

Abstract
Factor XIII A (FXIIIA) is a member of the transglutaminase enzyme family that cross-links both intra- and extracellular protein substrates. To prevent undesired cross-linking, FXIIIA is expressed as an inactive zymogen and exists intracellularly as an A2 homodimer. In plasma, FXIII A2 is complexed with two protective factor XIII B subunits (A2B2) that dissociate upon activation of the zymogen. Based on limited experimental data, activated FXIII was considered a dimer of two catalytically active A subunits. However, accumulating but indirect evidence has suggested activation may lead to a monomeric state instead. In the present study, we employed analytical ultracentrifugation (AUC) to directly explore the oligomerization state of zymogen as well as active FXIIIA in solution. We first confirmed that the zymogen was a FXIIIA2 dimer. When we activated FXIIIA non-proteolytically (by high mM Ca2+), the protein dissociated to monomers. More importantly, FXIIIA incubation with its physiological partner, the protease thrombin, led to a monomeric state as well. AUC studies of partially cleaved FXIIIA further suggested that thrombin cleavage of a single activation peptide in a zymogen dimer is sufficient to weaken inter-subunit interactions, initiating the transition to monomer. The enzymatic activity of the thrombin-cleaved species was higher than non-proteolytically activated enzyme, suggesting that displacement of the activation peptide renders the FXIIIA more accessible to substrates. Thus, results provide evidence that FXIII undergoes a change in oligomerization state as part of the activation process, and emphasize the role of the activation peptide in preventing FXIIIA catalytic activity.
Keywords: Factor XIII, transglutaminase, oligomerization state, analytical ultracentrifugation, size exclusion chromatography
Graphical Abstract Text
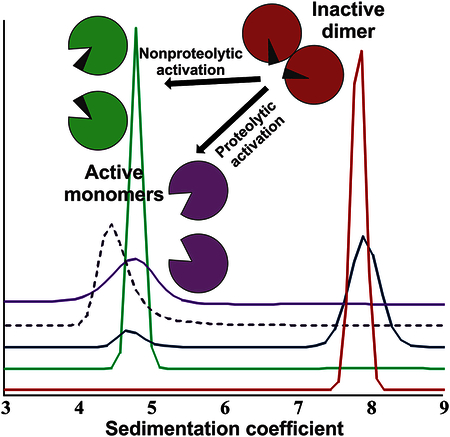
Alteration in oligomeric assemblies has been emerging as a regulatory mechanism for many proteins. Numerous well-studied enzymes require self-association to fulfill their catalytic function. By contrast, the protein of interest in the current project, Factor XIII, is inactive in its oligomeric form. Dissociation of dimeric zymogen is demonstrated here as a critical step in Factor XIII activation.
INTRODUCTION
Factor XIII (FXIII) plays a critical role in blood coagulation by covalently crosslinking fibrin γ- and α-chains into a fibrin clot along with inhibitors of fibrinolysis. The significance of FXIII in hemostasis is well recognized, and evidence is growing for the involvement of this enzyme in other physiological events, such as wound healing, bone tissue formation, and even signaling processes [1–3]. FXIII is found in plasma mostly as a heterotetramer of two catalytic A-subunits, and two carrier B-subunits (A2B2, ~325 kDa, 14–28 mg/L [4]), and ~1% as an A2-homodimer (~166 kDa) [5]. The intracellular form, for example in platelets (46–82 fg/platelet [6]), is observed exclusively as A2 [1]. Both plasma and cellular FXIII oligomers are stabilized by non-covalent inter-subunit interactions.
The A-subunit of FXIII (FXIIIA, Fig. 1A) is a transglutaminase (protein glutamine:amine γ-glutamyltransferase, EC 2.3.2.13) that is expressed as an inactive zymogen form. The activation paths are different for plasma- and intracellular FXIII (Fig. 1B). In a final stage of blood coagulation, thrombin cleaves the N-terminal 4 kDa activation peptides on the A-subunits of plasma FXIII A2B2. Then in the presence of low mM Ca2+, the B-subunits dissociate from the A-subunits exposing catalytically active transglutaminase [7]. In contrast, intracellular FXIIIA2 undergoes slow non-proteolytic activation by Ca2+ [8–10]. In vitro, FXIIIA2 can be activated by thrombin in essentially the same manner as plasma FXIII A2B2, but without involvement of the B-subunits. Cleavage of a single activation peptide in the dimeric zymogen FXIIIA2 has been reported to promote exposure of active sites on both subunits, yielding full enzymatic activity of such a “single-cleaved” dimer [11]. In addition, both plasma and cellular FXIII can be activated in the presence of non-physiologically high (>50 mM) Ca2+ [12]. Ca2+ has been demonstrated to be crucial for transglutaminase activity, whereas other divalent cations are much less effective (Ca2+>Sr2+>Ba2+>Mg2+) [13]. Furthermore, the binding of Ca2+ stabilizes FXIIIA preventing its premature degradation by proteases [14].
Fig. 1. Structure and activation of the A-subunit of FXIII.
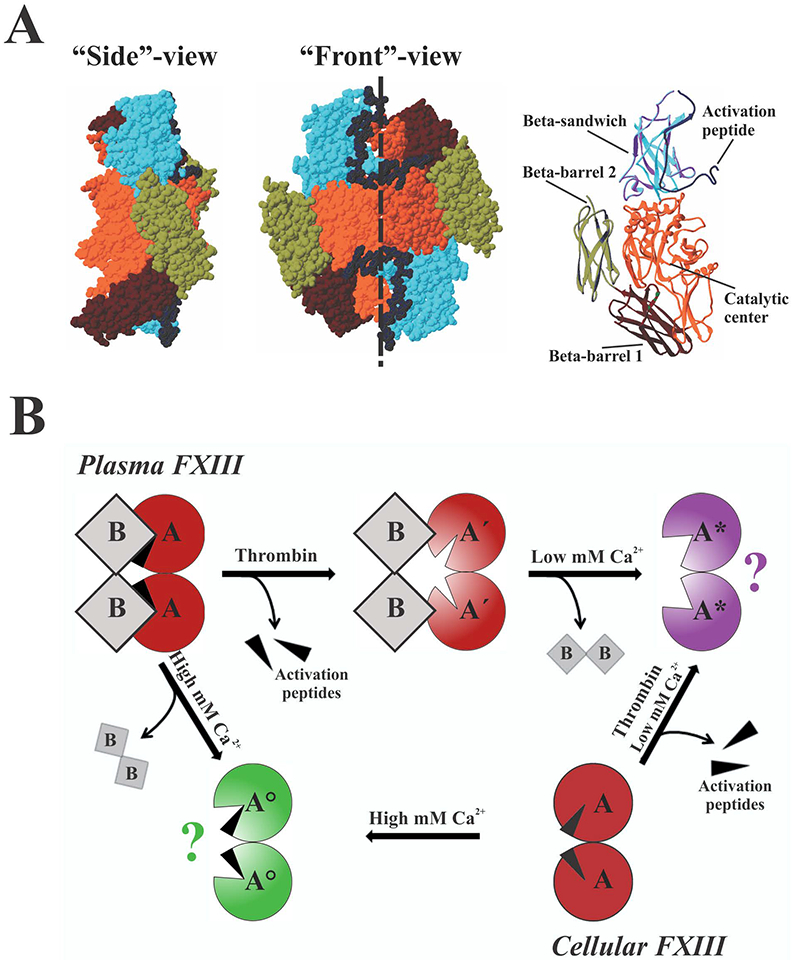
(A) Zymogen FXIIIA2 (PDB 1F13) is formed by two identical subunits oriented in a head-to-tail fashion (shown as space-filling models, dashed line denotes homodimer interface), with the activation peptide of each subunit blocking access to the active site of the neighboring subunit. Structural domains of the left subunit are highlighted in a ribbon representation. (B) Both plasma and cellular FXIII can be activated by thrombin in the presence of low mM Ca2+ or non-proteolytically by high (>50 mM) concentration of Ca2+. Aʹ - FXIIIA after cleavage of the activation peptide; A* - FXIIIA activated by thrombin in the presence of Ca2+; A° - non-proteolytically activated FXIIIA; question marks denote unknown oligomerization state. A more detailed description can be found in the text.
Although both active sites in FXIIIA2 are exposed upon activation by thrombin, previous inhibition studies [11, 12, 15] revealed that only one mole of radiolabeled iodoacetamide (IAA) was incorporated per two moles of thrombin-activated A-subunits (half-of-the-sites reactivity), thus negative cooperativity was proposed for the catalytic action of FXIIIA. Interestingly, no large differences between the zymogen and thrombin-activated forms were observed in early X-ray crystal studies [16, 17]. However, with a FXIIIA-catalyzed reaction where two polypeptide chains are cross-linked, significant rearrangements in the enzyme would be required to bind substrate and to release the product. Indeed, solution based hydrogen-deuterium exchange and chemical modification experiments indicated conformational alterations with opening of the FXIIIA dimer interface upon activation [18–20]. Consistent with those conclusions, non-proteolytically activated FXIIIA that had been covalently inhibited was later crystallized with an exposed active site and proposed to be monomeric [21]. In addition, recent in silico steered molecular dynamics simulations have suggested a weakening of inter-subunit interactions in both non-proteolytically and proteolytically activated FXIIIA [22].
In the present work, we address the oligomeric state of the A-subunits of FXIII in a solution environment. We demonstrate that the dimeric FXIIIA zymogen dissociates upon non-proteolytic activation. For the first time to our knowledge, direct experimental evidence is presented for the monomeric state of thrombin-activated FXIIIA. Our results suggest that the cleavage of one activation peptide in a zymogen FXIIIA2 weakens inter-subunit interactions, likely leading to subsequent dissociation of the dimer. Furthermore, displacement of the activation peptide from the A-subunits of FXIII provides better accessibility of the enzyme to its substrates.
RESULTS
Activated FXIIIA species experience significant conformational rearrangement compared to the zymogen form.
In an attempt to probe FXIIIA for possible conformational rearrangements in solution upon activation, we performed intrinsic tryptophan fluorescence measurements (Fig. 2A). To activate FXIIIA proteolytically (by cleavage of the activation peptide), the zymogen was incubated with thrombin in the presence of 4 mM Ca2+. Non-proteolytic activation was achieved in the presence of 100 mM Ca2+. An overall fluorescence decrease was observed for activated FXIIIA forms relative to the zymogen, suggesting an increase in polarity of the environment surrounding some of the tryptophan residues in the FXIIIA molecule. Thus, we speculated that these tryptophan residues became more solvent-exposed, indicating conformational rearrangements in the A2-homodimer. As a control, FXIIIA was also treated with 100 mM Mg2+, and no effect on intrinsic fluorescence of the protein was detected.
Fig. 2. FXIIIA undergoes conformational rearrangements upon activation.
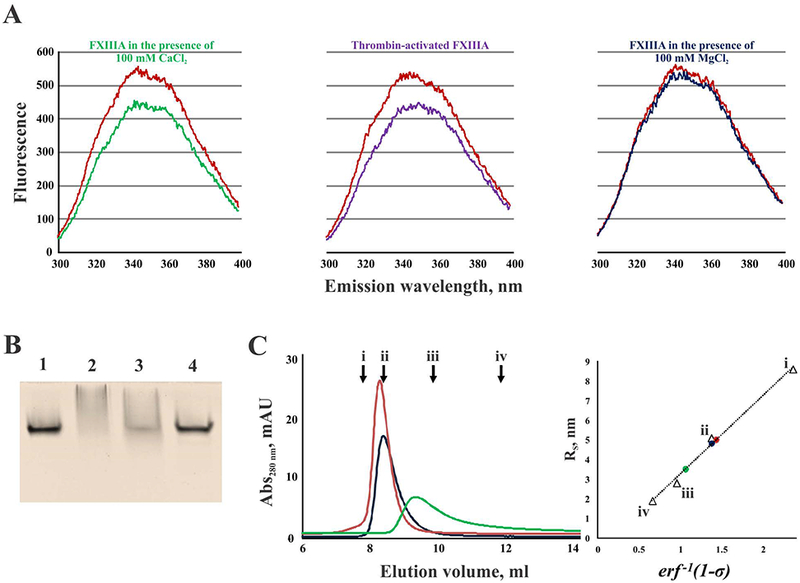
(A) fluorescence emission spectra of FXIIIA (500 nM) in the presence of 100 mM CaCl2 (green), 100 mM MgCl2 (dark blue), and thrombin-activated FXIIIA in the presence of 4 mM CaCl2 (purple). Control zymogen FXIIIA is shown in red; (B) nPAGE of FXIIIA: lane 1 – zymogen FXIIIA; lane 2 – FXIIIA activated by thrombin in the presence of 4 mM CaCl2; lane 3 – FXIIIA treated with 100 mM CaCl2; lane 4 – FXIII treated with 100 mM MgCl2; (C) Elution profiles of FXIIIA under different conditions (left): 100 μL of 2 μM FXIIIA was applied to a SEC column and elution profiles were recorded for the zymogen form (red), FXIIIA in the presence of 100 mM CaCl2 (green), and FXIIIA in the presence of 100 mM MgCl2 (dark blue). Arrows indicate peak elution volumes (Ve) of the calibration standards (i – thyroglobulin, 670 kDa, Ve = 7.80 ml; ii – bovine γ-globulin, 158 kDa, Ve = 8.40 ml; iii – chicken ovalbumin, 43 kDa, Ve = 9.87 ml; iv – equine myoglobin, 17 kDa, Ve = 11.89 ml); calculation of Stokes radii (RS) for FXIIIA forms based on their partition coefficients (σ): inverse error function of 1-σ (erf −1 (1-σ)) was plotted versus Stokes radii (RS) for the calibration standards (triangles, i – thyroglobulin, RS = 8.6 nm; ii – bovine γ-globulin, RS = 5.1 nm; iii – chicken ovalbumin, RS = 2.8 nm; iv – equine myoglobin, RS = 1.9 nm). Using the calibration line, the RS-values were calculated for FXIIIA samples (circles): zymogen (red, RS = 5.0 nm), FXIIIA treated with 100 mM MgCl2 (dark blue, RS = 4.8 nm), and FXIIIA in the presence of 100 mM CaCl2 (green, RS = 3.5 nm).
To further assess possible dissociation of FXIIIA upon activation, a series of non-denaturing polyacrylamide gel electrophoresis (nPAGE) experiments was conducted. A significant change in FXIIIA electrophoretic mobility was visible upon activation by thrombin (Fig. 2B, lane 2) and by 100 mM Ca2+ (Fig. 2B, lane 3). However, these conditions caused the protein to spread along a migration path (especially in thrombin-activated FXIIIA), yielding no well-defined bands on the gel. By contrast, the migration of FXIIIA treated with 100 mM Mg2+ (Fig. 2B, lane 4) was identical to that of the zymogen form (Fig. 2B, lane 1).
Finally, FXIIIA was analyzed by size exclusion chromatography (SEC, Fig. 2C). Both zymogen and 100 mM Mg2+-treated FXIIIA had similar elution profiles. Their Stokes radii were determined to be 5.0 nm and 4.8 nm, respectively. In the presence of 100 mM Ca2+, FXIIIA eluted after the zymogen form (Fig. 2C), and its Stokes radius was 3.5 nm. These observations are consistent with the possibility of dissociation of FXIII A2-homodimer upon non-proteolytic activation.
FXIIIA2 dissociates into monomers upon activation.
Although fluorescence measurements, nPAGE, and SEC results indicated significant perturbations in FXIIIA upon activation, the oligomerization states of the enzyme forms remained inconclusive. In contrast, analytical ultracentrifugation (AUC) provided distinct resolution of the FXIIIA species. Representative results of sedimentation velocity analyses are summarized in Table 1 and Fig. 3. We confirmed the dimeric state of FXIIIA zymogen in solution (Fig. 3, trace i). The sedimentation coefficient of ~7.9 S was consistent with the molecular weight of ~160 kDa. A frictional ratio of ~1.3 indicated a slightly asymmetric molecule. In the presence of 100 mM Mg2+, 90% of the protein existed in the dimeric form (Fig. 3, trace iii). A profound change in sedimentation properties was observed in the presence of 100 mM Ca2+ (Fig. 3, trace ii). This non-proteolytically activated FXIIIA migrated uniformly with a sedimentation coefficient of ~4.8 S, consistent with the molecular weight ~80 kDa, thus indicating a monomeric state. A more detailed analysis of the non-proteolytically activated FXIIIA oligomeric state is presented later in this manuscript, along with activity studies.
Table 1. Hydrodynamic parameters of FXIIIA.
Sedimentation coefficients (in Svedberg units, S) and frictional ratios were obtained by analyzing sedimentation velocity data using the Sedfit software. Observed s-values were corrected to s20,w as described in Experimental procedures. For each experimental condition, data are presented for two independently made samples.
| Sample | s20,w, S | Frictional ratio |
|---|---|---|
| FXIIIA zymogen | 7.91 7.85 |
1.32 1.35 |
| FXIIIA with 100 mM Ca2+ | 4.83 4.80 |
1.38 1.31 |
| FXIIIA with 100 mM Mg2+ | 7.93 7.73 |
1.35 1.35 |
| FXIIIA thrombin-activated in presence of 4 mM Ca2+ | 4.57 4.70 |
1.53 1.37 |
Fig. 3. Representative sedimentation velocity analyses of FXIIIA.
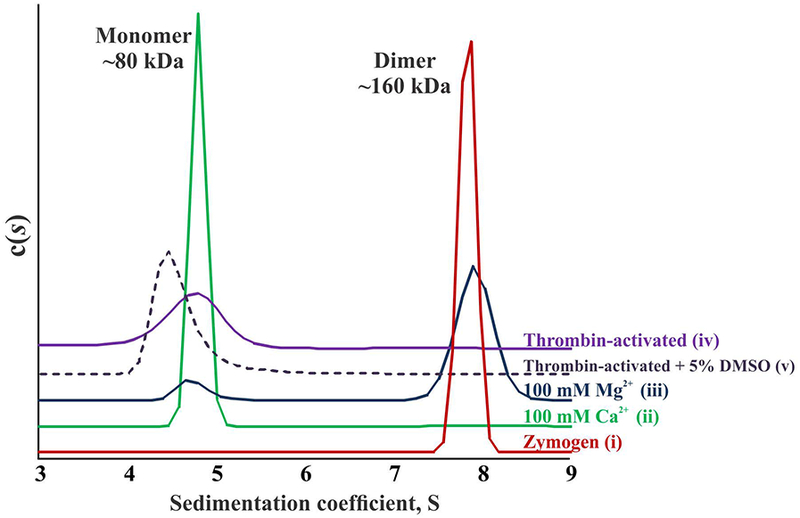
(i) – FXIIIA zymogen in the borate buffer; (ii) – FXIIIA incubated in the presence of 100 mM CaCl2; (iii) – FXIIIA incubated in the presence of 100 mM MgCl2; (iv) – FXIIIA cleaved by thrombin in the presence of 4 mM CaCl2; (v) – same as iv, with addition of 5% DMSO. For all conditions, loading concentration of FXIIIA was 2 μM. Experiments were performed with two independent samples for each condition. Sedimentation coefficient (s20,w) is in Svedberg units (S). c(s) is the concentration distribution as a function of s20,w.
AUC of thrombin-activated FXIIIA revealed a monomer with a sedimentation coefficient of ~4.6 S (Fig. 3, trace iv). The lower s-value of proteolytically activated FXIIIA compared to the 100 mM Ca2+-activated enzyme is consistent with the loss of a 4 kDa activation peptide in the cleaved protein. Although no significant differences could be seen between the frictional ratios of thrombin-cleaved FXIIIA compared to non-proteolytically activated enzyme (Table 1), the broader peak of the thrombin-activated form (Fig. 3, trace iv) was suggestive of a more flexible conformation of FXIIIA after activation peptide cleavage. In addition, as previously observed [15, 23], thrombin-activated FXIIIA tends to aggregate over the course of a few hours, causing its low abundance in solution and thus making it difficult to analyze by both SEC and AUC. Whereas non-denaturing treatment with sodium deoxycholate or dithiothreitol did not resolve this issue, we did find that the addition of 5% DMSO facilitates solubility of the active FXIIIA (Fig. 3, trace v). Although this co-solvent decreases the apparent FXIIIA sedimentation coefficient, suggesting an altering of FXIIIA shape, the 5% DMSO does not cause dissociation of zymogen FXIIIA2 (data not shown) nor does it affect the enzyme catalytic activity (see activity studies below).
Previously published kinetic studies by Hornyak et al. [11] have suggested that only one activation peptide has to be cleaved on the FXIIIA2 dimer to expose the active sites in both subunits in the presence of Ca2+. To assess the oligomerization state of this “single-cleaved” FXIIIA, activation conditions were adjusted so that approximately half of the A-subunits were cleaved by thrombin (Fig. 4A). A series of samples was prepared, and some of them were supplemented with 5% DMSO. Representative AUC results for these “half-cleaved” samples are depicted on Fig. 4C. From a quantitative analysis of sedimentation velocity data of one of the samples (without DMSO), 48% of the protein appeared as a monomer, 42% was a dimer, and 10% formed a large aggregate. Another sample was supplemented with 5% DMSO, and no aggregation was observed yielding 62% monomer and 38% dimer. Assuming random cleavage by thrombin, the resulting distribution of species in these samples (Fig. 4B) would be ~ 25% A2 (non-cleaved zymogen) : 50% AʹA (“single-cleaved”) : 25% Aʹ (“double-cleaved”). Hence, the “double-cleaved” species would constitute ~25% of monomers in the samples. Our observation of 48% (with no DMSO) and 62% (in the presence of 5% DMSO) monomeric protein can be accounted for by assuming dissociation of a major portion of the “single-cleaved” FXIIIA dimers.
Fig. 4. Analysis of monomer-dimer distribution of FXIIIA partially activated by thrombin in the presence of 4 mM CaCl2.
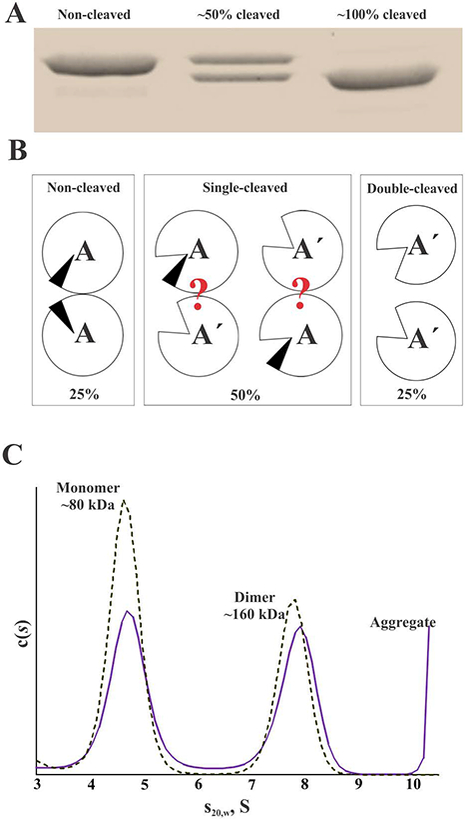
The activation conditions were adjusted so that approximately half of the A-subunits were cleaved. (A) SDS-PAGE (8% gel), monitoring cleavage of FXIIIA by thrombin; (B) theoretic distribution of non-cleaved and cleaved species in the “half-cleaved” samples of FXIIIA. Question marks denote unknown oligomerization state of the “single-cleaved” species. (C) representative c(s) distribution in the 2 μM FXIIIA “half-cleaved” samples with (dashed line) and without (solid line) addition of 5% DMSO. AUC experiments were performed as two replicates for each condition (no DMSO versus 5% DMSO).
To quantitatively assess inter-subunit interactions in FXIIIA, we performed a series of sedimentation equilibrium AUC experiments. Representative distribution profiles obtained for the zymogen and activated forms at three different rotor speeds are presented in Fig. 5. Global fitting of experimental data to a monomer-dimer equilibrium model yielded apparent dissociation constants (KD) for dimerization of FXIIIA. As expected, interactions of the A-subunits in the zymogen form were relatively tight (KD = 8 nM). Much higher KD-values (weaker interactions) were observed upon activation: 90 mM for non-proteolytically activated and 220 mM for thrombin-activated FXIIIA. Obtained at much lower centrifugal force than that used in the sedimentation velocity experiments, sedimentation equilibrium results provide critical support that activated FXIIIA is likely to exist in monomeric form at physiological concentrations.
Fig. 5. Analysis of monomer-dimer equilibrium of FXIIIA forms.
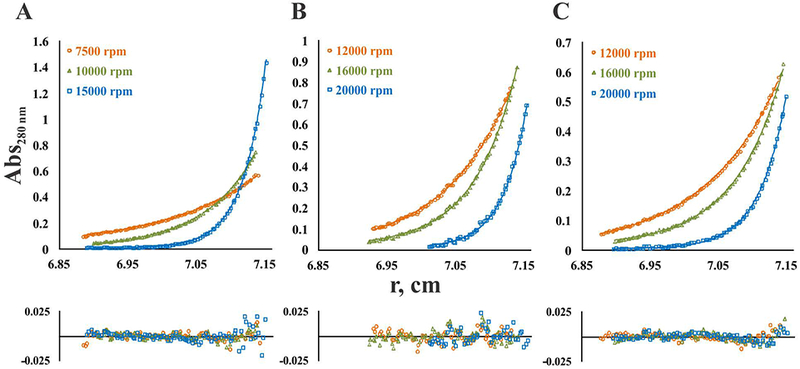
Sedimentation equilibrium profiles are presented for single loading concentration: (A) 2 μM zymogen FXIIIA; (B) 4μM FXIIIA in the presence of 100 mM CaCl2; (C) 4 μM FXIIIA activated by thrombin in the presence of 4 mM CaCl2. Experimental data (open symbols) obtained at each rotor speed (denoted on the figure panels) were fitted to a monomer-dimer equilibrium model (solid lines). Residuals are provided below the fitted data. Apparent KD-values were 8 nM for the zymogen FXIIIA, 90 mM for FXIIIA in the presence of 100 mM CaCl2, and 220 mM for the thrombin-activated FXIIIA. Global molecular weight values were 158,424 Da for zymogen, 86,801 for CaCl2-activated FXIIIA and 77,025 for thrombin-activated FXIIIA. Monte-Carlo error analysis indicated an average error for zymogen (A) of ±30 Da in molecular weight and ±0.03 for logKa, for CaCl2-activated FXIII (B) of ±40 Da in molecular weight and ±0.003 for logKa, and for thrombin-activated FXIII (C) of ±60 Da in molecular weight and ±0.002 for logKa.
Activated FXIIIA remains monomeric upon binding of substrate analogs.
To study possible structural rearrangements in the active FXIIIA upon binding substrate analogs, thrombin-activated enzyme was subjected to alkylation of the active site C314 by the thiol reagent IAA. As another test candidate, a peptide inhibitor K9-DON (1LGPG-(DON)-SLVIG10), where glutamine is replaced with its isostere 6-diazo-5-oxo-norleucine [18] was used to target the FXIIIA catalytic site region. These two covalent FXIIIA-inhibitor adducts resemble an acyl-enzyme intermediate transiently formed during the transglutaminase reaction. In the AUC experiments, both FXIIIA-IAA and FXIIIA-K9-DON complexes appeared as monomers with no significant differences in the sedimentation properties compared to thrombin-activated free enzyme (Fig. 6).
Fig. 6. Sedimentation velocity analysis of covalently inhibited FXIIIA.
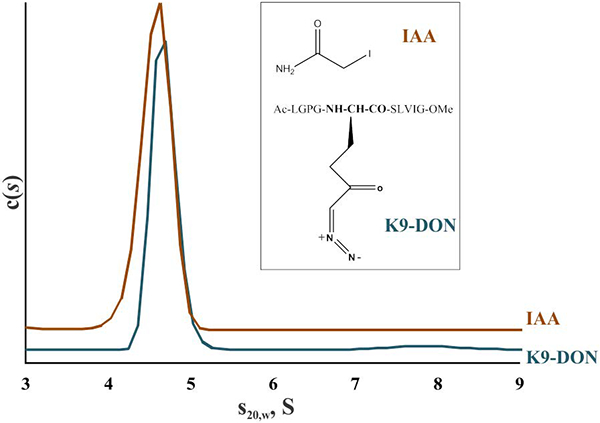
2 μM FXIIIA was activated by thrombin in the presence of 4 mM CaCl2 and incubated with inhibitors IAA or K9-DON to form covalent adducts with the active site C314 residue. Samples of inhibited FXIIIA were supplemented with 5% DMSO and subjected to AUC. Structures of IAA and K9-DON are depicted in an insert. Two independent samples were analyzed for each condition.
Comparison of catalytic activities of proteolytically and non-proteolytically activated FXIIIA.
Transglutaminase activity of FXIIIA under different activation conditions was compared using a coupled spectrophotometric assay (Fig. 7). In AUC studies, 5% DMSO facilitated solubility of thrombin-cleaved FXIIIA, however this co-solvent caused a slight decrease in apparent sedimentation coefficient of the protein (Fig. 3). Thus the conformation of FXIIIA could be affected by DMSO and hence, catalytic activity. However, as seen from Fig. 7 (ii), addition of 5% DMSO did not affect transglutaminase activity of the thrombin-cleaved FXIIIA. The activity of samples with half of the A-subunits cleaved by thrombin (Fig. 7, iii) was ~75% of that observed with the fully-cleaved FXIIIA. Enzyme activated in the presence of 100 mM Ca2+ yielded ~25% activity (Fig. 7, iv) as compared to the thrombin-activated FXIIIA. Finally, in the presence of control 100 mM Mg2+ no detectable activity was observed (Fig. 7, v), consistent with previous conclusions that among divalent cations, Mg2+ was the least efficient in supporting FXIIIA transglutaminase activity [19].
Fig. 7. FXIIIA catalytic activity studies.
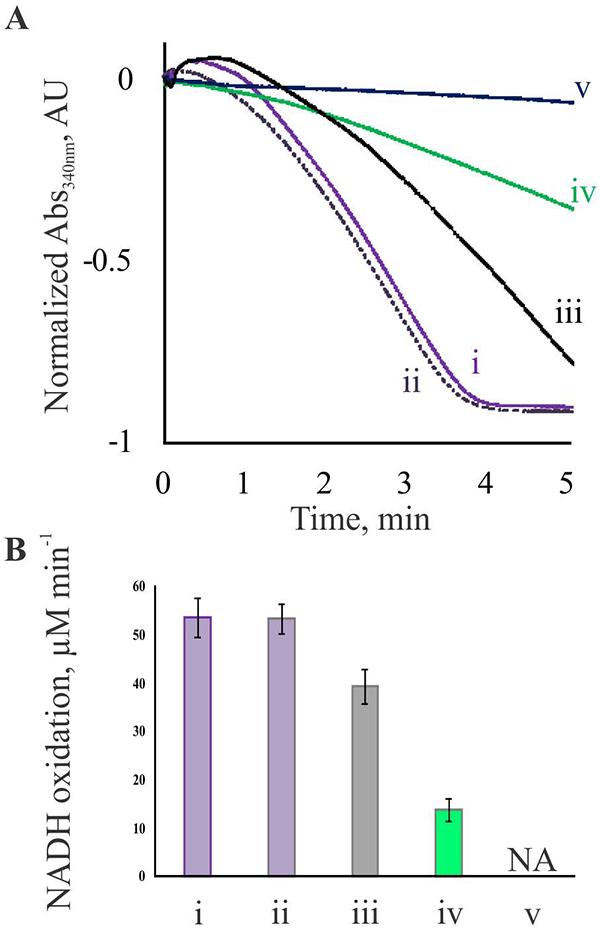
Transglutaminase activity of FXIIIA was determined via a coupled spectrophotometric assay, as described under Experimental Procedures. (A) Representative absorbance curves monitoring FXIIIA-mediated ammonia release followed by oxidation of NADH. (i) – FXIIIA fully cleaved by thrombin in the presence of 4 mM CaCl2; (ii) – same as (i), with addition of 5% DMSO; (iii) – FXIIIA sample with ~50% of the A-subunits cleaved by thrombin in the presence of 4 mM CaCl2; (iv) – FXIIIA incubated in the presence of 100 mM CaCl2; (v) – FXIIIA incubated in the presence of 100 mM MgCl2. (B) transglutaminase activity of FXIIIA is presented by the rate of ammonia-dependent NADH oxidation. Measurements were performed in triplicates for each condition outlined in part A. NADH oxidation rate in the presence of 100 mM MgCl2 was essentially the same as that in absence of FXIIIA. Values are presented as mean ± SD (n=3).
For a more detailed assessment of FXIIIA non-proteolytic activation, the protein was treated in the presence of different concentrations of Ca2+. The samples were then subjected to AUC. The resultant sedimentation velocity data were used to quantitate the amount of FXIIIA monomers depending on the concentration of Ca2+ and to plot a “titration curve” (Fig. 8, green line). As expected, increasing Ca2+ concentrations resulted in increase in the number of monomers. The sigmoidal shape of the “titration curve” indicated cooperative Ca2+ binding to FXIIIA, consistent with previous computational predictions [22]. Transglutaminase activity was also measured for each Ca2+ concentration (Fig. 8, green triangles). The most FXIIIA catalytic activity (~35%, compared to the thrombin-cleaved FXIIIA) was observed in the presence of 50 mM Ca2+, followed by a slight decline.
Fig. 8. FXIIIA dissociation and activity as a function of Ca2+ concentration.
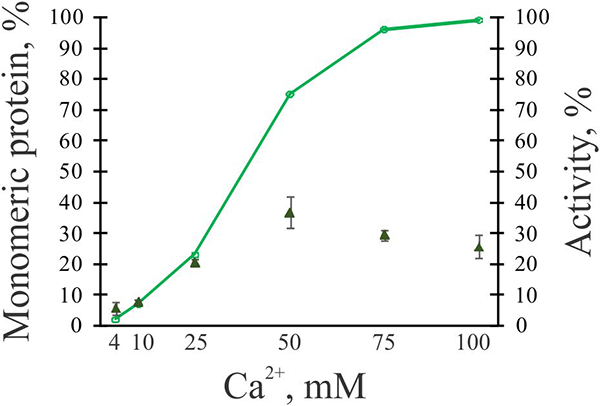
2 μM FXIIIA was incubated in the presence of 4–100 mM CaCl2 for 30 min at 37°C. Samples were then subjected to sedimentation velocity AUC, and quantitation of monomeric species was performed for each sample (green line). AUC runs were performed with at least two independently made samples for each condition studied. In parallel, transglutaminase activity of FXIIIA with different CaCl2 concentrations was determined using a coupled spectrophotometric assay (green triangles, values are presented as mean ± SD (n=3). The % activity is compared to thrombin-activated FXIIIA (100%)).
DISCUSSION
Oligomeric assemblies of proteins, particularly those of enzymes, attracted the attention of investigators as soon as it was realized that such assemblies regulate protein function [24]. With continuing advances in experimental techniques and instrumentation, the scope of protein oligomerization research has expanded significantly. It has been estimated [25] that 60% of enzymes form oligomers. Many of these enzymes exist as monomers but require self-association to fulfill the catalytic function. By contrast, the spotlight of the current project is on a protein that is inactive in its oligomeric form.
In this work, we evaluated the oligomerization state of the A-subunits of FXIII in relation to their catalytic activity under different conditions in a solution. Intrinsic tryptophan fluorescence profiles and electrophoretic mobilities of both proteolytically and non-proteolytically activated forms of FXIIIA were different from that of the control zymogen, suggesting significant perturbations to the protein upon activation. However, our attempt to resolve proteolytically and non-proteolytically activated FXIIIA using nPAGE encountered difficulties and did not yield defined protein bands on the gels. By contrast, similar experiments with homologous monomeric Transglutaminase 2 demonstrated two different forms. In nPAGE [26] as well as capillary electrophoresis [27] studies, the high mobility (compact conformation) and the low mobility (extended conformation) species were observed. These species were correlated with the crystal structures of inactive and active enzyme, correspondingly.
A reasonable explanation for our nPAGE results is that during separation in an electric field Ca2+ ions would be migrating in the opposite direction from the negatively charged protein, constantly “washing out” from the samples. Interestingly, the activation of FXIIIA by high concentrations of Ca2+ was previously shown to be a reversible process: loss of catalytic activity was observed upon removal of the Ca2+ ions from FXIIIA, and the protein could be re-activated again by addition of Ca2+ [28]. Thus nPAGE results in the current study indicate that presumably monomeric, activated FXIIIA might re-associate into zymogen-like dimers or higher-order aggregates upon loss of Ca2+ ions. Such re-associations have been proposed by Gupta et al. [22]. These phenomena observed in our nPAGE studies occurred specifically in the presence of Ca2+ but not Mg2+, adding to the evidence that Ca2+ is critical for maintaining the conformation of active FXIIIA.
SEC analysis indicated different sizes (Stokes radii) of zymogen FXIIIA and its activated form. However, FXIIIA is not an ideal globular protein (Fig. 1A), and we were cautious about correlating SEC data with molecular weights of FXIIIA forms. By contrast, the AUC approach provided information on the molecular weight and the shape of the protein in solution. The results of sedimentation velocity AUC experiments confirmed the dimeric state of zymogen FXIIIA2 and its asymmetric shape, in accord with existing crystal structures and ultracentrifugation studies by Bishop et al. [29]. FXIIIA activated proteolytically by thrombin in the presence of low (4 mM) Ca2+ appeared as a monomer. Compared to the non-proteolytically Ca2+-activated form, the broader sedimentation coefficient peak distribution of thrombin-cleaved FXIIIA indicated its more flexible conformation. Additionally, sedimentation equilibrium AUC provided an effective strategy to quantitatively characterize inter-subunit interactions of FXIIIA forms. The apparent dissociation constant for the zymogen FXIIIA was in the nanomolar range, whereas the value increased into the millimolar range for the activated FXIIIA. In platelets, an established reference interval for FXIII A2 is 46–82 fg/platelet [6]; given the mean platelet volume of 9 fL [30], the concentration of the A-subunits is 61–110 μM. The concentration of FXIII A2B2 in plasma is 14–28 mg/L [4], corresponding to 86–172 nM of the A-subunits. During a coagulation event, activated FXIII is mobilized at the clotting site and its local concentration may exceed regular plasma levels, however, the amount would not be expected to reach high millimolar values. Thus, the observed drastic weakening of inter-subunit interactions supports our concept of dissociation of FXIIIA upon activation at physiological concentrations. Since dissociation of the B-subunits from the A-subunits after cleavage of the activation peptide has been well documented [1, 2], the physiological presence of FXIIIB is not predicted to significantly affect post-cleavage rearrangements of FXIIIA in plasma.
An observation of monomeric, thrombin-activated FXIIIA is contrary to previously reported results. Chung and coworkers [15] performed AUC of both plasma (A2B2) and platelet (A2) FXIII using 50 mM sodium citrate buffer. A valid concern from the Chung study is that most of the added (10 mM) Ca2+ may have been chelated by citrate. Under their conditions, the dissociation of B2-dimer from the A2-dimer had occurred, but no evidence for the presence of monomeric FXIIIA either in plasma or platelet-derived enzyme was obtained, much like in our nPAGE efforts. In the present study, no chelating agents were used in the buffers, thus 4 mM Ca2+ present in the samples was available for binding and stabilizing monomeric thrombin-cleaved FXIIIA [14].
In an X-ray crystal study, Yee et al. [17] reported no significant changes in structure of FXIIIA after activation with thrombin. However, non-cleaved FXIIIA zymogen was present in their crystallographic samples, which was described as a ‘minor component’ (< 20%). Since thrombin-cleaved FXIIIA is prone to precipitation especially at the high concentrations (10 mg/ml) used, the Yee group may have actually crystallized the ‘minor component’, zymogen FXIIIA, instead of the thrombin-cleaved form.
In samples where half of the FXIIIA subunits were cleaved by thrombin, we observed ~75% activity compared to the “fully-cleaved” FXIIIA, in agreement with previously published data [29]. Intuitively, one would expect ~75% monomers in such a sample if all “single-cleaved” species became monomers (Fig. 4B). Yet our analysis revealed 48% (with no DMSO) and 62% (with 5% DMSO) monomeric protein, thus indicating that some of the “single-cleaved” species remained dimeric at the concentrations and solution conditions employed in this work. Our present results therefore indicate significant weakening of inter-subunit interactions in dimeric FXIIIA2 upon cleavage of a single activation peptide by thrombin.
Intracellular FXIIIA has been reported to undergo non-proteolytic activation by Ca2+ ions [9, 10]. To imitate this activation pathway in the current work, FXIIIA was treated with Ca2+. Sedimentation velocity experiments revealed increasing dissociation of FXIIIA dimer in the presence of increasing Ca2+ concentrations (Fig. 8). Intriguingly, the catalytic activity of non-proteolytically activated species was lower than that of thrombin-activated form. We found no previous reports comparing activities of plasma and cellular forms of FXIII and more in-depth evaluation of the catalytic behavior of non-proteolytically activated FXIIIA is needed to better understand this phenomenon. Our results do, however, suggest that with the activation peptide still present, the A-subunits of FXIII are less accessible to the substrate. The high mM concentrations of Ca2+ used in the current work to non-proteolytically activate FXIIIA within a reasonable time scale (30 min), would not be physiologically available. Thus, it is reasonable to expect much slower rate of non-proteolytic FXIIIA activation in vivo. In addition, a portion of the intracellular FXIIIA population may exist as non-dissociated species, or weak dimers as computationally predicted by Gupta et al. [22].
In earlier experiments employing radiolabeled IAA [11, 12, 15], it was found that only half of the possible catalytic sites underwent alkylation, thus half-of-the-sites reactivity was postulated for the enzyme, implying its dimeric state. Upon observing active FXIIIA as a monomer in the present study, we considered the possibility of re-association of the two active A-subunits upon binding an inhibitor to one of them. To test this possibility, activated FXIIIA was modified with IAA or the more substrate-like peptide K9-DON to form covalent adducts with the active site C314 that mimic the acyl-enzyme complex. No dimerization was observed in sedimentation velocity analyses of both inhibited FXIIIA species. While we have no reason to dispute previous findings of the Folk [15] and Shafer [11, 12] research groups on half-of-the-sites reactivity of FXIIIA, there is an obvious need for a detailed study of possible cooperativity in FXIIIA-mediated catalysis.
Our current findings not only contribute to understanding the structure and function relationship in FXIIIA, but also provide insight on the protein regulation. Dissociation of the homodimer is likely crucial for FXIIIA cross-linking activity since the active site of each A-subunit must be sufficiently accessible for binding of the two large polypeptide substrates, and, more importantly, for release of the cross-linked product. Upon local elevations of Ca2+ concentration (for example, during activation of platelets), the intracellular FXIIIA dissociates and exhibits cross-linking activity, while depletion of Ca2+ inactivates the enzyme, possibly causing re-association of the A-subunits or formation of higher-order aggregates. In plasma, upon initiation of the coagulation cascade, FXIII is cleaved by thrombin. In this case, low Ca2+ concentration is sufficient to cause the dissociation of enzyme. Subsequently, monomeric plasma FXIIIA is deactivated proteolytically [31, 32].
The dimeric state of the zymogen form may thus be viewed as a way to stabilize the protein in a physiological setting. In the FXIIIA dimer, the outer, solvent exposed parts are formed mainly by β-sheet structural elements that are less susceptible to proteolytic degradation, while the more vulnerable α-helical structures are shielded within the dimer interface (Fig. 1A). At the same time, the active sites in the dimeric zymogen are buried, thus preventing undesirable protein crosslinking. Interestingly, a few naturally occurring mutants causative of severe FXIIIA deficiency [33–36] were predicted to be unable to dimerize and thus susceptible to proteolytic degradation.
To summarize briefly, current research provides experimental insight into FXIIIA oligomeric states in solution. For the first time to our knowledge, homodimeric FXIII A2 was demonstrated to dissociate as a part of the activation process, and inter-subunit interactions were quantitatively assessed. The study also highlights the role of the activation peptide in suppressing FXIIIA transglutaminase activity. This series of findings is intended to provide a better understanding of FXIIIA function as an intra- and extra-cellular transglutaminase.
EXPERIMENTAL PROCEDURES
Materials–
Recombinant FXIIIA expressed in Saccharomyces cerevisiae was a generous gift from the late Dr. Paul Bishop (Zymogenetics, USA). A stock solution of FXIIIA (40 μM) was made in deionized water. Concentration was determined from the absorbance at 280 nm using ε = 125710 M−1cm−1 (calculated using Protparam tool, www.expasy.org), and aliquots were stored at −80°C. Molar concentration of FXIIIA in this work refers to the A-subunits, as opposed to A2-dimers.
Bovine thrombin was purchased from Sigma Aldrich (USA). Thrombin inhibitor D-phenylalanyl-prolyl-arginyl chloromethyl ketone (PPACK) was purchased from Haematologic Technologies (USA). A glutamine-containing substrate peptide K9 (1LGPGQSKVIG10) was synthesized by New England Peptide (USA), stock solutions (15–30 mM) were made in deionized H2O, and accurate concentration was determined by quantitative amino acid analysis (AAA Service Laboratory, Damascus, OR). FXIIIA inhibitor peptide K9-DON [18] was synthesized by Zedira (Germany), and a stock solution (500 μM) was made in dimethyl sulfoxide (DMSO). All other reagents were of the highest purity available.
FXIIIA sample preparation for AUC–
All samples were prepared in a final volume of 500 μL for sedimentation velocity and 130 μL for sedimentation equilibrium experiments. To study the effect of Ca2+ concentration on FXIIIA, 2–6 μM zymogen was incubated in borate buffer (20 mM boric acid, 150 mM NaCl, pH 7.8) in the presence of 4–100 mM CaCl2 at 37°C for 30 min. As a negative control for a divalent metal cation, 100 mM MgCl2 was employed.
For proteolytic activation, FXIIIA (2–6 μM) was treated with bovine thrombin in the borate buffer containing 4 mM CaCl2. The cleavage progress was monitored by 8% SDS-PAGE with densitometry analysis using Gel Analyzer 2010 software (www.gelanalyzer.com). For complete activation peptide cleavage (all A-subunits), FXIIIA was incubated with 1.75–3.5 NIH units of thrombin at 37°C for 30 min. To achieve a limited amount of proteolysis (cleavage of approximately half of the A-subunits, the zymogen (2 μM) was incubated with 1.75 NIH units of thrombin for 6 min at 28°C. Thrombin cleavage reactions were stopped by the addition of 760 nM PPACK. After inhibition of thrombin, some samples were supplemented with 5% DMSO (see Results and Discussion).
For inhibition studies, FXIIIA (2 μM) activated by thrombin in the borate buffer (pH 7.8) with 4 mM CaCl2 was inhibited with five-fold molar excess of alkylating agent iodoacetamide (IAA) or five-fold molar excess of covalent inhibitor peptide K9-DON (1LGPG-(DON)-SLVIG10). All samples of inhibited FXIIIA contained 5% DMSO.
Control samples of zymogen FXIIIA (2 μM) were made in PBS (10 mM Phosphate, 150 mM NaCl, pH 7.4 [29] or in borate buffer.
AUC–
The oligomerization and hydrodynamic properties of zymogen as well as activated FXIIIA were studied by analytical ultracentrifugation. Samples were subjected to centrifugation within 2 h of preparation.
Sedimentation velocity experiments were carried out in a Beckman Coulter ProteomeLab XL-A analytical ultracentrifuge (Beckman Coulter Inc., USA) at 20°C and 50,000 rpm in standard 2 sector cells. Buffer density was determined on a Mettler/Paar Calculating Density Meter DMA 55A at 20.0°C and viscosity was measured using an Anton Parr AMVn Automated Microviscosimeter at 20°C. Data were analyzed with the program Sedfit (www.analyticalultracentrifugation.com) using the continuous c(s) distribution model. The partial specific volume of FXIIIA was calculated from the amino acid composition (0.734 mL/g) using the Protparam tool (web.expasy.org). Frictional ratios were calculated by Sedfit assuming no bound water. Experimental sedimentation coefficients were corrected to s20,w using the corrections based on the measured density and viscosity. Analytical runs were performed with at least two independent samples for each condition examined.
Sedimentation equilibrium experiments were carried out in standard 2 sector cells using 150 μL of buffer in the reference sector and 130 μL of sample in the sample sector. Three different rotor speeds were used and equilibrium was confirmed by observing the identical distribution of 280 nm absorbance in 3 successive scans at 2 hour intervals for each rotor speed. Data were analyzed using the program Sedphat (www.analyticalultracentrifugation.com). Global analysis was carried out using the monomer-dimer self association model and allowing the values of Ka, molecular weight and cell bottom and meniscus to float. Error analysis was performed using the Monte-Carlo error analysis feature of Sedphat with 1000 iterations.
Intrinsic tryptophan fluorescence measurements–
Fluorescent spectra were collected using a Perkin Elmer LS55 spectrofluorimeter. FXIIIA samples were prepared in the same manner as for the AUC. Prior to the measurements, each sample (500 μL) was transferred into a 4.0 ml quartz cuvette and diluted with corresponding buffer to give final volume 2.0 ml (thus giving final FXIIIA concentration of 500 nM). Samples were excited at 280 nm, and emission spectra at 300–400 nm were recorded. Fluorescence measurements were conducted with two independent samples for each condition studied.
nPAGE–
Samples of FXIIIA were prepared similarly to the AUC studies but in smaller volumes (20–100 μL). Samples were then supplemented with a loading buffer (0.5M tris, pH 6.8; 20% glycerol, 0.01% w/v bromophenol blue) in a 2:1 sample:buffer ratio. Electrophoretic separation was conducted at room temperature in a Tris-Glycine running buffer without SDS using 4% stacking and 7.5% resolving gels. During electrophoresis, the temperature of the running buffer did not elevate above 28°C. Gels were stained with Coomassie Blue. At least three independent samples were prepared for each condition studied, with essentially the same results.
SEC–
Size exclusion chromatography was performed using a 10 × 300 mm Superdex 75 analytical column (GE Healthcare) on an Akta FPLC (GE Healthcare). The column was calibrated with gel filtration standard from BioRad containing proteins of known Stokes radii (RS): bovine γ-globulin (150 kDa, RS = 5.1 nm), chicken ovalbumin (43 kDa, RS = 2.8 nm), and equine myoglobin (17 kDa, RS = 1.9 nm). Dextran Blue and vitamin B12 were used to determine the void column volume (V0) and the total volume (Vt), respectively. The BioRad standard protein mixture was separated in borate buffer. Samples of zymogen FXIIIA as well as 100 mM Ca2+ and 100 mM Mg2+ -treated FXIIIA were prepared as described for the sedimentation velocity AUC. Concerned about possible precipitation of the thrombin-activated FXIIIA on the analytical column, we did not subject this form to SEC analysis.
For each run, a 100 μL aliquot of 2 μM protein sample was applied to the column that had been pre-equilibrated with corresponding buffer. All protein separations were carried out at 0.5 ml/min, room temperature. Protein elution was monitored by absorbance at 280 nm. The peak elution volume (Ve) of each protein was converted to a partition coefficient (σ) using the equation
An inverse error function (erf −1) was calculated for (1- σ) [37] of each calibration standard and plotted against their Stokes radii (Fig. 2C). This calibration “curve” was then used to determine RS for different forms of FXIIIA.
Activity assays–
Transglutaminase activity of FXIIIA was determined using a standard coupled spectrophotometric assay as described elsewhere [18, 38, 39]. Briefly, during the transamidation reaction FXIIIA incorporates a primary amine (glycine ethyl ester) into a glutamine-containing K9-peptide with the release of ammonia. Glutamate dehydrogenase utilizes this ammonia along with NADH to convert α-ketoglutarate to glutamate, accompanied by a sigmoidal decrease in absorbance at 340 nm. The reaction velocity was determined as a slope at the steepest part of the curve. This value was corrected by subtraction of NADH oxidation rate observed in the absence of FXIIIA. Activity measurements were performed in triplicates for all conditions employed for AUC experiments.
Acknowledgements.
The authors thank K. Mouapi, R. Billur and L. Wagner for their critical discussions. This research was supported by a grant from the National Institutes of Health R15 HL120068.
Abbreviations:
- FXIII
blood coagulation factor XIII
- FXIIIA
the A-subunit of factor XIII
- nPAGE
non-denaturing polyacrylamide gel electrophoresis
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- DMSO
dimethyl sulfoxide
- SEC
size exclusion chromatography
- AUC
analytical ultracentrifugation
- IAA
iodoacetamide
Footnotes
Conflict of interest. The authors state that they have no conflict of interests.
REFERENCES
- 1.Komaromi I, Bagoly Z & Muszbek L (2011) Factor XIII: novel structural and functional aspects, J Thromb Haemost. 9, 9–20. [DOI] [PubMed] [Google Scholar]
- 2.Schroeder V & Kohler HP (2016) Factor XIII: Structure and Function, Semin Thromb Hemost. 42, 422–8. [DOI] [PubMed] [Google Scholar]
- 3.Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GV & Mehta K (2014) Transglutaminase regulation of cell function, Physiol Rev. 94, 383–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katona E, Haramura G, Karpati L, Fachet J & Muszbek L (2000) A simple, quick one-step ELISA assay for the determination of complex plasma factor XIII (A2B2), Thromb Haemost. 83, 268–73. [PubMed] [Google Scholar]
- 5.Katona E, Penzes K, Csapo A, Fazakas F, Udvardy ML, Bagoly Z, Orosz ZZ & Muszbek L (2014) Interaction of factor XIII subunits, Blood. 123, 1757–63. [DOI] [PubMed] [Google Scholar]
- 6.Katona EE, Ajzner E, Toth K, Karpati L & Muszbek L (2001) Enzyme-linked immunosorbent assay for the determination of blood coagulation factor XIII A-subunit in plasma and in cell lysates, J Immunol Methods. 258, 127–35. [DOI] [PubMed] [Google Scholar]
- 7.Lorand L & Konishi K (1964) Activation of the fibrin stabilizing factor of plasma by thrombin, Arch Biochem Biophys. 105, 58–67. [DOI] [PubMed] [Google Scholar]
- 8.Muszbek L, Polgar J & Boda Z (1993) Platelet Factor XIII becomes active without the release of activation peptide during platelet activation, Thromb Haemostasis. 69, 282–285. [PubMed] [Google Scholar]
- 9.Polgar J, Hidasi V & Muszbek L (1990) Non-proteolytic activation of cellular protransglutaminase (placenta macrophage factor XIII), Biochem J. 267, 557–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muszbek L, Haramura G & Polgar J (1995) Transformation of cellular factor XIII into an active zymogen transglutaminase in thrombin-stimulated platelets, Thromb Haemost. 73, 702–5. [PubMed] [Google Scholar]
- 11.Hornyak TJ, Bishop PD & Shafer JA (1989) Alpha-thrombin-catalyzed activation of human platelet factor XIII: relationship between proteolysis and factor XIIIa activity, Biochemistry. 28, 7326–32. [DOI] [PubMed] [Google Scholar]
- 12.Hornyak TJ & Shafer JA (1991) Role of calcium ion in the generation of factor XIII activity, Biochemistry. 30, 6175–82. [DOI] [PubMed] [Google Scholar]
- 13.Curtis CG, Brown KL, Credo RB, Domanik RA, Gray A, Stenberg P & Lorand L (1974) Calcium-dependent unmasking of active center cysteine during activation of fibrin stabilizing factor, Biochemistry. 13, 3774–80. [DOI] [PubMed] [Google Scholar]
- 14.Mary A, Achyuthan KE & Greenberg CS (1988) The binding of divalent metal ions to platelet factor XIII modulates its proteolysis by trypsin and thrombin, Arch Biochem Biophys. 261, 112–121. [DOI] [PubMed] [Google Scholar]
- 15.Chung SI, Lewis MS & Folk JE (1974) Relationships of the catalytic properties of human plasma and platelet transglutaminases (activated blood coagulation factor XIII) to their subunit structures, J Biol Chem. 249, 940–50. [PubMed] [Google Scholar]
- 16.Yee VC, Pedersen LC, Le Trong I, Bishop PD, Stenkamp RE & Teller DC (1994) Three-dimensional structure of a transglutaminase: human blood coagulation factor XIII, Proc Natl Acad Sci U S A. 91, 7296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yee VC, Pedersen LC, Bishop PD, Stenkamp RE & Teller DC (1995) Structural evidence that the activation peptide is not released upon thrombin cleavage of factor XIII, Thromb Res. 78, 389–397. [DOI] [PubMed] [Google Scholar]
- 18.Sabo TM, Brasher PB & Maurer MC (2007) Perturbations in factor XIII resulting from activation and inhibition examined by solution based methods and detected by MALDI-TOF MS, Biochemistry. 46, 10089–10101. [DOI] [PubMed] [Google Scholar]
- 19.Woofter RT & Maurer MC (2011) Role of calcium in the conformational dynamics of factor XIII activation examined by hydrogen-deuterium exchange coupled with MALDI-TOF MS, Arch Biochem Biophys. 512, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turner BT, Sabo TM, Wilding D & Maurer MC (2004) Mapping of factor XIII solvent accessibility as a function of activation state using chemical modification methods, Biochemistry. 43, 9755–9765. [DOI] [PubMed] [Google Scholar]
- 21.Stieler M, Weber J, Hils M, Kolb P, Heine A, Buchold C, Pasternack R & Klebe G (2013) Structure of active coagulation factor XIII triggered by calcium binding: basis for the design of next-generation anticoagulants, Angew Chem Int Ed Engl. 52, 11930–4. [DOI] [PubMed] [Google Scholar]
- 22.Gupta S, Biswas A, Akhter MS, Krettler C, Reinhart C, Dodt J, Reuter A, Philippou H, Ivaskevicius V & Oldenburg J (2016) Revisiting the mechanism of coagulation factor XIII activation and regulation from a structure/functional perspective, Sci Rep. 6, 30105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooke RD & Holbrook JJ (1974) The calcium-induced dissociation of human plasma clotting factor XIII, Biochem J. 141, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Traut TW (1994) Dissociation of enzyme oligomers: a mechanism for allosteric regulation, Crit Rev Biochem Mol Biol. 29, 125–63. [DOI] [PubMed] [Google Scholar]
- 25.Hashimoto K, Nishi H, Bryant S & Panchenko AR (2011) Caught in self-interaction: evolutionary and functional mechanisms of protein homooligomerization, Phys Biol. 8, 035007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinkas DM, Strop P, Brunger AT & Khosla C (2007) Transglutaminase 2 undergoes a large conformational change upon activation, PLoS Biol. 5, e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clouthier CM, Mironov GG, Okhonin V, Berezovski MV & Keillor JW (2012) Real-time monitoring of protein conformational dynamics in solution using kinetic capillary electrophoresis, Angew Chem Int Ed Engl. 51, 12464–8. [DOI] [PubMed] [Google Scholar]
- 28.Kristiansen GK & Andersen MD (2011) Reversible activation of cellular factor XIII by calcium, J Biol Chem. 286, 9833–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bishop PD, Teller DC, Smith RA, Lasser GW, Gilbert T & Seale RL (1990) Expression, purification, and characterization of human factor XIII in Saccharomyces cerevisiae, Biochemistry. 29, 1861–9. [DOI] [PubMed] [Google Scholar]
- 30.Demirin H, Ozhan H, Ucgun T, Celer A, Bulur S, Cil H, Gunes C & Yildirim HA (2011) Normal range of mean platelet volume in healthy subjects: Insight from a large epidemiologic study, Thromb Res. 128, 358–60. [DOI] [PubMed] [Google Scholar]
- 31.Hur WS, Mazinani N, Lu XJ, Britton HM, Byrnes JR, Wolberg AS & Kastrup CJ (2015) Coagulation factor XIIIa is inactivated by plasmin, Blood. 126, 2329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bagoly Z, Haramura G & Muszbek L (2007) Down-regulation of activated factor XIII by polymorphonuclear granulocyte proteases within fibrin clot, Thromb Haemost. 98, 359–67. [PubMed] [Google Scholar]
- 33.Ichinose A, Tsukamoto H, Izumi T, Yamazaki T, Togashi M, Takamatsu J, Saito H & Umeyama H (1998) Arg260-Cys mutation in severe factor XIII deficiency: conformational change of the A subunit is predicted by molecular modelling and mechanics, Br J Haematol. 101, 264–72. [DOI] [PubMed] [Google Scholar]
- 34.Maeda S, Zhang WG, Souri M, Yee VC & Ichinose A (2012) Impaired dimer assembly and decreased stability of naturally recurring R260C mutant A subunit for coagulation factor XIII, J Biochem. 152, 471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Souri M & Ichinose A (2001) Impaired protein folding, dimer formation, and heterotetramer assembly cause intra- and extracellular instability of a Y283C mutant of the A subunit for coagulation factor XIII, Biochemistry. 40, 13413–20. [DOI] [PubMed] [Google Scholar]
- 36.Zhang WG, Souri M & Ichinose A (2013) Proteosomal degradation of naturally recurring R260C missense and exon-IV deletion mutants of factor XIII A-subunit expressed in mammalian cells, Haemophilia. 19, 415–9. [DOI] [PubMed] [Google Scholar]
- 37.Ackers GK (1970) Analytical gel chromatography of proteins, Adv Protein Chem. 24, 343–446. [DOI] [PubMed] [Google Scholar]
- 38.Karpati L, Penke B, Katona E, Balogh I, Vamosi G & Muszbek L (2000) A modified, optimized kinetic photometric assay for the determination of blood coagulation factor XIII activity in plasma, Clin Chem. 46, 1946–55. [PubMed] [Google Scholar]
- 39.Fickenscher K, Aab A & Stuber W (1991) A photometric assay for blood coagulation factor XIII, Thromb Haemost. 65, 535–40. [PubMed] [Google Scholar]