9 (AAV9) vectors to deliver the NPC1 gene under the transcriptional control of the neuronal-specific (CamKII) or a ubiquitous (EF1a) promoter. The Npc1-/- mice that received a single dose of AAV9-CamKII-NPC1 as neonates (2.6 × 1011GC) or at weaning (1.3 × 1012GC), and the mice that received a single dose of AAV9-EF1a-NPC1 at weaning (1.2 × 1012GC), exhibited an increased life span, characterized by delayed weight loss and diminished motor decline. Cholesterol storage and Purkinje neuron loss were also reduced in the central nervous system of AAV9 treated Npc1-/- mice. Treatment with AAV9-EF1a-NPC1, as compared to AAV9-CamKII-NPC1, resulted in significantly increased survival (mean survival increased from 69 days to 166 and 97 days, respectively) and growth, and reduced hepatic-cholesterol accumulation. Our results provide the first demonstration that gene therapy may represent a therapeutic option for NPC1 patients and suggest that extraneuronal NPC1 expression can further augment the lifespan of the Npc1-/- mice after systemic AAV gene delivery.
Introduction
Niemann-Pick disease, type C (NPC) is a rare and fatal, autosomal recessive, neurodegenerative disease that can present in infants, children, or adults. As predicted by analysis of massively parallel sequencing data sets, the incidence in persons of Western European descent has been estimated at 1/90,000 (1). Approximately 95% of NPC patients harbour mutations in NPC1, a gene encoding a 1278 amino acid transmembrane protein important for intracellular cholesterol trafficking (2). NPC is genetically heterogeneous and rare forms of the disease can also be caused by mutations in NPC2, a gene encoding a small lysosomal cholesterol binding protein that transfers cholesterol to NPC1 (3,4). In cells harbouring mutations in either NPC1 or NPC2, there is impairment of low-density lipoprotein (LDL) cholesterol esterification, as well as reduced trafficking of unesterified cholesterol to the plasma membrane (5–8). Furthermore, NPC1 dysfunction causes marked late endosomal/lysosomal accumulation of unesterified cholesterol and glycosphingolipids, the latter especially prominent in neuronal tissue (2). Intracellular cholesterol storage can be detected by filipin staining (9).
NPC1 patients generally present with neurological degeneration and hepatosplenomegaly (enlargement of liver and spleen) in early childhood, although other clinical phenotypes are well-recognized (10). The classical presentation of NPC1, which is normally diagnosed in school-age children, consists of ataxia, vertical supranuclear gaze palsy (VSGP), gelastic cataplexy and intellectual regression. Seizures are common and neurological symptoms become disabling. Although there is one approved treatment for NPC1 (11), approved only outside the United States, fatal neurodegeneration is inevitable. The fact that many patients have disease onset in childhood makes the search for effective therapies urgent.
There are a number of animal models of NPC1 disease, including a well-characterized murine model, BALB/cNctr-Npc1m1N/J, originating from a spontaneous mutation in the Npc1 gene (12). Mice homozygous for the Npc1 mutant allele (Npc1-/-) are null, and fail to produce a functional protein. Of the additional NPC1 mouse models that have been characterized, the Npc1-/- mice display the most severe and rapidly progressing disease, which is characterized by weight loss, ataxia and consistent lethality within a particular NPC1 laboratory colony. The neuropathological manifestations seen in the Npc1-/- mice are very similar to what has been described in NPC1 patients, and include lipid and cholesterol accumulation in the neurons and glial cells of the CNS, with Purkinje cell necrosis in the cerebellum (13,14). Thus, these animals represent a very good model of human NPC1 disease caused by loss of function mutations in NPC1.
Overexpression of NPC1 under the control of the neuronal-specific prion promoter in Npc1-/-mice can prevent lethality and cerebellar Purkinje cell loss, suggesting that restoring metabolic function in neurons and Purkinje cells is sufficient to eliminate the neurodegeneration of NPC1 (15). Conditional deletion of Npc1 in cerebellar Purkinje cells alone leads to impairment of motor function associated with typical NPC1 disease progression, demonstrating the importance of cerebellar Purkinje cells (16), while the deletion of Npc1 in CNS neurons is sufficient to cause NPC1 disease (17). The use of embryonic NPC1 chimeras has established that restoration of Npc1 expression in as few as 30% of neurons is adequate for significant phenotypic correction (18). In total, the transgenic, conditional and embryonic stem cell data strongly support a CNS gene addition strategy as a treatment option for NPC1 disease, assuming a delivery platform could provide adequate neuronal transduction and transgene expression.
In the past few years, it has been firmly established that certain adeno-associated virus (AAV) serotypes, such as 9 and rh10, possess an inherent capability to cross the blood-brain barrier and to transduce cells in the central nervous system (CNS), especially neurons, after systemic delivery (19–21). For these reasons, recombinant AAV vectors have been successfully used to treat neurometabolic mouse models and even patients with childhood neurodegenerative diseases, such as Canavan disease (22) and neuronal ceroid lipofuscinosis (23). We therefore hypothesized that treatment of Npc1-/- mice with a neurotropic AAV vector, configured to robustly express a therapeutic NPC1 transgene, would significantly ameliorate the fatal disease course that accompanies the murine NPC1 phenotype. We constructed two AAV serotype 9 gene therapy vectors: one used the small neuronal-specific promoter, mouse calcium/calmodulin-dependent protein kinase II (CamKII), and the other, a miniaturized version of the human elongation factor 1 alpha (EF1a), to control the expression of the NPC1 gene. The systemic delivery of AAV9 to Npc1-/- mice, as neonates or at weaning, resulted in an improved clinical appearance, delayed weight loss, significantly increased life span, reduced cholesterol storage, and decreased cerebellar Purkinje cell degeneration compared to untreated Npc1-/- mice or those treated with AAV9 GFP reporter controls. These pre-clinical studies are the first to demonstrate the efficacy of AAV gene delivery as an option to treat NPC1 disease.
Results
AAV vectors
A series of therapeutic and reporter AAV vectors were constructed. We first designed an AAV vector that could both transduce and express the human NPC1 gene in neurons. Because the full-length NPC1 cDNA is 3.8 kb, a small neuronal-specific promoter, CamKII (375 bp), was selected given the size constraints on AAV vector packaging. The final 4.8 kilobase (kb) AAV-CamKII-NPC1 vector contained the 5’ and 3’ AAV2-derived inverted terminal repeats, which flanked the CamKII promoter, NPC1 cDNA and a polyadenylation signal. A related vector, AAV9-CamKII-GFP, contained the same promoter driving the expression of GFP and served as a reporter control.
Next, we created a vector that placed the NPC1 gene under the control of a ubiquitous promoter. The EF1a promoter was truncated to a 227 base-pair core fragment and tested in vitro for activity using a GFP reporter assay (not presented). The truncated EF1a promoter was then used to replace the CamKII promoter in the AAV-CamKII-NPC1 construct to generate a 4.7 kb AAV-EF1a-NPC1 vector. In parallel, the truncated EF1a promoter was placed in front of GFP to create an analogous control reporter, AAV-EF1a-GFP.
All transgenes were packaged with an AAV9 capsid to create AAV vectors capable of crossing the blood-brain barrier after systemic delivery. Both the AAV9-CamKII-NPC1 and AAV9-EF1a-NPC1 vectors were intended to widely transduce the CNS. However, the use of the CamKII promoter was intended to restrict NPC1 expression to neurons, including Purkinje cells, while the AAV9-EF1a-NPC1 vector was designed to allow pan-expression, as would be expected for vectors using the EF1a promoter to direct transgene expression.
AAV9-CamKII-GFP mediated transduction of neuronal populations in Npc1-/- mice
In order to define the tropism and expression pattern of the AAV9-CamKII vectors in vivo, a retro-orbital injection of 1.3×1012 GC of AAV9-CamKII-GFP into Npc1-/- mice at day of life (DOL) 23 was performed, with analysis of transgene expression 14 days later (DOL37). The expression pattern of the endogenous CamKII promoter in an adult wildtype mouse is shown in Fig. 1A (taken from the Allen Mouse Brain Atlas (24), (http://mouse.brain-map.org/experiment/show/68668030; date last accessed February 11, 2016). Immunohistochemical imaging of the AAV9-CamKII-GFP treated Npc1-/- mice taken at 9 weeks of age showed a remarkably similar expression pattern especially in the anterior aspect of the brain, with a strong GFP expression in the olfactory bulb, cerebral cortex, striatum and hippocampus, with a weaker GFP expression throughout the midbrain and hindbrain (Fig. 1B). In contrast to the endogenous expression pattern of CaMKII, sparse cerebellar expression of GFP was observed following AAV9-CamKII-GFP administration (Fig. 1B). All GFP-positive cells also labelled with NeuN, indicating that expression of the viral gene product occurred only in neurons (Fig. 1C). Higher magnification images of the cerebral cortex (Fig. 1D and E) and the CA3 field of the hippocampus (Fig. 1F and G) clearly show the neurospecific double-labelling (arrowheads) and neuronal morphology of the AAV9-CamKII-GFP transduced cells.
Figure 1.
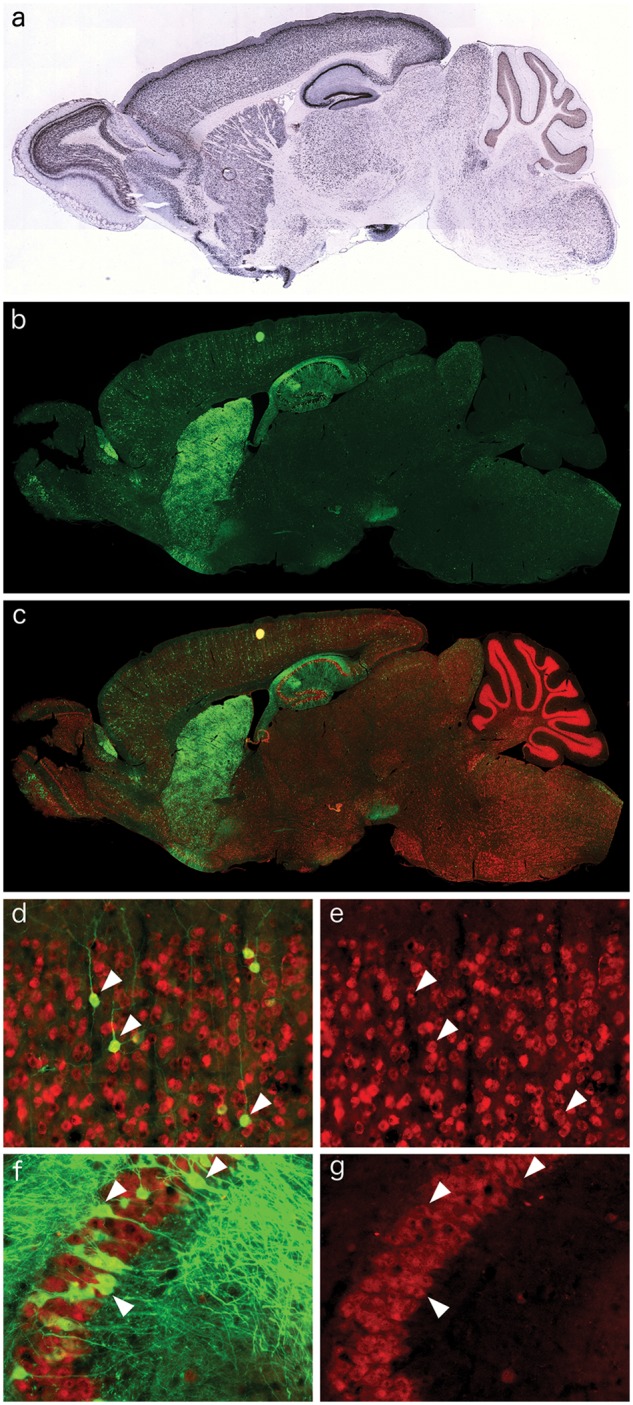
Neuronal distribution of AAV9-CamKII-GFP in the Npc1-/- mouse brain after retro-orbital injection. (A) Excerpt from the Allen Brain Atlas, demonstrating the neuronal expression pattern of CamKII in the wildtype mouse brain. (B) Immunofluorescence of AAV9-CamKII-GFP (green) in the Npc1-/- brain after retro-orbital injection. (C–G) Co-localization of the AAV9-CamKII-GFP signal with NeuN immunofluorescence (red), indicating incorporation of AAV9-CamKII-GFP into neuronal populations, including cortical pyramidal neurons (D, green removed in E to show double labelling, arrowheads) and CA3 hippocampal neurons (F, green removed in G to show double labelling, arrowheads).
AAV9-CamKII-NPC1 gene therapy improves survival and delays loss of motor function and weight decline of Npc1-/- mice
To test the efficacy of gene therapy as a treatment for NPC1 at different time points, Npc1-/- mice received retro-orbital injections of AAV9-CamKII-NPC1 during the neonatal period (n = 6; dose 2.6×1011 GC), or at weaning on DOL 20–25 (n = 9; dose 1.3×1012 GC). Another group of Npc1-/- mice (n = 6; dose 1.2×1012 GC) received AAV9-CamKII-GFP by retro-orbital injection at DOL 20–25 and served as treatment controls. Consistent with previous reports, the untreated Npc1-/- mice (n = 16) had a mean survival of 69 (± 3.1 S.D.) days, which was unaffected by AAV9-CamKII-GFP administration (Fig. 2A and B). In contrast, Npc1-/- mice that received AAV9-CamKII-NPC1 as neonates or at weaning exhibited an increased life span, with a mean survival of 97 (± 18.5 S.D.) and 103 (± 29.7 S.D.) days, respectively (P < 0.001, Log-rank (Mantel-Cox) test, Fig. 2A and B). Although formal strength, balance and gait measurements were not assayed, the AAV9-CamKII-NPC1 treated Npc1-/- mice at 8–9 weeks of life appeared to have grossly improved motor function compared to age-matched, untreated Npc1-/- mice or GFP treated controls (Supplementary Material, Video S1; available at ftp://ftp.nhgri.nih.gov/pub/manuscripts/NPC1/).
Figure 2.

Survival of Npc1-/- mice and growth following AAV9 treatment. (A) Kaplan-Meier Curve depicts survival of: Npc1-/- pups (n = 6) treated with 2.6 × 1011 GC of AAV9-CamKII-NPC1 between 1 and 3 days, Npc1-/- mice (n = 9) treated with 1.3 × 1012 GC of AAV9-CamKII-NPC1 between 20 and 25 days of life, Npc1-/- mice (n = 6) treated with 1.2 × 1012 GC of AAV9-CamKII-GFP between 20 and 25 days of life and untreated Npc1-/- mice (n = 16). (B) Survival data depicted as a vertical scatter plot to show survival distribution. (C) Week at which Npc1-/- mice reached peak weight. (D) Percentage weight change between weeks 6 and 9. **P < 0.01, ***P < 0.001, Log-ranked (Mantel Cox) test or t-test two-tailed.
While gene delivery improved both survival and mobility of Npc1-/- mice, it had no significant effect on mean weight between 4 and 9 weeks (data not shown). Therefore, the week at which the Npc1-/- mice achieved their maximal or peak weight was used to determine if gene therapy delayed or prevented the inevitable weight loss displayed by the untreated Npc1-/- mice (Fig. 2C). While both untreated (n = 16) and AAV9-CamKII-GFP treated Npc1-/- mice (n = 6) almost uniformly reach their peak weights at 6 weeks (Fig 2C), Npc1-/- pups (n = 6) or weanlings (n = 9) that received AAV9-CamKII-NPC1 reached peak weights at 8 weeks (P < 0.01 and P < 0.1, respectively). Percent weight change from 6 to 9 weeks [% weight (wt) change = (wt9weeks-wt6weeks)/wt6weeks x 100] was also compared between untreated, AAV9-CamKII-NPC1 and AAV9-CamKII-GFP treated Npc1-/- mice (Fig. 2D) because the weight decline of untreated mice began at 6 weeks, and most untreated mice did not survive beyond 9 weeks. Untreated and AAV9-CamKII-GFP treated Npc1-/- mice had a percent weight change of −17% and −23%, respectively, between 6 and 9 weeks. Relative to untreated Npc1-/- mice, AAV9-CamKII-NPC1 Npc1-/- pups treated as neonates or at weaning demonstrated significantly less weight loss [−3.8% (P < 0.02) and −2.3% (P < 0.001), respectively] over this same time period. Additionally, the mean AAV genome copy number was 0.22±0.07 vector copies per diploid genome in the brains of Npc1-/- AAV9-CamKII-NPC1 treated mice (n = 4) that range in age from 11 to 14 weeks.
AAV9-CamKII-NPC1 treatment increases NPC1 protein expression and reduces the intracellular accumulation of cholesterol in disease-affected brain regions
Layer V of the cerebral cortex (Layer V) and the CA3 pyramidal layer of the hippocampus (CA3) were chosen as representative brain structures to assess the effectiveness of AAV9-CamKII-NPC1 administration. The pyramidal neurons in these regions are prone to high levels of unesterified cholesterol accumulation (14), but show no evidence of neuronal death in this mouse model (25), negating cell loss as a confounding factor in our analysis. The polyclonal NPC1 antibody used for immunohistochemistry was capable of detecting endogenous mouse Npc1 protein as well as exogenously expressed human NPC1 protein. Immunohistochemical assessment of the resulting Npc1+/+ brain tissue at 9-weeks of age (Fig. 3A–F) showed the expected absence of unesterified cholesterol accumulation via filipin labelling, and expression of Npc1 protein in NeuN positive neurons throughout the forebrain, including the neocortical and hippocampal regions of interest (Layer V neurons, Fig. 3C and D; CA3 hippocampal neurons, Fig. 3E and F; graphical representation of filipin or NPC1 mean pixel intensity/area is depicted in Fig. 3S–V). The majority of the filipin signal in Npc1+/+ mice was found in myelin-rich structures such as the corpus callosum. The opposite was found in the PBS injected Npc1-/- mice (Fig. 3G–L). Almost no specific staining was detected with the NPC1 antibody, and many of the NeuN positive neurons throughout the forebrain exhibited high levels of unesterified cholesterol accumulation typical of late-stage NPC1 disease pathology, including neurons of the Layer V (Fig. 3I and J) and CA3 hippocampal neurons (Fig. 3K and L).
Figure 3.

Effect of AAV9-CamKII-NPC1 treatment in the CA3 hippocampus and layer V neocortex of Npc1-/- mice. (A–R) Immunohistochemical imaging of mouse Npc1 or human NPC1 protein (red) in the hippocampus and Layer V neocortex, co-stained with NeuN (green) and filipin (blue). (A–F) Endogenous Npc1 expression in the Npc1+/+ mouse, with NeuN stain removed in (B,D,F) to better show the neuronal Npc1 or NPC1 expression and magnified images of Layer V neocortex (C,D) and CA3 hippocampus (E,F). (G–L) Endogenous expression of Npc1 or NPC1 protein in the Npc1-/- mouse, with NeuN stain removed in (H,J,L) to better show the lack of Npc1 or NPC1 and high level of intracellular filipin inclusions, with magnified images of Layer V neocortex (I,J) and CA3 hippocampus (K,L). (M-R) NPC1 expression in the Npc1-/- mice injected with AAV9-CamKII-NPC1. NeuN stain removed in (N,P,R) to better show the presence of NPC1 in some neurons and the reduced level of intracellular filipin inclusions, with magnified images of Layer V neocortex (O,P), and CA3 hippocampus (Q,R). Quantification of filipin and Npc1 or NPC1 mean pixel intensity of the neuronal cell body in Layer V neocortical neurons (S, T) and CA3 hippocampal neurons (U, V), data expressed as mean ± S.E.M. A.U. = arbitrary units. * P < 0.05, **P < 0.01, ****P < 0.0001, one-way ANOVA with Tukey’s post-test.
Analysis of the AAV9-CamKII-NPC1 treated Npc1-/- mice (Fig. 3M–R) revealed an intermediate phenotype. While intracellular cholesterol accumulation was widespread throughout the brain, the average neuronal intracellular filipin intensity in Layer V (Fig. 3O,P,S) and in CA3 hippocampal neurons (Fig. 3Q,R,U) was significantly lower than observed in the untreated Npc1-/- mice (P < 0.05 and P < 0.01, respectively). This coincided with an NPC1 signal in AAV9-CamKII-NPC1 treated Npc1-/-neurons in Layer V (Fig. 3O,P,T) and in CA3 hippocampal neurons (Fig. 3Q,R,V). Although below Npc1+/+ neuron levels, this still represents a significant increase of NPC1 protein in AAV9-CamKII-NPC1 treated Npc1-/- mice when compared to the Npc1-/- vehicle controls (P <0.01 and P <0.0001 respectively, all quantifications in Fig. 3S–V).
Biochemical correction of Npc1-/- neurons following gene delivery
Having identified that AAV9-CamKII-NPC1 produced NPC1 protein and reduced cholesterol pathology after transduction in NPC1-affected regions, it was next determined whether the AAV-mediated expression of NPC1 was biochemically functional in a physiologically typical manner in Npc1-/- mice treated with AAV9 at day 23. Close inspection of the Layer V neuronal population in AAV9-CamKII-NPC1 treated mice revealed a punctate and perinuclear intracellular localization pattern for NPC1 protein, typical of a lysosomal distribution. In addition, Layer V neurons that lacked cholesterol accumulation were strongly NPC1-positive, while nearby weakly- or non-transduced neurons remained filipin positive (Fig. 4A and B).
Figure 4.

Biochemical correction of the cholesterol storage phenotype in neurons transduced with AAV9-CamKII-NPC1 in the Npc1-/- mouse. Immunohistochemical imaging of NPC1 protein levels (red) in Layer V neocortex (A,B) and CA3 of hippocampus (E,F), co-stained with filipin (blue) and NeuN (green, in A and E only). Arrows indicate neurons without appreciable NPC1 protein and high filipin staining. Arrowheads indicate neurons successfully transduced by the AAV9-CamKII-NPC1 with subsequent intense NPC1 staining and reduced filipin labelling. (C,D) NPC1 intensity of all Layer V neurons measured, plotted against filipin intensity. (G,H) NPC1 intensity of all CA3 hippocampal neurons measured, plotted against filipin intensity. Upper left quadrants in (D,H) indicate the percentage of Npc1-/- neurons corrected to control levels with AAV9-CamKII-NPC1 treatment. Imaged density of AAV-CamKII-GFP (I,J) and AAV9-CamKII-NPC1 (K,L) incorporation in the Layer V cortex (I,K) and CA3 hippocampus (J,L), with quantification in (M,N). ns = non-significant.
Plotting the NPC1 expression versus cholesterol accumulation of the Layer V neuron population analysed in AAV9-CamKII-NPC1 treated mice against those of control Npc1+/+ and Npc1-/- mice revealed that 21% (±5.71 S.E.M, n = 4) of Layer V neurons were biochemically corrected to normal levels (Fig. 4C and D). The same phenomenon was observed in the CA3 hippocampal neurons, where 31% (±1.58 S.E.M, n = 4) of this population in AAV9-CamKII-NPC1 treated mice was indistinguishable from normal, healthy neurons (Fig. 4E–H). There was a significant inverse correlation between filipin intensity and Npc1 intensity in both Layer V pyramidal neurons and CA3 hippocampal neurons (Pearson’s correlation coefficient of −0.2779 and −0.2880, respectively; P < 0.0001).
The percentage of successfully transduced neurons in the AAV9-CamKII-GFP treated Npc1-/- mice (Fig. 4I and J) and the percentage of biochemically-corrected neurons in the AAV9-CamKII-NPC1 treated Npc1-/- mice (Fig. 4K and L) were compared to assess whether the levels of biochemical correction directly matched the transduction pattern of the AAV-CamKII-GFP construct. Transduction of AAV9-CamKII-GFP was 14.9% (±0.60 S.E.M, n = 3) in LV neurons, and 27.7% (±0.75 S.E.M, n = 3) in CA3 neurons, not significantly different from that of AAV9-CamKII-NPC1 (quantification in Fig. 4M and N), indicating the level of biochemical correction observed in Npc1-/- neurons paralleled the transduction pattern of the AAV9-CamKII-GFP vector.
Delayed Purkinje cell loss in Npc1-/- mice treated with AAV9-CamKII-NPC1
Significant improvements in lifespan and physiological criteria are usually accompanied by a preservation of cerebellar Purkinje cells in Npc1-/- mice (Fig. 5). Following immunohistochemical evaluation of Purkinje cell density at 9 weeks of age in the experimental group, a significant delay was noted in the typical anterior-to-posterior loss of these neurons upon AAV9-CamKII-NPC1 treatment (Fig. 5E). In the Npc1+/+ control mice, Purkinje cell numbers remained at normal levels (Fig. 5A; 31.01, 26.94 and 29.51 cells/mm of Purkinje cell loss in lobules VI, VII and IX respectively), but the large-scale neuron loss was observed in Npc1-/- control mice (Fig. 5C; 2.43, 3.52 and 9.46 cells/mm of pcl in lobules VI, VII and IX respectively). While Purkinje cell loss had initiated in the anterior lobules I-V in AAV9-CamKII-NPC1 treated Npc1-/- mice (Fig. 5E), significantly more neurons remained when compared to the Npc1-/- control mice (Fig. 5C), with 9.37 cells/mm of the Purkinje cell layer remaining in lobule VI (P < 0.05), 9.71 cells/mm of Purkinje cells remaining in lobule VII (P < 0.05), and 17.22 cells/mm of Purkinje cells remaining in lobule IX (P <0.01) (quantifications in Fig. 5B,D,F). These data are suggestive of a secondary AAV9-CamKII-NPC1 gene therapy-mediated delay of Purkinje cell death and motor function decline.
Figure 5.

Delayed Purkinje cell death upon AAV9-CamKII-NPC1 treatment in the Npc1-/- mouse. Immunofluorescent calbindin staining of Purkinje cells (green) in Npc1+/+ mice (A), Npc1-/- mice without treatment (C), and Npc1-/- mice treated with AAV9-CamKII-NPC1 (E) at 9 weeks of age [I-X in image (A) = cerebellar lobule number]. (B,D,F) Quantification of Purkinje cell number (positive cells determined by presence of staining in Purkinje cell body and dendritic arbor or axon) in posterior cerebellar lobules. All data in bar-graphs expressed as mean ± S.E.M., * P < 0.05, ** P < 0.01, one-way ANOVA with Tukey’s post-test.
AAV9-EF1a-NPC1 gene therapy significantly improves survival and increases growth relative to AAV9-CamKII-NPC1 treatment
An AAV9 vector that utilized a ubiquitous promoter was next tested to determine whether AAV gene therapy designed to correct both neurons as well as other cell types in the brain and peripheral organs, such as the liver, might improve upon the efficacy that was observed with AAV9-CamKII-NPC1 gene delivery. Npc1-/- mice (n = 7) received 1.2×1012 GC of AAV9-EF1a-NPC1 at weaning, on 24 days of life, by retro-orbital injection. Compared to Npc1-/- mice (n = 9) treated with the similar dose (1.3×1012 GC) of the AAV9-CamKII-NPC1 vector at weaning, delivered in an identical fashion, the AAV9-EF1a-NPC1 treated Npc1-/- mice (n = 7) had a significant increase (P < 0.01) in survival, with a mean survival 166 (±89.2 S.D.) days (Fig. 6A). Relative to 9-week-old untreated Npc1-/- mice (n = 16), AAV9-EF1a-NPC1 treated Npc1-/- mice at twice this age (19 weeks old) displayed physical characteristics corresponding to improved motor function, where mice appeared to maintain their strength and coordination to walk and explore the home-cage with reduced signs of tremor (Supplementary Material, Video S2; available at ftp://ftp.nhgri.nih.gov/pub/manuscripts/NPC1/). Moreover, improved mobility was maintained over time as exemplified by an AAV9-EF1a-NPC1 treated Npc1-/- mouse recorded at 19, 33 and 38 weeks of age (Supplementary Material, Video S3; available at ftp://ftp.nhgri.nih.gov/pub/manuscripts/NPC1/).
Figure 6.

AAV9-EF1a-NPC1 treatment increase survival and growth in Npc1-/- mice. (A) Kaplan-Meier curve depicts survival of: untreated Npc1-/- mice (n = 16), Npc1-/- mice (n = 9) treated with 1.3 × 1012 GC of AAV9-CamKII-NPC1 between 20 and 25 days of life, and Npc1-/- mice (n = 7) treated with 1.2 × 1012 GC of AAV9-EF1a-NPC1 between 20 and 25 days of life. (B) Week at which Npc1-/- mice reached peak weight. (C) Percentage weight change between weeks 6 and 9. ** P < 0.01, ***P < 0.001, Log-ranked (Mantel Cox) test or t-test two-tailed. The arrow on the X-axis indicates the date nutragel was added to the pellet diet for two of the three mice.
The AAV9-EF1a-NPC1 treated Npc1-/- mice reached their mean peak weight at week 12, which was significantly delayed compared to the AAV9-CamKII-NPC1 treated animals (P = 0.02), who reached their mean peak weight at week 8 (Fig. 6B and C). The mean AAV genome copy number was 0.17±0.02 vector copies per diploid genome in the brains of Npc1-/- AAV9-EF1a-NPC1 treated mice (n = 3) at 9 weeks.
Hepatocerebral cholesterol storage is reduced in Npc1-/- mice after AAV9-EF1a-NPC1 gene therapy
To examine the extent of correction following treatment with the AAV9-EF1a-NPC1 vector, unesterified cholesterol storage was first assessed in the brain using filipin labelling (pons region of the brainstem was selected as an example). Immunohistochemistry for NPC1 was not possible because the polyclonal NPC1 antibody used in the previous AAV9-CamKII-NPC1 studies was unavailable, and four others from commercial vendors and academic investigators did not produce specific NPC1 staining in fixed tissue sections (not presented). While Npc1+/+ mice exhibit no CNS cholesterol storage (Fig. 7B), mice treated with AAV9-EF1a-NPC1 appeared to have greatly reduced filipin staining (Fig. 7D) compared to untreated Npc1-/- mice (Fig. 7C), and even less than that seen after AAV9-CamKII-NPC1 gene therapy (Fig. 7E). Further magnification of a region of interest allows for better visualization of this trend in filipin reduction (Fig. 7F–I). Although quantification of filipin was not performed, use of a previously published method (26) allowed three independent blinded observers to identify the experimental groups, with least to most filipin staining as follows: Npc1+/+ untreated < Npc1-/- EF1a < Npc1-/- CamKII < Npc1-/- untreated.
Figure 7.

Effect of AAV9-EF1a-NPC1 vs AAV9-CamKII-NPC1 treatment in brain of Npc1-/- mice. (A–E) Immunohistochemical imaging of cholesterol (visualized with filipin labelling and noted as white intracellular punctae) in the pons, as outlined by box in (A). Npc1+/+ mice exhibit no cholesterol storage (B) while abundance of cholesterol laden vesicles are apparent in Npc1-/- mice (C). Npc1-/- mice treated with AAV9-EF1a-NPC1 exhibit reduced cholesterol accumulation (D) and Npc1-/- mice treated with AAV9-CamKII-NPC1 show intermediate correction of pathology (E). (F–I) Magnification of region outlined by box in (C) for figures (B–E) to facilitate easier visual comparison of cholesterol storage between treatment groups. Scale bars = 100 µm.
Similarly, cholesterol accumulation in hepatocytes and Kupffer cells was studied in Npc1+/+ mice (Fig. 8A and E) and untreated Npc1-/- mice (Fig. 8B and F), as well as those that had received the AAV9-EF1a-NPC1 (Fig. 8C and G) or AAV9-CamKII-NPC1 (Fig. 8D and H) vectors. While mice treated with the AAV9-EF1a-NPC1 show hepatocytes free of cholesterol accumulation (Fig. 8C), the AAV9-CamKII-NPC1 treated mice resemble untreated Npc1-/- mice, with cholesterol storage prominent in both hepatocytes and Kupffer cells. Consistent with the correction of hepatic cholesterol accumulation conferred by the AAV9-EF1a-NPC1 vector, a Western blot of liver homogenate revealed robust NPC1 expression (Fig. 8I, Lane 3) that was not present in Npc1-/- mice that received the AAV9-CamKII-NPC1 vector (Fig. 8I, Lane 4).
Figure 8.

Effects of AAV9-EF1a-NPC1 vs AAV9-CamKII-NPC1 gene therapy on the liver of Npc1-/- mice. Immunohistochemical imaging of cholesterol (filipin, A–D) and Kupffer cells (CD68+, E–H). Npc1+/+ mice (A,E) exhibit no cholesterol accumulation or filipin staining (A) and Kupffer cells are minimal (E) whereas Npc1-/- mice without treatment (B,F) have extensive cholesterol accumulation (B) and increased abundance of Kupffer cells (F). AAV9-EF1a-NPC1 treated Npc1-/- mice (C,G) have hepatocytes free of cholesterol storage (C; e.g. white arrows) but demonstrate cholesterol storage in Kupffer cells (G), while AAV9-CamKII-NPC1 treated Npc1-/- mice show no correction of either cell type (D,H). Scale bar = 50 μm. (I) Western blot of liver homogenate from control and treated mice showing NPC1 protein (∼180 kDa) in the untreated Npc1+/+ and an AAV9-EF1a-NPC1 treated Npc1-/- mouse with Tubulin (50 kDa) serving as loading control.
Discussion
AAV-mediated gene delivery studies were carried out to determine whether gene addition might represent a possible treatment for NPC1 disease, a recessively inherited, progressive and lethal neurodegenerative disorder. A serotype 9 capsid was selected to encapsidate the viral transgene since this serotype has been shown to cross the blood brain barrier after systemic delivery, allowing transduction of neurons and glial cells. AAV9 vectors have been successfully used in other murine models of inherited neurological disease (27–32), and related serotypes have been given to humans without untoward effects (33). In fact, recent studies have demonstrated striking success with the use of a systemically delivered AAV9 vector to treat infants with spinal muscular atrophy type 1 (SMA1), a lethal neuromuscular disorder caused by anterior horn cell degeneration (34).
The large size of the NPC1 cDNA limited the selection of promoters for neurological and ubiquitous expression. The neuron-specific CamKII promoter was selected because of its small size and expression profile in cerebellar Purkinje cells, neurons which play a critical role in the pathogenesis of NPC1 disease. Likewise, a small but powerful ubiquitous promoter was desired, thus a miniaturized EF1a promoter was engineered into the AAV9 vector to enable broad expression.
Npc1-/- mice treated as neonates or at weaning AAV9-CamKII-NPC1 demonstrated significantly increased survival, delayed weight loss and preservation of normal motor activity relative to untreated or AAV9-CamKII-GFP injected Npc1-/- mice. No significant difference in efficacy was observed regardless of the timing of the AAV9 delivery. We find these results particularly encouraging because the delivery of AAV9 to weanling mice is analogous to treating a child with NPC1 and more accurately reflects the timing of NPC1 gene therapy in a clinical trial when compared to neonatal delivery.
Along with increased survival and improved clinical appearance of the treated Npc1-/- mice, immunohistochemisty for NPC1 protein and filipin staining confirmed that a subset of neurons showed persistent expression of NPC1 that was accompanied by a corresponding reduction of cholesterol storage. However, a small number of transduced Purkinje neurons were noted in the cerebellum, an area where Purkinje neuron loss parallels the progression of the NPC1 disease in the mouse model. Despite the relatively modest AAV9-CamKII-NPC1 transduction of the Purkinje neurons, the AAV9 treated mice had significantly improved survival and improved clinical appearance relative to untreated Npc1-/- mice, as well as delayed Purkinje cell degeneration as evidenced by calbindin staining and density counts.
Since the AAV9-CamKII-NPC1-treated Npc1-/- mice achieved a modest increase in life expectancy, the neuronal specific vector was reengineered to express the NPC1 gene under the transcriptional control of a miniaturized ubiquitous EF1a promoter in an attempt to further improve the efficacy of the gene therapy. Npc1-/- mice treated with the AAV9-EF1a-NPC1 vector showed a significant increase in life expectancy and growth relative to Npc1-/- mice treated with AAV9-CamKII-NPC. As the same AAV serotype was used to deliver both vector constructs, the only difference between the two vectors is likely the cell types that express NPC1 after gene therapy because the vector genome copy number analyses show approximately the same number of vector genomes in the brains of mutant mice treated with either vector. The improved survival of mice treated with AAV9-EF1a-NPC1 compared to AAV9-CamKII-NPC suggests that correction of non-neuronal cells in the brain such as glia, or perhaps the periphery, may be responsible for the increased therapeutic effect observed with AAV9-EF1a-NPC1. Indeed, this observation would be consistent with the results from transgenic-knock out models of NPC1, where mice were engineered to express NPC1 from a germ-line transgene under the control of the GFAP promoter and displayed greatly increased survival compared to non-transgenic littermates (35). Despite the possibility that the GFAP promoter used in the aforementioned studies might have residual expression in neurons, our observations that an AAV vector designed to widely express NPC align with the suggestion that extra-neuronal NPC1 activity might be beneficial to Npc1-/- mice. While many mouse studies and human patient observations prove the CNS symptoms are the most critical clinical determinants of progression, hepatic disease is well-recognized in NPC1 patients, either as a presenting symptom or later complication, and the systemic delivery of the AAV9-EF1a-NPC1 vector described here might enable simultaneous treatment of both CNS and hepatic manifestations of NPC1.
The fact that some of the Npc1-/- mice treated with AAV9-EF1a-NPC1 lived as long as Npc1-/- mice treated with cyclodextrin (36–38), with several gene therapy treated mice surviving past 30 weeks of age, might justify advancement of this vector to clinical trials after additional preclinical dose finding and route of delivery experiments are performed. Furthermore, the combination of systemically delivered AAV9-EF1a-NPC1 with other interventions such as the pharmaceutical excipient 2-hydroxypropyl-β-cyclodextrin (VTS-270, currently in Phase 3 clinical trial, NCT02534844) (36–39), histone deacetylase inhibitors (40,41), or even with a second, distinct CNS-specific AAV vector delivered intrathecally, might improve the effectiveness of gene therapy as a new treatment for NPC1, which, based on the results presented herein, represents a viable and efficacious therapeutic approach for NPC1 disease.
Materials and methods
AAV vector construction and production
The expression vector, pENN.AAV.CamKII0.4.eGFP.rBG (PL-C-PV1474) was obtained from the University of Pennsylvania Vector Core. This vector contains transcriptional control elements from the mouse calcium/calmodulin-dependent protein kinase II (CamKII) promoter, cloning sites for the insertion of a complementary DNA, and the rabbit β-globin polyA signal. Terminal repeats from AAV serotype 2 flank the expression cassette. The eGPF cDNA was excised from pENN.AAV.CamKII0.4.eGFP.rB plasmid and replaced with the human NPC1 cDNA. This newly created vector was called AAV-CamKII-NPC1. These AAV vectors were packaged into an AAV9 capsid, purified by cesium chloride centrifugation and titred by qPCR as previously described. The CamKII promoter was removed from the AAV-CamKII-NPC1 vector and replaced with a truncated EF1a promoter containing the first 227 base pairs of the sequences embedded in the full-length promoter. The truncated EF1a promoter was cloned into an eGFP expression vector and transfected into 293T cells to test for promoter activity. The truncated EF1a expressed GFP in 293T cells at levels similar to the full length EF1a promoter. The AAV9-CamKII-NPC1 and AAV9-EF1a-NPC1 vectors were produced by the Penn Vector Core at the University of Pennsylvania as previously described (42).
Animals
All animal work was done according to NIH-approved animal care and use protocols. Heterozygous Npc1+/- mice (BALB/cNctr-Npc1m1N/J strain) were bred to obtain control (Npc1+/+) and mutant (Npc1-/-) littermates. Mice were weighed weekly and untreated Npc1-/- mice were euthanized at ∼9 weeks of age, typically end-stage disease in our colony as determined by rapid weight loss and severe loss of motor function. The same criteria were used for evaluation of lifespan in AAV9-CamKII-NPC1 and AAV9-CamKII-GFP Npc1-/-treated groups.
Administration of AAV9
Neonatal Npc1-/- pups (1–3 days, n = 6) received a retro-orbital injection of 2.6×1011GC of AAV9-CamKII-NPC1 in a total volume of 10 µls. Weanling Npc1-/- mice (20–25 days) received a retro-orbital injection of 1.3×1012GC of AAV9-CamKII-GFP (n = 6) virus, AAV9-CamKII-NPC1 (n = 9) or 1.2×1012GC AAV9-EF1a-NPC1 (n = 7) virus in a total volume of 50 µls. Alternatively, Npc1-/- and Npc1+/+ received a sham injection of 50 µls of PBS at 23 days of age.
Western blotting
Liver tissue lysates prepared in RIPA buffer were quantified using the BCA assay. Equal amounts of protein (40 µg) were loaded onto 4–12% Bis-Tris SDS-polyacrylamide gels (Thermo Fischer Scientific) and separated via electrophoresis. Protein was transferred onto a nitrocellulose membrane (Life Technologies) and incubated for 1 h in a blocking solution containing TBSTween and LI-CORE Odyssey Blocking Buffer. The membrane was then incubated overnight with rabbit anti-NPC1 (1:1000, Abcam) and mouse anti-alpha-tubulin (1:1000 Millipore) as a loading control. Primaries were detected using Odyssey donkey anti-rabbit 680 (1:5000, LI-CORE Biosciences) and Odyssey donkey anti-mouse 800 (1:5000, LI-CORE Biosciences). Bands were imaged using the LI-CORE Odyssey Imaging System.
Vector genome copy number
Vector copy (VC) number was measured by quantitative real-time PCR analysis using a Taqman Probe for human NPC1 (Hs00264835_m1; ThermoFisher cat. #4331182). A standard curve was prepared, using serial dilutions of the AAV-CamKII-NPC1 plasmid. Genomic DNA was extracted from murine brain samples using DNeasy Kit (Qiagen) and 500 ng of DNA was used to determine the vector copy number of AAV.
Immunohistochemistry
AAV9-CamKII-GFP virus Npc1-/-mice (n = 3), AAV9-CamKII-NPC1 Npc1-/- mice (n = 4), control Npc1-/- mice (n = 5) and control Npc1+/+ mice (n = 3) were taken at 9 weeks of age for immunohistochemical analysis. Brain tissue was processed as described previously (38,43,44). Mice were euthanized by CO2 asphyxiation and transcardially perfused with 4% paraformaldehyde in phosphate buffer. The brains were post-fixed for 24 h then cryoprotected in 30% sucrose until the tissues sank. Brains were then cryostat-sectioned parasagittally (25 µm) and floating sections collected in phosphate buffered saline supplemented with 0.25% Triton-x100 (PBSt). The sections were incubated overnight at 4 °C with either rabbit anti-calbindin (1:3000, Swant), rabbit anti-NPC1 (45) (1:2000, generous gift of Dr. Daniel Ory, Washington University in St. Louis), mouse anti-NeuN (1:1000, Millipore) in PBSt, or rat anti-CD68 (1:1000, AbD Serotec) and the primaries detected using DyLight-488 goat anti-rabbit/mouse IgG or Alexa-594 anti-rabbit (1:1000 in PBSt, Vector Labs) or Alexa-488 anti-rat (1:300, ThermoFisher Scientific). Filipin (Polysciences Inc.) staining was performed at a final concentration of 50 µg/ml in PBSt. Sections were mounted and coverslipped with ProLong Gold mounting medium (Life Technologies).
Image analysis
Images of the whole cerebellum or hippocampal/neocortical region were taken using a Zeiss Axio Observer Z1 microscope fitted with an automated scanning stage, Colibri II LED illumination and Zeiss ZEN software using a high-res AxioCam MRm camera and a 10× or 20× objective. Each fluorophore channel was pseudo-coloured in ZEN, exported as TIFF, and analysed using the FIJI distribution of ImageJ, adjusting each channel for brightness and contrast in an identical manner across all experimental groups.
For CA3 hippocampal and Layer V neocortical neuron analysis, the area of a neuron was delineated according to the cell body size determined by NeuN staining and the mean pixel intensity (mpi) of the filipin and NPC1 stain within the cell recorded. Every 5th neuron was chosen at random along the CA3 or Layer V axis to obtain a representative neuronal population. 20 cells were counted in each region per section, and three sections counted per brain (60 cells per n; Npc1+/+ untreated n = 3, Npc1-/- untreated n =5, Npc1-/- AAV9-CamKII-NPC1 n = 4). To assess the level of biochemical correction in the AAV9-CamKII-NPC1 treated neuronal population, boundaries were set at the lower 2% of the filipin and NPC1 intensities of the Npc1-/- and Npc1+/+ control groups, respectively. Neurons with ‘lower than the disease-baseline’ levels of cholesterol storage together with ‘above wildtype-baseline’ levels of NPC1 expression were considered biochemically corrected.
To analyse the transduction of the AAV9-CamKII-GFP virus, the total number of NeuN positive neurons was measured in a set area of CA3 hippocampus or Layer V neocortex, and the percent of those cells double-labelled with GFP recorded (3 sections counted per brain, minimum of 72 cells counted in each section, total of 1086 hippocampal and 2367 neocortical neurons measured).
Purkinje cells were counted by measuring the number of calbindin positive Purkinje cell bodies with the recognizable dendritic tree or axonal projection still remaining within a given cerebellar lobule. The data were expressed as the number of Purkinje cells per mm of Purkinje cell layer:granule cell layer interface. The entire lobule was counted per section, with 3 sections counted per brain.
Statistical analysis
Results are expressed as means ±S.E.M. or ±S.D. and analysed for statistical significance by ANOVA, where P < 0.05 using Tukey’s post-test was considered significant or by Pearson’s correlation coefficient where P < 0.05 was considered significant. Kaplan-Meier survival curves were tested for significance using the Log-Rank Mantel-Cox test, where results were considered significant using a Bonferroni-corrected threshold of P < 0.0083 to account for multiple comparisons. All statistics were calculated using Graphpad Prism software.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
R.J.C, A.L.G, A.A.I, B.T.H, W.J.P, and C.P.V supported by the Intramural Research Program of the National Human Genome Research Institute; I.M.W., F.D.P supported by the Intramural Research Program of the National Institute of Child Health and Human Development; C.D.D. is supported by Support of Accelerated Research for Niemann-Pick C (Dana's Angels Research Trust, The Hide & Seek Foundation for Lysosomal Disease Research, Fight NPC). W.J.P and C.P.V. also acknowledge support from the Liferay Foundation and Ara Parseghian Medical Research Foundation (APMRF).
Conflict of Interest Statement. A patent has been filed by the NIH on behalf of C.P.V., R.J.C and W.J.P for the use of AAV gene therapy to treat NPC1 (US Provisional Patent no. 62/144, 702). C.D.D. is a member of the SAB for Galyatech but has not received shares, grants or consultancy fees.
Funding
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, the Intramural Research Program of the National Institute of Child Health and Human Development, Support of Accelerated Research for Niemann-Pick C (Dana's Angels Research Trust, The Hide & Seek Foundation for Lysosomal Disease Research, Fight NPC), Liferay Foundation and Ara Parseghian Medical Research Foundation (APMRF).
References
- 1. Wassif C.A., Cross J.L., Iben J., Sanchez-Pulido L., Cougnoux A., Platt F.M., Ory D.S., Ponting C.P., Bailey-Wilson J.E., Biesecker L.G., et al. (2016) High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med., 18, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vanier M.T. (2015) Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis., 38, 187–199. [DOI] [PubMed] [Google Scholar]
- 3. Naureckiene S., Sleat D.E., Lackland H., Fensom A., Vanier M.T., Wattiaux R., Jadot M., Lobel P. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science, 290, 2298–2301. [DOI] [PubMed] [Google Scholar]
- 4. Infante R.E., Radhakrishnan A., Abi-Mosleh L., Kinch L.N., Wang M.L., Grishin N.V., Goldstein J.L., Brown M.S. (2008) Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem., 283, 1064–1075. [DOI] [PubMed] [Google Scholar]
- 5. Wojtanik K.M., Liscum L. (2003) The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem., 278, 14850–14856. [DOI] [PubMed] [Google Scholar]
- 6. Pentchev P.G., Comly M.E., Kruth H.S., Vanier M.T., Wenger D.A., Patel S., Brady R.O. (1985) A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc. Natl Acad. Sci. U S A, 82, 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neufeld E.B., Cooney A.M., Pitha J., Dawidowicz E.A., Dwyer N.K., Pentchev P.G., Blanchette-Mackie E.J. (1996) Intracellular trafficking of cholesterol monitored with a cyclodextrin. J. Biol. Chem., 271, 21604–21613. [DOI] [PubMed] [Google Scholar]
- 8. Millard E.E., Gale S.E., Dudley N., Zhang J., Schaffer J.E., Ory D.S. (2005) The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. J. Biol. Chem., 280, 28581–28590. [DOI] [PubMed] [Google Scholar]
- 9. Vanier M.T., Gissen P., Bauer P., Coll M.J., Burlina A., Hendriksz C.J., Latour P., Goizet C., Welford R.W., Marquardt T., et al. (2016) Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol. Genet. Metab., 118:244–254. [DOI] [PubMed] [Google Scholar]
- 10. Patterson M.C., Hendriksz C.J., Walterfang M., Sedel F., Vanier M.T., Wijburg F. and NP-C Guidelines Working Group. (2012) Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol. Genet. Metab., 106, 330–344. [DOI] [PubMed] [Google Scholar]
- 11. Patterson M.C., Mengel E., Vanier M.T., Schwierin B., Muller A., Cornelisse P., Pineda M., Registry Investigators N.P.C. (2015) Stable or improved neurological manifestations during miglustat therapy in patients from the international disease registry for Niemann-Pick disease type C: an observational cohort study. Orphanet. J. Rare Dis., 10, 65.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Loftus S.K., Morris J.A., Carstea E.D., Gu J.Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M.A., Tagle D.A., et al. (1997) Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science, 277, 232–235. [DOI] [PubMed] [Google Scholar]
- 13. Sarna J.R., Larouche M., Marzban H., Sillitoe R.V., Rancourt D.E., Hawkes R. (2003) Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol., 456, 279–291. [DOI] [PubMed] [Google Scholar]
- 14. Zervas M., Dobrenis K., Walkley S.U. (2001) Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol., 60, 49–64. [DOI] [PubMed] [Google Scholar]
- 15. Loftus S.K., Erickson R.P., Walkley S.U., Bryant M.A., Incao A., Heidenreich R.A., Pavan W.J. (2002) Rescue of neurodegeneration in Niemann-Pick C mice by a prion-promoter-driven Npc1 cDNA transgene. Hum. Mol. Genet., 11, 3107–3114. [DOI] [PubMed] [Google Scholar]
- 16. Elrick M.J., Pacheco C.D., Yu T., Dadgar N., Shakkottai V.G., Ware C., Paulson H.L., Lieberman A.P. (2010) Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet., 19, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu T., Shakkottai V.G., Chung C., Lieberman A.P. (2011) Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Genet., 20, 4440–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ko D.C., Milenkovic L., Beier S.M., Manuel H., Buchanan J., Scott M.P. (2005) Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet., 1, 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. (2009) Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol., 27, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gray S.J., Matagne V., Bachaboina L., Yadav S., Ojeda S.R., Samulski R.J. (2011) Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther., 19, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manfredsson F.P., Rising A.C., Mandel R.J. (2009) AAV9: a potential blood-brain barrier buster. Mol. Ther., 17, 403–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McPhee S.W., Janson C.G., Li C., Samulski R.J., Camp A.S., Francis J., Shera D., Lioutermann L., Feely M., Freese A., et al. (2006) Immune responses to AAV in a phase I study for Canavan disease. J. Gene Med., 8, 577–588. [DOI] [PubMed] [Google Scholar]
- 23. Worgall S., Sondhi D., Hackett N.R., Kosofsky B., Kekatpure M.V., Neyzi N., Dyke J.P., Ballon D., Heier L., Greenwald B.M., et al. (2008) Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum. Gene Ther., 19, 463–474. [DOI] [PubMed] [Google Scholar]
- 24. Lein E.S., Hawrylycz M.J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A.F., Boguski M.S., Brockway K.S., Byrnes E.J., et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature, 445, 168–176. [DOI] [PubMed] [Google Scholar]
- 25. Pressey S.N., Smith D.A., Wong A.M., Platt F.M., Cooper J.D. (2012) Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol. Dis., 45, 1086–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davidson C.D., Fishman Y.I., Puskas I., Szeman J., Sohajda T., McCauliff L.A., Sikora J., Storch J., Vanier M.T., Szente L., et al. (2016) Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol., 3, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., et al. (2010) Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol., 28, 271–274. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Gong Y., Mu D., Prabhakar S., Moser A., Musolino P., Ren J., Breakefield X.O., Maguire C.A., Eichler F.S. (2015) Adenoassociated virus serotype 9-mediated gene therapy for x-linked adrenoleukodystrophy. Mol. Ther., 23, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walia J.S., Altaleb N., Bello A., Kruck C., LaFave M.C., Varshney G.K., Burgess S.M., Chowdhury B., Hurlbut D., Hemming R., et al. (2015) Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol. Ther., 23, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Passini M.A., Bu J., Richards A.M., Treleaven C.M., Sullivan J.A., O'Riordan C.R., Scaria A., Kells A.P., Samaranch L., San Sebastian W., et al. (2014) Translational fidelity of intrathecal delivery of self-complementary AAV9-survival motor neuron 1 for spinal muscular atrophy. Hum. Gene Ther., 25, 619–630. [DOI] [PubMed] [Google Scholar]
- 31. Foust K.D., Salazar D.L., Likhite S., Ferraiuolo L., Ditsworth D., Ilieva H., Meyer K., Schmelzer L., Braun L., Cleveland D.W., et al. (2013) Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther., 21, 2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garg S.K., Lioy D.T., Cheval H., McGann J.C., Bissonnette J.M., Murtha M.J., Foust K.D., Kaspar B.K., Bird A., Mandel G. (2013) Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci., 33, 13612–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. (2014) Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl J. Med., 371, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L., Kissel J.T., Prior T.W., Miranda C.J., Lowes L., Alfano L., et al. (2016), In American Society of Gene and Cell Therapy 19th Annual Meeting, Washington, D.C., in press.
- 35. Zhang M., Strnatka D., Donohue C., Hallows J.L., Vincent I., Erickson R.P. (2008) Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1-/- mice. J. Neurosci. Res., 86, 2848–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu B., Li H., Repa J.J., Turley S.D., Dietschy J.M. (2008) Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res., 49, 663–669. [DOI] [PubMed] [Google Scholar]
- 37. Liu B., Turley S.D., Burns D.K., Miller A.M., Repa J.J., Dietschy J.M. (2009) Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl Acad. Sci. U S A, 106, 2377–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davidson C.D., Ali N.F., Micsenyi M.C., Stephney G., Renault S., Dobrenis K., Ory D.S., Vanier M.T., Walkley S.U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One, 4, e6951.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vite C.H., Bagel J.H., Swain G.P., Prociuk M., Sikora T.U., Stein V.M., O'Donnell P., Ruane T., Ward S., Crooks A., et al. (2015) Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl Med., 7, 276ra226.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pipalia N.H., Cosner C.C., Huang A., Chatterjee A., Bourbon P., Farley N., Helquist P., Wiest O., Maxfield F.R. (2011) Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl Acad. Sci. U S A, 108, 5620–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alam M.S., Getz M., Haldar K. (2016) Chronic administration of an HDAC inhibitor treats both neurological and systemic Niemann-Pick type C disease in a mouse model. Sci. Transl Med., 8, 326ra323.. [DOI] [PubMed] [Google Scholar]
- 42. Lock M., Alvira M., Vandenberghe L.H., Samanta A., Toelen J., Debyser Z., Wilson J.M. (2010) Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene Ther., 21, 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams I.M., Carletti B., Leto K., Magrassi L., Rossi F. (2008) Cerebellar granule cells transplanted in vivo can follow physiological and unusual migratory routes to integrate into the recipient cortex. Neurobiol. Dis., 30, 139–149. [DOI] [PubMed] [Google Scholar]
- 44. Williams I.M., Wallom K.L., Smith D.A., Al Eisa N., Smith C., Platt F.M. (2014) Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol.Dis., 67, 9–17. [DOI] [PubMed] [Google Scholar]
- 45. Millard E.E., Srivastava K., Traub L.M., Schaffer J.E., Ory D.S. (2000) Niemann-pick type C1 (NPC1) overexpression alters cellular cholesterol homeostasis. J. Biol. Chem., 275, 38445–38451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.