

Abstract
Sulfur mustard (SM) is a chemical warfare agent that causes chronic airway remodeling. This study’s objective was to assess for changes to the bronchiolar epithelium after SM exposure to explain its contribution to chronic airway remodeling.
Materials and methods: Adult male rats were exposed to a sublethal dose of SM inhalation (1.0–1.2 mg/kg) for 50 min. Histological sections of the bronchiolar epithelium were analyzed for changes using hematoxylin and eosin, trichrome, and immunofluorescent staining for acetylated tubulin (AT) and club cell secretory protein (CCSP). CCSP in bronchoalveolar lavage fluid was assessed using western blot. A bromodeoxyuridine (BRDU) assay was used to assess for epithelial proliferation, and real-time PCR measured changes in Notch mRNA expression.
Results: SM caused significant proximal bronchiolar epithelial injury with epithelial denudation, loss of acetylated tubulin and CCSP staining, and reduced bronchoalveolar lavage fluid CCSP levels. bromodeoxyuridine (BRDU) + staining of proximal bronchiolar epithelial cells was not increased, but staining was increased in the distal bronchiolar epithelium. One month after injury, the proximal bronchiolar epithelium was not fully repaired. Significant collagen deposition surrounded proximal bronchioles with luminal obstruction, consistent with bronchiolitis obliterans. These changes corresponded with a downregulation of Notch1, Notch3, and Hes1 mRNA expressions.
Conclusions: This study demonstrates that SM exposure resulted in severe proximal airway epithelial injury, persistent morphological changes, impaired epithelial proliferation and, ultimately, bronchiolitis obliterans. These changes occurred at the same time that the Notch signaling genes were downregulated. Thus, the lung epithelium and the Notch signaling pathway may be worthy targets for the prevention of chronic airway remodeling after SM inhalation injury.
Keywords: sulfur mustard, bronchiolitis obliterans, epithelium, Notch, proliferation, regeneration
Sulfur mustard (SM) is a common chemical warfare agent. Exposure affects multiple organs, including the skin, gastrointestinal, nervous system, and respiratory system (Somani and Babu, 1989; Willems, 1989). Airway injury is the principal cause of mortality after SM exposure. In humans, acute SM inhalation at high concentrations has caused cardiorespiratory failure, bronchial obstruction, plastic bronchitis, pulmonary edema, and in a few extreme cases, death (Ghanei et al., 2011, 2012; Saber et al., 2012; Willems, 1989).
The majority of humans exposed to SM have long-term symptoms with productive cough, dyspnea, and sputum production (Ghanei et al., 2008, 2010; Zarchi et al., 2004). Radiologic findings include air trapping and, in some cases, bronchiectasis on high-resolution chest tomography (Ghanei et al., 2010). Bronchiectasis is a chronic lung disease state characterized by permanently damaged, thickened, and enlarged bronchial airways. However, the predominant histopathology on lung biopsy on patients exposed to SM was bronchiolitis obliterans with impaired significant epithelial injury (Beheshti et al., 2006; Ghanei et al., 2008). Bronchiolitis obliterans is a progressive and debilitating chronic lung disease state, affecting the conducting airways, resulting in a permanent and slowly progressing fixed airway obstruction. When bronchiolitis obliterans is severe, the airways become permanently scarred, resulting in decreased pulmonary function, and sometimes also chronic respiratory failure.
As a significant majority of subjects biopsied showed histopathological changes of the airway epithelium, Ghanei et al. (2011) used immunohistochemistry to characterize specific epithelial changes with basal cell markers of cytokeratin 5 (CK5) and p63 in histological sections. In those biopsies with findings of constrictive bronchiolitis or bronchiolitis obliterans, the mean number of cells positive for CK5 and p63 were significantly reduced, suggestive of a reduced regenerative capacity of the bronchial epithelium adjacent to areas of pathologic bronchiolitis obliterans.
Studies in animal models after SM exposure have recapitulated some of the findings from human pathology. Using guinea pigs with full body exposure to SM, Calvet et al. (1994) assessed for histologic changes of the proximal tracheal epithelium, in the acute phase (5 days) and the subacute phase (14 days) of repair after SM. The guinea pig’s tracheal epithelium showed sloughing and edema acutely. In the subacute phase, the epithelium had not returned to normal with an absence of secretory cells and cuboid-shaped ciliated cells with few attached cilia. In a subsequent study by the same group using intratracheal delivery of SM (0.3 mg/kg), the tracheal epithelium had not fully recovered 1 month after exposure (Calvet et al., 1996). These results suggest that SM causes both acute and persistent epithelial injury with impaired tracheal epithelial differentiation.
Allon et al. (2009) further assessed for changes in the tracheal epithelium 1 month after injury at various concentrations of SM exposure. Results demonstrated that changes in the tracheal epithelium were dose-dependent. At higher concentrations of SM, a large percentage of the trachea lacked an intact epithelial layer with only a small percentage remaining stratified. Additionally, at higher concentrations of SM, the number of ciliated cells and goblet cells were also reduced. Thus, the epithelium’s ability to repair after SM inhalation was impaired in a dose-dependent manner.
Despite advances in animal models studying SM, the mechanism of how SM inhalation contributes to impaired epithelial proliferation and differentiation remains poorly understood. Additionally, most of these prior studies have only assessed changes in the more proximal trachea and not in the bronchiolar epithelium where the predominance of long-term airway remodeling occurs.
This study’s objectives were to assess for changes to the proximal and distal bronchiolar epithelium in both the acute and chronic phases of injury after SM exposure, and to suggest one mechanistic pathway involved in epithelial proliferation and differentiation by assessing for changes in mRNA expression levels of the Notch signaling pathway, a known developmental pathway involved in epithelial regeneration after injury.
MATERIALS AND METHODS
Animal research approval
The study protocol was approved by University of Colorado’s Institutional Animal Care and Use Committee (IACUC).
Animals
Male Sprague-Dawley rats weighing 250–300 g (Charles River Laboratory, Wilmington, Massachusetts) were used in this study, maintained in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited animal care facility. All animals were provided commercially certified rat chow and water ad libitum. Rats were quarantined for 7 days following arrival to allow for acclimatization, and their health status was monitored daily. In conducting the research described in this report, the investigators adhered to the “Guide for Care and Use of Laboratory Animals” (1996).
Chemicals and reagents
SM (2,20-dichlorodiethyl sulfide; 4 mM, vapor inhalation) was made at the University of Colorado Research facilities.
SM inhalation exposure
SM inhalation exposure was performed at University of Colorado Denver, Denver, Colorado. For all studies performed, rats were exposed to SM inhalation using a vapor generator-based model as previously described in Anderson et al. (2000). Briefly, SM was diluted in 100% ethanol, and a dose of 1.0 or 1.2 mg/ml was placed into the water-jacketed (37 °C) glass vaporization chamber for exposure. Survival time was measured for each animal as the time from exposure to euthanasia. All animals were exposed to a single, acute 50-min exposure of SM vapor. A dose-response study for SM inhalation injury in rats to validate the current doses was performed by our lab (McGraw MD, et al., White CW, Veress LA; article in transcription for publication). The time to 50% mortality in the rats was 6 days (LD50 at 6 days is 1.0 mg/kg SM; Supplementary Figure 1). Of the animals who survived the acute injury, a second, more gradual, decline in mortality occurred after 14 days with a progressive decline in oxygen saturations, weight, and lung function, as well as histological evidence of both pulmonary fibrosis and bronchiolitis obliterans weeks after the initial exposure.
Euthanasia criteria
Animals were euthanized at scheduled time points (1, 2, 7, 14, 21, or 28 days), or when euthanasia criteria was met due to severity of illness, as per University of Colorado IACUC guidelines. Animals were evaluated once daily. To meet euthanasia criteria, an animal required an oxygen saturation <70% and clinical score >7, as previously described (Veress et al., 2013), or if weight loss >30% occurred. If animals were found dead, their lungs were not included in epithelial analysis due to tissue degradation concerns. Epithelial analysis was only performed on animals that were euthanized at scheduled time points. For euthanasia, a cocktail of ketamine (75 mg/kg)/xylazine (7.5 mg/kg)/acepromazine (1.5 mg/kg) was administered by intraperitoneal injection. Once unresponsive to toe-pinch maneuver, the animal was euthanized via diaphragmatic puncture, followed by exsanguination. After careful clamping of the left main stem bronchus with a bulldog clamp, right lung lavage was performed with two 2.5 ml washes of sterile 0.9% normal saline. Collected lavage fluid was centrifuged at 1000 rpm for 10 min and aliquots of the lavage fluid supernatant were frozen at −80°C until use. Right lung was then perfused free of blood, using sterile 0.9% normal saline delivered into the pulmonary artery via a perfusion pump. Right lung was then tied off via suture at the right main stem, followed by removal and snap freezing of all right lobes. After removal of left bronchus clamp, the left lung lobe was intratracheally fixed in 4% PFA at 20 cm H2O for 30 min, then removed and placed in same fixative, and refrigerated.
Histology
Slides were stained with hematoxylin and eosin (H&E) or Masson’s trichrome, and evaluated by light microscopy. H&E and Masson’s trichrome staining were performed as per manufacturer’s instructions on paraffin embedded whole lung sections (kit No. AR173, Dako, Carpenteria, California).
Airway nomenclature was followed based on published criteria (Lee et al., 2011; Paige et al., 1997). A terminal, or distal, bronchiole was defined as the single-layered epithelium present 1–3 generations proximal to the first alveolar out pocketing (airway generations 20–24), at a luminal diameter of <300 μm in a 12-week-old rat. A proximal bronchiole was defined as the single-layered epithelium present 3–6 generations proximal to the terminal bronchiole (airway generations 14–20), at a luminal diameter of 300–500 µm in a 12-week-old rat. These locations in the bronchial tree were selected based off of: the location of bronchiolitis obliterans in this model (McGraw et al. White CW, Veress LA; article in transcription for publication); the single epithelial layer; and the distribution of basal and club cells, 2 known resident progenitor cells of the lung epithelium (Hogan et al., 2014; Rock et al., 2011).
In trichrome-stained lung sections, all airways were assessed for collagen deposition within walls. Those airways that stained positive for collagen (blue) and/or fibrin (pink), were considered positive for airway fibrosis.
Immunofluorescence
Serial 5-mm sections were obtained from blocks; mounted on silane-coated glass slides; deparaffinized with xylene; dehydrated in graded alcohol; and processed for immunofluorescence. Cells containing acetylated tubulin (AT) and club cell secretory protein (CCSP) were localized in lung sections with respective mouse monoclonal antibody raised against rat AT (1:1000, Abcam, Cambridge, Massachusetts) and goat polyclonal antibody raised against rat CCSP (1:2500; Santa Cruz Biotechnology, Santa Cruz, California). Rabbit IgG (1:1000; Dako) was also used as a negative control. Briefly, sections were washed in PBS buffer and blocked with 10% normal BSA in PBS for 1 h at room temperature. Slides were then rinsed and incubated sequentially at 4 °C with primary antibody overnight. After PBS rinse, the slides were counterstained with AlexaFluor secondary immunofluorescent antibody (diluted 1:1000; ThermoFisher Scientific, Rockford, Illinois, USA) and mounted with DAPI Fluoromount-G (Southern Biotechnology, Birmingham, Alabama) followed by cover slide.
Bromodeoxyuridine assay
We measured bromodeoxyuridine (BRDU) incorporation using a BRDU proliferation protocol for animals 1, 2, 14, and 28 days after SM exposure. 6–8 animals were euthanized on each respective day with 3–4 exposed animals and 3–4 naïve, matched controls. Briefly, animals were injected 2 h prior to euthanasia with 10 mg/l of BRDU. Lung sections were fixed and incubated with a mouse antiBRDU monoclonal antibody (1:320; Abcam) overnight at 4 °C. Cells were washed 3 times in TBS-T and counterstained with AlexaFluor secondary immunofluorescent antibody (1:1000; ThermoFisher Scientific) for 30 min at room temperature. The cells were washed 3 times, and mounted with DAPI Fluoromount (Southern Biotechnology).
The number of BRDU positive cells was counted in both proximal and distal bronchioles. A total of 10 bronchioles, 5 proximal, and 5 distal, were assessed for each slide section. For each bronchiole, the circumferential luminal length was measured for standardization. The total number of BRDU positive cells was normalized to 1000 µm of bronchiolar luminal surface length.
Western blot
Analysis of CCSP in bronchoalveolar lavage fluid (BALF) was performed on samples collected at 1, 7, 14, 21, and 28 days after SM exposure. 5 animals’ lavage samples were analyzed at each postexposure time point and compared with naïve control animals’ lavages for quantification. Total protein of each sample (Pierce BCA assay kit, Thermo Scientific, Waltham, Massachusetts) was measured to normalize each BALF sample loading to 5 µg total protein. Sample values appearing below the lower limit of detection for each assay were considered to be 0 pg./ml/mg total protein. Albumin was used as a standard control measured by Ponceau staining (Rancourt et al., 2014).
BALF was added to 5× Laemmli buffer (1:4) and (5 µg total protein) loaded onto SDS-polyacrylamide gels. After transfer, membranes were incubated overnight with primary antibody recognizing goat antiCCSP (1:20 000; gift from Dr Susan Reynolds). Blots were washed, followed by incubation with a rabbit antigoat secondary antibody (1:10 000; Southern Biotechnology). Experiments were performed under reducing conditions.
RNA processing
Total RNA was extracted from right lower lung lobes using a modified version of the TRIzol method (InVitrogen, Carlsbad, California), in which RNA was purified directly from the aqueous phase by Rneasy purification (Rneasy MinElute RNA purification kit, Qiagen, Valencia, California). RNA integrity was determined by running an aliquot of each RNA sample on an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, California). The concentration was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, Delaware). RNA samples accepted for further processing met 3 quality control criteria: (1) A260/A280 ratio between 1.7 and 2.3; (2) concentration between 0.1 and 1 mg/ml; and (3) Agilent electropherogram displaying 2 distinct peaks corresponding to the 28S and 18S ribosomal RNA bands at a ratio of 0.5 or greater with minimal or no degradation.
Double-stranded cDNA was synthesized from 500 µg total RNA using the Reverse Transcriptase cDNA Synthesis Kit with gDNA Wipeout, followed by quantification of the biotin-labeled cDNA yield by spectrophotometry. All kits were purchased from QIAGEN.
Real-time PCR was carried out using the Power SYBR Green PCR master mix (4367659; Applied Biosystems, Inc.) on an Applied Biosystems 7500 System with associated technology (Thermo Fisher Scientific, Rockford, Illionois, USA). Hypoxanthine phosphoribosyltransferase 1 (HPRT1) was used as the endogenous control as both beta-actin and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were significantly altered after SM exposure in lung homogenate (Cabiati et al., 2012). Primers for Notch ligand, receptors, and downstream effector genes, including Delta-like ligand1 (DLL1), Jagged1 (Jag1), Notch1, Notch2, Notch3, Notch4, Hes1, Hes5, and Hey1, were obtained from Genecopoeia Inc., Maryland, USA. All primer sequences are listed in Supplementary Table 1. The Ct value of each sample was normalized to HPRT1 as the endogenous control gene and the relative quantification was calculated by the 2−ΔΔCt method.
Statistical analysis
Prism 6.0 software (GraphPad, La Jolla, California) was used, with Kurskal-Wallis 1-way analysis of variance (ANOVA) followed by Tukey’s post hoc analysis, unless otherwise indicated. All mean values are reported with SEM. A P < .05 was considered significant.
RESULTS
Histopathology of Conducting Airways
To characterize changes to the airway's epithelium after SM exposure, we analyzed histological sections of both proximal and distal bronchiolar epithelium of the conducting airways (Figure 1). In a naïve rat, the proximal bronchiolar epithelium consisted of a single layer of epithelium with a uniform underlying basement membrane (Figure 1A). Two days after SM exposure (Figure 1B), the epithelium was patchy with areas of complete epithelial denudation. In areas without denudation, the epithelium was flattened with squamous morphology and lacking cilia. This was in stark contrast to the typical columnar, ciliated epithelium of a naïve rat proximal bronchiole (Figure 1A, inset).
FIG. 1.

Histological sections of proximal and distal conducting airways. (size bar: 200 µm.) Inset: Higher magnification of epithelial layer. (size bar: 50 µm.) A, Naïve rat section with the proximal bronchiolar epithelium with a single layer of epithelium with a uniform underlying basement membrane. B, Two days after SM exposure, the epithelium was patchy with areas of complete epithelial denudation and flattened, squamous-appearing epithelium without cilia. C, Two weeks after exposure, areas of proximal epithelium appeared with persistent areas of denudation and metaplastic, squamous epithelium. The surrounding mesenchyme was disorganized with significant inflammatory infiltrates. D, One month after SM injury, the epithelial layer of the proximal bronchioles was still not restored with epithelial sloughing (black arrow).
Two weeks after exposure (Figure 1C), areas of proximal epithelium were not repaired with continued areas of denudation and metaplastic, squamous-appearing epithelium. Beneath the epithelium, the mesenchyme was disorganized, with significant inflammatory infiltrates. One month after injury (Figure 1D), during the chronic phase of repair, the proximal bronchiolar epithelium was not restored with areas of persistent epithelial sloughing (black arrow; Figure 1D). Below the epithelial layer, the surrounding mesenchyme was also thickened. Trichrome staining of the proximal bronchioles 21 days after SM exposure confirmed significant collagen deposition surrounding the injured proximal bronchiolar epithelium (Figure 2). These findings of surrounding collagen deposition with a metaplastic or denuded epithelium as well as a narrowed airway lumen were pathologically consistent with bronchiolitis obliterans. Thus, in the proximal conducting airway, SM inhalation caused significant epithelial injury with patchy epithelial denudation. Weeks after the initial injury, the epithelium’s integrity was not restored with patchy areas of denudation and subepithelial collagen deposition.
FIG. 2.
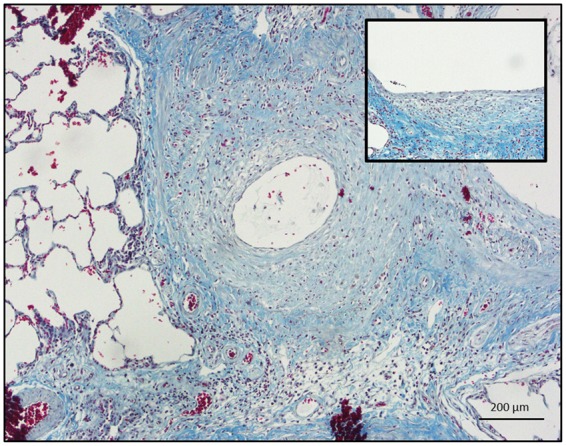
Trichrome staining of a proximal rat bronchiole 21 days after SM exposure with significant collagen deposition surrounding an injured and denuded proximal epithelium (size bar: 200 µm.). Inset: Higher magnification demonstrating a patchy and squamous-appearing epithelium overlying dense, subepithelial collagen (blue) staining (size bar: 50 µm).
Immunofluorescent Staining of Proximal and Distal Bronchiolar Epithelium
We performed immunofluorescent staining for common epithelial markers, including AT and CCSP, to further characterize the morphological changes seen on H&E. In naïve rat controls, the proximal bronchiolar epithelium was well organized. The majority of cells stained positive for AT but with a few, sporadic cells positive for CCSP (Figure 3A). Two days after SM exposure, there was near complete loss of staining for both AT and CCSP (Figure 3B). Fourteen and twenty-eight days after the initial exposure, staining for AT and CCSP remained absent with areas of complete denudation (Figs. 3C and D, respectively).
FIG. 3.

Immunofluorescent staining for common epithelial markers, including AT (red) and CCSP (green) of proximal bronchiolar epithelium (size bar: 100 µm). A, Naïve rat proximal bronchiolar epithelium with the majority of cells staining positive for AT but with a few, sporadic cells positive for CCSP. B, Two days after SM exposure, there was near complete loss of staining for both AT and CCSP. C, Fourteen days after SM injury, staining for ciliated and club cells remained absent with areas of complete denudation. D, Twenty-eight days after SM injury, staining of the proximal epithelium remained absent for AT and CCSP.
Due to the severe and persistent changes seen in the proximal bronchiolar epithelium, we assessed the more distal bronchiolar epithelium for additional morphological changes. In naïve controls, the distal bronchiolar epithelium showed well-organized staining for AT and CCSP (Figure 4A). In comparison to the proximal bronchiolar epithelium, the distal bronchioles showed an increase in CCSP positive cells. Two days after SM exposure, the distal epithelium showed normal architectural integrity, but with complete loss of CCSP staining (Figure 4B). AT staining remained in the distal bronchioles 2 days after injury, suggestive of the persistence of ciliated cells. Fourteen days after injury, CCSP staining returned, but was less uniform (Figure 4C). By day 28 postexposure, the epithelium’s structure appeared similar to naïve bronchioles with uniform AT and CCSP staining (Figure 4D). Thus, different from the more proximal epithelium, the distal bronchiolar epithelium’s structure remained intact, with a loss of CCSP staining, but persistence of AT staining, within days of SM exposure. Weeks after injury, the number of CCSP positive cells returned to levels equivalent to naïve controls.
FIG. 4.

Immunofluorescent staining for common epithelial markers, including AT (red) and CCSP (green), of distal bronchiolar epithelium (size bar: 100 µm). A, In naïve control rats, the distal bronchiolar epithelium showed well organized staining with both ciliated and CCSP. B, Two days after SM, the distal epithelium showed normal architectural integrity and persistent AT staining, but with complete loss of CCSP staining. C, Fourteen days after injury, CCSP staining returned, but was less uniform than naïve control. D, Twenty-eight days postexposure, the epithelium’s structure appeared similar to naïve bronchioles with uniform AT and CCSP staining.
Western Blot of BALF CCSP
Since club cells are known to function both as early progenitor cells as well as differentiated secretory cells, BALF was assessed using Western blot for CCSP quantification (Figure 5A bottom line) to see if the CCSP positive cells were functioning as differentiated secretory cells after injury. Albumin was used as the positive standard control (Figure 5A top line). The amount of CCSP was quantified at each sequential time point by comparing the amount of BALF CCSP relative to albumin on postexposure day to the amount of BALF CCSP relative to albumin in naïve control (Figure 5B).
FIG. 5.

A, Western blot of BALF for CCSP quantification (bottom line). Albumin was used as the positive standard control (top line). B: Quantification of change in BALF CCSP normalized to albumin on postexposure day to amount of BALF CCSP normalized to naïve control. CCSP was significantly reduced on day 1 (0.03 ± 0.13 vs. 1.00 ± 0.03; P < .0001) and remained reduced on day 7 (0.29 ± 0.12; P < .0001) and day 14 (0.51 ± 0.13; P < .01) after SM exposure. By day 21 postexposure, CCSP returned to levels equivalent to that of naïve controls (0.97 ± 0.13; P > .05). By day 28 postexposure, BALF CCSP was increased in exposed animals compared with controls (1.38 ± 0.13 vs 1.00 ± 0.03; P < .05).
When compared with naïve controls, CCSP was significantly reduced on day 1 (0.03 ± 0.13 vs 1.00 ± 0.03; P < .0001) and remained reduced on day 7 (0.29 ± 0.12; P < .0001) and day 14 (0.51 ± 0.13; P < .01) after SM exposure. By day 21 postexposure, CCSP returned to equivalent levels to that of naïve controls (0.97 ± 0.13; P > .05). By day 28 postexposure, BALF CCSP was increased in exposed animals compared with controls (1.38 ± 0.13 vs 1.00 ± 0.03; P < .05, respectively). Thus, within the first 2 weeks after SM exposure, the amount of secreted CCSP relative to total albumin in BALF was reduced, suggestive of impaired secretory function of club cells after SM exposure. Within 3 weeks of injury, the amount of CCSP relative to albumin was equivalent to naïve controls, but by 1 month, the amount of BALF CCSP was increased, suggestive of increased secretion by the distal bronchiolar club cells.
Epithelial Proliferation—BRDU Staining
As the morphology and functionality of the proximal and distal conducting airways differed significantly after injury, we performed BRDU staining to assess the bronchiolar epithelium’s ability to proliferate. We assessed BRDU staining at day 1, 2, 14, and 28 after SM exposure in both the proximal and distal conducting airway. In the proximal bronchioles, there was no significant increase in the number of proliferative epithelial cells per 1000 µm of epithelial length on days 1 or 2 after exposure compared with naïve controls (day 1: 4.4 ± 0.3; day 2: 5.1 ± 1.0; Naïve: 2.6 ± 0.5; P > .05, Figure 7A). Beneath the non-proliferative proximal bronchiolar epithelium, BRDU staining was present in the adjacent, subepithelial mesenchyme on day 1 and 2 postexposure (Figure 6A). At days 14 and 28 after SM exposure, the number of proliferative, BRDU positive epithelial cells was equivalent to naïve controls (day 14: 5.0 ± 0.5; day 28: 3.8 ± 0.5; P > .05).
FIG. 7.

Number of BRDU positive epithelial cells per 1000 µm of epithelial length in proximal (A) and distal (B) bronchiolar epithelium. 5 proximal and distal bronchioles were assessed for each animal with 3–4 animals at each time point (naïve, days 1, 2, 14, and 28). (A) There was no significant increase in the number of BRDU positive epithelial cells per 1000 µm of the proximal bronchiolar epithelial length on days 1 or 2 after exposure compared with naïve controls (Naïve: 2.6 ± 0.5; day 1: 4.4 ± 0.3; day 2: 5.1 ± 1.0; day 14: 5.0 ± 0.5; day 28: 3.8 ± 0.5; P > .05). (B) The number of BRDU+ epithelial cells was significantly increased in the distal bronchiolar epithelium on day 1 after injury compared with naïve controls (14.2 ± 0.9 vs 3.4 ± 0.4; P = .0001) and remained significantly elevated on day 2 after injury (7.0 ± 0.7 vs 3.4 ± 0.4; P = .01), but was equivalent to naïve controls on day 14 and 28 (day 14: 3.3 ± 0.4; day 28: 3.8 ± 0.4; P > .05).
FIG. 6.
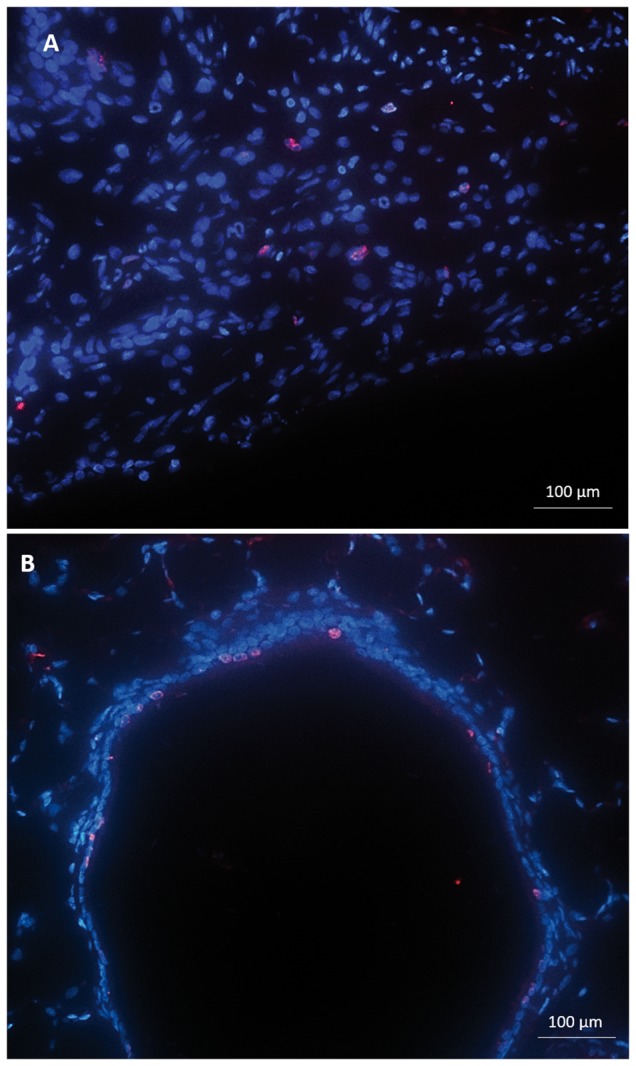
Immunofluorescent staining for BRDU (pink) and DAPI nuclear (blue) staining at day 1 after SM exposure in both the proximal (A) and distal conducting airway (B). (Size bar: 100 µm). (A) In the proximal bronchioles, there was no significant increase in the number of proliferative epithelial cells per 1000 µm of epithelial length. BRDU staining was present in the adjacent, subepithelial mesenchyme. (B) In the distal bronchioles, there was a significant increase in number of proliferative epithelial cells per 1000 µm of epithelial length.
In contrast, the number of proliferative, BRDU+ epithelial cells was significantly increased in the distal bronchiolar epithelium on day 1 after injury compared with naïve controls (14.2 ± 0.9 vs 3.4 ± 0.4; P = .0001, Figs. 6B and 7B). The number of BRDU+ cells remained significantly elevated on day 2 after injury (7.0 ± 0.7 vs 3.4 ± 0.4; P = .01, Figure 7B). By day 14 and 28 after SM exposure, the number of proliferative, BRDU positive epithelial cells in the distal bronchioles was equivalent to naïve controls (day 14: 3.3 ± 0.4; day 28: 3.8 ± 0.4; P > .05). Additionally, there was no appreciable increase in the number of adjacent, subepithelial BRDU+ cells at any time point in the distal bronchioles. Collectively, these results demonstrate a lack of proliferation present in the proximal bronchial epithelium, but with a significant increase in distal bronchiolar epithelial proliferation after SM inhalation injury. Furthermore, in proximal areas lacking epithelial proliferation, there was adjacent subepithelial proliferation, suggestive of compensatory proliferation of the mesenchyme.
mRNA Notch Signaling
One developmental pathway known to assist with epithelial proliferation and differentiation after lung injury is the Notch signaling pathway (Hogan et al., 2014; Mori et al., 2015; Rock et al., 2011; Tsao et al., 2008, 2016; Vaughan et al., 2015; Xing et al., 2012). Relative quantification of mRNA expression levels of certain Notch ligands, receptors, and downstream effector genes were assessed sequentially after SM exposure.mRNA expression of 2 Notch receptor genes, Notch 1 and Notch 3, and 1 downstream effector, Hes1, were found to be upregulated within the first 2 days of SM exposure (Figure 8). At days 1 and 2 postexposure, Notch 1 levels were upregulated compared with normalized naïve controls (2.27 ± 0.40 and 1.74 ± 0.43, respectively, Figure 8A). Gene expression levels of Notch 1 receptor progressively declined after day 2. By day 14, Notch1 levels were downregulated, and differed significantly from days 1 to 2 (0.30 ± 0.38, P < .001 and P < .05, respectively). Levels remained significantly downregulated at days 21 and 28 compared with day 1 expression levels (0.35 ± 0.38, P < .001; 0.41 ± 0.40, P < .05).
FIG. 8.

Relative quantification of RNA expression of 2 Notch receptor genes, Notch1 (A), Notch3 (B), and 1 downstream effector gene, Hes1 (C). The Ct value of each sample was normalized to HPRT1 as the endogenous control gene and relative quantification was calculated by the 2−ΔΔCt method. (A) On days 1 and 2 postexposure, Notch 1 levels were upregulated (2.27 ± 0.40 and 1.74 ± 0.43, respectively), but downregulated on days 14, differing significantly from day 1 and 2 (0.30 ± 0.38, P < .001 and P < .05). RNA expression levels remained significantly downregulated at days 21 and 28 compared with day 1 expression levels (0.35 ± 0.38, P < .001; 0.41 ± 0.40, P < .05). (B) Receptor Notch3 RNA levels were significantly upregulated on day 2 postexposure (1.84 ± 0.24, P < .05) but downregulated on day 7 (0.60 ± 0.22, P < .001), day 14 (0.53 ± 0.24, P < .001), day 21 (0.32 ± 0.22, P < .0001), and day 28 (0.53 ± 0.24, P < .01) compared with day 2. (C) Hes1 levels were significantly upregulated on days 1 and 2 (2.66 ± 0.45, P < .05 and 3.03 ± 0.45, P < .01. By day 7, Hes1 RNA levels were downregulated (0.73 ± 0.51) and remained significantly downregulated at days 14, 21, and 28 (P < .05) compared with days 1 and 2.
Receptor Notch3 mRNA was also significantly upregulated on day 2 postexposure (1.84 ± 0.24, P < .05; Figure 8B), but was also significantly downregulated on day 7 (0.60 ± 0.22, P < .001), day 14 (0.53 ± 0.24, P < .001), day 21 (0.32 ± 0.22, P < .0001), and day 28 (0.53 ± 0.24, P < .01) compared with day 2.
Similar changes were present in the downstream effector gene Hes1 mRNA expression levels. At days 1 and 2, Hes1 levels were significantly upregulated (2.66 ± 0.45, P < .05 and 3.03 ± 0.45, P < .01; Figure 8C). By day 7, Hes1 RNA levels were downregulated (0.73 ± 0.51) and remained significantly downregulated at days 14, 21, and 28 (P < .05) compared with days 1 and 2. The Notch ligand, Jag1, showed similar trends with a modest upregulation on days 1 and 2 after injury and downregulation thereafter, but these changes were not statistically significant. Other ligands, receptors and downstream effector genes, including DLL1, Notch2, Notch4, Hey1, Hes5, were assessed but did not show statistically significant changes in mRNA levels following SM exposure.
Collectively, Notch1, Notch3, and Hes1 mRNA levels were upregulated within the first 2 days of injury. The upregulation of these genes occurred on the same days when proliferation of the distal bronchiolar epithelium and adjacent proximal mesenchyme was significantly increased by BRDU. More importantly, RNA gene expressions of Notch1, Notch3, and Hes1 were downregulated at time points when the proximal bronchiolar epithelium remained denuded and unrepaired without significant proliferation and a squamous morphology, suggestive of impaired epithelial regeneration after SM exposure.
DISCUSSION
This study’s purpose was to assess for persistent changes to the proximal and distal bronchiolar epithelium, during the subacute and chronic phases of repair, following a single SM inhalation exposure to explain some of the histological changes of the conducting airways. Furthermore, we assessed RNA gene expression of the Notch signaling pathway, a known pathway involved in epithelial proliferation and differentiation, to help explain the impaired epithelial proliferation and differentiation seen after SM inhalation injury.
In the proximal bronchiolar epithelium, SM inhalation resulted in direct epithelial injury with denudation and morphological changes. Weeks after the initial injury, the proximal bronchiolar epithelium was not fully repaired with a metaplastic, squamous-appearing epithelium compared with naïve control. In other areas of the proximal bronchiolar epithelium, areas of complete denudation persisted without noticeable recovery.
Using immunofluorescent staining, ciliated and club cell markers, AT and CCSP, were absent 1 month after the initial injury in the proximal bronchiolar epithelium. Some areas showed return of these common epithelial markers, but the majority of the proximal airway remained without these cell markers. Subepithelial fibrosis was also more common in areas of impaired epithelial regeneration.
After chlorine gas exposure, O’Koren et al. (2013) demonstrated the development of ‘obliterative bronchiolitis-like’ pathology in the proximal trachea following acute inhalation exposure. These authors showed that fibrosis developed in areas of epithelial denudation. Results from this paper were similar to this article in that fibrosis was present in areas of significant epithelial injury. The injury from chlorine gas exposure was more proximal in the trachea while the inhalation injury from SM was present in the more distal bronchioles where bronchiolitis obliterans develops pathologically. The location of the inhaled injury along the bronchial tree was relative to each gas’s solubility and reactivity. Chlorine gas is more soluble in water than SM and is a highly reactive gas forming hypochlorous acid (Bracher et al., 2012; White and Martin, 2010). Both of these properties contribute to the more proximal airway injury. In contrast, SM is extremely insoluble in aqueous solution; is highly lipophilic; and requires ethanol for vaporization (White et al., 2016). SM’s insolubility in water and lower reactivity than chlorine supports the bronchiolar distribution of inhalation injury of this study.
Using conditional knock-out and transgenic mice, Perl et al. (2011) demonstrated the development of subepithelial fibrosis after a recurrent airway injury to epithelial progenitor cells. Fibrosis developed when the variant club cells, a known epithelial progenitor cell, were depleted. These results suggested that depletion of specific airway progenitor cells contributes to the development of subepithelial fibrosis.
Here, we show the development of bronchiolitis obliterans in the proximal bronchioles. This pathology developed in areas of the conducting airway where the epithelium was most severely injured. In this proximal location of the conducting airway, the number of BRDU+ positive cells was not increased, suggestive that the epithelium’s ability to proliferate was impaired. In the adjacent mesenchyme, BRDU+ cells were increased, suggestive of a compensatory, mesenchymal proliferation to support the injured epithelium. Similar results were demonstrated by Calvet et al. (1996) where the number of proliferating cells of the tracheal epithelium were reduced after intratracheal SM exposure. Differences between these prior studies and our results are that the prior studies used direct intratracheal delivery of topical SM while this study used vaporized SM. Furthermore, these authors did not publish their assessment of the bronchiolar epithelium. In this study, ciliated and club cell staining were not present in the proximal epithelium 1 month after exposure, suggesting that not only proliferation but also differentiation was impaired.
In the distal bronchiolar epithelium, the epithelium remained intact. There was a loss of CCSP staining likely due to immediate secretion of CCSP after SM exposure. The loss of CCSP staining on the distal bronchiolar epithelium corresponded temporally with a decrease in CCSP concentration in BAL fluid. Two weeks after injury, CCSP levels in BALF remained decreased even though the number of CCSP positive cells present within the distal bronchiolar epithelium on immunofluorescent analysis was equivalent to naïve controls.
The number of proliferative epithelial cells was significantly increased in the distal bronchiolar epithelium on days 1 and 2 after injury. Even though CCSP staining was decreased by immunofluorescence and by western blot in BALF, the distal epithelium was still able to proliferate. Thus, unlike the more severely injured proximal bronchiolar epithelium, the distal epithelium retained the ability to proliferate after SM inhalation exposure.
Multiple studies have shown that variant club cells function as progenitor cells in the distal, conducting airways (Cardoso et al., 1993; Guha et al., 2014; Perl et al., 2011; Xing et al., 2012). When variant club cells proliferate, they express CCSP, but may not be terminally differentiated. Thus, despite expressing CCSP, the cells may not function as differentiated secretory cells. Likely, the CCSP-stained cells that were present on histology were not functional in secreting CCSP up to 2 weeks after injury as the amount of CCSP present in BALF remained significantly reduced. Supplementation with CCSP may assist with recovery from acute inhalation injury and prevention of further airway injury. One limitation of this study is that we did not identify specifically the progenitor cell contributing to proliferation of the distal bronchioles. Future studies using co-staining with BRDU, CCSP, and other cell markers of resident progenitor cells are needed to better identify which specific cells contribute to proliferation of the distal bronchiolar epithelium after SM exposure.
The Notch signaling pathway is one developmentally conserved pathway previously shown to assist with basal cell proliferation (Mori et al., 2015; Tsao et al., 2011) as well as the lung epithelium’s terminal differentiation (Guha et al., 2014; Hogan et al., 2014; Mori et al., 2015; Rock et al., 2011; Tsao et al., 2008, 2009, 2016; Xing et al., 2012). We assessed for changes in mRNA expression levels of the Notch signaling pathway to explain the regional differences in recovery of the conducting airway’s epithelium. In the first 48 h after injury, mRNA levels of 2 Notch receptors, Notch 1 and Notch 3, as well as the downstream effector gene, Hes1, were significantly upregulated. The increase in expression levels of Notch 1, 3, and Hes1 was on days 1 and 2 after SM exposure when the number of proliferating, BRDU+ cells of the distal bronchiolar epithelium was increased. Thus, there was a temporal relation between an upregulation of Notch mRNA expression when the distal bronchiolar epithelium was proliferating.
At days 7, 14, 21, and 28 after SM injury, Notch1, Notch3, and Hes1 gene expressions were downregulated. During these days, the proximal epithelium was unrepaired with a squamous, single-layered epithelium. Tilley et al. (2009) showed previously the downregulation of certain Notch receptors and ligands, specifically Notch3, in human bronchial epithelial samples of patients with COPD and chronic smoke exposure. Similar to these findings, we demonstrated the downregulation of certain Notch receptors, specifically Notch1 and Notch3, after significant inhalation, epithelial injury. Additionally, we showed the downregulation of Hes1, a downstream effector gene controlled by the Notch signaling pathway. When Hes1 is upregulated, cells are prevented from terminal differentiation. When these genes are downregulated, cells differentiate and lose potency. We speculate that after SM exposure, the Notch signaling pathway is downregulated resulting in aberrant differentiation of the proximal epithelium. Hence, changes seen in the proximal bronchiolar epithelium after SM exposure were similar to the morphological changes seen in histological samples of patients with COPD and chronic smoke exposure with a squamous and dysmorphic epithelium (Staudt et al., 2014; Tilley et al., 2009).
This study used whole lung homogenate to assess for changes in the Notch signaling pathway. Whole lung homogenate is a combination of all cell types from both the lung parenchyma and airway. Thus, we cannot conclude that changes in the Notch signaling pathway were from the airway epithelium alone. Future studies using cell culture or microdissection of the airway epithelium may help delineate the changes seen in the Notch signaling pathway from the airway epithelium from other cell types found in whole lung homogenate.
In this study, we used SM doses of 1.0–1.2 mg/kg. At a SM dose of 1.0 mg/kg, mortality occurred in a biphasic distribution (Kaplan-Maier curve; Supplementary Figure 1) with 50% of deaths occurring within the first 7 days after SM exposure. Of those animals who survived the acute injury, a second, more gradual, decline in survival occurred after 14 days (McGraw et al. and White and Veress, unpublished data). At lower doses of SM, the second decline in survival did not occur, and there was no histological evidence of BO at these dosages. Thus, a second limitation of this study was that assessment of the bronchiolar epithelium was not performed at lower doses of SM exposure. Future studies designed to assess the bronchiolar epithelium at lower SM doses may provide further insight into the airway epithelium’s ability to proliferate and differentiate in the absence of bronchiolitis obliterans.
In conclusion, SM exposure resulted in severe, proximal bronchiolar epithelial injury with patchy denudation, loss of club and ciliated cell markers, and impaired proximal epithelial proliferation with aberrant differentiation. In the more distal bronchiolar epithelium, there was a loss of CCSP that corresponded with a drop in BALF CCSP concentration, but with a compensatory increase in epithelial proliferation and upregulation of certain Notch RNA signals that assist with epithelial repair.
Weeks after the initial SM exposure, the proximal bronchiolar epithelium was not fully repaired with persistent patchy areas of denudation, squamous metaplasia, and the development of subepithelial fibrosis. In areas of severe epithelial injury, there was a loss of structural integrity and the development of bronchiolitis obliterans. These changes corresponded temporally with a downregulation of certain Notch signals, specifically Notch1, Notch3, and Hes1, all of which have been shown to be downregulation in the conducting airways of humans chronically exposed to toxic inhalants such as cigarette smoke. Thus, targeting of the airway epithelium and Notch signaling pathway after SM inhalation injury may be a worthy pursuit for the prevention of fibrosis generation and chronic airway remodeling.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
This work was supported by the CounterACT Program, National Institutes of Health (NIH), Office of the Director, and the National Institute of Environmental Health Sciences (NIEHS) (U54-ES027698-01 to C.W.W. and L.A.V.; University of Colorado Denver). M.D.M is partially supported by NIH T32HL 007670-27 (awarded to S.H.A; University of Colorado Denver).
Supplementary Material
REFERENCES
- Allon N., Amir A., Manisterski E., Rabinovitz I., Dachir S., Kadar T. (2009). Inhalation exposure to sulfur mustard in the guinea pig model: Clinical, biochemical and histopathological characterization of respiratory injuries. Toxicol. Appl. Pharmacol. 241, 154–162. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Byers SL, Vesely KR. 2000. Treatment of sulfur mustard (HD)-induced lung injury. J Appl Toxicol. 20 Suppl 1, S129–S132. [DOI] [PubMed] [Google Scholar]
- Beheshti J., Mark E. J., Akbaei H. M., Aslani J., Ghanei M. (2006). Mustard lung secrets: Long term clinicopathological study following mustard gas exposure. Pathol. Res. Pract. 202, 739–744. [DOI] [PubMed] [Google Scholar]
- Bracher A., Doran S. F., Squadrito G. L., Postlethwait E. M., Bowen L., Matalon S. (2012). Targeted aerosolized delivery of ascorbate in the lungs of chlorine-exposed rats. J. Aerosol. Med. Pulm. Drug Deliv. 25, 333–341. doi:10.1089/jamp.2011.0963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabiati M., Raucci S., Caselli C., Guzzardi M. A., D’Amico A., Prescimone T., Del Ry S. (2012). Tissue-specific selection of stable reference genes for real-time PCR normalization in an obese rat model. J. Mol. Endocrinol. 48, 251–260. [DOI] [PubMed] [Google Scholar]
- Calvet J. H., Coste A., Levame M., Harf A., Macquin-Mavier I., Escudier E. (1996). Airway epithelial damage induced by sulfur mustard in guinea pigs, effects of glucocorticoids. Hum. Exp. Toxicol. 15, 964–971. [DOI] [PubMed] [Google Scholar]
- Calvet J. H., Jarreau P. H., Levame M., D'Ortho M. P., Lorino H., Harf A., Macquin-Mavier I. (1994). Acute and chronic respiratory effects of sulfur mustard intoxication in guinea pig. J. Appl. Physiol. (1985) 76, 681–688. [DOI] [PubMed] [Google Scholar]
- Cardoso W. V., Stewart L. G., Pinkerton K. E., Ji C., Hook G. E., Singh G., Plopper C. G. (1993). Secretory product expression during Clara cell differentiation in the rabbit and rat. Am. J. Physiol. 264(6 Pt 1), L543–L552. [DOI] [PubMed] [Google Scholar]
- Ghanei M., Chilosi M., Mohammad Hosseini Akbari H., Motiei-Langroudi R., Harandi A.A., Shamsaei H., Tazelaar H.D. (2011). Use of immunohistochemistry techniques in patients exposed to sulphur mustard gas. Patholog. Res. Int. 2011, 659603. doi:10.4061/2011/659603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanei M., Ghayumi M., Ahakzani N., Rezvani O., Jafari M., Ani A., Aslani J. (2010). Noninvasive diagnosis of bronchiolitis obliterans due to sulfur mustard exposure: Could high-resolution computed tomography give us a clue?. Radiol. Med. 115, 413–420. doi:10.1007/s11547-010-0503-6 [DOI] [PubMed] [Google Scholar]
- Ghanei M., Harandi A. A., Tazelaar H. D. (2012). Isolated bronchiolitis obliterans: High incidence and diagnosis following terrorist attacks. Inhal. Toxicol. 24, 340–341. [DOI] [PubMed] [Google Scholar]
- Ghanei M., Tazelaar H. D., Chilosi M., Harandi A. A., Peyman M., Akbari H.M., Mohammadi A. (2008). An international collaborative pathologic study of surgical lung biopsies from mustard gas-exposed patients. Respir. Med. 102, 825–830. doi:10.1016/j.rmed.2008.01.016 [DOI] [PubMed] [Google Scholar]
- Guha A., Vasconcelos M., Zhao R., Gower A.C., Rajagopal J., Cardoso W. V. (2014). Analysis of Notch signaling-dependent gene expression in developing airways reveals diversity of Clara cells. PLoS One 9, e88848.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B. L., Barkauskas C. E., Chapman H. A., Epstein J. A., Jain R., Hsia C. C., Niklason L., Calle E., Le A., Randell S. H., et al. (2014). Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D., Srirama P. K., Wallis C., Wexler A. S. (2011). Postnatal growth of tracheobronchial airways of Sprague-Dawley rats. J, Anat, 218, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M., Mahoney J. E., Stupnikov M. R., Paez-Cortez J. R., Szymaniak A. D., Varelas X., Herrick D. B., Schwob J., Zhang H., Cardoso W. V. (2015). Notch3-Jagged signaling controls the pool of undifferentiated airway progenitors. Development 142, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Koren E. G., Hogan B. L., Gunn M. D. (2013). Loss of basal cells precedes bronchiolitis obliterans-like pathological changes in a murine model of chlorine gas inhalation. Am. J. Respir. Cell Mol. Biol. 49, 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige R., Wong V., Plopper C. (1997). Dose-related airway-selective epithelial toxicity of 1-nitronaphthalene in rats. Toxicol. Appl. Pharmacol. 147, 224–233. [DOI] [PubMed] [Google Scholar]
- Perl A. K., Riethmacher D., Whitsett J. A. (2011). Conditional depletion of airway progenitor cells induces peribronchiolar fibrosis. Am. J. Respir. Crit. Care Med. 183, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancourt R. C., Ahmad A., Veress L. A., Rioux J. S., Garlick R. B., White C. W. (2014). Antifibrinolytic mechanisms in acute airway injury after sulfur mustard analog inhalation. Am. J. Respir. Cell Mol. Biol. 51, 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock J. R., Gao X., Xue Y., Randell S. H., Kong Y. Y., Hogan B. L. (2011). Notch-dependent differentiation of adult airway basal stem cells. Cell Stem Cell 8, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber H., Saburi A., Ghanei M. (2012). Clinical and paraclinical guidelines for management of sulfur mustard induced bronchiolitis obliterans; from bench to bedside. Inhal. Toxicol. 24, 900–906. [DOI] [PubMed] [Google Scholar]
- Somani S. M., Babu S. R. (1989). Toxicodynamics of sulfur mustard. Int. J. Clin. Pharmacol. Ther. Toxicol. 27, 419–435. [PubMed] [Google Scholar]
- Staudt M. R., Buro-Auriemma L. J., Walters M. S., Salit J., Vincent T., Shaykhiev R., Crystal R. G. (2014). Airway Basal stem/progenitor cells have diminished capacity to regenerate airway epithelium in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 190, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley A. E., Harvey B. G., Heguy A., Hackett N. R., Wang R., O’Connor T. P., Crystal R. G. (2009). Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am. J. Respir. Crit Care Med. 179, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao P. N., Chen F., Izvolsky K. I., Walker J., Kukuruzinska M. A., Lu J., Cardoso W. V. (2008). Gamma-secretase activation of notch signaling regulates the balance of proximal and distal fates in progenitor cells of the developing lung. J. Biol. Chem. 283, 29532–29544. doi:10.1074/jbc.M801565200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao P. N., Matsuoka C., Wei S. C., Sato A., Sato S., Hasegawa K., Morimoto M. (2016). Epithelial Notch signaling regulates lung alveolar morphogenesis and airway epithelial integrity. Proc. Natl. Acad. Sci. U.S.A. 113, 8242–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao P. N., Vasconcelos M., Izvolsky K. I., Qian J., Lu J., Cardoso W. V. (2009). Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development 136, 2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao P. N., Wei S. C., Wu M. F., Huang M. T., Lin H. Y., Lee M. C., Lin K. M., Wang I. J., Kaartinen V., Yang L. T., Cardoso W. V. (2011). Notch signaling prevents mucous metaplasia in mouse conducting airways during postnatal development. Development 138, 3533–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan A. E., Brumwell A. N., Xi Y., Gotts J. E., Brownfield D. G., Treutlein B., Tan K., Tan V., Liu F. C., Looney M. R., et al. (2015). Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 517, 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veress L. A., Hendry-Hofer T. B., Loader J. E., Rioux J. S., Garlick R. B., White C. W. (2013). Tissue plasminogen activator prevents mortality from sulfur mustard analog-induced airway obstruction. Am J Respir Cell Mol Biol. 484, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C. W., Martin J. G. (2010). Chlorine gas inhalation: Human clinical evidence of toxicity and experience in animal models. Proc. Am. Thorac. Soc. 7, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C. W., Rancourt R. C., Veress L. A. (2016). Sulfur mustard inhalation: Mechanisms of injury, alteration of coagulation, and fibrinolytic therapy. Ann. N. Y. Acad. Sci. 1378, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems J. L. (1989). Clinical management of mustard gas casualties. Ann Med Milit Belg 3S, 1–61. [Google Scholar]
- Xing Y., Li A., Borok Z., Li C., Minoo P. (2012). NOTCH1 is required for regeneration of Clara cells during repair of airway injury. Stem Cells 30, 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarchi K., Akbar A., Naieni K. H. (2004). Long-term pulmonary complications in combatants exposed to mustard gas: A historical cohort study. Int. J. Epidemiol. 33, 579–581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.