

Abstract
IFN-γ has been found to be robustly important to disease pathogenesis in both idiopathic and induced models of murine lupus. In transgenic mice, over production of IFN-γ in the skin results in an inflammatory response and autoimmunity. This suggests that localized exposure to environmental factors that induce autoimmunity may be associated with expression of an IFN-γ-dependent inflammatory response. Using murine mercury-induced autoimmunity (mHgIA), the severity of inflammation and proinflammatory cytokine expression, including the cellular source of IFN-γ, were assessed at the site of subcutaneous exposure and in secondary lymphoid organs. Exposure induced a localized chronic inflammation comprising both innate and adaptive immune cells but only CD8+ T and NK cells were reduced in the absence of IFN-γ. IFN-γ+ cells began to appear as early as day 1 and comprised both resident (γδ T) and infiltrating cells (CD8+ T, NKT, CD11c+). The requirements for inflammation were examined in mice deficient in genes required (Ifng, Il6) or not required (Casp1) for mHgIA. None of these genes were essential for induction of inflammation, however IFN-γ and IL-6 were required for exacerbation of other proinflammatory cytokines. Additionally, lack of IFN-γ or IL-6 impacted expression of genes regulated by either IFN-γ or type I IFN. Significantly, both IFN-γ and IL-6 were required for increased expression of IRF-1 which regulates IFN stimulated genes and is required for mHgIA. Thus IRF-1 may be at the nexus of the interplay between IFN-γ and IL-6 in exacerbating a xenobiotic-induced inflammatory response, regulation of interferon responsive genes and autoimmunity.
Keywords: environmental autoimmunity, mercury, interferon, interleukin 6, inflammation, mouse
Interferons have significant effects on the pathogenesis of autoimmunity (Pollard et al., 2013) with recent studies demonstrating that type I interferons (IFN-α/β) contribute to both innate and adaptive (auto)immune processes (Theofilopoulos et al., 2005). The type II interferon, IFN-γ, has also been shown to be a major player in idiopathic autoimmune responses (Pollard et al., 2013). IFN-γ levels are increased in peripheral blood mononuclear cells of SLE patients (Harigai et al., 2008) and correlate with disease activity (Lit et al., 2007) and lupus nephritis (Masutani et al., 2001). Numerous studies of murine models of spontaneous lupus have supported the importance of IFN-γ in systemic autoimmune disease. Indeed IFN-γ has been found to be important for disease across all murine lupus models (Pollard et al., 2013) and overproduction of IFN-γ in transgenic mice leads to a lupus-like systemic autoimmunity characterized by anti-dsDNA autoantibodies and glomerulonephritis (Seery et al., 1997, 2001). Genetic deficiency of IFN-γR in autoimmune-prone NZB/W F1 (Haas et al., 1998) and MRL-Faslpr (Hron and Peng, 2004) mice protects from disease. Non-IFN cytokines also contribute to SLE (Ohl and Tenbrock, 2011) particularly IL-6 which is increased in SLE patients with active disease (Chun et al., 2007) and is linked with autoantibody production (Grondal et al., 2000). Levels of IL-6 and its soluble receptor are also increased in lupus prone mice (Suzuki et al., 1993) and IL-6 deficiency ameliorates development and severity of disease (Cash et al., 2010).
Exposure of humans to the heavy metal mercury is associated with features of autoimmunity (Crowe et al., 2016), including autoantibodies and increases in proinflammatory cytokines in gold miners (Gardner et al., 2010) and fish consumers (Nyland et al., 2011) in Amazonian Brazil. Mercury exposure results in a localized inflammatory response in murine mercury-induced autoimmunity (mHgIA) sensitive but not resistant mice (Toomey et al., 2014). This inflammatory response includes elevation of IFN-γ, IL-1β, and TNF-α and is regulated in part by cathepsin B (Toomey et al., 2014). mHgIA is abrogated by the absence of IFN-γ and its receptor (Kono et al., 1998; Pollard et al., 2012). Attempts to demonstrate elevated IFN-γ in the spleen or blood (Hemdan, 2008) have been unsuccessful, although elevation has been found in mesenteric lymph nodes (Haggqvist and Hultman, 2001) and skin (Toomey et al., 2014). Little is known about the role of other cytokines such as IL-17 or TNF-α but IL-6 is required for disease expression (Havarinasab et al., 2009).
Recent studies have suggested that inflammatory cytokine profiles can differ between healthy individuals, those who are ANA positive, and those who have SLE (Slight-Webb et al., 2016). In particular serum levels of proinflammatory cytokines, including IFN-γ, increase as the extent of autoimmunity moves from ANA-negative individuals to those with SLE. Temporal examination of the relationship between development of SLE and the serum presence of inflammatory mediators and autoantibodies shows that inflammatory mediators such as IL-6 and IFN-γ are present in the serum before autoantibodies (Lu et al., 2016). Additionally, elevation of IFN-γ and autoantibodies occurs before elevations in type I IFNs and SLE diagnosis (Munroe et al., 2016). It is unclear how these processes are initiated in human patients, however, animal model studies suggest that localized over production of IFN-γ in the skin can lead to systemic autoimmunity resembling SLE (Seery et al., 1997). To examine the relationship between a localized inflammatory response, IFN-γ expression, and autoimmunity we exploited the requirements of mHgIA for both exposure-induced inflammation (Toomey et al., 2014) and dependence on IFN-γ (Pollard et al., 2012). The mercury-induced local inflammatory response resulted in production of IFN-γ in a variety of cell types. Although the initiation of the inflammatory response was not dependent upon IFN-γ, IL-6, or caspase 1, the expression of the proinflammatory cytokines IL-1β, IL-4, and TNF-α was affected by their absence. In particular IFN-γ and IL-6 were required for the expression of each other. In addition, the expression of genes regulated by either IFN-γ or type I IFN was reduced by the absence of either IFN-γ or IL-6. This interplay between IFN-γ and IL-6 highlights the importance of these two cytokines in regulating the severity of the localized inflammation necessary for mHgIA.
MATERIALS AND METHODS
Animals . B10.S, B10.S-Ifng−/−, B10.S-Il6−/−, B10.S-Casp1−/−, and DBA/2 mice were as described previously (Kono et al., 1998; Pollard et al., 2004; Toomey et al., 2014). Breeding and maintenance were performed under specific pathogen-free conditions at The Scripps Research Institute Animal Facility (La Jolla, CA). Experiments were carried out with 6- to 12-week old animals. Animal rooms were kept at 68–72 °F and 60%–70% humidity, with a 12 h/12 h light–dark cycle, and sterilized cages were replaced each week with fresh water and food to which the mice had access ad libitum. All procedures were approved by The Scripps Research Institute Institutional Animal Care and Use Committee (IACUC).
Induction and assessment of murine mercury-induced autoimmunity (mHgIA).Mice were injected subcutaneously with HgCl2 (Mallinckrodt Baker Inc., Phillipsburg NJ) as previously described (Kono et al., 1998). The HgCl2 dose used is consistent with occupational exposure (Barregard et al., 1999). Controls received PBS alone. Use of mercuric chloride was approved by The Scripps Research Institute Department of Environmental Health and Safety.
Skin histology. Mice were sacrificed at different time points and 1.34 cm2 piece of skin centered on the site of PBS or HgCl2 injection was excised by punch biopsy and either fixed for 24 h in formalin and embedded in paraffin or snap-frozen in Tissue-Tek OCT (Sakura Finetek). Paraffin-embedded sections (6 μm) were stained in hematoxylin and eosin. Frozen sections (6 μm) were stained with 50 nM red MitoTracker CMXRos (Molecular Probe, Invitrogen, Carlsbad, CA) and mounted with Vectashield mounting medium containing DAPI and observed under an inverted fluorescent microscope (Zeiss Axiocam).
Assessment of inflammatory changes observed in skin sections stained with hematoxylin and eosin included determination of inflammatory cell infiltration, fibroblast proliferation, myocyte necrosis, and regeneration were recorded under blinded conditions by E.J. Each of the inflammatory changes were graded on a scale of 0–5. Grade 1—minimal: A histopathologic change ranging from inconspicuous to barely noticeable but so minor, small, or infrequent as to warrant no more than the least assignable grade. Grade 2—mild: A histopathologic change that is a readily noticeable but not a prominent feature of the tissue and/or may be considered to be of no functional consequence. Grade 3—moderate: A histopathologic change that is a prominent but not a dominant feature of the tissue and/or may be considered to have limited impact on organ function. Grade 4—marked: A histopathologic change that is a dominant feature and may be considered to cause significant tissue or organ dysfunction. Grade 5—severe: A histopathologic change that is an overwhelming component of the tissue and/or may represents an end-stage feature of the tissue (ie, total organ failure). The severity of inflammation elicited by HgCl2 exposure was determined as the sum total of scores for inflammatory cell infiltration, fibroblast proliferation, myocyte necrosis and regeneration.
RNA isolation and reverse transcription.RNA was extracted from skin biopsied at the site of HgCl2 injection, or whole spleen and cervical lymph nodes using Trizol reagent (Invitrogen). Integrity of RNA was determined by RIN analysis using an Agilent 2100 Bioanlayzer (Agilent, Santa Clara, CA). Only samples with RIN values >7.5 were reverse transcribed. RNA was transcribed using random hexamers, dNTPs, RNase inhibitor (RNaseOUT, Invitrogen), and SuperScript III reverse transcriptase (Invitrogen). Control PCR reactions were carried out using the β-actin primers: Forward 5’-TGGAATCCTGTGGCATCCATGAAACT-3’ and reverse 5’-TGTAAAACGCAGCTCAGTAACAGTCCG-3’ and products were separated using a 1.5% agarose gel and visualized by ethidium bromide.
Quantitative real- time PCR. The cDNA levels of genes listed in Supplementary Table 1 were measured by quantitative real-time PCR (qPCR) using the QuantiTect SYBR Green PCR kit (Qiagen, Valencia, CA) with QuantiTect validated primer sets (Qiagen, Supplementary Table 1). All PCR reactions were performed using the 7500 Fast Real-Time PCR System (Applied Biosystems). Melting curve analysis was performed for each primer set to ensure amplification specificity. Corresponding standard curves were added in each PCR reaction. The housekeeping gene GAPDH was used to normalize data to cDNA inputs. Results are expressed as copy numbers of target gene per copy numbers of GAPDH.
For our earliest experiments, specific primers and probes for IL-1β, IL-2, IL-4, IL-6, TNF-α, and IFN-γ with FAM reporters and BHQ-1 quenchers (Integrated DNA Technologies) were used. Reactions for each sample were performed in duplicate as follows: 100 ng primers, 0.2 mM dNTPs, 4 mM MgCl2, 0.625 U Amplitaq gold (Applied Biosystems), 1× Amplitaq gold reaction buffer, and 0.5 μl cDNA in 25 μl total volume. Reactions were performed using an iCycler iQ machine (Bio-Rad). Corresponding standard curves ranging from 100 to 0.0001 attomoles/well were added in each PCR reaction. Confirmation of PCR specificity was done by subcloning PCR products using the pGEM-T Easy vector kit (Promega, Madison, WI). Plasmid inserts were sequenced using an ABI Prism 3100 Genetic Analyzer, and analyzed using Sequencing Analysis version 3.7 (Applied Biosystems). IFN-γ, IL-2, and IL-4 transcript levels were expressed relative to cyclophilin A levels for spleen and lymph node samples. IL-1β, IL-2, IL-4, IL-6, TNF-α, and IFN-γ transcript levels were expressed as attomoles per μg of RNA in skin samples.
Spleen cells isolation and stimulation. B10.S and B10.S-Ifng−/− mice were sacrificed and spleens were placed in RPMI1640 (Invitrogen) containing 10% FBS. Spleen single cell suspensions were obtained by disrupting the tissue between two pre-cleaned frosted slides. Red blood cells were removed by 10 min incubation at room temperature in red blood cell lysis buffer (eBioscience, San Diego, CA). Debris and dead cells were removed with lympholyte-M (Cedarlane Laboratories, Burlington, NC). Cells were counted and seeded at 2× 106 cells in RPMI1640 containing 10% FBS/ml and allowed to rest for 4 h. Stimulation was performed for 8 h with 40 ng/ml of PMA (Fisher Scientific, Pittsburgh, PA), 2 μg/ml of ionomycin (MP Biomedicals, Solon, OH), and 1 μl/ml of Golgi Plug (BD Biosciences) before analysis by flow cytometry.
Isolation and stimulation of skin cells. B10.S and B10.S-Ifng−/− mice were sacrificed and hair around injection site was removed by using hair remover (Nair, Church & Dwight Co, Princeton, NJ). A 1.34 cm2 piece of skin centered on the site of PBS or HgCl2 injection was then excised and incubated in a 0.3% trypsin/glucose/NaCl/KCl solution containing 0.1% deoxyribonuclease I (MP Biomedicals) for 1 h at 37 °C. Cell suspension was collected, filtered through a sera-separa column (Evergreen, Los Angeles, CA) and placed on ice. Two additional rounds of extraction were performed as described above. Cells were then washed, counted and seeded at 1× 106 cells/ml in RPMI1640 containing 10% FBS for 6 h. Stimulation was performed for 8 h with 40 ng/ml of PMA (Fisher Scientific), 2 μg/ml of ionomycin (MP Biomedicals), and 1 μl/ml of Golgi Plug (BD Biosciences) before analysis by flow cytometry.
Flow cytometry analysis. To assess cell subsets and IFN-γ production, spleen, and skin single-cell suspensions, stimulated as described above, were first stained with the following surface markers (all from BD Pharmingen, San Diego, CA, unless otherwise specified): CD45R/B220 (FITC, RA3-6B2), γδ TCR (FITC, GL3), NK1.1 (FITC, PK136), I-Ap (FITC, 7-16.17), CD3ε (PerCP, 145-2C11), CD4 (PerCP, RM4-5), CD8a (PerCP, 53-6.7), CD19 (PerCP-Cy5.5, 1D3), CD3ε (APC, 145-2C11), CD11c (APC, HL3), CD11a (FITC, M17/4 eBioscience), CD49f (PE, GoH3), and F4/80 (APC, CI:A3-1, Invitrogen). For detection of intracellular IFN-γ, cells, activated and stained as described above, were then fixed and permeabilized in Cytofix/Cytoperm solution (BD Pharmingen) for 20 min at 4 °C, washed twice with Perm/Wash buffer (BD Pharmingen) and stained with PE-conjugated anti-IFN-γ (XMG1.2, eBioscience) for 45 min at 4 °C in Perm/Wash buffer. Fluorescence acquisition was done using a dual laser FACSCalibur flow cytometer using CELLQuest Pro software (BD Biosciences). Dead cells were excluded on the basis of forward and side light scatter. Analysis was performed with FlowJo V6.4.3 software (Tree Star, Ashland, OR).
Statistical analysis. Unless otherwise noted, all data is expressed as mean and standard error. Statistical analysis was done using GraphPad Software (V5.02), San Diego, CA. Un-paired, two-tailed Mann–Whitney U test was used for comparisons between PBS and HgCl2 exposure within a strain and one-way analysis of variance (ANOVA) followed by Tukey multiple comparison test used for comparison between PBS or HgCl2 exposed groups. P < .05 was considered significant.
RESULTS
Mercury Exposure Leads to Chronic Inflammation
Histological examination of the skin from HgCl2 exposed mice showed macroscopic lesions, accompanied in most cases by mild scab formation and skin erythema, appearing after the first injection of mercury. These features tended to disappear during the second week (data not shown). Compared to PBS injected mice, the skin from mercury injected mice showed significant thickening, hardening and erythema at the site of injection. Examination of hematoxylin and eosin, and DAPI and mitotracker, stained sections on day 3 revealed epidermal thickening (Figs. 1A–C). Increased thickness and fibrosis of the dermis were also observed along with mononuclear cell infiltrates (Figs. 1A and B). The hypodermis of mercury-injected mice was characterized by infiltration of mononuclear cells and thickening of the subcutaneous adipocyte layer (Figs. 1A and B). Moreover, cell infiltrates seemed to highly disorganize the hypodermis compared to PBS controls. Similar observations were made at days 7 and 28 (data not shown). These histological studies reveal that mercury induces a chronic inflammation of the skin overlaying the site of injection provoking considerable changes in the structure of the different skin layers and the recruitment of mononuclear cells.
Figure 1.
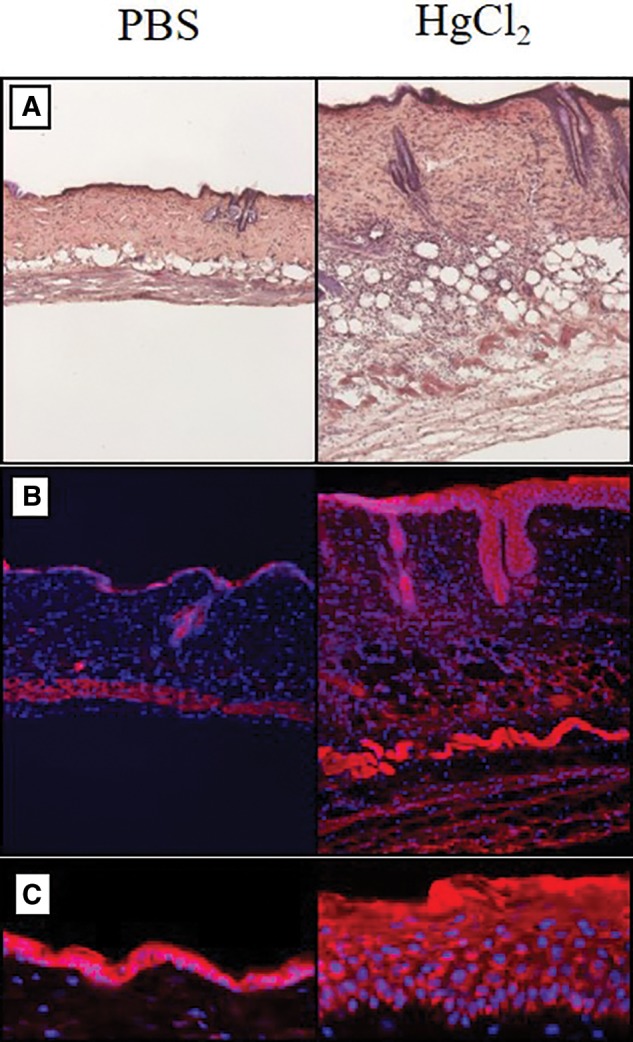
Histological analysis of inflammatory response in the skin. mHgIA sensitive B10.S mice were injected subcutaneously with PBS or 40 μg HgCl2 in PBS and skin harvested 3 days later. A, Hematoxylin and eosin staining of paraffin embedded skin sections (×10 magnification), and B, Red MitoTracker CMXRos and DAPI staining of snap-frozen skin sections (×10 magnification) showing the increased thickness and fibrosis of the dermis in mercury-exposed compared to PBS control animals. C, Red MitoTracker CMXRos and DAPI staining of snap-frozen skin sections showing thickening of the epidermis in mercury-exposed compared to PBS control animals (×32 magnification).
Localized Inflammation is Associated With IFN-γ Production at the Site of Exposure
Although localized inflammation (Toomey et al., 2014) and IFN-γ (Kono et al., 1998; Pollard et al., 2012) are necessary for mHgIA, the relationship between inflammation and IFN-γ expression has not been investigated. To identify the site and kinetics of IFN-γ production, flow cytometric analysis of intracellular IFN-γ was done using cells isolated from skin biopsies overlying the site of mercury injection and splenocytes at different time points. Mercury exposure produced a significant increase in the percentage of skin cells producing IFN-γ as early as 1 day after the first injection, peaking at day 7 (Figure 2); at any time, less than 1.7% of skin cells were producing IFN-γ in PBS treated control B10.S mice. No change in the percent of IFN-γ producing cells could be observed in the spleen of HgCl2 treated mice compared to PBS control animals (Figure 2). These results argue that the presence of IFN-γ+ cells in mHgIA occurs preferentially at the site of exposure.
Figure 2.

IFN-γ expression in the skin of mercury-exposed mice. B10.S were injected subcutaneously with PBS or 40 μg HgCl2 in PBS twice/week for up to 28 days. Skin and spleen cells were isolated at the indicated times and treated for 8 h with PMA, ionomycin and Golgi plug. IFN-γ production in both skin and spleen cells was assessed by intracellular FACS staining. Top: Representative FACS profile (left panel) for intracellular expression of IFN-γ in the skin after 7 days of PBS (shaded histogram) or mercury (bold histogram) treatment. The average of viable IFN-γ positive cells in the skin of PBS or mercury-treated animals for each indicated times are summarized (right panel). Bottom: Representative FACS profile (left panel) for intracellular expression of IFN-γ in the spleen after 7 days of PBS (shaded histogram) or mercury (bold histogram) treatment. The average of viable IFN-γ positive cells in the spleen of PBS or mercury-treated animals for each indicated times are summarized (right). Data (mean ± SEM) are from 4 mice in each group. Statistical significance was assigned based on un-paired two tailed Mann–Whitney U test comparing PBS- and mercury-treated mice. *P values <.05.
IFN-γ Is Necessary but Not Essential to Mononuclear Cell Changes in the Skin following Mercury Exposure
Cell surface marker analysis identified increases in a number of mononuclear cells following mercury exposure in B10.S mice during the 28 day exposure period (Supplementary Figure 1). Examination of cell populations at day 7 (Figure 3) revealed increases in all cell types except γδ T cells in the skin of mercury-treated animals. No recruitment of keratinocytes, defined as CD11a negative and CD49f positive-cells (Chen et al., 2004), was observed throughout the entire experiment (data not shown).
Figure 3.

HgCl2-induced mononuclear cell infiltration. B10.S and B10.S-Ifng−/−mice were injected with PBS (clear column) or 40 μg HgCl2 (filled column) in PBS twice/week. Cells from the skin overlying the site of injection were isolated and stained with specific cell surface markers for CD8+ T, CD4+ T, NKT, B, F4/80+, CD11c+, NK, and γδ T cells and expressed as percent of viable cells. Values are the mean ± SEM for 4 PBS- and 4 mercury-treated mice. Statistical significance was assigned by un-paired, two-tailed Mann–Whitney U test was used for comparisons between PBS and HgCl2 exposure within a strain and one-way analysis of variance (ANOVA) with Tukey post-test used for comparison between PBS or HgCl2 exposed groups.
To assess the role played by IFN-γ in mononuclear cell infiltration, flow cytometry was also used to identify the cells recruited to the site of mercury injection after 7 days of treatment of B10.S-Ifng−/− mice (Figure 3). Absence of IFN-γ reduced the increase in mononuclear cells following 7 days of mercury treatment from 10% to 11% of viable skin cells in B10.S to around 6% in B10.S-Ifng−/− mice. This decrease can be explained in part by reduction in CD8+ T cells and NK cells (Figure 3).
Mercury treatment did not affect the presence of NKT cells in the skin of B10.S-Ifng−/− mice. This discrepancy might be explained by the high number of NKT cells present in the skin of B10.S-Ifng−/− which was significantly higher than that in B10.S mice (P < .0001). Mercury increased the percent of B cells and F4/80+ cells in the skin of B10.S-Ifng−/− mice compared to PBS controls. The number of CD11c+ cells was not affected by mercury treatment probably because, as for NKT cells, B10.S-Ifng−/− mice possessed a greater percentage of CD11c+ cells in their skin. The number of γδ T cells present in the skin was similar in wild-type and B10.S-Ifng−/− mice and mercury had no effect on γδ T cells recruitment. Taken together these data indicate that IFN-γ contributes to the severity of the inflammatory response in the skin following mercury exposure although its absence does not abrogate infiltration of some inflammatory cells particularly B cells and F4/80+ cells.
Innate and Adaptive Immune Cells Are Responsible for IFN-γ Production at the Site of Exposure
IFN-γ is produced by cells of the innate and adaptive immune systems (Boehm et al., 1997), but it is unclear which IFN-γ+ cells are essential for mHgIA. Almost all cell types showed increases in IFN-γ+ cells during the 28-day exposure (Supplementary Figure 2). The percentage of IFN-γ producing cells was increased for all 3 lymphocyte subpopulations with 40% of CD8+ T cells being IFN-γ+ at day 7 compared to 22% of CD4+ T cells and 20% of B cells (Figure 4) but only CD8+ T cells were significantly increased above PBS control. The percentage of NKT cells producing IFN-γ was significantly higher in mercury-treated animals as was the percentage of CD11c+ cells. Neither F4/80+ nor NK cells had a significant increase in IFN-γ+ cells after mercury exposure. IFN-γ producing γδ T cells were higher in mercury-treated animals but no more than 20% were ever IFN-γ+. Thus only a subgroup of cell types (CD8+ T, NKT, CD11c+) that were increased in the skin also showed significantly increased IFN-γ expression while a small percentage of resident γδ T cells became IFN-γ producers.
Figure 4.

Recruitment of IFN-γ producing cells. B10.S mice were injected with PBS (clear column) or 40 μg HgCl2 (filled column) in PBS twice/week. Cells from the skin overlying the site of injection were isolated and stained with specific cell surface markers for CD8+ T, CD4+ T, NKT, B, F4/80+, CD11c+, NK, and γδ T cells and expressed as percent of IFN-γ+ cells. Values are the mean ± SEM for 4 PBS- and 4 mercury-treated mice. Statistical significance was assigned as described above and in Materials and Methods.
Tissue Inflammation Does Not Require IFN-γ or IL-6
In a previous study, we found that increased cathepsin B activity was associated with local inflammation following HgCl2 exposure (Toomey et al., 2014). However, deficiency of genes required for mHgIA, such as Ifng and Il6, was not linked to reduced cathepsin B activity. This observation together with the finding in Figure 3 suggested that inflammation at the site of exposure may not be totally dependent upon the presence of genes required for mHgIA. To test this hypothesis wild-type B10.S, B10.S-Ifng−/−, B10.S-Il6−/−, and B10.S-Casp1−/− mice were injected subcutaneously with HgCl2 on days 1 and 4 and skin overlying the injection site harvested on day 7, fixed and stained in hematoxylin and eosin. The main finding in PBS controls was minor inflammation of the subcutaneous tissue, panniculus carnosus and dermis, characterized by infiltration by macrophages admixed with a few lymphocytes and occasionally neutrophils and plump fibroblasts. Additionally hyperkeratosis with or without hyperplasia was reported in the epidermis. Microscopic findings observed in the skin after injection of HgCl2, were characterized by a more severe inflammation of the subcutaneous tissue, panniculus carnosus and dermis compared to the PBS controls. There was a considerable increase in the number of macrophages and neutrophils, and plump fibroblasts were more frequently and more abundantly observed. Additional findings were necrosis of the subcutaneous tissue and the muscle fibers of the panniculus carnosus, with occasional multinucleated muscle fiber (regeneration). Generally the hyperkeratosis was more severe and the concurrent hyperplasia was more constantly observed in the epidermis overlying the injection site. In general mercury exposure resulted in marked expansion of the dermis and subcutaneous tissue by macrophages, neutrophils, proliferating fibroblasts, and a few lymphocytes irrespective of the lack of IFN-γ, IL-6, or caspase 1 (Figure 5). Although it is worth mentioning that the increase in neutrophils within the inflammatory infiltrate observed in the B10.S and Ifng−/− mice occurred to a much lesser extent in Il6−/− and Casp1−/− mice. In contrast DBA/2 mice which do not develop mHgIA (Hultman et al., 1992) and show little evidence of mercury-induced inflammation (Toomey et al., 2014) did not develop the same level of infiltrates (see Supplementary Figure 3). While separate assessment of inflammatory cell infiltration, fibroblast proliferation, and myocyte necrosis and regeneration show some differences between strains (Supplementary Figure 4), the sum total of the severity of inflammation elicited by HgCl2 exposure showed no difference between wild-type and mice deficient in Ifng, Il6, or Casp1 (Figure 6). These observations show that the initiation of inflammation at the site of exposure is not dependent upon IFN-γ or IL-6 even though they are required for expression of mHgIA.
Figure 5.

Presence of tissue inflammation does not require IFN-γ or IL-6. B10.S, B10.S-Ifng−/−, B10.S-Il6−/−, and B10.S-Casp1−/− mice were injected with PBS or 40 μg HgCl2 in PBS twice/week and skin overlying the injection site excised and embedded in paraffin. Paraffin sections (6 μm) were stained in hematoxylin and eosin. Left column: PBS treated mice, showing minimal expansion of the dermis and subcutaneous tissue. Middle column: HgCl2-treated mice, showing marked expansion of the dermis and subcutaneous tissue by macrophages, neutrophils, proliferating fibroblasts and a few lymphocytes irrespective of the lack of IFN-γ, IL-6 or caspase 1. Right column: High power field showing lymphocytes (red arrow), neutrophils (green arrow), macrophages and giant cells (black arrow) and proliferating fibroblasts (blue arrow). Magnification; white bar 50 μm, black bar 20 μm.
Figure 6.

Overall severity of inflammation was not influenced by lack of Ifng, Il6, or Casp1. B10.S, B10.S-Ifng−/−, B10.S-Il6−/−, and B10.S-Casp1−/− mice were injected with PBS (clear circle) or 40 μg HgCl2 (filled circle) in PBS twice/week and skin overlying the injection site excised and embedded in paraffin. Paraffin sections (6 μm) were stained in hematoxylin and eosin and the severity of inflammation determined as the sum of inflammatory cell infiltration, fibroblast proliferation, and myocyte necrosis and regeneration. See Supplementary Figure 4 for details of breakdown of inflammatory components. Values are the mean ± SEM for 4–8 PBS and 4–8 mercury-treated mice. Statistical significance was assigned as described above and in Materials and Methods.
IFN-γ and IL-6 Contribute to Proinflammatory Cytokine Expression at the Site of Exposure
In addition to increased cathepsin B activity, HgCl2-induced inflammation is also associated with expression of proinflammatory cytokines (Pollard et al., 2011; Toomey et al., 2014). To determine if IFN-γ is required for such expression and whether such expression is limited to the site of exposure, we examined cytokine expression in the skin, draining lymph nodes and spleen of wild-type B10.S and B10-S-Ifng−/− mice (Supplementary Table 2). In the skin of wild-type mice only small amounts of IL-2 and IL-4 could be found, compared to pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1β. In secondary lymphoid organs production of IL-4 was increased, however, IL-2 production was not significantly affected. Expression of IL-2 and IL-4 in skin, spleen and lymph nodes of B10.S-Ifng−/− mice was similar to that of wild-type for both PBS and mercury exposure. This was in contrast to other pro-inflammatory cytokines in the skin which were increased by mercury exposure, although the amounts detected were lower than found in wild-type B10.S by 3-, 5-, and 3.7-fold for IL-6, IL-1β, and TNF-α, respectively.
Because IL-6 (Havarinasab et al., 2009) is required for mHgIA, we asked if its absence has similar effects as IFN-γ deficiency on the production of proinflammatory cytokines at the site of exposure. HgCl2 exposure of wild-type B10.S mice elicited significant increases in IL-1β, IL-4, IL-6, and TNF-α compared to PBS controls (Figure 7). mHgIA sensitive B10.S-Casp1−/− mice also had significant increases in the same cytokines. Levels of mercury-induced IL-1β and IL-2 were not significantly different between the strains. In contrast IL-4 and TNF-α levels were significantly reduced in B10.S-Ifng−/−, B10.S-Il6−/−, and B10.S-Casp1−/− mice compared to wild-type mice. B10.S-Ifng−/− mice had modest increases in IL-1β, IL-4, and IL-6 but these were not significantly elevated above PBS controls. However, the level of IL-6 in B10.S-Ifng−/− mice was significantly reduced compared to wild-type. IL-4 was found increased and IL-2 decreased in B10.S-Il6−/− mice after exposure (Figure 7). Interestingly, the endogenous levels of IL-1β and IL-2 in B10.S-Il6−/− mice were significantly increased above the other strains (P < .01). Thus, although there were differences in the levels of expression, both wild-type B10.S and caspase-1 deficient mice exhibited mercury-induced increases in proinflammatory cytokines. In contrast, absence of IFN-γ and IL-6 adversely affected expression. Furthermore, the absence of IFN-γ reduced IL-6 expression supporting a suspected regulatory linkage between these 2 cytokines (Faggioli et al., 1997).
Figure 7.

Lack of Ifng and Il6 reduces proinflammatory cytokine expression. B10.S, B10.S-Ifng−/−, B10.S-Il6−/−, and B10.S-Casp1−/− mice were injected with PBS (open column) or 40 μg HgCl2 (filled column) in PBS twice/week and skin overlying the injection site excised and RNA isolated, reverse transcribed and Il1b, Il2, IL4, Il6, and Tnfa determined by real-time PCR. Values were normalized by total mRNA levels using GADPH as a housekeeping gene. Values are the mean ± SEM for 7–8 PBS- and 8–10 mercury-treated mice. Statistical significance was assigned as described above and in Materials and Methods.
IL-6 Is Required for IFN-γ and Interferon-Regulated Gene Expression
While regulation of IL-6 by IFN-γ, especially in combination with IL-1β, has been described (McLoughlin et al., 2003), less is known about the regulation of IFN-γ by IL-6 (Scheller et al., 2011). B10.S-Il6−/− mice showed no mercury-induced increase in IFN-γ and although B10.S-Casp1−/− mice had increased IFN-γ compared to PBS controls, the level was significantly reduced compared to wild-type B10.S. Failure to elicit increased IFN-γ expression in IL-6 deficient mice may impact expression of IFN-γ regulated genes and may help explain the dependence of mHgIA on IL-6 (Havarinasab et al., 2009). Examination of several genes primarily regulated by IFN-γ revealed this to be the case. Irf1, which is required for mHgIA (Pollard et al., 2012), although increased in HgCl2-exposed wild-type and B10.S-Casp1−/− mice was not increased in either B10.S-Ifng−/− or B10.S-Il6−/− mice (Figure 8). However, the level of Irf1 in B10.S-Casp1−/− mice was significantly reduced compared to that of wild-type. Similar results were found with Ifr8 and Fcgr1 (Figure 8). In contrast, HgCl2 increased expression of Msr1 in all strains although the levels in gene-deficient mice were less than that found in wild-type mice. Cxcl10 was increased in B10.S although this was significantly less than that in B10.S-Casp1−/− mice. Thus mercury-induced expression of IL-6 regulates not only IFN-γ but also genes predominantly regulated by IFN-γ.
Figure 8.

Lack of Ifng and Il6 affects expression of IFN-γ regulated genes. B10.S, B10.S-Ifng−/−, B10.S-Il6−/−, and B10.S-Casp1−/− mice were injected with PBS (open column) or 40 μg HgCl2 (filled column) in PBS twice/week and skin overlying the injection site excised and RNA isolated, reverse transcribed and IFN-γ, Irf1, Irf8, Fcgr1, Msr1, and Cxcl10 determined by real-time PCR. Values were normalized by total mRNA levels using GADPH as a housekeeping gene. Values are the mean ± SEM for 7–8 PBS- and 8–10 mercury-treated mice. Statistical significance was assigned as described above and in Materials and Methods.
Type I IFNs are also required for systemic autoimmunity (Kono et al., 2013), therefore we asked if expression of genes predominantly regulated by type I IFN are also influenced by the absence of IL-6. Expression of Ifi44, Rsad2, and Isg15 were increased in wild-type and B10.S-Casp1−/− but not B10.S-Il6−/− mice compared to their respective PBS controls (Figure 9). Only Ifi44 was increased in B10.S-Ifng−/− mice. Mercury exposure increased expression of Rsad2 and Isg15 in B10.S-Casp1−/− and Ifi44 in B10.S-Il6−/−mice over that of wild-type B10.S. However B10.S-Il6−/− mice had endogenous levels of Ifi44, Rsad2, Isg15 as well as Cxcl10 (Figure 7) that were elevated above those of the other strains (P < .05–.01) but these levels were not changed by mercury exposure. Expression of a number of other genes, including Mx1, Gbp2b, Ifnb, Ifna4, Ido1, Lcn2, Ap3b1, Bcl6, and Arid5a, were not significantly changed by mercury exposure in B10.S mice (data not shown). These findings reveal that lack of IFN-γ adversely affected HgCl2-induced expression of genes regulated by type I IFN and that although lack of IL-6 increases endogenous expression of these genes, mercury exposure did not lead to increased expression. Thus, deficiency of either IFN-γ or IL-6 adversely affects the ability of HgCl2 to increase expression of genes regulated by both IFN-γ and type I IFN.
Figure 9.

Lack of Ifng and Il6 affects expression of type I IFN regulated genes. B10.S, B10.S-Ifng−/−, B10.S-Il6−/− and B10.S-Casp1−/− mice were injected with PBS (open column) or 40 μg HgCl2 (filled column) in PBS twice/week and skin overlying the injection site excised and RNA isolated, reverse transcribed and Ifi44, Rsad2, and Isg15 determined by real-time PCR. Values were normalized by total mRNA levels using GADPH as a housekeeping gene. Values are the mean ± SEM for 7–8 PBS- and 8–10 mercury-treated mice. Statistical significance was assigned as described above and in Materials and Methods.
DISCUSSION
We have demonstrated that IFN-γ, its receptor and the primary IFN-γ response gene Irf1 are required for the development of mHgIA (Kono et al., 1998; Pollard et al., 2012) and that mHgIA is linked to an inflammatory response characterized by increased expression of IFN-γ and other proinflammatory cytokines (Toomey et al., 2014). In this study, we show that IFN-γ is up-regulated both at the protein and mRNA levels at the site of HgCl2 exposure, but not in the spleen or draining lymph nodes. Subcutaneous mercury exposure resulted in a dramatic cellular infiltration of the dermis and thickening of the epidermis within days. The infiltrate was characterized by the presence of numerous cell types and production of proinflammatory cytokines, with many cell types producing IFN-γ. However, the initiation of this inflammation was not dependent upon IFN-γ or IL-6, another cytokine required for mHgIA (Havarinasab et al., 2009). Rather IFN-γ and IL-6 were required for increased expression of proinflammatory cytokines as well as being required for the expression of each other. This mutual requirement was demonstrated by the observation that IFN-γ and IL-6 affected the HgCl2-induced expression of genes regulated by IFN-γ and type I IFN. Thus the localized expression of IFN-γ appears to be important for the severity of proinflammatory cytokine responses which includes an interplay with IL-6 in the regulation of genes expressed in response to interferons.
The importance of cutaneous IFN-γ in the development of systemic autoimmune disease has been demonstrated in female mice with epidermal transgenic expression of IFN-γ. Although these mice show no evidence of IFN-γ in their blood they do develop systemic autoimmunity characterized by dsDNA and histone autoantibodies and glomerulohephritis (Seery et al., 1997). In contrast, transgenic IFN-γ expression in extracutaneous sites, such as pancreatic islets and neuromuscular junction, does not mediate the induction of systemic autoimmunity (Seery, 2000). The skin of mice transgenic for cutaneous IFN-γ shows histological similarity with the skin overlying the site of mercury injection including inflammation, severe erythema, thickening of the epidermis and dermal mononuclear infiltration (Carroll et al., 1997). We show that the presence of IFN-γ positive cells occurs as early as 24 h after the first mercury treatment and peaks as a percentage of viable cells after 7 days before decreasing but continuing to remain significantly higher than in PBS controls. The data suggest that the chronic localized inflammation caused by repeated administration of mercury in the skin of B10.S mice may closely recapitulate the constitutive over-expression of IFN-γ in transgenic mice expressing IFN-γ in the epidermis (Carroll et al., 1997).
The skin contains a number of resident immune cells including CD8+ and CD4+ T cells, γδ T cells, NKT cells, macrophages, and dendritic cells whose activation contributes to localized inflammatory responses (Nestle et al., 2009). Many of these cell types were increased at the site of mercury exposure but only some had increased IFN-γ+ cells. Although the increase in CD8+ T cells depended significantly upon the presence of IFN-γ, their involvement in the development of mHgIA is unlikely to be essential because mice lacking β2-microglobulin, and thus functional CD8+ T cells, still develop mHgIA (Pollard et al., 2011). Significant production of IFN-γ was also detected in innate-like lymphocytes such as NKT cells and γδ T cells. In mHgIA, activation of NKT cells has been shown to exacerbate autoimmunity without increasing IFN-γ (Vas et al., 2008); however, measurement of IFN-γ was done using splenocytes which, as found in this study and previously (Kono et al., 1998), is not the appropriate tissue. NKT cells can stimulate NK cells to produce IFN-γ (Carnaud et al., 1999). Thus the early activation of NKT cells to IFN-γ producers found in our study may explain the subsequent appearance of increased NK cells found after 3 days of mercury exposure. The appearance of IFN-γ producing CD11c+ cells as early as 1 day after the first mercury injection could also influence production of IFN-γ by NK cells (Walzer et al., 2005). However, there was only a modest increase in IFN-γ+ NK cells in response to HgCl2. γδ T cells were not increased at the site of mercury injection but produced significant amounts of IFN-γ in response to mercury supporting the observation that activation of epidermal γδ T cells leads to the production of IFN-γ (Boismenu et al., 1996).
In the absence of IFN-γ, localized inflammation at the site of exposure was significantly reduced but not eliminated. In particular, there were reductions in CD8+ T cells and NK cells compared to mercury exposed wild-type mice, however both B and F4/80+ cells were significantly increased and there was a modest increase in CD4+ T cells in IFN-γ deficient mice compared to PBS controls. Other cell types (CD11c+, NKT, γδ T) were largely unchanged in the absence of IFN-γ. Histological examination of the skin revealed that the presence of inflammatory cells such as macrophages as well as proliferating fibroblasts was not dependent upon genes required (Ifng, Il6) (Kono et al., 1998) or not required (Casp1) (Pollard et al., 2012) for mHgIA. Although this study does not identify the events or types of cells that initiate the inflammatory response elicited by HgCl2, especially in the absence of IL-6, a previous study implicates increased expression of cathepsin B as a major contributor to the localized inflammation induced by HgCl2 even in the absence of IFN-γ, IL-6, or caspase 1 (Toomey et al., 2014).
The requirement for IFN-γ was also reflected in expression of proinflammatory cytokines. Mercury-induced increases in IL-4, IL-6, and TNF-α, were dependent upon IFN-γ. Mercury also elicited a significant increase in IL-1β but although this was decreased in the absence of IFN-γ, the difference was not significant. Importantly, with the exception of IL-4 which is not required for mHgIA (Kono et al., 1998), increases in cytokine expression were limited to the site of exposure rather than secondary lymphoid organs. Initial experiments of cytokine mRNA expression in the skin (Supplementary Table 2) were expressed relative to total RNA. Although this is a less sensitive approach to normalization of gene expression than use of the housekeeping gene GAPDH (Bustin et al., 2005), this study did reveal increases in cytokine expression in the absence of IFN-γ, particularly for IL-6 and TNF-α. However, these levels were always lower than that found in wild-type mice. Subsequent experiments using GAPDH confirmed these results with the exception that cytokine expression in IFN-γ null mice, although increased by HgCl2 exposure, were not significantly above PBS controls.
Mercury-exposure had little effect on proinflammatory cytokine expression, including IFN-γ, in the absence of IL-6 with the exception of an increase in IL-4 and a decrease in IL-2. This reduced expression of proinflammatory mediators, particularly IFN-γ, likely contributes to the importance of IL-6 not only to mHgIA (Havarinasab et al., 2009) but also idiopathic models of systemic autoimmunity (Cash et al., 2010). IFN-γ and IL-6 signal through the JAK/STAT pathway to regulate gene expression with IFN-γ preferentially activating STAT1 and IL-6 activating STAT3 (Hu and Ivashkiv, 2009). Although this may result in opposing functional roles (Qi et al., 2013), there is also considerable overlap. In particular both induce overlapping sets of genes, including IRF-1 as well as several acute phase proteins, presumably by activation of different transcription factors which bind common DNA sequences (Yuan et al., 1994). Regulation of IRF-1, which is required for mHgIA (Pollard et al., 2012), by either IFN-γ or IL-6 would significantly impact expression of IFN regulated genes including those regulated by type I IFN (Schmitz et al., 2007). IRF-1 is also involved in the IFN-γ regulated expression of IL-6 together with TNF-α activated p65-NF-κB (Faggioli et al., 1997) or via co-activation of TLR3 and TLR7 pathways (Liu et al., 2015). IFN-γ may also drive IL-6 production in combination with IL-1β (Faggioli et al., 1997). This is supported by our studies and confirms the observation that deficiency of IFN-γ reduces both IL-6 and IL-1β (McLoughlin et al., 2003). However, we also found that absence of caspase 1, which cleaves pro-IL-1β (Dinarello, 2007), while reducing both IL-1β and IFN-γ, did not affect the level of HgCl2-induced IL-6. Casp1 null mice also retained HgCl2-induced increase in IRF-1, suggesting that expression of mHgIA in these mice (Pollard et al., 2012) may be due to IL-6 regulation of IRF-1 (Yuan et al., 1994) and subsequent expression of IFN regulated genes. In composite, these studies point to induction of IRF-1 as a pivotal event in inflammatory responses required for development of mHgIA.
Deficiency of IFN-γ dramatically affected the HgCl2-induced expression of genes regulated predominantly by IFN-γ itself (Ifr1, Irf8, Fcgr1, Msr1, Cxcl10) or type I IFN (If144, Rsad2, Isg15). This is not entirely unexpected as there is extensive overlap in the hundreds of genes regulated by interferon types I and II (Pollard et al., 2013). This is supported by our observation that, type I IFNs (Ifnb, Ifna4) were not significantly increased by HgCl2 exposure, suggesting that IFN-γ was the major IFN affecting gene regulation.
The observation that HgCl2-induced expression of IFN-γ was reduced in the absence of IL-6 was unexpected given that IL-6 has been shown to negatively regulate IFN-γ (Diehl et al., 2000). Recent studies have suggested that both IFN-γ and IL-6 can be co-regulated by the RNA binding protein Arid5a and the ribonuclease Regnase1 (Akira, 2013; Zaman et al., 2016). Arid5a stabilizes mRNA of IL-6 and T-bet, a transcription factor that regulates IFN-γ expression by T cells, thereby augmenting expression of these cytokines. In contrast Regnase-1 cleaves IL-6 and IL-12p40 mRNA and deficiency of Regnase-1 results in an aggressive form of systemic autoimmunity (Akira, 2013). Regnase 1 deficiency is also associated with increased IL-6, IL-12p40 and IFN-γ. Although it is unlikely that Regnase 1 directly affects the stability of IFN-γ mRNA, its effects on IL-12 likely impacts IFN-γ expression. Although we found no effect of HgCl2 on Arid5a expression in wild-type, Ifng null and Il6 null mice, further examination of the role of Arid5a and Regnase1 is warranted in mHgIA given their importance to regulation of IFN-γ, IL-6 and autoimmunity.
The interplay between IFN-γ and IL-6 described in this study covers only the first week of HgCl2 exposure when development of autoimmunity is not fully established. While activated/memory CD44high CD4+ T cells can be found after 1 week (Cauvi et al., 2007), development of autoantibodies occurs within 1–2 weeks (Havarinasab et al., 2007) and renal deposits of immunoglobulin and complement around 4 weeks (Hultman and Enestrom, 1988). There is considerable evidence that IFN-γ and IL-6 both contribute significantly to steps leading to autoimmunity. For example, both cytokines contribute to germinal center and follicular helper T (Tfh) cell function including induction of Bcl-6 (Sweet et al., 2012). Significantly, it is B cell intrinsic IFN-γ receptor signaling that regulates autoimmune responses in germinal centers (Jackson et al., 2016).
There is growing evidence that co-ordinate expression of these 2 cytokines is important in the development of autoimmunity. A combination of IL-5, IL-6, and IFN-γ levels distinguishes preclinical SLE from healthy individuals (Lu et al., 2016), although IL-6 appears less predictive when considered alone (Slight-Webb et al., 2016). Additionally, IL-6 and type I IFN regulated genes may have a pathogenic linkage in dermatomyositis (Bilgic et al., 2009). Interestingly, this study identified Cxcl10 (IP-10) as a type I IFN regulated chemokine, however, CXCL10 levels are reduced in SLE flowing anti-IFN-γ treatment (Welcher et al., 2015) confirming the difficulty in identifying interferons responsible for expression of individual genes. As therapeutic regulation of IFN-γ and IL-6 are attempted in systemic autoimmunity it will be of interest to determine if such approaches effect the functions of both cytokines and whether a combined approach is more beneficial.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENT
This is publication number 20594-MEM from The Scripps Research Institute.
FUNDING
This work was supported by the National Institute of Health [grants ES007511, ES021464 and ES022625 to K.M.P.]. C.B.T. was supported by a National Institute of Environmental Health Sciences Supplement to Support High School and Undergraduate Research Experiences [grant ES007511-S1], an Amylin Pharmaceuticals Research Scholarship and a Julia Brown Research Scholarship from the University of California, San Diego, CA.
REFERENCES
- Akira S. (2013). Regnase-1, a ribonuclease involved in the regulation of immune responses. Cold Spring Harb. Symp. Quant. Biol. 78, 51–60. [DOI] [PubMed] [Google Scholar]
- Barregard L., Sallsten G., Conradi N. (1999). Tissue levels of mercury determined in a deceased worker after occupational exposure. Int. Arch. Occup. Environ. Health 72, 169–173. [DOI] [PubMed] [Google Scholar]
- Bilgic H., Ytterberg S. R., Amin S., McNallan K. T., Wilson J. C., Koeuth T., Ellingson S., Newman B., Bauer J. W., Peterson E. J., et al. (2009). Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. 60, 3436–3446. [DOI] [PubMed] [Google Scholar]
- Boehm U., Klamp T., Groot M., Howard J. C. (1997). Cellular responses to interferon-gamma. Annu. Rev. Immunol. 15, 749–795. [DOI] [PubMed] [Google Scholar]
- Boismenu R., Hobbs M. V., Boullier S., Havran W. L. (1996). Molecular and cellular biology of dendritic epidermal T cells. Semin. Immunol. 8, 323–331. [DOI] [PubMed] [Google Scholar]
- Bustin S. A., Benes V., Nolan T., Pfaffl M. W. (2005). Quantitative real-time RT-PCR–a perspective. J. Mol. Endocrinol. 34, 597–601. [DOI] [PubMed] [Google Scholar]
- Carnaud C., Lee D., Donnars O., Park S. H., Beavis A., Koezuka Y., Bendelac A. (1999). Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 163, 4647–4650. [PubMed] [Google Scholar]
- Carroll J. M., Crompton T., Seery J. P., Watt F. M. (1997). Transgenic mice expressing IFN-gamma in the epidermis have eczema, hair hypopigmentation, and hair loss. J. Invest. Dermatol. 108, 412–422. [DOI] [PubMed] [Google Scholar]
- Cash H., Relle M., Menke J., Brochhausen C., Jones S. A., Topley N., Galle P. R., Schwarting A. (2010). Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: The IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J. Rheumatol. 37, 60–70. Research Support, Non-U.S. Govt. [DOI] [PubMed] [Google Scholar]
- Cauvi D. M., Cauvi G., Pollard K. M. (2007). Reduced expression of decay-accelerating factor 1 on CD4+ T cells in murine systemic autoimmune disease. Arthritis Rheum. 56, 1934–1944. [DOI] [PubMed] [Google Scholar]
- Chen L., Martinez O., Overbergh L., Mathieu C., Prabhakar B. S., Chan L. S. (2004). Early up-regulation of Th2 cytokines and late surge of Th1 cytokines in an atopic dermatitis model. Clin. Exp. Immunol. 138, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun H. Y., Chung J. W., Kim H. A., Yun J. M., Jeon J. Y., Ye Y. M., Kim S. H., Park H. S., Suh C. H. (2007). Cytokine IL-6 and IL-10 as biomarkers in systemic lupus erythematosus. J. Clin. Immunol. 27, 461–466. [DOI] [PubMed] [Google Scholar]
- Crowe W., Allsopp P. J., Watson G. E., Magee P. J., Strain J. J., Armstrong D. J., Ball E., McSorley E. M. (2016). Mercury as an environmental stimulus in the development of autoimmunity - A systematic review. Autoimmun. Rev. doi:10.1016/j.autrev.2016.09.020. [DOI] [PubMed] [Google Scholar]
- Diehl S., Anguita J., Hoffmeyer A., Zapton T., Ihle J. N., Fikrig E., Rincon M. (2000). Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity 13, 805–815. [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. (2007). Interleukin-18 and the pathogenesis of inflammatory diseases. Semin. Nephrol. 27, 98–114. [DOI] [PubMed] [Google Scholar]
- Faggioli L., Merola M., Hiscott J., Furia A., Monese R., Tovey M., Palmieri M. (1997). Molecular mechanisms regulating induction of interleukin-6 gene transcription by interferon-gamma. Eur. J. Immunol. 27, 3022–3030. [DOI] [PubMed] [Google Scholar]
- Gardner R. M., Nyland J. F., Silva I. A., Ventura A. M., de Souza J. M., Silbergeld E. K. (2010). Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: A cross-sectional study. Environ. Res. 110, 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondal G., Gunnarsson I., Ronnelid J., Rogberg S., Klareskog L., Lundberg I. (2000). Cytokine production, serum levels and disease activity in systemic lupus erythematosus. Clin. Exp. Rheumatol. 18, 565–570. [PubMed] [Google Scholar]
- Haas C., Ryffel B., Le Hir M. (1998). IFN-gamma receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice. J. Immunol. 160, 3713–3718. [PubMed] [Google Scholar]
- Haggqvist B., Hultman P. (2001). Murine metal-induced systemic autoimmunity: Baseline and stimulated cytokine mRNA expression in genetically susceptible and resistant strains. Clin. Exp. Immunol. 126, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigai M., Kawamoto M., Hara M., Kubota T., Kamatani N., Miyasaka N. (2008). Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J. Immunol. 181, 2211–2219. [DOI] [PubMed] [Google Scholar]
- Havarinasab S., Bjorn E., Ekstrand J., Hultman P. (2007). Dose and Hg species determine the T-helper cell activation in murine autoimmunity. Toxicology 229, 23–32. [DOI] [PubMed] [Google Scholar]
- Havarinasab S., Pollard K. M., Hultman P. (2009). Gold- and silver-induced murine autoimmunity–requirement for cytokines and CD28 in murine heavy metal-induced autoimmunity. Clin. Exp. Immunol. 155, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemdan N. Y. (2008). The role of interleukin-12 in the heavy metal-elicited immunomodulation: Relevance of various evaluation methods. J. Occup. Med. Toxicol. 3, 25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hron J. D., Peng S. L. (2004). Type I IFN protects against murine lupus. J. Immunol. 173, 2134–2142. [DOI] [PubMed] [Google Scholar]
- Hu X., Ivashkiv L. B. (2009). Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity 31, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultman P., Bell L. J., Enestrom S., Pollard K. M. (1992). Murine susceptibility to mercury. I. Autoantibody profiles and systemic immune deposits in inbred, congenic, and intra-H-2 recombinant strains. Clin. Immunol. Immunopathol. 65, 98–109. [DOI] [PubMed] [Google Scholar]
- Hultman P., Enestrom S. (1988). Mercury induced antinuclear antibodies in mice: Characterization and correlation with renal immune complex deposits. Clin. Exp. Immunol. 71, 269–274. [PMC free article] [PubMed] [Google Scholar]
- Jackson S. W., Jacobs H. M., Arkatkar T., Dam E. M., Scharping N. E., Kolhatkar N. S., Hou B., Buckner J. H., Rawlings D. J. (2016). B cell IFN-gamma receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J. Exp. Med. 213, 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono D. H., Baccala R., Theofilopoulos A. N. (2013). TLRs and interferons: A central paradigm in autoimmunity. Curr. Opin. Immunol. 25, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono D. H., Balomenos D., Pearson D. L., Park M. S., Hildebrandt B., Hultman P., Pollard K. M. (1998). The prototypic Th2 autoimmunity induced by mercury is dependent on IFN-gamma and not Th1/Th2 imbalance. J. Immunol. 161, 234–240. [PubMed] [Google Scholar]
- Lit L. C., Wong C. K., Li E. K., Tam L. S., Lam C. W., Lo Y. M. (2007). Elevated gene expression of Th1/Th2 associated transcription factors is correlated with disease activity in patients with systemic lupus erythematosus. J. Rheumatol. 34, 89–96. [PubMed] [Google Scholar]
- Liu Q., Zhu Y., Yong W. K., Sze N. S., Tan N. S., Ding J. L. (2015). Cutting edge: Synchronization of IRF1, JunB, and C/EBPbeta activities during tlr3-TLR7 cross-talk orchestrates timely cytokine synergy in the proinflammatory response. J. Immunol. 195, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Munroe M. E., Guthridge J. M., Bean K. M., Fife D. A., Chen H., Slight-Webb S. R., Keith M. P., Harley J. B., James J. A. (2016). Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J. Autoimmun. doi:10.1016/j.jaut.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani K., Akahoshi M., Tsuruya K., Tokumoto M., Ninomiya T., Kohsaka T., Fukuda K., Kanai H., Nakashima H., Otsuka T., et al. (2001). Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum. 44, 2097–2106. [DOI] [PubMed] [Google Scholar]
- McLoughlin R. M., Witowski J., Robson R. L., Wilkinson T. S., Hurst S. M., Williams A. S., Williams J. D., Rose-John S., Jones S. A., Topley N. (2003). Interplay between IFN-gamma and IL-6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J. Clin. Invest. 112, 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munroe M. E., Lu R., Zhao Y. D., Fife D. A., Robertson J. M., Guthridge J. M., Niewold T. B., Tsokos G. C., Keith M. P., Harley J. B., et al. (2016). Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis. doi:10.1136/annrheumdis-2015-208140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestle F. O., Di Meglio P., Qin J. Z., Nickoloff B. J. (2009). Skin immune sentinels in health and disease. Nat. Rev. Immunol. 9, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyland J. F., Fillion M., Barbosa F. Jr., Shirley D. L., Chine C., Lemire M., Mergler D., Silbergeld E. K. (2011). Biomarkers of methylmercury exposure immunotoxicity among fish consumers in Amazonian Brazil. Environ. Health Perspect. 119, 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohl K., Tenbrock K. (2011). Inflammatory cytokines in systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 432595 (Review). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K. M., Arnush M., Hultman P., Kono D. H. (2004). Costimulation requirements of induced murine systemic autoimmune disease. J. Immunol. 173, 5880–5887. [DOI] [PubMed] [Google Scholar]
- Pollard K. M., Cauvi D. M., Toomey C. B., Morris K. V., Kono D. H. (2013). Interferon-gamma and Systemic Autoimmunity. Discov. Med. 16, 123–131. [PMC free article] [PubMed] [Google Scholar]
- Pollard K. M., Hultman P., Toomey C. B., Cauvi D. M., Hoffman H. M., Hamel J. C., Kono D. H. (2012). Definition of IFN-gamma-related pathways critical for chemically-induced systemic autoimmunity. J. Autoimmun. 39, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K. M., Hultman P., Toomey C. B., Cauvi D. M., Konoc D. H. (2011). beta2-microglobulin is required for the full expression of xenobiotic-induced systemic autoimmunity. J. Immunotoxicol. 8, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y. F., Huang Y. X., Wang H. Y., Zhang Y., Bao Y. L., Sun L. G., Wu Y., Yu C. L., Song Z. B., Zheng L. H., et al. (2013). Elucidating the crosstalk mechanism between IFN-gamma and IL-6 via mathematical modelling. BMC Bioinformatics 14, 41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller J., Chalaris A., Schmidt-Arras D., Rose-John S. (2011). The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 1813, 878–888. [DOI] [PubMed] [Google Scholar]
- Schmitz F., Heit A., Guggemoos S., Krug A., Mages J., Schiemann M., Adler H., Drexler I., Haas T., Lang R., et al. (2007). Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur. J. Immunol. 37, 315–327. [DOI] [PubMed] [Google Scholar]
- Seery J. P. (2000). IFN-gamma transgenic mice: Clues to the pathogenesis of systemic lupus erythematosus? Arthritis Res. 2, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seery J. P., Carroll J. M., Cattell V., Watt F. M. (1997). Antinuclear autoantibodies and lupus nephritis in transgenic mice expressing interferon gamma in the epidermis. J. Exp. Med. 186, 1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seery J. P., Cattell V., Watt F. M. (2001). Cutting edge: Amelioration of kidney disease in a transgenic mouse model of lupus nephritis by administration of the caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-(beta-o-methyl)-fluoromethylketone. J. Immunol. 167, 2452–2455. [DOI] [PubMed] [Google Scholar]
- Slight-Webb S., Lu R., Ritterhouse L. L., Munroe M. E., Maecker H. T., Fathman C. G., Utz P. J., Merrill J. T., Guthridge J. M., James J. A. (2016). Autoantibody-positive healthy individuals display unique immune profiles that may regulate autoimmunity. Arthritis Rheumatol. 68, 2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Yasukawa K., Saito T., Narazaki M., Hasegawa A., Taga T., Kishimoto T. (1993). Serum soluble interleukin-6 receptor in MRL/lpr mice is elevated with age and mediates the interleukin-6 signal. Eur. J. Immunol. 23, 1078–1082. [DOI] [PubMed] [Google Scholar]
- Sweet R. A., Lee S. K., Vinuesa C. G. (2012). Developing connections amongst key cytokines and dysregulated germinal centers in autoimmunity. Curr. Opin. Immunol. 24, 658–664. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos A. N., Baccala R., Beutler B., Kono D. H. (2005). Type I interferons (alpha/beta) In Immunity And Autoimmunity. Annu. Rev. Immunol. 23, 307–335. [DOI] [PubMed] [Google Scholar]
- Toomey C. B., Cauvi D. M., Hamel J. C., Ramirez A. E., Pollard K. M. (2014). Cathepsin B regulates the appearance and severity of mercury-induced inflammation and autoimmunity. Toxicol. Sci. 142, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vas J., Mattner J., Richardson S., Ndonye R., Gaughan J. P., Howell A., Monestier M. (2008). Regulatory roles for NKT cell ligands in environmentally induced autoimmunity. J. Immunol. 181, 6779–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzer T., Dalod M., Robbins S. H., Zitvogel L., Vivier E. (2005). Natural-killer cells and dendritic cells: “l'union fait la force”. Blood 106, 2252–2258. [DOI] [PubMed] [Google Scholar]
- Welcher A. A., Boedigheimer M., Kivitz A. J., Amoura Z., Buyon J., Rudinskaya A., Latinis K., Chiu K., Oliner K. S., Damore M. A., et al. (2015). Blockade of interferon-gamma normalizes interferon-regulated gene expression and serum CXCL10 levels in patients with systemic lupus erythematosus. Arthritis Rheumatol. 67, 2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Wegenka U. M., Lutticken C., Buschmann J., Decker T., Schindler C., Heinrich P. C., Horn F. (1994). The signalling pathways of interleukin-6 and gamma interferon converge by the activation of different transcription factors which bind to common responsive DNA elements. Mol. Cell Biol. 14, 1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman M. M., Masuda K., Nyati K. K., Dubey P. K., Ripley B., Wang K., Chalise J. P., Higa M., Hanieh H., Kishimoto T. (2016). Arid5a exacerbates IFN-gamma-mediated septic shock by stabilizing T-bet mRNA. Proc. Natl. Acad. Sci. U.S.A. 113, 11543–11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.