

Abstract
Background:
We have previously reported that epoxyeicosatrienoic acid (EET) has multiple beneficial effects on renal and adipose tissue function, in addition to its vasodilatory action; it increases insulin sensitivity and inhibits inflammation. In an examination of the signaling mechanisms by which EET reduces renal and peri-renal fat function, we hypothesized that EET ameliorates obesity-induced renal dysfunction by improving sodium excretion, reducing the sodium-chloride cotransporter NCC, lowering lood pressure, and enhancing mitochondrial and thermogenic gene levels in PGC-1α dependent mice.
Methods:
EET-agonist treatment normalized glucose metabolism, renal ENaC and NCC protein expression, urinary sodium excretion and blood pressure in obese (db/db) mice. A marked improvement in mitochondrial integrity, thermogenic genes, and PGC- 1α-HO-1-adiponectin signaling occurred. Knockout of PGC-1α in EET-treated mice resulted in a reversal of these beneficial effects including a decrease in sodium excretion, elevation of blood pressure and an increase in the pro-inflammatory adipokine nephroblastoma overexpressed gene (NOV). In the elucidation of the effects of EET on peri-renal adipose tissue, EET increased adiponectin, mitochondrial integrity, thermogenic genes and decreased NOV, i.e. “Browning’ peri-renal adipose phenotype that occurs under high fat diets. Taken together, these data demonstrate a critical role of an EET agonist in the restoration of healthy adipose tissue with reduced release of inflammatory molecules, such as AngII and NOV, thereby preventing their detrimental impact on sodium absorption and NCC levels and the development of obesity-induced renal dysfunction.
Keywords: hypertension, epithelial sodium channel, sodium excretion, sodium-chloride cotransporter, mitochondrial function, insulin sensitivity, obesity
INTRODUCTION
In 2014 the World Health Organization determined that, worldwide, more than 1.9 billion individuals were overweight and another 600 million obese; numbers that have more than doubled since 1980. Obesity has become a challenge to global health care systems. In addition to type 2 diabetes mellitus, kidney dysfunction is considered to be a cardiovascular risk factor in obese individuals (1, 2). Chronic obesity and diabetes are not only major contributors to renal dysfunction but are manifested in increased risk of cardiovascular disease and high morbidity and mortality (1, 3). A strong positive correlation exists between obesity, body mass index and the risk of renal failure (1, 4, 5). Diabetes and obesity-related elevated blood pressure and kidney injury can arise through several mechanisms. Adipocytes are considered a major source of angiotensinogen that leads to elevations in plasma angiotensin II concentrations and systolic blood pressure, effects that are prevented in mouse models of angiotensinogen deficiency (6, 7). An abundance of plasma angiotensin II leads to increased levels of vasoconstrictors (6, 8), ultimately leading to an increase in inflammatory cytokines such as NOV, causing vasoconstriction, an increase in systolic blood pressure, ROS formation (7, 9), mitochondrial dysfunction, hyperglycemia, and increased body weight (10). NOV is a pro-inflammatory molecule expressed at elevated levels in adipose tissue and elevation of NOV is attributed to increases in obesity and plasma triglycerides (7, 11). Ablation of NOV resulted in decreased obesity, improved glucose tolerance and insulin sensitivity (7, 12).
In addition to obesity-diabetes-induced hyperglycemia, and elevated insulin levels resulting from insulin insensitivity, visceral obesity results in intrarenal adipose tissue accumulation and physical compression of the kidneys (2) as well as lipotoxicity. Inflammation, oxidative stress (7) and activation of the renal renin-angiotensin system(13).
Reactive oxygen species (ROS) and oxidative stress, a result of an increase in adipocyte release of Ang II and a decrease in the levels of HO-1, plays an important role in obesity-induced renal injury and mitochondrial dysfunction and fragmentation (14–18). Increased levels of HO-1 decrease ROS, ameliorate renal dysfunction, increase PGC-1α and decrease blood pressure through modulation of NADPH oxidase activity (7, 9, 17, 19). Further, the suppressive effect of increased HO-1 expression on Ang II-induced hypertension and renal injury has been reported (5, 18, 20). HO-1- derived CO and bilirubin to reduce vascular reactivity and blood pressure through vasodilation (14, 20–22). Both carbon monoxide and bilirubin exert protective effects on the kidney following injury (23, 24) and bilirubin prevents endothelial dysfunction which is a hallmark of cardiovascular complications (23). Global overexpression of human HO- 1 using a lentiviral probe results in the lowering of blood pressure in spontaneously hypertensive rats (24). Gene targeting of HO-1 in the thick ascending loop of Henle reduced angiotensin II (Ang Independent hypertension, an effect that was associated with a reduction in the expression of medullary NKCC2 (25). Both humans and mice deficient in HO-1 display vascular, renal and organ damage (26–28). Ablation of HO-1 in adipose tissue causes an increase in inflammation and inhibition of mitochondrial function (29). Further, obesity-mediated reduction in HO-1 expression or activity augments adipose tissue dysregulation, which is abated by upregulation of the heme- HO system (30, 31).
We previously demonstrated that endothelium-specific targeting of human CYP2J2-derived EET attenuates adiposity and renal dysfunction in mice fed a high salt diet(32). Moreover, we have evidence of a synergistic interplay between HO-1 and epoxygenase(s), whereby the expression of one is promoted by the other (7, 33). Furthermore, interplay between HO-1 and CYP-derived EETs modulates the adipocyte phenotype by regulating peroxisome proliferator-activated receptor gamma coactivator- 1 alpha (PGC-1α) (a transcription factor known to regulate mitochondrial biogenesis and insulin sensitivity) which plays a critical role in the regulation of mitochondrial function manifested by decreased lipid accumulation and mitochondrial production of ROS, and increased adiponectin (34, 37). Our present hypothesis that EET is a positive regulator of HO-1, PGC-1α and adiponectin production, leading to activation of a signaling pathway which controls mitochondrial function, decreases formation of NOV, sodium retention, NCC channel and which may impact the metabolic phenotype thereby normalizing obesity-associated hypertension in mice fed a high fat diet. A corollary of this is possible in that a deficit in the functionality of this regulatory pathway that occurs by blocking PGC-Ια may facilitate the development of obesity-associated hypertension. The apparent crosstalk between adipose tissue hypertrophy and hyperplasia with increased inflammation, AngII and NOV levels and decreased adiponectin mitochondrial function levels, leads to the increased in sodium absorption, NCC, and the development of obesity- and inflammation-induced renal dysfunction. We report that EET-agonist treatment increases mitochondrial integrity and PGC-1α- HO-1 levels with a concomitant normalization of blood pressure that positively impact urinary sodium excretion, decreases plasma Ang II levels as well as NCC, SPAK and ENaC activity in the kidneys of obese mice.
MATERIALS AND METHODS
Animal Protocols
Four-week-old db/db mice from Jackson Laboratories (Bar Harbor, ME) were used for this investigation. Mice were divided into 3 treatment groups following a 16- week acclimatization period. The mice were fed regular chow (Harlan, Teklad Lab Animal Diets, Indianapolis, IN) and were treated as follows: Group 1) control, Group 2) injected intraperitoneally with EET-A twice/week for 8 weeks with a dose of 1.5 mg/100g of body weight as previously described (35), and Group 3) received EET-A as above and also 2 bolus injections of 40–70 × 106 TU/mouse in 80–100 μl PGC-1α (sh) lentivirus (Dharmacon, Lafayette, CO) into the retro orbital vein. At the end of the experiment, mice were euthanized, assessed for fat content and total body and kidney weights were measured. All animal experiments followed the NYMC IACUC institutionally approved protocol in accordance with the NIH guidelines.
Fasting Blood Glucose, Glucose Tolerance Testing and Urinary Sodium Excretion
Fasting blood glucose, glucose tolerance, and urinary sodium were measured from tail blood following a 6-h fast. Blood pressure was determined as described previously (7, 36). Urine was collected and sent to Antech diagnostics (Fountain Valley, CA) for urinary sodium excretion and urinary protein measurements.
Determination of Oxygen Consumption
The db/db mice groups were allowed to acclimatize in the oxygen consumption chambers for a total of 3 weeks. Adaptation periods for the 3 week duration were executed in 2 hour increments, three times a week. The Oxylet gas analyzer and air flow unit (Oxylet; Panlab-Bioseb, Vitrolles, France) were used to determine mouse oxygen consumption (VO2). Each mouse was placed individually in the machine and VO2, VCO2 and respiratory quotient (RQ) was calculated as VCO2/VO2. (7)
Western Blot Analysis and RNA Analysis
For protein expression analyses, kidney tissues were lysed in RIPA lysis buffer supplemented with protease and phosphatase inhibitors (Complete™Mini and PhosSTOP™, Roche Diagnostics, Indianapolis, IN as previously described (29, 37). NOV serum protein levels were measured by Western Blot and were normalized to transferrin protein. β-actin was used as loading control for western blotting analysis of tissues.
Statistical Analysis
Data are expressed as means ± S.E.M. Bonferroni’s post-test analysis for multiple comparisons was used to calculate the significance of mean value differences using one-way analysis of variance. The null hypothesis was rejected at p<0.05.
RESULTS
Effect of EET Agonist on Systemic Metabolic Parameters and Peri-renal Fat Deposition
We examined the effect of EET-A on visceral and peri-renal adipose tissue deposition. Figures 1A and B depict body weight at the commencement of treatment and at the experimental end-point relative to the weight at the start of the treatment (20 weeks of age). After 3 weeks on EET-A, one group of mice was injected with lentiviral particles for silencing of PGC-1α. Over the 8 weeks of the experiment, control mice exhibited on average a weight gain of 0.99 g/week with an end-point mean body weight of 63.9 ± 1.3 g. In contrast, mice treated with EET-A lost on average 0.93 g/week and weighed 45.2 ± 3.8 g at the end of the study. However, the EET-A-mediated weight reduction was not only reversed, but the rate of weight gain was fully restored to that of the control group after administration of Ln- PGC-Ια (sh) despite continued EET-A- treatment and at the end of the experiment these mice weighed 55.0 ± 1.1 g.
Figure 1.
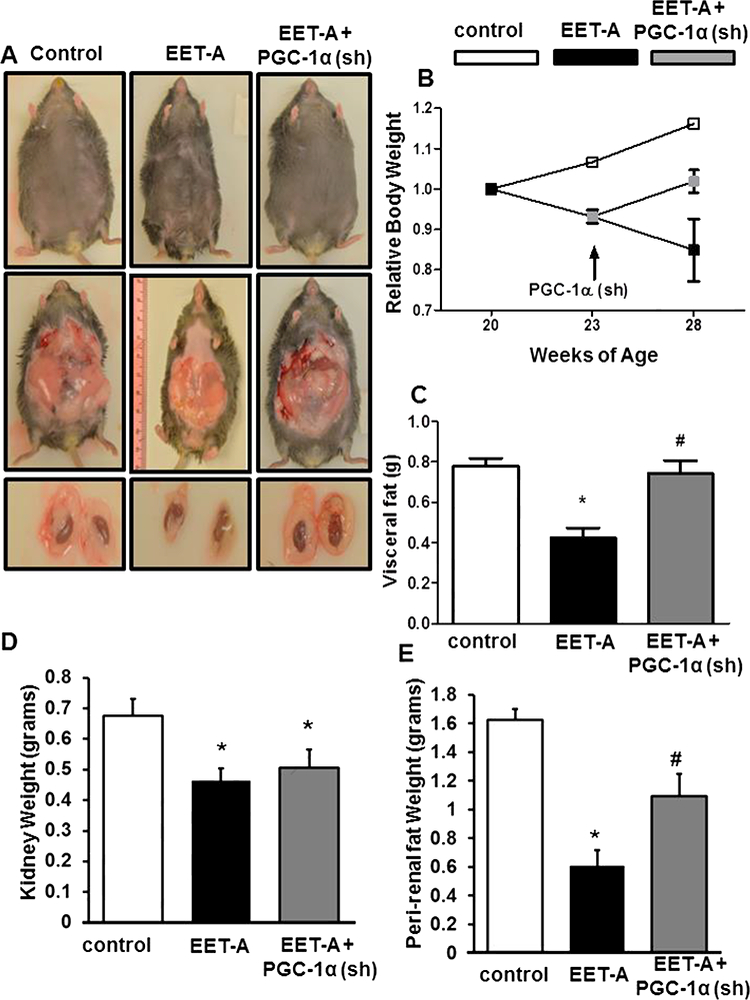
Effect of EET-A treatment on body weight, fasting blood glucose, glucose tolerance, and adipose tissue deposition in db/db mice. (A) Representative photographs showing appearance of body, abdominal adipose tissues, kidney with peri-renal adipose tissue from each group of mice, (B) relative body weight, (C) visceral fat weight, (D) kidney fat, (E) peri-renal fat weight. Db/db control (white bars/boxes), db/db treated with EET-A (black bars/boxes), and PGC-1α-deficient db/db mice treated with EET-A (grey bars/boxes). Results are mean ± SE, n=6, *p<0.05 vs db/db control, #p<0.05 vs db/db mice treated with EET-A.
As seen in Figures 1C, D and E, EET-A reduced visceral, kidney and peri-renal fat weights in db/db mice (p<0.05). The reduced accumulation of adipose tissue in EET- A-treated mice can be seen visually when compared with control db/db mice through both external bulk (Figure 1A top row) and internal adipose tissue deposition (Figure 1A middle row). EET-A reduced peri-renal fat deposition as compared to db/db control mice (Figure 1A bottom row). External dimensions as well as internal adipose tissue content and peri-renal fat tissue deposition in the EET-A-PGC-Ια (sh) group paralleled the adiposity observed in control db/db mice. Silencing PGC-Ια when combined with EET- agonist administration reduced the effect of EET-agonist on kidney weight (Figure 1D) and on peri-renal adipose tissue deposition (Figure 1E).
EET Agonist decreases Systolic Blood Pressure, Fasting Blood Glucose and Glucose Intolerance.
Systolic blood pressure was lower in EET-A-treated db/db mice but not in db/db mice that were deficient in PGC-1α (Figure 2A). As seen in Figure 2B, the fasting blood glucose level in the control group was 573.7 ± 15.9 mg/dL, compared with the group treated with EET-A with a level of 187.7 ± 34.9 mg/dL, (p<0.05). That this EET-A- mediated effect was PGC-1a-dependent was evidenced in the EET-A-PGC-1α (sh) group where the fasting blood glucose level was increased to 308.6 ± 2.6 mg/dL (p<0.05) as compared with the mice treated with EET-agonist alone. EET-A lowered glucose levels in glucose-loaded db/db mice and this effect was reversed in db/db mice that were also treated with PGC-1α (sh) lentivirus (Figure 2C). Food intake decreased in mice treated with EET-A, an effect that was reversed when also treated with PGC-1α (sh) lentivirus (Figure 2D).
Figure 2.
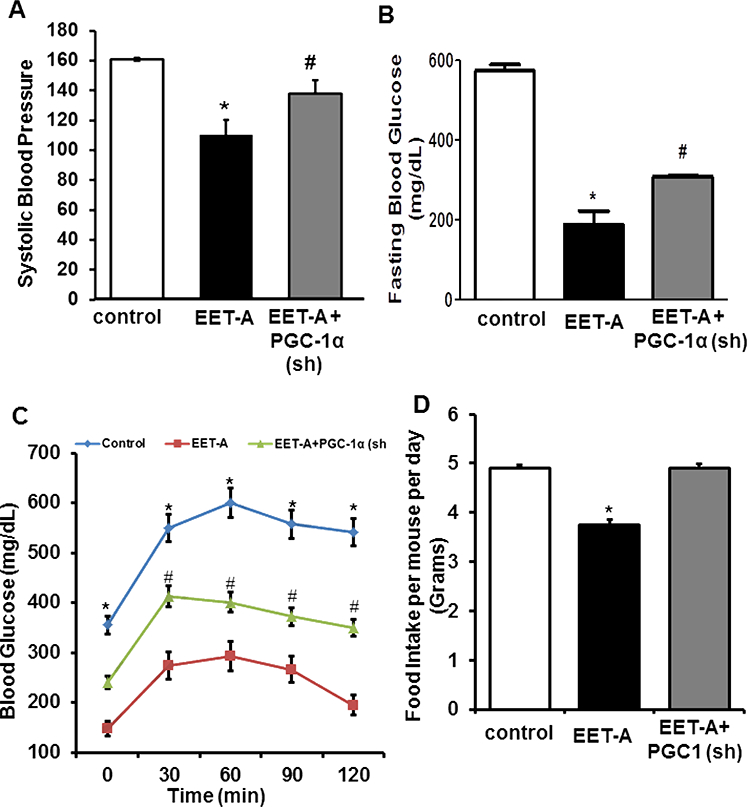
Effect of EET-A treatment on (A) systolic blood pressure, (B) fasting blood glucose levels and (C) glucose tolerance (D) the food intake per mouse per day. Db/db control (white bars/boxes), db/db treated with EET-A (black bars/boxes), and PGC-1α- deficient db/db mice treated with EET-A (grey bars/boxes). Results are mean ± SE, n=6, *p<0.05 vs db/db control, #p<0.05 vs db/db mice treated with EET-A.
EET Agonist increases Oxygen Consumption and decreases the Respiratory Quotient.
To determine whether an effect of EET-A on whole body oxygen consumption is mediated through PGC-Ια expression, we compared VO2, VCO2 and RQ (Figure 3A-C). VO2 and VCO2 increased following EET-A treatment in db/db mice (p<0.05) (Figure 3A and B). Moreover, VO2 and VCO2 levels in EET-A-PGC-Ια (sh) mice were lowered by comparison with EET-A treated db/db mice (Figure 3A and B). RQ was decreased by EET-A compared with control db/db mice, but returned to levels of control animals in db/db mice administered PGC-1α (sh) (Figure 3C).
Figure 3.
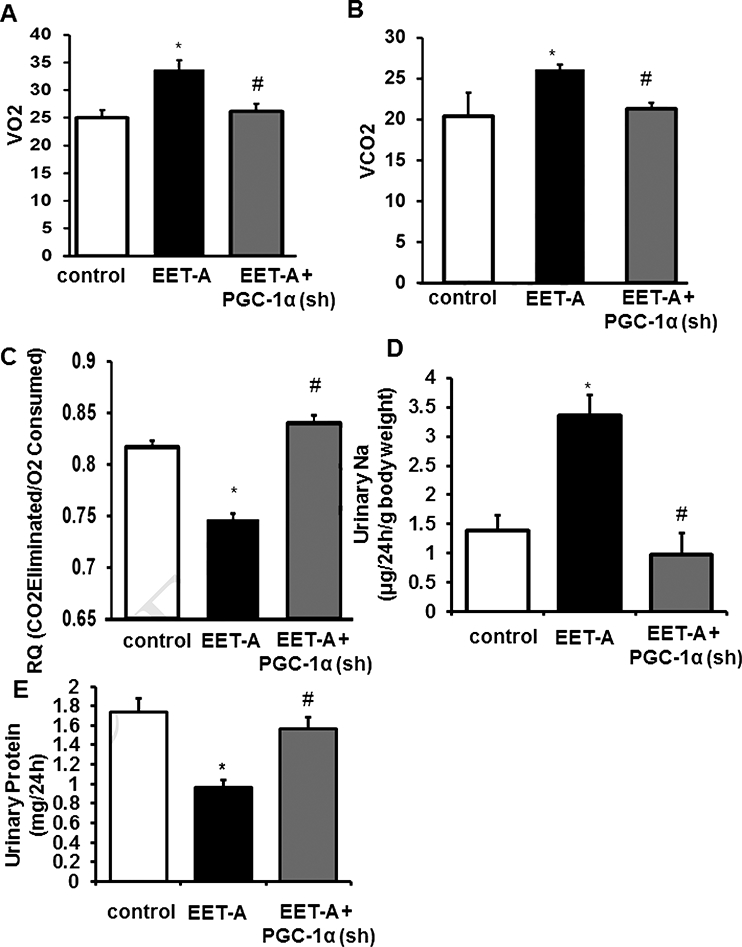
Db/db mice were fed regular chow (Harlan, Teklad Lab Animals Diets, Indianapolis, IN) and following a 16-week acclimatization period were divided into 3 treatment groups. Group 1 was control. Groups 2 and 3 were injected intraperitoneally with EET-A twice/week for 8 weeks with a dose of 1.5 mg/100g of body weight as previously described (7). The mice in Group 3 were also given 2 bolus injections of 4070 × 106 TU/mouse in 80–100 μΙ PGC-1α (sh) lentiviruses (Dharmacon, Lafayette, CO) into the retro orbital vein. At the end of the experiment, VO2 (A) and VCO2 (B) levels (mL/min/Kg body weight) were determined using the Oxylet gas analyzer. RQ (the respiratory quotient) was calculated as VCO2/VO2 (C). Effect of EET-A treatment on (D) Urinary Sodium normalized to body weight, (E) Protein, Db/db treated with EET-A (black bars/boxes), and PGC-1a-deficient db/db mice treated with EET-A (grey bars/boxes). Results are mean ± SE, n=6, *p<0.05 vs db/db control, #p<0.05 vs db/db mice treated with EET-A alone.
EET Agonist Decreases Urinary Sodium, and Urinary Protein
EET-agonist increased (p<0.05) urinary sodium excretion, an effect that was reversed in PGC-1α-deficient mice (Figure 3D). Urinary protein content was decreased (p<0.05) by EET-agonist and returned to control levels in PGC-1α-deficient mice (Figure 3E).
Effect of EET Agonist on SPAK, ENaC, NCC, and NKCC2 Protein Levels.
EET-A decreased (p<0.05) the expression of SPAK by nearly 50% as compared with the levels observed in control db/db mice (Figure 4A and B) and this reduction was reversed (p<0.05) in Ln-PGC-1α (sh) db/db mice (Figure 4A and B). The same effect of EET-A was observed for both ENaC (Figure 4A, C, F, and G) and NCC (Figure 4A and D) protein levels, and for both ENaC and NCC the EET-A-mediated effect was reversed in PGC-1α-deficient mice (Figure 4A, C and D). EET-A did not affect NKCC2 expression, but in PGC-1α-deficient mice treated with EET-A NKCC2 expression was elevated as compared with EET-A-treated mice (Figure 4A, and E).
Figure 4.
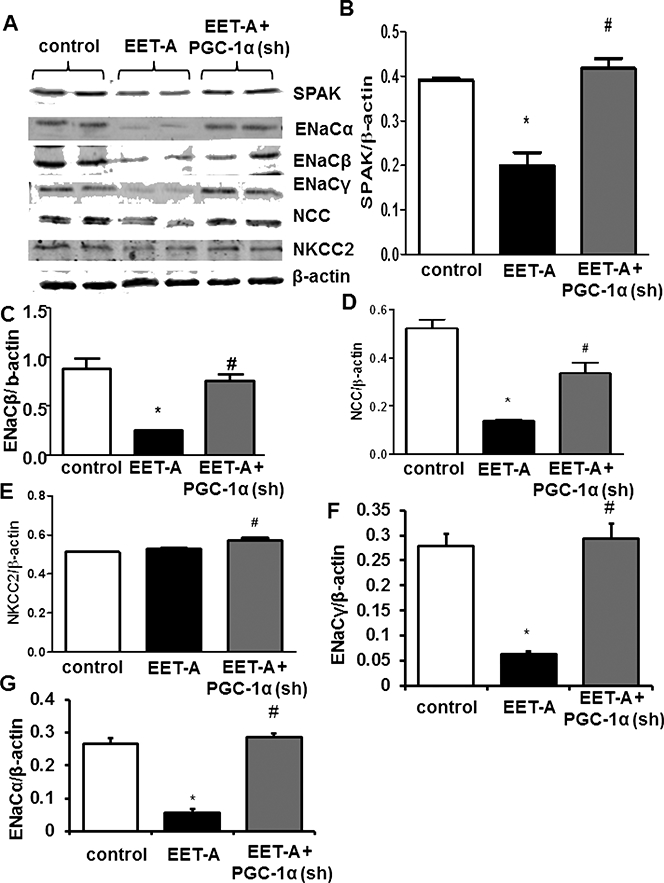
Effect of EET-A treatment on natriuresis and sodium reabsorptive protein expression in kidneys of db/db mice. Representative Western Blots (A), and densitometry analysis of (B) SPAK, (C) ENaCβ, (D) NCC, (E) NKCC2, (F) ENaC, and (G) ENaCα in kidney tissues of mice. Db/db control mice (white bars), db/db treated with EET-A (black bars), and PGC-1α-deficient db/db mice treated with EET-A (grey bars). Results are mean ± SE, n=6, *p<0.05 vs db/db control, #p<0.05 vs db/db mice treated with EET-A.
Effect of EET Agonist on PGC-1α, HO-1, SIRT1, Adiponectin, pAMPK/AMPK, Mitochondrial Fusion-Fission Proteins and NOV.
EET-A treatment led to a 2-fold upregulation of PGC-1α as compared with db/db control mice (Figure 5A and B). This increase in PGC-1α levels by EET-A was prevented in Ln-PGC-Ια (sh) mice (Figure 5A and B). EET-A increased HO-1 levels 3fold (p<0.05) as compared with control db/db mice (p<0.05) (Figure 5A and C). Ln-PGC- 1α (sh) prevented the EET-A-mediated increase in HO-1 protein expression (Figure 5A and C). EET-A-treated mice expressed >2-fold higher (p<0.05) SIRT1 levels compared to control db/db mice (p<0.05), while PGC-1α (sh) lentivirus prevented this effect (Figure 5A and D). Furthermore, EET-A increased the expression of adiponectin more than 3-fold (p<0.05) as compared with control db/db mice (Figure 5A and E). The effect of EET-A on adiponectin levels was diminished in PGC-1α(sh) lentivirus-treated db/db mice (Figure 5A and E). Because phosphorylation of AMPK-AKT increased vascular function we examined levels of pAMPK and pAKT, Figure 5F, EET-A-increased (p<0.05) renal pAMPK/AMPK by ~40% as compared with db/db control mice. In PGC- 1α-deficient mice the EET-A-mediated effect on pAMPK/AMPK was completely abrogated and the relative level of AMPK phosphorylation was even lower than that observed in control db/db mice.
Figure 5.
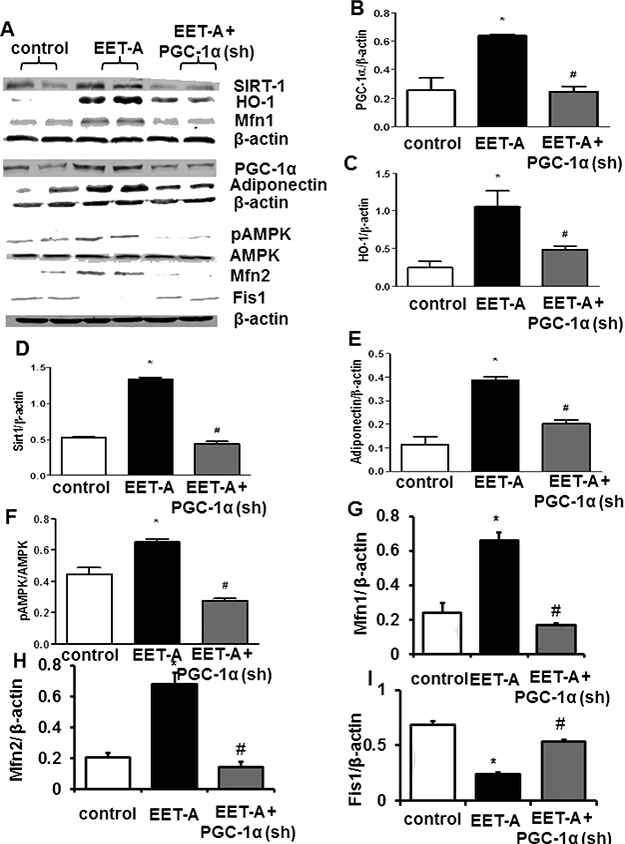
Effect of EET-A treatment on PGC-1α, HO-1, SIRT1, adiponectin, Mfn1, Mfn2, Fis1, and NOV in kidney tissue of db/db mice. (A) Representative Western Blots and densitometry analysis of (B) PGC-1α, (C) HO-1, (D) SIRT1, (E) adiponectin, (F) pAMPK/AMPK, (G) Mfn1, (H) Mfn2, (I) Fis1. Db/db control mice (white bars), db/db treated with EET-A (black bars), and PGC-1α-deficient db/db mice treated with EET-A (grey bars). Results are mean ± SE, n=6, *p<0.05 vs db/db control, #p<0.05 vs db/db mice treated with EET-A.
To assess the mitochondrial fusion-fission proteins we measured Mfn1, Mfn2 and Fis1 levels in kidney of db/db mice. As seen in Figure 5G, the Mfn1 levels in EET-A treated mice were elevated (p<0.05) about 5-fold as compared with control db/db mice and this EET-A-mediated effect was completely abrogated in PGC-1α-deficient mice (Figure 5G). The Mfn2 ratio (Figure 5H) followed a pattern identical to the Mfn1 and Mfn2 ratios which were higher (p<0.05) in EET-A treated mice when compared with both control and PGC-1α deficient mice treated with EET-A (Figure 5H). Fis1 levels decreased (p<0.05) in EET-A-treated mice; this decrease was prevented in PGC-1α- deficient mice (Figure 5I).
EET-agonist causes reduction of pre-renal and renal tissues of NOV.
We examined whether peri-renal adipose tissue and renal levels of NOV were increased in obese mice. Renal tissue expressed lower levels of NOV compared to perirenal adipose tissue (<0.05). EET-agonist decreased NOV levels in both renal and peri- renal fat in a PGC-1α-dependent manner. Decreased levels of PGC-1α reversed the ability of EET-agonist to reduce NOV in both peri-renal and renal tissue (Figure 6A-D). EET agonist treatment decreased plasma And II levels (Figure 6E).
Figure 6.
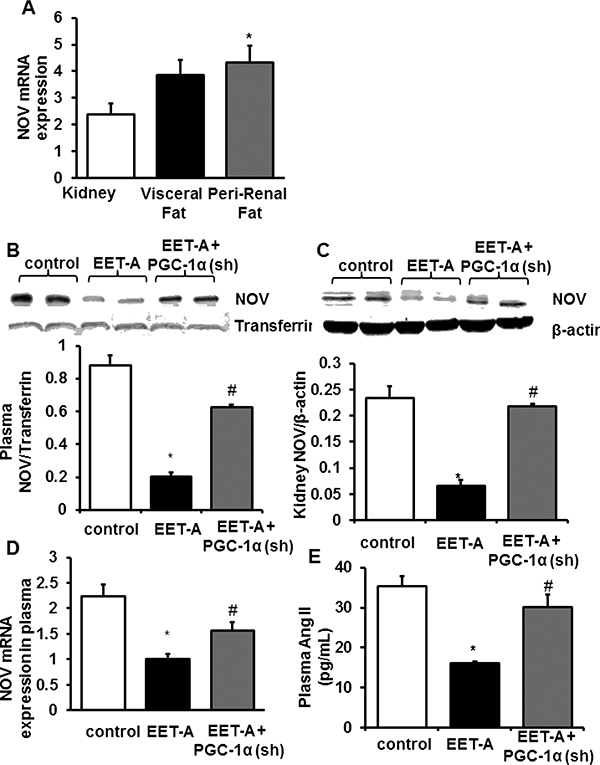
Tissue expression of NOV and EET agonist effect on plasma and kidney NOV levels and plasma Angiotensin 2. (A) The mRNA expression of NOV in kidney, visceral fat, and peri-renal fat. (B) Representative western blots, densitometry analysis of NOV in plasma of db/db control mice, db/db treated with EET-A mice, and PGC-1α- deficient. (C) Representative western blots, densitometry analysis of NOV in Kidney of db/db control mice, db/db treated with EET-A mice, and PGC-1a-deficient. (D) mRNA expression of NOV in plasma of db/db control mice, db/db treated with EET-A mice, and PGC-1α-deficient. (E) Plasma Angiotensin II. Results are means ± SE, n=6, *p<0.05 vs db/db control, # p<0.05 vs db/db mice treated with EETA /.
DISCUSSION
In the present study we demonstrate that EET agonist administered to obese mice with diabetes has substantial beneficial effects including improved mitochondrial function, and increased glucose and fat metabolism. We demonstrate an obesity-mediated increase in the pro-inflammatory adipokine NOV and plasma angiotensin II are attenuated by an EET-agonist. Our results show improved renal function and urinary sodium excretion. Prior studies indicate that EET agonists improve heart function and cardiomyopathy in obese mice by decreasing NOV (7). It is not yet clear whether increases in inflammatory markers such as NOV significantly upregulated ENaC and blood pressure. In addition, EET agonist decreased both NCC and ENaC protein expression in association with enhanced urinary sodium excretion as well as having major effects on glucose and fat metabolism in db/db mice manifested by the decrease in metabolic parameters and increased adiponectin. In adipose tissue, we found that the decrease in peri-renal fat by EET agonist treatment reduced the physical compression and stress of the kidney which may be involved in an observed decrease in blood pressure and likely also lead to a reduction of inflammatory markers other than AngII and NOV. This is important as obesity and associated chronic inflammation go hand in hand contributing to kidney disease (1, 4, 5). Mechanistically, the effects of the EET-agonist appear to be dependent on PGC-1α levels as these perturbations were prevented by reduction in PGC-1α levels.
Increased expression and activation of the thiazide-sensitive NCC accompanied by increased sodium reabsorption has been reported (1, 5, 38–40). The bumetanide- sensitive sodium-potassium- 2- chloride cotransporter (NKCC2), the amiloride-sensitive epithelial sodium channel (ENaC) and sodium-chloride cotransporter (NCC) all have a role in sodium reabsorption, but only the expression and activity of NCC and ENaC appear to be affected in diabetes and obesity (1, 5, 38–41).
Obesity and hyperinsulinemia are known factors that increase the expression of NKCC2, ENaC, NCC and the NCC-activating kinase, SPAK, (42–44) all of which are involved in renal sodium reabsorption. EET-agonist reduced levels of SPAK, NCC and ENaC, consistent with reduced sodium reabsorption and urinary sodium excretion. Importantly, this effect of the EET-agonist on various channels is dependent on PGC-1α levels. Our results confirm previous observations that EET reduced ENaC activity and lowered blood pressure in high-salt diet induced hypertension in mice (45). Indapamide (a thiazide-like diuretic which inhibits NCC) can stimulate the release of EETs (46). In agreement with the current result that EET improved sodium excretion and may involve a reduction of NCC and ENaC. Our findings are relevant as indapamide improved both endothelial function and metabolic parameters in patients with type 2 diabetes as compared with the prototypical NCC inhibitor hydrochlorothiazide (47). Thus, EETs contribute to the beneficial cardiovascular effects seen and improved metabolic function with the clinical use of indapamide. Further, our results indicate that the EET-agonist not only interferes with NCC expression and fat and glucose metabolism, but also ENaC expression. ENaC inhibition either directly by amiloride, or secondary to mineralocorticoid receptor antagonism with spironolactone or eplerenone, has beneficial cardiovascular effects (48).
The improvement of insulin receptor phosphorylation in EET agonist treated db/db mice is evidenced by lowered fasting blood glucose levels, increased glucose tolerance and decreased body weight, peri-renal fat and inflammatory adipokines. Chronic abdominal adiposity is associated with increased levels of Ang II which alone leads to an increase in systolic blood pressure (38, 39) while the pro-inflammatory cytokines NOV contribute to the development of diabetic cardiomyopathy (7).
The beneficial effects of the EET-agonist were abolished in PGC-α deficient db/db mice suggesting a seminal role for the EET-mediated increases in PGC-1a (7). In agreement with this finding mice lacking PGC-1α in adipose tissue and fed a high fat diet develop insulin resistance and have increased levels of circulating lipid (40). An increase in PGC-Ια expression results in higher levels of the mitochondrial proteins, Mfn1 and Mfn2, which is consistent with the role of PGC-1α as a major regulator of mitochondrial function (41). Consistent with our observations, the EET-agonist increased VO2 indicating increased mitochondrial function and whole body metabolism, this effect was prevented in PGC-Ια deficient mice. These positive effects of the EET- agonist were associated with restored mitochondrial and thermogenic gene levels and reprograming of the peri-renal fat to a brite phenotype, thereby increasing the PGC-1α- mediated increase in HO-1 that positively impacts renal and adipocyte function by decreasing the levels of ROS (49). The concept that EET-HO-1-PGC-1α exerts a counter-regulatory influence on high fat intake is an important advance in our understanding of EET-agonist function (reduces sodium retention, NCC Channel and NOV levels), for it reveals a new role of this interplay in peri-renal adipose and renal function that improves obesity-induced hypertension. The current report does not address the possibility that the hypertensive phenotype displayed by obese mice fed a high fat diet is mechanistically related to upregulation of the renin-angiotensin system of adipocytes, which previous reports have linked to the pathogenesis of obesity-induced hypertension.(50–52). However, the present study expands both the breadth and depth of our understanding of the actions of EET in cardiovascular disease and provides important new mechanistic information on the possible crosstalk between healthy adipose tissue, renal function and normotension with a focus on adiponectin, NOV, AngII and sodium transport.
Figure 7.
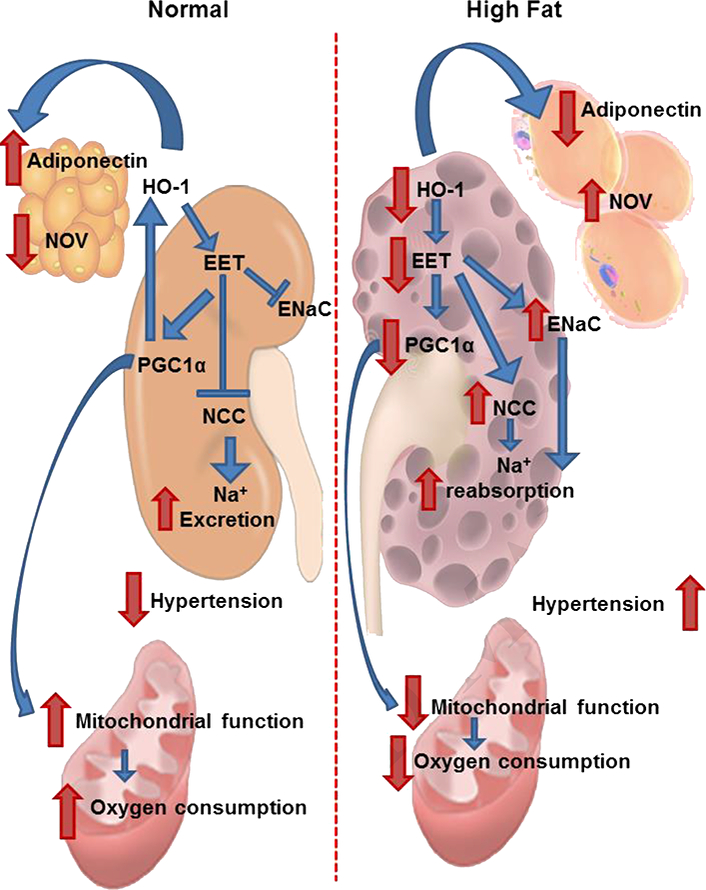
Schematic description of the EET-PGC-1α-HO-1 interplay, increased mitochondrial biogenesis and function. The EET-A-mediated activation of PGC-1α and HO-1 leads to increased oxygen consumption, increased mitochondrial fusion, reduced visceral adipose tissue deposition that together with increased insulin sensitivity contributes to reduced sodium retention. These factors in concert lead to a lowering of blood pressure and an improvement in renal and cardiovascular function.
HIGHLIGHTS.
EET-agonist reduces sodium retention and decreased both NCC and ENaC proteins.
EET agonist improvement of insulin receptor phosphorylation and decreased perirenal fat and inflammatory adipokines
EET-Agonist reprograming of the perirenal fat to a brite phenotype.
EET-agonist intervention mitochondrial and thermogenic gene levels
ACKNOWLEDGMENTS
We thank Mrs. Jennifer Brown and Mrs. Gail Anderson for their editorial assistance in preparing the manuscript.
SOURCE OF FUNDING
This work was supported by National Institutes of Health grant (HL34300 to NGA).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
EET Enhances Renal Function in Obese Mice Resulting in Restoration of Mfn1/2 -HO-1 Signaling, and Decrease in Hypertension through Inhibition of Sodium Chloride Co-Transporter.
DISCLOSURES
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Maric C, Hall JE. Obesity, metabolic syndrome and diabetic nephropathy. Contrib Nephrol 2011;170:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall ME, do Carmo JM, da Silva AA, Juncos LA, Wang Z, Hall JE. Obesity, hypertension, and chronic kidney disease. Int J Nephrol Renovasc Dis 2014;7:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Zeeuw D, Ramjit D, Zhang Z, Ribeiro AB, Kurokawa K, Lash JP, et al. Renal risk and renoprotection among ethnic groups with type 2 diabetic nephropathy: a post hoc analysis of RENAAL. Kidney Int 2006;69:1675–82. [DOI] [PubMed] [Google Scholar]
- 4.Gelber RP, Kurth T, Kausz AT, Manson JE, Buring JE, Levey AS, et al. Association between body mass index and CKD in apparently healthy men. Am J Kidney Dis 2005;46:871–80. [DOI] [PubMed] [Google Scholar]
- 5.Csongradi E, Storm MV, Stec DE. Renal Inhibition of Heme Oxygenase-1 Increases Blood Pressure in Angiotensin II-Dependent Hypertension. Int J Hypertens 2012;2012:497213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moreno C, Maier KG, Hoagland KM, Yu M, Roman RJ. Abnormal pressure natriuresis in hypertension: role of cytochrome P450 metabolites of arachidonic acid. Am J Hypertens 2001;14:90S–7S. [DOI] [PubMed] [Google Scholar]
- 7.Cao J, Singh SP, McClung J, Joseph G, Vanella L, Barbagallo I, et al. EET Intervention on Wnt1, NOV and HO-1 Signaling Prevents Obesity-Induced Cardiomyopathy in Obese Mice. Am J Physiol Heart Circ Physiol 2017;313:H368–H380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Messer-Letienne I, Bernard N, Roman RJ, Sassard J, Benzoni D. Cytochrome P 450 arachidonate metabolite inhibition improves renal function in Lyon hypertensive rats. Am J Hypertens 1999;12:398–404. [DOI] [PubMed] [Google Scholar]
- 9.Singh SP, Bellner L, Vanella L, Cao J, Falck JR, Kappas A, et al. Downregulation of PGC-1alpha Prevents the Beneficial Effect of EET-Heme Oxygenase-1 on Mitochondrial Integrity and Associated Metabolic Function in Obese Mice. J Nutr Metab 2016;2016:Article ID 9039754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agarwal R Proinflammatory effects of oxidative stress in chronic kidney disease: role of additional angiotensin II blockade. Am J Physiol Renal Physiol 2003;284:F863–F869. [DOI] [PubMed] [Google Scholar]
- 11.Bodiga S, Zhang R, Jacobs DE, Larsen BT, Tampo A, Manthati VL, et al. Protective actions of epoxyeicosatrienoic acid: dual targeting of cardiovascular PI3K and KaTP channels. J Mol Cell Cardiol 2009;46:978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boudina S, Bugger H, Sena S, O’Neill BT, Zaha VG, Ilkun O, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009;119:1272–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fowler J, Johnson N, Haroldson T, Brintnall J, Herrera J, Katz S, et al. Regulated renin release from 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 2009;296:E1383–E1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaide J-I, Zhang F, Wei Y, Jiang H, Yu C, Wang WH, et al. Carbon monoxide of vascular origin attenuates the sensitivity of renal arterial vessels to vasoconstrictors. J Clin Invest 2001;107:1163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayer A, Zarjou A, Agarwal A, Stocker R. Heme Oxygenases in Cardiovascular Health and Disease. Physiol Rev 2016;96:1449–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 2008;60:79–127. [DOI] [PubMed] [Google Scholar]
- 17.Chan EC, Dusting GJ, Liu GS, Jiang F. Redox mechanisms of the beneficial effects of heme oxygenase in hypertension. J Hypertens 2017;32:1379–86. [DOI] [PubMed] [Google Scholar]
- 18.Lever JM, Boddu R, George JF, Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxidants and Redox Signaling 2016;25:165–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Idris-Khodja N, Ouerd S, Trindade M, Gornitsky J, Rehman A, Barhoumi T, et al. Vascular smooth muscle cell peroxisome proliferator-activated receptor ? protects against endothelin-1-induced oxidative stress and inflammation. J Hypertens 2017;35:1390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandrashekar K, Lopez-Ruiz A, Juncos R, Nath K, Stec DE, Vera T, et al. The Modulatory Role of Heme Oxygenase on Subpressor Angiotensin II- Induced Hypertension and Renal Injury. Int J Hypertens 2012;2012:392890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosick PA, AlAmodi AA, Hankins M, Stec D. Chronic treatment with a carbon monoxide releasing molecular revereses dietary induced obesity in mice. Adipocyte 2016;5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mustafa MR, Johns EJ. The role of haem oxygenase in renal vascular reactivity in normotensive and hypertensive rats. J Hypertens 2001;19:1105–11. [DOI] [PubMed] [Google Scholar]
- 23.Jansen T, Hortmann M, Oelze M, Opitz B, Steven S, Schell R, et al. Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J Mol Cell Cardiol 2010;49:186–95. [DOI] [PubMed] [Google Scholar]
- 24.Sabaawy HE, Zhang F, Nguyen X, Elhosseiny A, Nasjletti A, Schwartzman M, et al. Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension 2001;38:210–5. [DOI] [PubMed] [Google Scholar]
- 25.Stec DE, Ishikawa K, Sacerdoti D, Abraham NG. The emerging role of heme oxygenase and its metabolites in the regulation of cardiovascular function. Int J Hypertens 2012;2012:593530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase- 1 deficiency. J Clin Invest 1999;103:129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radhakrishnan N, Yadav SP, Sachdeva A, Pruthi PK, Sawhney S, Piplani T, et al. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J Pediatr Hematol Oncol 2011;33:74–8. [DOI] [PubMed] [Google Scholar]
- 28.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A 1997;94:10919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh SP, Grant I, Meissner A, Kappas A, Abraham NG. Ablation of adipose-HO-1 expression increases white fat over beige fat through inhibition of mitochondrial fusion and of PGC1alpha in female mice. Horm Mol Biol Clin Investig 2017;31. [DOI] [PubMed] [Google Scholar]
- 30.Sodhi K, Puri N, Hyun KD, Hinds TD Jr., Stechschulte LA, Favero G, et al. PPAR-delta binding to heme oxygenase 1 promoter prevents angiotensin II induced adipocyte dysfunction in goldblatt hypertensive rats. Int J Obes (Lond) 2013. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Hinds TD Jr., Sodhi K, Meadows C, Fedorova L, Puri N, Kim DH, et al. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity (Silver Spring) 2014;22:705–12. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Abraham NG, Sodhi K, Silvis AM, Vanella L, Favero G, Rezzani R, et al. CYP2J2 targeting to endothelial cells attenuates adiposity and vascular dysfunction in mice fed a high-fat diet by reprogramming adipocyte phenotype. Hypertension 2014;64:1352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham NG, Junge JM, Drummond GS. Translational Significance of Heme Oxygenase in Obesity and Metabolic Syndrome. Trends Pharmacol Sci 2016;37:17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 2003;24:78–90. [DOI] [PubMed] [Google Scholar]
- 35.Sodhi K, Puri N, Inoue K, Falck JR, Schwartzman ML, Abraham NG. EET agonist prevents adiposity and vascular dysfunction in rats fed a high fat diet via a decrease in Bach 1 and an increase in HO-1 levels. Prost Other Lipid Mediat 2012;98:133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther 2009;331:906–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waldman M, Bellner L, Vanella L, Schragenheim J, Sodhi K, Singh SP, et al. Epoxyeicosatrienoic Acids Regulate Adipocyte Differentiation of Mouse 3T3 Cells, Via PGC-1alpha Activation, Which Is Required for HO-1 Expression and Increased Mitochondrial Function. Stem Cells Dev 2016;25:1084–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A, et al. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol 2012;302:R244–R251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eagan BM, Greene EL, Goodfriend TL. Insulin resistance and cardiovascular disease. Am J Hypertens 2001;14:116S–25S. [DOI] [PubMed] [Google Scholar]
- 40.Kleiner S, Mepani RJ, Laznik D, Ye L, Jurczak MJ, Jornayvaz FR, et al. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc Natl Acad Sci U S A 2012;109:9635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN, et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ Res 2016;118:1960–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan O, Riazi S, Hu X, Song J, Wade JB, Ecelbarger CA. Regulation of the renal thiazide-sensitive Na-Cl cotransporter, blood pressure, and natriuresis in obese Zucker rats treated with rosiglitazone. Am J Physiol Renal Physiol 2005;289:F442–F450. [DOI] [PubMed] [Google Scholar]
- 43.Davies MR, Gleich K, Katerelos M, Lee M, Mount PF, Power DA. The Thiazide Sensitive Co-Transporter Promotes the Development of Sodium Retention in Mice with Diet-Induced Obesity. Kidney Blood Press Res 2015;40:509–19. [DOI] [PubMed] [Google Scholar]
- 44.Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, et al. SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol 2013;24:407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Capdevila JH, Pidkovka N, Mei S, Gong Y, Falck JR, Imig JD, et al. The Cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. J Biol Chem 2014;289:4377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma F, Lin F, Chen C, Cheng J, Zeldin DC, Wang Y, et al. Indapamide lowers blood pressure by increasing production of epoxyeicosatrienoic acids in the kidney. Mol Pharmacol 2013;84:286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vinereanu D, Dulgheru R, Magda S, Dragoi GR, Florescu M, Cinteza M, et al. The effect of indapamide versus hydrochlorothiazide on ventricular and arterial function in patients with hypertension and diabetes: results of a randomized trial. Am Heart J 2014;168:446–56. [DOI] [PubMed] [Google Scholar]
- 48.Stier CT Jr., Koenig S, Lee DY, Chawla M, Frishman WH Aldosterone and aldosterone antagonism in cardiovascular disease: focus on eplerenone (Inspra). Heart Dis 2003;5:102–18. [DOI] [PubMed] [Google Scholar]
- 49.Singh SP, Schragenheim J, Cao J, Falck JR, Abraham NG, Bellner L. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat 2016;125:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boustany CM, Bharadwaj K, Daugherty A, Brown DR, Randall DC, Cassis LA. Activation of the systemic and adipose renin-angiotensin system in rats with diet-induced obesity and hypertension. Am J Physiol Regul Integr Comp Physiol 2004;287:R943–R949. [DOI] [PubMed] [Google Scholar]
- 51.Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin- angiotensin system. Curr Hypertens Rep 2008;10:93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, et al. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol 2012;32:1392–9. [DOI] [PMC free article] [PubMed] [Google Scholar]