

Summary
The Cancer Genome Atlas (TCGA) cancer genomics dataset includes over ten-thousand tumor-normal exome pairs across 33 different cancer types, in total >400 TB of raw data files requiring analysis. Here we describe the Multi-Center Mutation Calling in Multiple Cancers (MC3) project, our effort to generate a comprehensive encyclopedia of somatic mutation calls for the TCGA data to enable robust cross-tumor-type analyses. Our approach accounts for variance and batch effects introduced by the rapid advancement of DNA extraction, hybridization-capture, sequencing, and analysis methods over time. We present best practices for applying an ensemble of seven mutation-calling algorithms with scoring and artifact filtering. The dataset created by this analysis includes 3.5 million somatic variants and forms the basis for PanCan Atlas papers. The results have been made available to the research community along with the methods used to generate them. This project is the result of collaboration from a number of institutes and demonstrates how team science drives extremely large genomics projects.
Graphical abstract
The MC3 project is a variant calling of over 10,000 cancer exome samples from 33 cancer types. Over 3 million somatic variants were detected using 7 different methods developed from institutions across the United States. These variants formed the basis for the PanCan Atlas papers.
Introduction
The cost of sequencing is dropping rapidly while the costs of computing and data storage are dropping more slowly in comparison (Stein, 2010), making it difficult to deploy core analysis on raw data in genomics cohorts. It is often too expensive for individual labs to each use a one-off method on all their data. A more efficient approach is to design, test and develop cohort-wide analysis by multi-lab consortiums with results that can be shared with a larger group of analysts. Scaling computational systems and genomic analysis to work for these large data sets requires the coordination of many institutions, many experiments, and many computational techniques. Aside from logistical problems, there are several technical issues that encumber large-scale analyses, revealing unmet needs: 1) deployment of reproducible computing methods in new computing environments 2) the ability to deploy methods without manual intervention 3) the biases of single methods and the need for consensus and 4) the large amount of noise and false positives that come from data including both germline sequencing, heterogeneous tumor sequencing, and low variant allele fraction of observed reads.
There are a number of cancer genomics projects working to do analysis on increasingly large datasets (Table 1) (Barretina et al., 2012; Brunner and Graubert, 2018; Campbell et al., 2017; Hartmaier et al., 2017; Turnbull, 2018; 2017). The Cancer Genome Atlas (TCGA), for example, was a massive effort in multi-center cooperation, computational tool development, and collaborative science. However, the protocols and tools for identifying and characterizing tumor sequence variants evolved over time and were not uniformly applied across the project. When somatic variant callers were first compared–early in the TCGA timeline (2012)–a surprisingly large number of unique calls were identified for each method(Kim and Speed, 2013). To address some of these preliminary issues, TCGA organized multi-center mutation calling (MC2), which focused on consensus call sets of calling efforts from the Broad, UCSC, Washington University, and Baylor. By the conclusion of the MC2 effort simply moving these data from one site to another became a daunting task–let alone correcting for potential batch effects or caller-specific biases. Although the MC2 produced high-quality calls within each tumor-specific analysis working group (AWG), there were still differences in the callers, parameters, and filters used from project to project. Another effort of large-scale sequencing aggregation was implemented at the Broad Institute, in the effective deployment of the Firehose system (https://gdac.broadinstitute.org/), which automatically ran a suite of tools, designed at the Broad, to perform variant calling on all TCGA samples. While these data addressed consistency across tumor-types, these data were not amenable to custom design by groups outside of the Broad. In 2014 the ICGC-TCGA Somatic Mutation Calling DREAM challenge (Ewing et al., 2015) created an open leaderboard to benchmark variant calling methods from groups around the world. The DREAM challenge identified methods with a large variety of techniques and performance profiles. However, no large-scale genomic calling effort had yet deployed a robust set of these methods in a uniform fashion.
Table 1.
Large Cohort Cancer Genomics projects
| Project | Method | Sample count (approx.) |
|---|---|---|
| TCGA MC3 | Exome | 10,000 |
| GENIE | 44 Gene Panel | 19,000 |
| ICGC PCAWG | Whole Genome | 2,800 |
| 100,000 Genomes Project | Whole Genome | Projected: 100,000 |
| CCLE | Exome | 950 |
| Target | Exome | 700 |
| Foundation Medicine | 306 Gene Panel | 18,000 |
To drive analysis outside of these silos, TCGA organized the Multi-Center Mutation Calling in Multiple Cancers (MC3) project, which has developed pipelines to uniformly apply many mature tools across the TCGA sequencing project. The combination of both cloud computing power, policy changes, and improved variant calling software made this effort possible. The result is an open science collaboration across multiple institutions, designed to translate brittle custom-coded methods deployed at individual sequencing centers into portable, robust methods that enable reproducibility, transparency, and shareability with the broader research community. The software methods for this endeavor have been made publicly available, along with the datasets that it created.
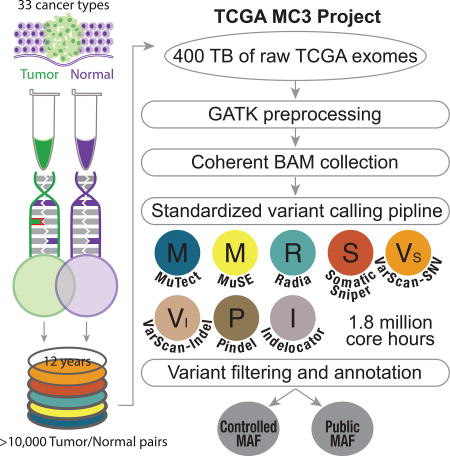
In this paper, we describe the various challenges and considerations of building standardized genomic analysis pipelines that can be deployed in mass to tens of thousands of samples, we also highlight some lessons learned, and considerations of performance when looking across widely varied cohorts. The resulting dataset, compiled in Mutation Annotation Format (MAF, https://wiki.nci.nih.gov/display/TCGA/Mutation+Annotation+Format+(MAF)+Specification), represents several million core-hours of computational time on over 400 TB of short read data using the current state-of-the-art variant calling and filtering methods. The MAF file represents over 20 million variants produced across approximately 10,000 tumor-normal pairs from 33 cancer types using 7 variant callers. This form of collaborative science, driven by a consortium of researchers across multiple institutions, is needed as the amount of genomic data continues to increase. The data generated by this work has formed the basis of the somatic exome variant analysis presented in the other papers from the TCGA PanCanAtlas project. More detailed analysis of the characteristics of the data and their biological implications will be discussed in other papers, such as 'Comprehensive Discovery and Characterization of Driver Genes and Mutations in Human Cancers', 'The Immune Landscape of Cancer', and the ‘DNA Damage Repair' (TCGA network et al., update citations after reviews)
Results
Cloud Deployment and Reproducibility
The MC3 project in support of the TCGA PanCanAtlas is the result of a number of institutions collaborating to provide resources and methods. In many cases, the project was able to utilize newly developed systems to deploy compute in ways that were not previously possible. These systems included custom written management scripts, institutional work management platforms and cloud-based systems. Alignment, GATK processing and variant calling for MuTect(Cibulskis et al., 2013) and Indelocator(Chapman et al., 2011) were run on the Broad's Firehose system. Additional GATK Indel realignment and base quality score recalibration (BQSR) was done on over 1000 tumor normal pairs on the University of California Santa Cruz cluster. Processed files were stored at the CGHub system. Over a four week period, using almost 1.8 million core-hours, 400TB of data was processed for variant calling using the Pindel(Ye et al., 2009, 2015), MuSE(Fan et al., 2016), Radia(Radenbaugh et al., 2014), Varscan(Koboldt et al., 2012) and SomaticSniper(Larson et al., 2012) pipelines on the DNANexus systems. OxoG scores for samples were calculated on the Institute for Systems Biology (ISB) Cancer Genomics Cloud (CGC) and validation data were processed using the Broad Firecloud platform.
The majority of the pipelines built for this project were designed to be deployed in multiple computing environments. To ensure reproducibility, the methods described in this paper have been implemented using modern workflow technologies, which are showing rapid adoption. In this model, the workflow is described using: i) a software container- a packaging system that simplifies deployment of the runtime environment, includes exact software dependencies and all features to run the program; ii) the tool wrappers - for each tool utilized, the command line argument to be invoked is described as a set of defined inputs, outputs, and parameters that can be used by a workflow engine to be scheduled and managed; iii) a pipeline description - a document that describes how all the tools fit together, the different parameters that should be modified, and required inputs. For distribution, the MC3 pipeline is described in the Common Workflow Language (CWL) format with the required software packages deployed using Docker software containerization technology. Docker provides methods to package a program and all of its dependencies. These container images can be shipped to any Linux machine, whether cloud-based or bare metal. Then the packaged tools can be easily run in new environments with minimal configuration. This workflow implementation is written using open standards which are easy to distribute and allow other researchers to replicate, modify and extend this analysis to their own data. Results are publically available from the NCI Genomic Data Commons (GDC) and include protected Variant Call Format (VCF) file releases as well as a filtered, open-access TCGA MC3 MAF release that contains only highest-confidence somatic mutations. These data will enable further PanCanAtlas efforts and, more generally, cancer research on TCGA data.
MC3 Variant Calling Strategy and Comparison to AWG MAFs
The MC3 effort used seven variant calling methods with proven performance (Figure 1) including Indelocator, MuSE, MuTect, Pindel, RADIA, SomaticSniper and VarScan (VarScan calls both indels and SNPs). Additionally, a collection of filtering methods were applied. These methods were applied to 10,510 tumor/normal pairs from 33 cancer types in the TCGA collection of whole exome sequencing data. This produced nearly 20 million variants. Definitions of controlled and open-access release of genomic variants for the TCGA data allows somatic variants that occur in exonic regions in open-access files (https://tcga-data.nci.nih.gov/docs/publications/tcga/datatype.html). Variants called in non-exonic regions, such as introns, 5' or 3' untranslated regions are restricted to controlled-access release. Additionally, somatic variants at sites that lacked sufficient normal depth coverage, or variants found in the panel of normals, were filtered from open-access since they were considered to be possible germline variants. Using these criteria, the full set of variants was narrowed down to an open-access file of around 4 million variants. A majority of downstream PanCanAtlas analyses was based on this subset of variants.
Figure 1. Workflow for mutation detection and filtering.

This workflow diagram reflects the internal design of the mutation calling pipeline. Squares in the flowchart represent files, and circles indicate processes. When colored, analysis was performed using the BROAD Firehose pipeline. Aligned input files were analyzed by 7 different variant callers using author recommended parameters to generate VCF files. All VCF files were merged and VEP annotated using vcf2maf tool. Processes flanking vcf2maf processes illustrate when filters were integrated. Finally, a separate set of annotation files were included and considered for variant and sample selection in the controlled and the public release of the annotated mutations file.
To gauge complementarity with previous efforts of calling mutations across many of these same tumor types, we compared the new set of calls to the MAF published as part of the first TCGA PanCan12 project for twelve tumor cancer types in 2013 (http://www.nature.com/tcga/). The PanCan12 MAF was created by collecting the variants from each separate TCGA AWG without any attempt at unification and includes data from a number of TCGA projects beyond the original PanCan12 set, including Pancreatic adenocarcinoma (PAAD) and Skin Cutaneous Melanoma (SKCM). We found that the new MC3 MAF had 1,079,216 variants in the PanCan12 MAF set of samples, while the PanCan12 MAF has 804,571. Among these calls, 717,326 variants are shared between the two sets (Figure S1). Thus, the MC3 project captured 89.5% of the original calls while increasing the size of the call set by 25%. The largest deviation was the PAAD project, which only saw 33% of the original variants and is likely due to poor tumor purity (see the PAAD marker paper for more details about somatic mutation calling efforts for this cancer type(Raphael et al., 2017)). Conversely, HNSC, SKCM, BRCA, BLCA, COAD/READ, and UCEC had greater than 90% of the original variants rediscovered by the MC3 effort (Figure S2).
For some cancer types, tumor cells profuse into the normal, causing issues in the identification of somatic variants. The best example of this is acute myeloid leukemia (LAML), which affects blood and bone marrow. Normal tissue samples (skin biopsies) from LAML patients often contain blood enriched with tumor cells. This can cause variant calling programs to mislabel somatic mutations as germline. The MC3 pipeline is conservative, attempting to remove all false positive germline calls. Much of the original MAF created by the TCGA LAML AWG was derived by manual interventions, including Sanger sequencing data not included as part of the TCGA data catalog, to recover variants that would have otherwise been uncalled. As a result, the open-access MC3 call set only recovered 44% of the variants called in the original MAF (Figure S1).
Effects of Somatic Filtering for Open-Access Release
To conform to release guidelines for open-access data in TCGA, the MC3 efforts took significant steps to remove potential germline calls as well as non-exonic variants. To accomplish this, filters were used against the flags that marked low normal depth coverage, non-exonic sites, sites outside of capture kit, sites marked by the Broad Panel of Normals, samples marked as being contaminated by ContEst, and variants that were only called by a single caller. The controlled-access MAF file contains 22,485,627 variants from 10,510 tumor samples and is comprised of 13,044,511 SNV events and 9,441,116 indels. The open-access MAF file contains 3,600,963 variants from 10,295 tumors with 3,427,680 SNV events and 173,283 indels. We observed that skin and lung cancers (SKCM, LUSC, and LUAD) had the largest median number of SNV per sample, consistent with previous publications(Akbani et al., 2015; Collisson et al., 2014; Hammerman et al., 2012) (Figure 2).
Figure 2. Distribution of mutations in controlled and open-access mutation files.

Two panels show mutation load for each sample in the dataset for SNVs (above) and indels (below). Each dot of the sorted scatter plots shows the total number of mutations pre- and post-filtering per sample. Total mutation counts are separated by total number SNVs (blue) and indels (red) per samples. Lighter colors indicate pre-filtered mutations from the controlled-access MAF, and deeper colors indicate post-filtered (PASS only) mutations from the open-access MAF. Cancers are ordered by the median number of post-filtered SNVs per tissue. Furthermore, samples are sorted by increasing number of total mutation count for SNV and indel plots respectively. Samples removed during post-filtering are also shown i.e. LAML and OV in lighter colors without an accompanying pair and are sorted accordingly. The total number of samples for each cancer type is displayed under each cancer label. Finally, Y-axis limits were placed from 0–50,000 for clarity. This resulted in the removal of 14 hypermutator samples from SNV plot and 10 hypermutator samples from the indel plot.
We plotted the proportion at which each of the different filters were found on variants in the three different datasets (the full call set, the open-access data set, and the set of variants used for validation) to show the reasons for differences in variant counts in the different sets (Figure 3). The most notable shift is the number of variants (over 60%) found in the full call set that were marked by the 'NonExonic' and 'bitgt' filters, which remove variants by genomic regions rather than technical reasons. These sites don't qualify for open-access release and may not be equally covered by all of the variant calling methods. Additionally, the Broad Panel of normals flagged almost 30% of the calls in the full set, which were also removed in accordance with TCGA data release policies.
Figure 3. Description of the filters implemented in controlled and open-access mutation files.

A) Filter flags (as displayed in MAF) and a brief description of their purpose. B) Variant counts in the open-access MAF by filter were processed using an UpSetR plot(Conway et al., 2017). The following filters were globally applied to the Open-access MAF: ‘ndp’, ‘NonExonic’, ‘bitgt’, ‘pcadontuse’, ‘contest’, ‘broad_PoN_v2’, and ‘badseq’. Thus, zero variants in the open-acess MAF were annotated with these flags. The inverted bar chart allows for the interpretation co-occurring filters at the variant level. For example, 304,602 variants were labeled with ‘wga’ alone, whereas, 2,455 variants were annotated with both ‘wga’ and ‘common_in_exac’. The connected dots indicate which of filter flags are assessed. C) UpSetR plot indicates the co-occurrence of filters with variants of the controlled MAF (same as B). D) The proportion and frequency of filters for both the open and controlled datasets are displayed. Additionally, validation flag counts and proportions are shown. The set of validation calls has a higher percentage of PASS calls, reflecting its bias toward higher quality variant calls. Filter flags are separated into samples level filters and variant level filters. See also Figure S4.
To further illustrate the importance of filtering on biological findings, we performed significantly mutated gene (SMG) analysis using both MutSig2CV(Lawrence et al., 2013) and MuSiC2(Dees et al., 2012) for all KIRC variants present in the controlled-access MAF compared to those present in the open-access MAF and marked as 'PASS' in the annotation. Typically this method of SMG analysis, using raw mutation calls, is performed in order to quickly identify sequencing and technical artifacts. Using the stringent p-value cutoff for both tools, MutSig2CV (P-Value < 3.5e-5) and MuSiC2 (P-value < 1e-7) each identified 10 SMGs using ‘PASS’ variants from the open-access MAF. Seven of these gen overlapped between MutSig2CV and MuSiC2 TP53, PTEN, VHL, SETD2, PBRM1, BAP1, MTOR. MutSig2CV uniquely identified TCEB1, PIK3CA, and ATM, and MuSiC2 uniquely identified ERBB4, SLITRK6, and KDM5C after long gene filtering. The complete set of variants from the controlled MAF yielded many more SMG hits (MutSig2CV = 1203, and MuSiC2 = 321). The noise introduced by the unfiltered variant calls made the identification of real SMG signals very difficult.
Performance Evaluation of MC3 Variant Calling by Experimental Validation
In order to evaluate calling performance, the TCGA project performed targeted deep sequencing on select variants for the purpose of validation for individual cancer papers. Selection of these variants were made by the original tumor-specific AWGs, and was not performed specifically to validate MC3 efforts This targeted sequencing included 3,128 samples with validation of a wide range of selected genes and was used for MC3 validation. This set of sequences included 33 samples with more than 500 targets genes and a median of 4 genes per sample. Variants from uterine corpus endometrial carcinoma (UCEC) comprised almost 28% of the sites and esophageal carcinoma (ESCA) 23% of the sites in this targeted validation dataset (Figure S3). Additionally, whole genome sequencing (WGS) was also run providing additional orthogonal data to use for validation. WGS data was available for a subset of 1059 samples, and provided a median of 126 validation sites per sample. Some methods, such as MuTect deployed by the Broad Firehose, only called variants within a region defined by the sequence capture kit definitions, even if additional sequencing was available. Because of this, sites marked by the ‘bitgt’ filter, which marked non-common-capture regions, were removed from the validation data set to provide consistent statistics when comparing across methods.
Because sites for targeted validation were selected from the most likely SMG candidates in the TCGA AWGs, rather than a random sampling of data, the validation data does not represent a robust benchmarking dataset. Every site involved in the targeted validation was called at least once by one of the variant calling methods. Because there is no background sampling, such as random sites not called by any of the methods, the false negative rate neglects sites not called by any method. Sites related to false positive germline signal would have been filtered before validation selection, and also not been part of validation efforts. Additionally, validation sites would be biased toward less complex and smaller events, which would impact performance evaluation of sites that are more difficult to characterize using targeted sequencing. We were able to partially manage this effect by including additional validation sites from samples where orthogonal WGS had also been performed. We should also note that the majority of validation data was generated using a similar sequence technology, therefore systematic errors such as those that several of the filters attempt to address will appear as erroneous filtering events. This particularly affects PoN filters. When comparing the subset of sites validated by targeted sequencing against WGS based validation the rate of these types of events doesn't seem to be very large. Given the profiles of filters among the datasets we see in Figure 4, the validation data does not mirror the characteristics of the full call sets. Despite these limitations, the validation data set does provide extensive data about the relative performance of callers and filters (Table S2).
Figure 4. Validation statistics of mutations calls.

While these results reflect validation of resequenced samples, technical artifacts may still be present because orthogonal technology was not implemented. A) Overview of the Mutations validation process. Symbols are used to illustrate how mutations predictions were assessed. Values shown in under ‘Predicted mutations’ are not mutually exclusive. Exclamation marks under ‘True negative’ and ‘False negative’ denotes the logical negation or not. B) The composition of variants with overlapping callers. Starting with any caller and increasing to require more callers to agree on a site. This is done for both SNVs (left) and indels (right). C) The composition of validation status for calls from each independent caller for both SNPs (left) and indels (right). D) The composition of validation status for pairs of callers. Panels B, C, and D all have a truncated y-axis, all values below indicate true positives mutation status. Omitted, as illustrated in panel A, reflects the limitations of assessing mutation predictions when validations does incorporate all possible events. E) The composition of validation status for each of the filter flags. See also Figure S3. See also Figure 3 and Tables S2 and S3.
As seen in Figure 4, meta calling methods, such as 'two caller intersection', are able to quickly eliminate false positives. This has been noted previously in other studies(Goode et al., 2013). The 'two-caller' rule for the set of five SNP callers finds more valid sites than any specific combination of two callers(Table S3). This draws on the wisdom of crowd principle(Costello and Stolovitzky, 2013). The two caller intersection is much less effective for indel calling methods, as it causes an increase in false negatives due to its conservative nature. We see general trends, such as MuTect and MuSE detecting the largest number of true positive sites among the validation variants surveyed. Somatic Sniper had the lowest number of detected sites, omitting the largest number of validated sites, but at the same time, it had the smallest number of false positive validated sites.
We observed many tool specific patterns pertaining to mutation identification (Figure 5). Most calls that passed all the filters were supported by all 5 callers. For SNP calls, MuSE and MuTect have the highest pair-wise agreement. They each also have the largest number of unique calls. For indel callers, Pindel makes the most calls, but over 130K of the variants were found in two samples, suggesting there may be characteristics of these samples that skew the numbers. Only a small fraction of indel calls made by all three callers.
Figure 5. Intersection of mutation calls across variant prediction software.

The top bar-plot indicates intersection size. More specifically, one or more tools called each variant. This plot provides the number of variants that are uniquely called by one tool (a single point) or the numbers of variants called by many tools (2 or more points). The bottom left plot indicates the set size. The linked points below display the intersecting sets of interest or which tools called variants. A) PASS only mutations from the controlled MAF are shown. B) Tools designed to call indels are displayed in a similar fashion to plot A. Only indels with greater than 3 supporting reads are displayed in this plot. Additionally, two samples were removed from these plots that represent extreme hypermutators (TCGA-D8-A27V, and TCGA-EW-A2FV).
Discussion
The previous paradigm of genomic research was that groups downloaded data, ran methods on their own, and then provided results to the community, representing a 'results-oriented' approach. Under this model, it became extremely difficult for external groups to reproduce calculations or apply new methods to new datasets. However, with the advent of cloud technologies such as computational virtualization and containerization systems, there is now the ability to capture computational methods in a way that can be run on external compute systems. This change allows for a 'methods-oriented' strategy in which the collaborating institutions provide shareable algorithms to be run on the data, rather than processing it themselves. The MC3 is a showcase for a methods-oriented project, collecting reproducible code for methods from collaborators and deploying them uniformly to data on the cloud.
Through collaboration, open science, and improved resources the MC3 effort overcame lingering artifacts from previous cancer-type specific analyses and reflects a true PanCancer set of somatic mutations. Many lessons were learned, or re-confirmed, while leveraging multi-institutional expertise: 1) While many methods have a public facing software on GitHub or clouds resources, default parameters were often insufficient. Achieving the best performance required additional input from the original authors. 2) Some tumor types, such as liquid tumors, require different strategies of variant calling and filtering to obtain an optimal set of mutations. 3) Providing annotation generated by various filtering methods, as opposed to generating files with fixed removal of possible artifacts allows for flexible usage of the mutation call set. 4) Using reproducible code and methods based approaches are essential as datasets increase in size and complexity. 5) Meta-calling methods, that utilize the results of multiple methods, can provide more robust results than single methods. 6) Multiple precautions and filters were needed to protect potential germline leakage of patient data into public facing, open-access data. These lessons learned allowed for customizable strategies based on algorithmic objectives or biological inquiries.
This organization of coherent variant calling for 10,000 genomes was a multi-year process. However, there were a number of technical advances that occurred during this time-frame, and these technologies will make utilization of cloud resources much more accessible for researchers going forward. While this effort was informed by the DREAM challenge(Ewing et al., 2015), many of the methods selected were based on best practices of the original TCGA AWGs. Ideally, future variant calling and filtering efforts should use a robust benchmarking effort to scan the various combinations of callers, filters, and parameters and evaluate which callers and filters are optimal for different tumor types and contexts. The lessons learned from this project should inform the design of a new somatic mutation calling pipeline having an end-to-end FASTQ-to-filtered-MAF file workflow with complete containerization in a single cloud. Resources such as the TCGA catalog form the backbone of reference data sets that can be used as a point of comparison in new research projects. But those comparisons are only useful if the analysis is applied consistently. Thus, when pipelines are applied to large datasets, the methods should be made available alongside the resultant data so that other groups can apply them to their own experimental data.
The PanCanAtlas project encompasses many research goals. For this reason, a one-size-fits-all approach would not cover the different types of analyses. An example of this would be the problems of driver gene discovery vs heterogeneity analysis. A high-confidence caller with lower false positive profiles is better geared for driver gene discovery, because the removal of false positive noise helps to better identify significant recurring patterns. Once the significant driver genes have been identified, a second pass over the mutation set can find lower confidence calls that could provide additional examples of the gene of interest. In contrast, heterogeneity analysis, which looks for variants that occur in fractions of the population, works much better with very sensitive algorithms because these variants, with potentially low variant allele fractions, may be filtered out by more stringent methods. Therefore, it was appropriate to include called variants and provide mechanisms for doing additional filtering that was appropriate to the analysis. These steps, in accordance to the TCGA open-access release guidelines, resulted in the collection of 3 mutation annotation format (MAF) files: a controlled-access MAF, an open-access MAF, and a validation MAF. Each of these MAF files have distinct properties which are compared and contrasted here.
The multi-center mutation calling in multiple cancer (MC3) effort reflects three objectives of large-scale data generation in an age of open science: collaboration, consensus, and consistency. First, multi-center collaboration combined efforts and expertise from multiple academic institutions. Second, mutation calling was performed using an array of 7 mutation-callers developed by the adopted by different TGCA analysis centers. We show consensus calling outperforms single algorithms in both sensitivity and validation status. Finally, the use of consistent methods for calling across multiple-cancers enhances the utility of this resource in future efforts to contrast the molecular makeup across tumors. The results of this effort provide integral components necessary for future efforts in somatic variant calling.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
All data associated with this project will be made available via the NCI's GDC data portal, source code will be made available on GitHub and docker containers on the Docker and Quay docker repos. Questions can be directed to the contact author at ellrott@ohsu.edu
METHOD DETAILS
Sample List Creation
The MC3 sample list was extensively verified to make sure that poor quality samples were removed, and that for every donor the best tumor and normal samples were paired. To this end, a number of rules were applied to remove samples and identify appropriate sequence data which BAM files fit pipeline specifications as well as identify samples with available sequencing information that required preprocessing prior running analysis.
The list of rules applied included:
Exclude redacted samples - A number samples in the TCGA had been removed or flagged over the course of the TCGA project for various reasons.
Exclude non-HG19 aligned files - Earlier samples from the TCGA project were aligned with older genome builds, including HG17 and HG18. Rather than attempt to back-port variant calling platforms to older genomes and lift-over the variants to new genome builds, these samples were eliminated from the resource pool when building the sample list. In many cases the data from these files had been realigned by the Broad Firehose platform as part of their efforts in various tumor specific working groups.
Preferentially select Broad genome build - In cases where a samples sequencing data had been run through multiple alignment pipelines, the Broad pipeline was preferentially selected to eliminate variance. In most cases when there was a multiple pipeline runs, the Broad pipeline was run to update the alignments to an HG19 genome build.
Reassign samples without GATK co-cleaning - Co-cleaning refers to the process of applying the GATK IndelRealignment to both the tumor and normal samples of an individual. This process is also accompanied by running Base Quality Score Recalibration (BQSR). While complete realignment of sequences was not required for inclusion in the MC3 analysis, it was required that the GATK co-cleaning process has been applied. Because this step was part of the Broad pipeline, any sample selected fit this requirement, thus the previous rule. In cases where a sample was not co-cleaned and had not already been realigned as part of the Broad pipeline, a the co-cleaning was done and the new sequences stored in a special project at CGHub.
Exclude non-Illumina sequenced samples -A small number of samples in the TCGA cohort had been sequenced with other technologies including ABI SOLiD and 454 for validation sequencing. To reduce artifacts and maintain consistency, these sample were eliminated from the list.
Exclude FFPE samples - Most of the TCGA samples were derived from fresh frozen samples, but a subset of samples were derived from Formalin-Fixed Paraffin-Embedded samples. These samples may have experienced more DNA damage and had different error profiles in mutation calling. This rule results in the removal of 97 samples.
Matched genome build string - While HG19 alignment was required for sample inclusion, there was in fact a number of different genome versions, including 'HomoSapien19' 'WustlBuild1' and others. These genome build were all based on HG19, but contained various patches. Genome patches add additional sequencing information to the assembly, without disrupting the chromosome coordinates. But while these multiple patches were allowed, for a tumor and normal sample to be matched the genome build title had to match, to eliminate the possibility of sequence patches being misidentified as somatic mutations.
Prefer Native DNA pairs over WGA pairs over Native+WGA - There is a number of earlier TCGA samples which were sequences with Whole Genome Amplification. Because of the technical artifacts associated with this technique, in cases where there was sequencing done without WGA, those samples were preferentially selected.
Prefer samples with matching RNA-Seq - We selected samples that had quality measures based on RNA-Seq.
Usually prefer latest plate - Operating on the principle that any later sequencing effort would have been triggered by issues in the earlier runs, the latest run from a sample was chosen.
Prefer pairs sequenced at the same center - Sometime tumor normal pairs were sequenced at multiple centers. We selected for samples tested at the same center. This step was not adjusted based on Contest or Oxog scores.
Tumor contamination estimates using Contest - Samples were removed if the Contest score estimated more than 4% contamination from another participant.
Spurious sequence artifacts: BadSeq - 6 samples were removed if they appeared to be affected by systematic sequencing artifacts. Systematic insertions or deletions were identified at the same base pair location in each of the reads in the both the forward and reverse strands. These artifacts have been previously reported(Ye et al., 2015).
Given these rules, the sample selection algorithm is as follows:
Pick best bam within aliquot + original sequencing center. This involves apply all hard filters and picking samples with a preference toward BAMs processed via the Broad pipeline or the MC3 secondary co-cleaning pipeline.
Pick best set of BAMs within an individual. First selecting the most “popular” build, using Broad-aligned or number-of-native as tiebreakers, and avoid selecting WGA samples. Some overrides were applied in these step, ie selecting Baylor-aligned native samples vs Broad-aligned WGA samples.
Pare back the aliquots within the individual. First drop non-paired samples and select one aliquot per sample.
The final white list consisted of 11,069 tumor-normal pairs for 10,486 participants. In cases where more than one pair was selected for a participant, all of the pairs were analyzed for mutations, but all but one were tagged as 'nonpreferredpair', based on criteria like preferring a primary to a metastatic tumor sample, and for solid tumor types preferring a blood to a tissue normal sample.
Variant Calling and Filtering Strategies
For the variant calling step, seven methods were applied, five covering Single Nucleotide Variant (SNV) calling and three covering short Insertion Deletion (INDEL) events, with Varscan 2 providing both types of analysis. Parameters used for these tools are found in Table S1.
MuTect (SNV) - This method at the Broad Institute(Cibulskis et al., 2013) uses a Bayesian classifier that allows it to identify low-read/low-allele fraction somatic mutations, while maintaining a high specificity. It was one of the top performing methods in the SMC-DNA DREAM challenge(Ewing et al., 2015).
Varscan 2 (SNV/INDEL) - Developed by Daniel Koboldt, Washington University, the algorithm uses heuristic and statistical approaches in its algorithm to detect germline, somatic and loss of hertezinocity. It can calculate SNV, Indel and CNA events(Koboldt et al., 2012).
Indelocator (INDEL) - Developed by the Broad team(Chapman et al., 2011) uses read count and alignment quality information to detect indel events found in tumor alignments.
Pindel (INDEL) - Developed by Kai Ye et al. at Washington University is used to identify medium size insertion and large deletion events. Pindel also generates complex variant calls that accurately reflect the genomic alterations even around substitution sites(Ye et al., 2009, 2015).
SomaticSniper (SNV) - Developed by David Larson et al. at Washington University, this method compares the tumor and normal bams to find differences using the samtools MAQ genotype likelihood model to make alteration calls(Larson et al., 2012).
RADIA (SNV) - Developed by Radenbaugh et al at University of California in Santa Cruz, RADIA stands for RNA and DNA Integrated Analysis. It augments it mutation calls using RNA-Seq samples from the same tumor making it possible to make mutation calls when there is lower DNA allelic frequencies. RADIA was applied using matched RNA when available(Radenbaugh et al., 2014).
MuSE (SNV) - Developed by Fan et al at Baylor College of Medicine and MD Anderson (Fan et al., 2016), uses a markov substitution model which characterizes the evolution of the allelic composition of the tumor and normal tissues at each reference base and is tuned for sensitivity. It further adopts a sample-specific error model that reflects the underlying tumor heterogeneity to improve the overall accuracy. Uses a Markov substitution model to calls mutations. MuSE was another method that scored very well in the SMC-DNA DREAM Challenge.
Default parameters for programs were used as much as possible, however in a number of instances non-default parameters for particular programs were used, based on discussions with tool authors or analysis that had utilized the tool in institutional pipelines (Table S1). These selections were based on empirical knowledge gained by observing effects on small cohorts.
In the process of sample collection, DNA amplification and short read sequencing, there are a number of events that could induce noise and create false mutation patterns. Though callers are tuned to remove some classes of systematic sequence error, it is often necessary to impose additional post hoc filters. In some cases the techniques are already embedded in some of the mutation calling programs themselves, but to maintain consistency these filters were applied across all calls uniformly. We applied several common filters employed by major sequencing centers during the TCGA. The filtering steps do not increase sensitivity, they only remove calls, so sensitivity can only be decreased in this phase. Since false positive somatic events can be highly misleading for downstream research, maintaining high specificity of the call set using post hoc filters is crucial. The final call set was filtered to identify cohort level artifacts and was subject to extensive variant, subject, and cohort level QC. In sum, 22,485,627 putative variants were identified and 2,907,335 high confidence mutations were retained after filtering.
To provide filtering, 8 methods were utilized. The final two filtering methods are not necessarily designed to increase accuracy, Some of the variant calls marked by these methods may be correct, but were removed from the public open-access release in accordance with TCGA data access tiers.
Broad PON V2 - (MAF tag: broad_PoN_v2) One of the most effective filters of false-positive, contamination, and germline variants is a Panel-of-Normals (PoN) (Hess et al., in preparation) filter. This filter postulates that if a variant is called or detected in a set of control (often non-tumor normal samples) then it is very unlikely that the variant is actually a somatic variant in any given tumor sample.
Common In ExAC - The Exome Aggregation Consortium (ExAC) publishes germline variants and recurrent artifacts seen in exome sequencing of over 60K unrelated individuals from across seven subpopulations(Lek et al., 2016). As implemented in vcf2maf v1.6.11, this filter tags variants with a non-reference allele count >16 in at least one subpopulation of the non-TCGA subset of ExAC v0.3.1, unless ClinVar flags it as pathogenic. AC=16 (for SF3B1:K700) was the highest value observed among known somatic events detected in the normal blood of older individuals due to clonal hematopoiesis.
OxoG - (MAF tag: oxog) The 8-Oxoguanine (OxoG) DNA lesion is a common sequence artifact caused by excessive oxidation during sequence library preparation(Costello et al., 2013). The DetOxoG tool was used to identify and flag likely OxoG error variants.
ContEst - (MAF tag: contest) This program predicts levels of contamination. Contamination coefficient produced by this method is used as a coefficient in the MuTect pipelines, and samples with a value greater than 4% were removed from the analysis.
StrandBias - (MAF tag: StrandBias) Implemented post MAF production and more appropriately identified as a mutation bias artifact, the StrandBias filter tags low-VAF G>T from samples sequenced at Washington University such that the number of untagged G>T variants equals the number of C>A variants within a sample. VAF cutoffs are set on a sample by sample basis such that the number of tagged G>T variants (with lowest VAF) maintains balanced untagged G>T mutations and C>A variant counts. This was implemented because of strong disparities between G>T and C>A mutation counts in samples sequenced at Washington University.
Normal Depth - (MAF tag: ndp) To avoid miscalling germline variants at least 8 reads in the normal sample in non-dbSNP sites and at least 19 reads in dbSNP sites.
Capture Kit - (MAF tag: 'bitgt') The filter represented a simple process of intersecting all mutations calls with the subset of the genome that intersected with all of the capture kits used by the different sequencing centers. During PCR small fractions of non-targeted sequences could be amplified and during alignment reads could have been placed in incorrect locations in the genome. This leads to low read coverage areas in non-targeted section of the genome to be included in the BAM file. If the variant calling program sweeps across of the the reads, it may produce calls using these off target reads, and create calls.
NonExomic - (MAF tag: NonExonic) As part of the NCI/NHGRI mutation release process, non-exonic mutations must be verified with orthogonal sequencing before they can be released publicly. The exon definitions were derived from the GAF 4.0 definition, which was based on Gencode 19 Basic.
Merger of Mutation Calls
Mutations were called by each of the callers and stored in VCF format. Following initial calling, variants from each caller were merged by allele with the exception of calls from Pindel. For alleles not involving Pindel, we extracted and averaged coverage metrics across all callers asserting the presence of a mutation and combined the various callers into the calling center column in the resulting MAF file. As Pindel generates complex variant calls we allowed Pindel to supersede allele representations from other callers. Any allele intersecting a Pindel call by position was discarded and the Pindel call was modified to add the other caller to the calling center column. We annotated these by placing a “star” next to the caller ID to signify that the caller may not have represented the allele in the same way.
Workflow Deployment
The various components of this part of the MC3 computational task took place at multiple sites using different technologies and computational resources.
UCSC NCI Cluster - A computational cluster, associated with the CGHub, was utilized to perform GATK co-cleaning on a subset of sequence files that had not been previously processed. This dataset represent approximately 1600 BAM files. The results of this run were stored on CGHub until its close in July 2016.
DNA Nexus - The primary set of computations, related to running the core set of variant calling pipelines as run on DNANexus's cloud platform. Over a four-week period approximately 1.8 million core-hours of computational time were used to process 400 TB of data on the DNAnexus Platform to yield reproducible results. This resulted in the 500GB of VCF files representing all detected variants.
Broad Firehose - The Broad Firehose is a system to deploy automated pipeline analysis on all the TCGA data. The somatic variant calling pipeline includes ContEst, MuTect, and Indelocator, and was run using an SGE cluster of 200 nodes. In addition, the OxoG filter was applied at this stage, and were also later applied to the calls from the other callers.
Institute of Systems Biology. These validation runs were deployed on the Institute of Systems Biology Cloud pilot. One this system, the OxoG variant filtering step was run on all variant data. Also, the WheelJack validation data genotyping algorithm was run on all samples with available validation data.
SMG Performance Analysis
MutSig2CV and MuSiC2 were performed on subsets of the data based on different filtering criteria. The results of this analysis resulted in drastically different results when taking filtered for raw variant calls. KIRC was selected because of its unique set of driver mutations compared to other tissues (PBRM1 and VHL) and it is often associate with few SMGs. Variants for the raw variants were assembled for the unfiltered MAF. MutSig2CV consists of 3 statistical tests, including mutation abundance, local clustering, and conservation. SMGs from MutSig2CV were defined as genes with a q-value <= 0.1. MuSiC2 analysis calculates SMGs using mutation abundance compared to background mutations rate calculations. Convolutions of multiple transition and transversion rates were used to calculate p-values. Strict p-value cutoffs of 1e-7 were used in defining SMGs for MuSiC2. SMGs were further filtered using the MuSiC2 long gene filter. This is a MuSiC2 specific long gene filter systematically increases the p-value threshold for larger genes until it no longer indicates a correlation between p-value and gene size. If the larger gene doesn't reach the new threshold it is subsequently removed from the SMG list. This was not applied to MutSig2CV output. Filtered variants were processed using “pass-only” variants from the public facing, open-access MAF. The same parameters from the above were applied resulting in a reduced number of SMGs in KIRC. No hypermutators were removed for this analysis.
Mutation Validation
The Broad 'Mutation Validator' pipeline was used to identify validation evidence at variant sites using alternate sequencing runs. Mutation Validator provides validation evidence at sites of candidate SNVs or INDELs from read pileups across multiple data-types including WXS, WGS, Targeted Validation, and RNA. The algorithm for each validation followed the step:
Collect pileup for each allele (A<C<G<T, INS,DEL) at candidate sites from each validation data type.
Parse normal sample for each data type to estimate maximum noise alternate allele fraction. If datatype has no normal sample (eg. RNA-seq) then use exome to estimate noise. Use binomial conditional distribution field to calculate the 99% upper limit alt count in the tumor at this noise allele fraction. This upper limit is the threshold validation read count “min_val_count“ in the tumor sample. The minimum “min_val_count“ for any data type is 2.
The power to validate the mutation is calculated using the hypergeometric cumulative probability distribution to project the observed tumor alternate allele fraction from the exome onto the coverage of the validation data type with a minimum of “min_val_count“ alt supporting reads. If power is less than 0.95, disregard this site+data type as unpowered.
If the normal sample for a given validation datatype has an allele fraction exceeding 0.2 for SNVs or 0.1 for INDELS, label the site+data type as “validation_judgement”=2 (germline).
If not germline, and if the tumor validation datatype alternate read count is at least “min_val_count“ (from step 2) then label the site+datatype as “validation_judgement=1 (somatic).
Otherwise, set “validation_judgement”=0 (not validated).
Using this method 7,680,483 candidate variants processed by mutation validator (1,476,028 DEL, 603,637 INS, 5,600,818 SNP). The sites within the target region (bitgt) created a set of 1,352,467 variants having 95% power in either rna, targeted, wgs, or lowpass validation data. Validation rules at sites with power in targeted or wgs data.
QUANTIFICATION AND STATISTICAL ANALYSIS
Effects of Cancer Type on Mutation Callers
When observing the total number of mutations per sample, separated by cancer type, we identified that mutation calling consistency differs by cancer type. Specifically, within single nucleotide events THYM, and PAAD, KICH and UVM tumors varied greatly between sample when compared to the total number of unique variants identified per sample. Such inconsistencies are likely attributable to various pathological reasons that yield low purity biopsies. For instances, when comparing to purity estimates (Figure S2), THYM and PAAD samples had the lowest purity estimates (ABSOLUTE (syn7870168) median 39.0% and 39.7% respectively).
OxoG Events
The oxidation of guanine to 8-oxoguanine, known as the OxoG event, affects a subset of TCGA samples. It can be caused by heat, contamination and physical forces on the DNA. This mutation causes G to T and C to A substitutions in the reads. To filter for this event, an OxoQ score is calculated, which describes the probability of an entire sample being affected by OxoG events. If this OxoQ value is above a threshold, then the sample is run through the OxoG filter which examines the original BAM file reads to determine if G to T and C to A mutations are real or created by the OxoG artifact.
Per Gene Filtering Effects
Per gene counts were generated based on the number of variants found in the MC3 controlled-access and open access files. The genes with the largest disparity of variant counts between the two populations were assessed (Figure S4A). Additionally, significant cancer genes found as part of the original PanCan12 project were highlighted(Kandoth et al., 2013) in this analysis (Figure S4B).
Indel Realignment and BQSR
In order to remove biases in the alignment protocols, a process called 'co-cleaning' was deployed, as part of the GATK best practices(McKenna et al., 2010), on each tumor normal pair. This processing step is composed of two analysis and adjustments that are run in the BAM files prior to variant calling. The first step, local realignment uses reads from both the tumor and normal, thus the 'co-cleaning', and utilizes this information to disambiguate potential areas of misalignment. The tumor and normal are co-analyzed so that arbitrary decisions can be made cohesively. Areas with small insertions and deletions in the initial alignments were realigned using all reads from an individual, including reads from both the tumor and normal samples. This additional joint information help to eliminate false positive SNPs caused be misaligned reads, particularly at the 3’ end. There has been a noted performance increase in downstream variant calling process for both indel and SNV calling. Pindel incorporates a similar process internally and thus doesn't require it, but for consistency all variant calling methods were based on the same co-cleaned BAMs.
The second step of co-cleaning is Base Quality Score Recalibration (BQSR). BQSR tweaks the quality score so that it represents a calibrated probability. This step is especially important for BAMs with a wide range of quality scores, as is common with older sequence data.
Co-cleaning had already been applied to all sequence alignments produced by the Broad since 2012, but for a subset of the TCGA cohort, totaling almost 50% of the pairs, the co-cleaning process was applied on samples already uploaded to the CGHub resource. Approximately 35% of the samples required full realignment. These secondary BAMs represented analysis products of the MC3 effort, and totalled almost 150TB. This processing was carried out at the Broad Institute and UCSC.
Variant Calling
The next phase in the MC3 process was variant calling followed by filtering. In the variant calling step, pairs of BAM files were run through programs developed from multiple institutions and the results of the putative variant calls were stored as Variant Call Format (VCF) files. The filtering steps, with the notable exception of the OxoG filter, use information stored in the VCF files produced by the different callers and produce a secondary filtered mutation file (usually VCF or MAF). This is an important detail for analysis and job scheduling. A pair of TCGA exome BAM files can average 10–30GB, while the average VCF file, filtered for somatic variants is a few hundred kilobytes. Many analysts employ a strategy of calling-then-filtering, ie create a set of putative variant calls and then applying filters as secondary steps downstream to remove false positives. If any information is required from the BAM file, it means that scheduling the analysis on the variant calls on 10K exomes would require accessing over 300TB of files. But if all of the filtering can be done only using the initial VCF file, the data requirements become tractable for doing analysis on a single machine. This strategy allows tuning of filtering methods, parameters and strategies but removes the complexity and logistical issues of obtaining the BAM files.
Variant Merger
We merged variants based on allelic location except in the case of Pindel calls, where we merged variants by proximity to Pindel calls. The additional merge criteria for Pindel calls was required because Pindel generates complex variant calls that other callers are incapable of generating. Complex variants are simultaneous indel and substitution mutations in cis. This merger process created 14,241 complex indel sites that included merged calls from SNP callers in the full MAF file, and 3,611 sites in the filtered open-access file. Finally, in order to generate consensus metrics, such as variant and reference allele counts, we averaged them across all callers that yielded a specific call.
Panel-of-Normals filter
In the case of systematic false positive variants, as the cohort becomes larger the likelihood that one of the PoN samples will also contain the systematic false positive increases. By statistical chance it is possible to miss germline variants in low coverage regions because the variant is not detected in the normal, the PoN reduces the rate of germline calls because it effectively increases sequence depth at these locations by leveraging the control cohort. Although the PoN filter is an effective way to remove germline variants, most of the variants that it flags are, in fact, recurrent sequencing artifacts.
Across the entire cohort the number of germline SNP events for every site where totals and if a SNP occurs in a number of samples above a threshold, it was determined that it was more likely that a mutational event was not recognized as a germline event, rather than a genuine somatic event.
One of the the most effective filters encoded the expected distribution of alternate allele read counts at every genomic position, based on a large panel of 8000 TCGA normals (PoN). A somatic variant call is tagged by this filter if its observed readcount is consistent with the PoN, based on a likelihood test. This allows calls with many supporting reads to be retained, if they occur at a site with low allele-fraction (AF) sequencing noise in the PoN. To remove germline events or high AF artifacts, all somatic call at a site with recurrently high AF across the PoN are removed.
For each genomic position, the PoN encodes the distribution of alt read counts across all TCGA normals. For each mutation call, we compute a score that its observed read counts are consistent with the PoN; if this score is above a certain threshold, the site gets flagged. Thus, if a site recurrently harbors low-level sequencing noise in the PoN and it is called at low allelic fraction, it is flagged, whereas a call with many supporting reads at the same locus would be left alone. Likewise, a common germline site would have recurrently high allelic fractions across the PoN; if a call at that site has similarly high AF, it gets flagged.
A full description of the PoN filter follows. Each genomic position’s histogram comprises six bins:
1 : alt read count >= 1 and alt fraction >= 0.1%
2 : alt read count >= 2 and alt fraction >= 0.3%
3 : alt read count >= 3 and alt fraction >= 1%
4 : alt read count >= 3 and alt fraction >= 3%
5 : alt read count >= 3 and alt fraction >= 20%
6 : alt read count >= 10 and alt fraction >= 20%
For a given position, denote the vector of bin counts h⃗. For each variant call, we represent its allelic fraction as a beta distribution parameterized by its alternate and reference read counts (to account for numerical uncertainty when converting read counts to allelic fraction):
and then slicing the beta distribution's PDF according to the alt. fraction bins encoded by the PoN, i.e.
Finally, we compute a score for this position by weighting each element of $\vec f$ by its corresponding histogram bin counts:
The units of this score are arbitrary. We found empirically that a cutoff of log10(S) ≥ −2.5 works well, determined by decreasing the score cutoff (thereby increasing the aggressiveness of the filter) until it started removing recurrently called sites (≥3 patients) listed in the COSMIC database. Because some COSMIC sites are themselves recurrent artifacts, manual review was necessary to exclude those from the list of true positives.
Restricting to Target/Coding Exons
While there are whole genome sequences that are part of the TCGA catalogue, the MC3 project targeted exome sequences. During PCR small fractions of non-targeted sequences could be amplified and during alignment reads could have been placed in incorrect locations in the genome. This leads to low read coverage areas in non-targeted section of the genome to be included in the BAM file. If the variant calling program sweeps across of the reads, it may produce calls using these off target reads, and create calls. To filter these non-target calls out, a BED file of the intersection of capture kit locations and applied to the variant calls to remove variant calls from non-target/non-exon regions. This target filter was applied across all samples, even on samples where other targeting panels may have been used because 1) not all capture kit targeting data was universally available and well annotated to sequences and 2) to simply cohort mutation significance analysis. The disadvantages of the capture kit based filtering strategy was that 170 CDS altering MC3 calls in MSK IMPACT's 410 cancer genes, that fall outside the Broad BED. The key misses are TERT promoter hits, truncations in putative tumor-suppressor CIC, splice alterations in the frequently rearranged CRLF2, and a cluster of events in the 5' end of FOXP1.
The exone definitions were derived from the GAF 4.0 definition, which was based on Gencode 19 Basic. The exome capture was based on the Broad Target Bed.
Minimum 3 supporting reads for Pindel indel calls
Some of the filtering parameters in Pindel were recently reconfigured to allow it to detect complex indel events. Complex indel events involve both the insertion and deletion of nucleotides in a mutation site(Ye et al., 2015). This increased ability of Pindel resulted in a number of false positive indel being included as part of the initial MC3 call set. To combat this, a minimum of three supporting reads were required to support a Pindel call, otherwise it was filtered out.
Minimum Indelocator indel size
For analyses in this manuscript we restricted Indelocator calls to indels size greater than or equal to 4 alleles.
Annotation
Additional annotations were added from COSMIC(Forbes et al., 2015), dbGaP(Sherry et al., 2001), ExAC(Lek et al., 2016), and Ensembl(Aken et al., 2016) using Variant Effect Predictor (VEP)(McLaren et al., 2016) and other custom built annotation tools including the normal depth of coverage filter and strand bias filters. The final call set was filtered to identify cohort level artifacts and was subject to extensive variant, subject, and cohort level QC.
DATA AND SOFTWARE AVAILABILITY
Data has been made available at the NCI's Genomic Data Commons. Result MAF files of the MC3 dataset is available in two different versions, the open-access and controlled-access data files. Additionally, intermediate files, such as the original called VCF and annotation marking files have been made available.
All pipelines and software developed as part of this project have been made available in https://github.com/OpenGenomics/mc3
Reference Files and intermediate result files have been made available at https://gdc.cancer.gov/about-data/publications/mc3-2017
Supplementary Material
Figure S2: Overlap of mutation calls with analysis working group MAFs at the time of Pancan12, 2013. Related to STAR Methods. Top) Proportion bar plots indicate the percentage intersection of previous efforts with MC3 variants (teal) and the percentage of variants that are unique to previous variant calling efforts (red). Bottom) Proportion bar plot indicates the percentage of MC3 variants that overlap with variants produced by previous efforts (teal) and the percentage of variants that are unique to the MC3 MAFs (blue).
Figure S3: Tumor purity scores calculated by ABSOLUTE, are shown in box-plot format. Related to STAR Methods. Cancer types on the y-axis are sorted by increasing median purity estimates.
Figure S4: Composition of validation data. Related to Figure 3. (A) Composition of the Variant Allele Fraction (VAF) of mutations in the validation set, the full mutation call set and the filtered open-access data set. Validation data has a clear bias toward lower VAF mutations, selected for validation because they were harder to call. (B) The composition of the validation data by cancer type. Most of the calls come from UCEC, COAD, and LUAD.
Figure S5: The effects of filtering on mutation counts by gene. Related to Figure 2. Mutation count analysis was performed for the pre- and post-filtering mutations using the “PASS” filter flag. Variants used for this analysis were restricted to the exonic regions only. (A) The height of each bar represents the total number of called mutations for each gene and is split by “PASS” calls and not “PASS” calls. The top genes 50 genes with the largest difference (‘Not passed’ minus ‘passed’) are plotted in order according to increasing gene length. (B) This panel is identical to panel (A) but is subset to 50 cancer genes identified by Kandoth et. al 2013.
Exome sequencing based variant calls from 10,000 individuals
Samples from 33 cancer types
Variants from: MuTect, MuSE, VarScan2, Radia, Pindel, Somatic Sniper, Indelocator
Acknowledgments
The authors would like to acknowledge contributions of all of the members of the TCGA network. This work was funded by grants from to the UC Santa Cruz Genomics Institute and supported by the NIH NHGRI (grant no U54HG007990), NCI (U24CA143858) and NCI ITCR (grant no R01CA180778); Oregon Health and Science University from the NCI (U24CA210957, U24CA143799); MD Anderson grants from NCI (R01CA183793, U24CA210950, U24CA210949, U24CA143883, CA150252, U24CA143845) and Keck Center of the Golf Coast Consortia for the Cancer Biology Training Program CPRIT (RP140113); NCI and NHGRI grants to McDonnell Genome Institute at Washington University (U24CA211006, U01HG006517 and U54HG003079); NCI grants to the Institute of Systems Biology (U24CA143835); NHGRI grants to the Broad Institute (U54HG003067); NCI and NHGRI grants to the Baylor College of Medicine (U54HG003273, U24CA143843); NCI grants to MSKCC (U24CA143840); NCI grants to MD Anderson (U24CA143845, P30CA016672, U24CA143883); NCI grants to University of North Carolina (U24CA143848); NCI grants to the Van Andel Research Institute (U24CA143882); NCI grants to Partners Healthcare (U24CA144025); NCI grants to Harvard (U24CA143867); NCI grants to BC Cancer Foundation (U24CA143866)
Additionally the author would like to thank DNAnexus for the compute processes they provided for the variant calling; OxoG calculations in this study was performed on the Institute for Systems Biology-Cancer Genomics Cloud (ISB-CGC), a pilot project of the National Cancer Institute (under contract number HHSN261201400007C) and the Broad Institute for validation data processing.
Appendix
Secondary author list:
Amy Blum, Samantha J. Caesar-Johnson, John A. Demchok, Ina Felau, Melpomeni Kasapi, Martin L. Ferguson, Carolyn M. Hutter, Heidi J. Sofia, Roy Tarnuzzer, Peggy Wang, Zhining Wang, Liming Yang, Jean C. Zenklusen, Jiashan (Julia) Zhang, Sudha Chudamani, Jia Liu, Laxmi Lolla, Rashi Naresh, Todd Pihl, Qiang Sun, Yunhu Wan, Ye Wu, Juok Cho, Timothy DeFreitas, Scott Frazer, Nils Gehlenborg, Gad Getz, David I. Heiman, Jaegil Kim, Michael S. Lawrence, Pei Lin, Sam Meier, Michael S. Noble, Gordon Saksena, Doug Voet, Hailei Zhang, Brady Bernard, Nyasha Chambwe, Varsha Dhankani, Theo Knijnenburg, Roger Kramer, Kalle Leinonen, Yuexin Liu, Michael Miller, Sheila Reynolds, Ilya Shmulevich, Vesteinn Thorsson, Wei Zhang, Rehan Akbani, Bradley M. Broom, Apurva M. Hegde, Zhenlin Ju, Rupa S. Kanchi, Anil Korkut, Jun Li, Han Liang, Shiyun Ling, Wenbin Liu, Yiling Lu, Gordon B. Mills, Kwok-Shing Ng, Arvind Rao, Michael Ryan, Jing Wang, John N. Weinstein, Jiexin Zhang, Adam Abeshouse, Joshua Armenia, Debyani Chakravarty, Walid K. Chatila, Ino de Bruijn, Jianjiong Gao, Benjamin E. Gross, Zachary J. Heins, Ritika Kundra, Konnor La, Marc Ladanyi, Augustin Luna, Moriah G. Nissan, Angelica Ochoa, Sarah M. Phillips, Ed Reznik, Francisco Sanchez-Vega, Chris Sander, Nikolaus Schultz, Robert Sheridan, S. Onur Sumer, Yichao Sun, Barry S. Taylor, Jioajiao Wang, Hongxin Zhang, Pavana Anur, Myron Peto, Paul Spellman, Christopher Benz, Joshua M. Stuart, Christopher K. Wong, Christina Yau, D. Neil Hayes, Joel S. Parker, Matthew D. Wilkerson, Adrian Ally, Miruna Balasundaram, Reanne Bowlby, Denise Brooks, Rebecca Carlsen, Eric Chuah, Noreen Dhalla, Robert Holt, Steven J.M. Jones, Katayoon Kasaian, Darlene Lee, Yussanne Ma, Marco A. Marra, Michael Mayo, Richard A. Moore, Andrew J. Mungall, Karen Mungall, A. Gordon Robertson, Sara Sadeghi, Jacqueline E. Schein, Payal Sipahimalani, Angela Tam, Nina Thiessen, Kane Tse, Tina Wong, Ashton C. Berger, Rameen Beroukhim, Andrew D. Cherniack, Carrie Cibulskis, Stacey B. Gabriel, Galen F. Gao, Gavin Ha, Matthew Meyerson, Steven E. Schumacher, Juliann Shih, Melanie H. Kucherlapati, Raju S. Kucherlapati, Stephen Baylin, Leslie Cope, Ludmila Danilova, Moiz S. Bootwalla, Phillip H. Lai, Dennis T. Maglinte, David J. Van Den Berg, Daniel J. Weisenberger, J. Todd Auman, Saianand Balu, Tom Bodenheimer, Cheng Fan, Katherine A. Hoadley, Alan P. Hoyle, Stuart R. Jefferys, Corbin D. Jones, Shaowu Meng, Piotr A. Mieczkowski, Lisle E. Mose, Amy H. Perou, Charles M. Perou, Jeffrey Roach, Yan Shi, Janae V. Simons, Tara Skelly, Matthew G. Soloway, Donghui Tan, Umadevi Veluvolu, Huihui Fan, Toshinori Hinoue, Peter W. Laird, Hui Shen, Wanding Zhou, Michelle Bellair, Kyle Chang, Kyle Covington, Chad J. Creighton, Huyen Dinh, HarshaVardhan Doddapaneni, Lawrence A. Donehower, Jennifer Drummond, Richard A. Gibbs, Robert Glenn, Walker Hale, Yi Han, Jianhong Hu, Viktoriya Korchina, Sandra Lee, Lora Lewis, Wei Li, Xiuping Liu, Margaret Morgan, Donna Morton, Donna Muzny, Jireh Santibanez, Margi Sheth, Eve Shinbrot, Linghua Wang, Min Wang, David A. Wheeler, Liu Xi, Fengmei Zhao, Julian Hess, Elizabeth L. Appelbaum, Matthew Bailey, Matthew G. Cordes, Li Ding, Catrina C. Fronick, Lucinda A. Fulton, Robert S. Fulton, Cyriac Kandoth, Elaine R. Mardis, Michael D. McLellan, Christopher A. Miller, Heather K. Schmidt, Richard K. Wilson, Daniel Crain, Erin Curley, Johanna Gardner, Kevin Lau, David Mallery, Scott Morris, Joseph Paulauskis, Robert Penny, Candace Shelton, Troy Shelton, Mark Sherman, Eric Thompson, Peggy Yena, Jay Bowen, Julie M. Gastier-Foster, Mark Gerken, Kristen M. Leraas, Tara M. Lichtenberg, Nilsa C. Ramirez, Lisa Wise, Erik Zmuda, Niall Corcoran, Tony Costello, Christopher Hovens, Andre L. Carvalho, Ana C. de Carvalho, José H. Fregnani, Adhemar Longatto-Filho, Rui M. Reis, Cristovam Scapulatempo-Neto, Henrique C.S. Silveira, Daniel O. Vidal, Andrew Burnette, Jennifer Eschbacher, Beth Hermes, Ardene Noss, Rosy Singh, Matthew L. Anderson, Patricia D. Castro, Michael Ittmann, David Huntsman, Bernard Kohl, Xuan Le, Richard Thorp, Chris Andry, Elizabeth R. Duffy, Vladimir Lyadov, Oxana Paklina, Galiya Setdikova, Alexey Shabunin, Mikhail Tavobilov, Christopher McPherson, Ronald Warnick, Ross Berkowitz, Daniel Cramer, Colleen Feltmate, Neil Horowitz, Adam Kibel, Michael Muto, Chandrajit P. Raut, Andrei Malykh, Jill S. Barnholtz-Sloan, Wendi Barrett, Karen Devine, Jordonna Fulop, Quinn T. Ostrom, Kristen Shimmel, Yingli Wolinsky, Andrew E. Sloan, Agostino De Rose, Felice Giuliante, Marc Goodman, Beth Y. Karlan, Curt H. Hagedorn, John Eckman, Jodi Harr, Jerome Myers, Kelinda Tucker, Leigh Anne Zach, Brenda Deyarmin, Hai Hu, Leonid Kvecher, Caroline Larson, Richard J. Mural, Stella Somiari, Ales Vicha, Tomas Zelinka, Joseph Bennett, Mary Iacocca, Brenda Rabeno, Patricia Swanson, Mathieu Latour, Louis Lacombe, Bernard Têtu, Alain Bergeron, Mary McGraw, Susan M. Staugaitis, John Chabot, Hanina Hibshoosh, Antonia Sepulveda, Tao Su, Timothy Wang, Olga Potapova, Olga Voronina, Laurence Desjardins, Odette Mariani, Sergio Roman-Roman, Xavier Sastre, Marc-Henri Stern, Feixiong Cheng, Sabina Signoretti, Andrew Berchuck, Darell Bigner, Eric Lipp, Jeffrey Marks, Shannon McCall, Roger McLendon, Angeles Secord, Alexis Sharp, Madhusmita Behera, Daniel J. Brat, Amy Chen, Keith Delman, Seth Force, Fadlo Khuri, Kelly Magliocca, Shishir Maithel, Jeffrey J. Olson, Taofeek Owonikoko, Alan Pickens, Suresh Ramalingam, Dong M. Shin, Gabriel Sica, Erwin G. Van Meir, Hongzheng Zhang, Wil Eijckenboom, Ad Gillis, Esther Korpershoek, Leendert Looijenga, Wolter Oosterhuis, Hans Stoop, Kim E. van Kessel, Ellen C. Zwarthoff, Chiara Calatozzolo, Lucia Cuppini, Stefania Cuzzubbo, Francesco DiMeco, Gaetano Finocchiaro, Luca Mattei, Alessandro Perin, Bianca Pollo, Chu Chen, John Houck, Pawadee Lohavanichbutr, Arndt Hartmann, Christine Stoehr, Robert Stoehr, Helge Taubert, Sven Wach, Bernd Wullich, Witold Kycler, Dawid Murawa, Maciej Wiznerowicz, Ki Chung, W. Jeffrey Edenfield, Julie Martin, Eric Baudin, Glenn Bubley, Raphael Bueno, Assunta De Rienzo, William G. Richards, Steven Kalkanis, Tom Mikkelsen, Houtan Noushmehr, Lisa Scarpace, Nicolas Girard, Marta Aymerich, Elias Campo, Eva Giné, Armando López Guillermo, Nguyen Van Bang, Phan Thi Hanh, Bui Duc Phu, Yufang Tang, Howard Colman, Kimberley Evason, Peter R. Dottino, John A. Martignetti, Hani Gabra, Hartmut Juhl, Teniola Akeredolu, Serghei Stepa, Dave Hoon, Keunsoo Ahn, Koo Jeong Kang, Felix Beuschlein, Anne Breggia, Michael Birrer, Debra Bell, Mitesh Borad, Alan H. Bryce, Erik Castle, Vishal Chandan, John Cheville, John A. Copland, Michael Farnell, Thomas Flotte, Nasra Giama, Thai Ho, Michael Kendrick, Jean-Pierre Kocher, Karla Kopp, Catherine Moser, David Nagorney, Daniel O’Brien, Brian Patrick O’Neill, Tushar Patel, Gloria Petersen, Florencia Que, Michael Rivera, Lewis Roberts, Robert Smallridge, Thomas Smyrk, Melissa Stanton, R. Houston Thompson, Michael Torbenson, Ju Dong Yang, Lizhi Zhang, Fadi Brimo, Jaffer A. Ajani, Ana Maria Angulo Gonzalez, Carmen Behrens, Jolanta Bondaruk, Russell Broaddus, Bogdan Czerniak, Bita Esmaeli, Junya Fujimoto, Jeffrey Gershenwald, Charles Guo, Alexander J. Lazar, Christopher Logothetis, Funda Meric-Bernstam, Cesar Moran, Lois Ramondetta, David Rice, Anil Sood, Pheroze Tamboli, Timothy Thompson, Patricia Troncoso, Anne Tsao, Ignacio Wistuba, Candace Carter, Lauren Haydu, Peter Hersey, Valerie Jakrot, Hojabr Kakavand, Richard Kefford, Kenneth Lee, Georgina Long, Graham Mann, Michael Quinn, Robyn Saw, Richard Scolyer, Kerwin Shannon, Andrew Spillane, Jonathan Stretch, Maria Synott, John Thompson, James Wilmott, Hikmat Al-Ahmadie, Timothy A. Chan, Ronald Ghossein, Anuradha Gopalan, Douglas A. Levine, Victor Reuter, Samuel Singer, Bhuvanesh Singh, Nguyen Viet Tien, Thomas Broudy, Cyrus Mirsaidi, Praveen Nair, Paul Drwiega, Judy Miller, Jennifer Smith, Howard Zaren, Joong-Won Park, Nguyen Phi Hung, Electron Kebebew, W. Marston Linehan, Adam R. Metwalli, Karel Pacak, Peter A. Pinto, Mark Schiffman, Laura S. Schmidt, Cathy D. Vocke, Nicolas Wentzensen, Robert Worrell, Hannah Yang, Marc Moncrieff, Chandra Goparaju, Jonathan Melamed, Harvey Pass, Natalia Botnariuc, Irina Caraman, Mircea Cernat, Inga Chemencedji, Adrian Clipca, Serghei Doruc, Ghenadie Gorincioi, Sergiu Mura, Maria Pirtac, Irina Stancul, Diana Tcaciuc, Monique Albert, Iakovina Alexopoulou, Angel Arnaout, John Bartlett, Jay Engel, Sebastien Gilbert, Jeremy Parfitt, Harman Sekhon, George Thomas, Doris M. Rassl, Robert C. Rintoul, Carlo Bifulco, Raina Tamakawa, Walter Urba, Nicholas Hayward, Henri Timmers, Anna Antenucci, Francesco Facciolo, Gianluca Grazi, Mirella Marino, Roberta Merola, Ronald de Krijger, Anne-Paule Gimenez-Roqueplo, Alain Piché, Simone Chevalier, Ginette McKercher, Kivanc Birsoy, Gene Barnett, Cathy Brewer, Carol Farver, Theresa Naska, Nathan A. Pennell, Daniel Raymond, Cathy Schilero, Kathy Smolenski, Felicia Williams, Carl Morrison, Jeffrey A. Borgia, Michael J. Liptay, Mark Pool, Christopher W. Seder, Kerstin Junker, Larsson Omberg, Mikhail Dinkin, George Manikhas, Domenico Alvaro, Maria Consiglia Bragazzi, Vincenzo Cardinale, Guido Carpino, Eugenio Gaudio, David Chesla, Sandra Cottingham, Michael Dubina, Fedor Moiseenko, Renumathy Dhanasekaran, Karl-Friedrich Becker, Klaus-Peter Janssen, Julia Slotta-Huspenina, Mohamed H. Abdel-Rahman, Dina Aziz, Sue Bell, Colleen M. Cebulla, Amy Davis, Rebecca Duell, J. Bradley Elder, Joe Hilty, Bahavna Kumar, James Lang, Norman L. Lehman, Randy Mandt, Phuong Nguyen, Robert Pilarski, Karan Rai, Lynn Schoenfield, Kelly Senecal, Paul Wakely, Paul Hansen, Ronald Lechan, James Powers, Arthur Tischler, William E. Grizzle, Katherine C. Sexton, Alison Kastl, Joel Henderson, Sima Porten, Jens Waldmann, Martin Fassnacht, Sylvia L. Asa, Dirk Schadendorf, Marta Couce, Markus Graefen, Hartwig Huland, Guido Sauter, Thorsten Schlomm, Ronald Simon, Pierre Tennstedt, Oluwole Olabode, Mark Nelson, Oliver Bathe, Peter R. Carroll, June M. Chan, Philip Disaia, Pat Glenn, Robin K. Kelley, Charles N. Landen, Joanna Phillips, Michael Prados, Jeff Simko, Jeffry Simko, Karen Smith-McCune, Scott VandenBerg, Kevin Roggin, Ashley Fehrenbach, Ady Kendler, Suzanne Sifri, Ruth Steele, Antonio Jimeno, Francis Carey, Ian Forgie, Massimo Mannelli, Michael Carney, Brenda Hernandez, Benito Campos, Christel Herold-Mende, Christin Jungk, Andreas Unterberg, Andreas von Deimling, Aaron Bossler, Joseph Galbraith, Laura Jacobus, Michael Knudson, Tina Knutson, Deqin Ma, Mohammed Milhem, Rita Sigmund, Andrew K. Godwin, Rashna Madan, Howard G. Rosenthal, Clement Adebamowo, Sally N. Adebamowo, Alex Boussioutas, David Beer, Thomas Giordano, Anne-Marie Mes-Masson, Fred Saad, Therese Bocklage, Lisa Landrum, Robert Mannel, Kathleen Moore, Katherine Moxley, Russel Postier, Joan Walker, Rosemary Zuna, Michael Feldman, Federico Valdivieso, Rajiv Dhir, James Luketich, Edna M. Mora Pinero, Mario Quintero-Aguilo, Carlos Gilberto Carlotti, Jr., Jose Sebastião Dos Santos, Rafael Kemp, Ajith Sankarankuty, Daniela Tirapelli, James Catto, Kathy Agnew, Elizabeth Swisher, Jenette Creaney, Bruce Robinson, Carl Simon Shelley, Eryn M. Godwin, Sara Kendall, Cassaundra Shipman, Carol Bradford, Thomas Carey, Andrea Haddad, Jeffey Moyer, Lisa Peterson, Mark Prince, Laura Rozek, Gregory Wolf, Rayleen Bowman, Kwun M. Fong, Ian Yang, Robert Korst, W. Kimryn Rathmell, J. Leigh Fantacone-Campbell, Jeffrey A. Hooke, Albert J. Kovatich, Craig D. Shriver, John DiPersio, Bettina Drake, Ramaswamy Govindan, Sharon Heath, Timothy Ley, Brian Van Tine, Peter Westervelt, Mark A. Rubin, Jung Il Lee, Natália D. Aredes, Armaz Mariamidze, Anant Agrawal, Jaeil Ahn, Jordan Aissiou, Dimitris Anastassiou, Jesper B. Andersen, Jurandyr M. Andrade, Marco Antoniotti, Jon C. Aster, Donald Ayer, Matthew H. Bailey, Rohan Bareja, Adam J. Bass, Azfar Basunia, Oliver F. Bathe, Rebecca Batiste, Oliver Bear Don't Walk, Davide Bedognetti, Gloria Bertoli, Denis Bertrand, Bhavneet Bhinder, Gianluca Bontempi, Dante Bortone, Donald P. Bottaro, Paul Boutros, Kevin Brennan, Chaya Brodie, Scott Brown, Susan Bullman, Silvia Buonamici, Tomasz Burzykowski, Lauren Averett Byers, Fernando Camargo, Joshua D. Campbell, Francisco J. Candido dos Reis, Shaolong Cao, Maria Cardenas, Helio H.A. Carrara, Isabella Castiglioni, Anavaleria Castro, Claudia Cava, Michele Ceccarelli, Shengjie Chai, Kridsadakorn Chaichoompu, Matthew T. Chang, Han Chen, Haoran Chen, Hu Chen, Jian Chen, Jianhong Chen, Ken Chen, Ting-Wen Chen, Zhong Chen, Zhongyuan Chen, Hui Cheng, Hua-Sheng Chiu, Cai Chunhui, Giovanni Ciriello, Cristian Coarfa, Antonio Colaprico, Lee Cooper, Daniel Cui Zhou, Aedin C. Culhane, Christina Curtis, Patrycja Czerwińska, Aditya Deshpande, Lixia Diao, Michael Dill, Di Du, Charles G. Eberhart, James A. Eddy, Robert N. Eisenman, Mohammed Elanbari, Olivier Elemento, Kyle Ellrott, Manel Esteller, Farshad Farshidfar, Bin Feng, Camila Ferreira de Souza, Esla R. Flores, Steven Foltz, Mitchell T. Frederick, Qingsong Gao, Carl M. Gay, Zhongqi Ge, Andrew J. Gentles, Olivier Gevaert, David L. Gibbs, Adam Godzik, Abel Gonzalez-Perez, Marc T. Goodman, Dmitry A. Gordenin, Carla Grandori, Alex Graudenzi, Casey Greene, Justin Guinney, Margaret L. Gulley, Preethi H. Gunaratne, A. Ari Hakimi, Peter Hammerman, Leng Han, Holger Heyn, Le Hou, Donglei Hu, Kuan-lin Huang, Joerg Huelsken, Scott Huntsman, Peter Hurlin, Matthias Hüser, Antonio Iavarone, Marcin Imielinski, Mirazul Islam, Jacek Jassem, Peilin Jia, Cigall Kadoch, Andre Kahles, Benny Kaipparettu, Bozena Kaminska, Havish Kantheti, Rachel Karchin, Mostafa Karimi, Ekta Khurana, Pora Kim, Leszek J. Klimczak, Jia Yu Koh, Alexander Krasnitz, Nicole Kuderer, Tahsin Kurc, David J. Kwiatkowski, Teresa Laguna, Martin Lang, Anna Lasorella, Thuc D. Le, Adrian V. Lee, Ju-Seog Lee, Steve Lefever, Kjong Lehmann, Jake Leighton, Chunyan Li, Lei Li, Shulin Li, David Liu, Eric Minwei Liu, Jianfang Liu, Rongjie Liu, Yang Liu, William J.R. Longabaugh, Nuria Lopez-Bigas, Li Ma, Wencai Ma, Karen MacKenzie, Andrzej Mackiewicz, Dejan Maglic, Raunaq Malhotra, Tathiane M. Malta, Calena Marchand, R. Jay Mashl, Sylwia Mazurek, Pieter Mestdagh, Chase Miller, Marco Mina, Lopa Mishra, Younes Mokrab, Raymond Monnat, Jr., Nate Moore, Nathanael Moore, Loris Mularoni, Niranjan Nagarajan, Aaron M. Newman, Vu Nguyen, Michael L. Nickerson, Akinyemi I. Ojesina, Catharina Olsen, Sandra Orsulic, Tai-Hsien Ou Yang, James Palacino, Yinghong Pan, Elena Papaleo, Sagar Patil, Chandra Sekhar Pedamallu, Shouyong Peng, Xinxin Peng, Arjun Pennathur, Curtis R. Pickering, Christopher L. Plaisier, Laila Poisson, Eduard Porta-Pardo, Marcos Prunello, John L. Pulice, Charles Rabkin, Janet S. Rader, Kimal Rajapakshe, Aruna Ramachandran, Shuyun Rao, Xiayu Rao, Benjamin J. Raphael, Gunnar Rätsch, Brendan Reardon, Christopher J. Ricketts, Jason Roszik, Carlota Rubio-Perez, Ryan Russell, Anil Rustgi, Russell Ryan, Mohamad Saad, Thais Sabedot, Joel Saltz, Dimitris Samaras, Franz X. Schaub, Barbara G. Schneider, Adam Scott, Michael Seiler, Sara Selitsky, Sohini Sengupta, Jose A. Seoane, Jonathan S. Serody, Reid Shaw, Yang Shen, Tiago Silva, Pankaj Singh, I.K. Ashok Sivakumar, Christof Smith, Artem Sokolov, Junyan Song, Pavel Sumazin, Yutong Sun, Chayaporn Suphavilai, Najeeb Syed, David Tamborero, Alison M. Taylor, Teng Teng, Daniel G. Tiezzi, Collin Tokheim, Nora Toussaint, Mihir Trivedi, Kenneth T. Tsai, Aaron D. Tward, Eliezer Van Allen, John S. Van Arnam, Kristel Van Steen, Carter Van Waes, Christopher P. Vellano, Benjamin Vincent, Nam S. Vo, Vonn Walter, Chen Wang, Fang Wang, Jiayin Wang, Sophia Wang, Wenyi Wang, Yue Wang, Yumeng Wang, Zehua Wang, Zeya Wang, Zixing Wang, Gregory Way, Amila Weerasinghe, Michael Wells, Michael C. Wendl, Cecilia Williams, Joseph Willis, Denise Wolf, Karen Wong, Yonghong Xiao, Lu Xinghua, Bo Yang, Da Yang, Liuqing Yang, Kai Ye, Hiroyuki Yoshida, Lihua Yu, Sobia Zaidi, Huiwen Zhang, Min Zhang, Xiaoyang Zhang, Tianhao Zhao, Wei Zhao, Zhongming Zhao, Tian Zheng, Jane Zhou, Zhicheng Zhou, Hongtu Zhu, Ping Zhu, Michael T. Zimmermann, Elad Ziv, and Patrick A. Zweidler-McKay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Kyle Ellrott led the dockerization of variant calling tools and wrote the manuscript. Matthew H. Bailey provided analysis and generated figures. Kyle R. Covington and Cyriac Kandoth developed the MAF conversion and variant annotation pipelines and provided analysis support. Gordon Saksena built sample sets, developed pipeline tools, and provided analysis support. Chip Stewart provided analysis for performance and validation data. Julian Hess provided Panel of Normals filter and description of its method. Michael McLellan conducted data analysis. Li Ding, Gad Getz, and David Wheeler developed project plan and provided guidance on data analysis and interpretation. Heidi Sofia and Carolyn Hutter managed the project. All authors revised the manuscript.
References
- Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, et al. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T, et al. The Ensembl gene annotation system. Database. 2016;2016 doi: 10.1093/database/baw093. baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner AM, Graubert TA. Genomics in childhood acute myeloid leukemia comes of age. Nat. Med. 2018;24:7–9. doi: 10.1038/nm.4469. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Getz G, Stuart JM, Korbel JO, Stein LD Net, - ICGC/TCGA Pan-Cancer Analysis of Whole Genomes. Pan-cancer analysis of whole genomes. bioRxiv 162784 2017 [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet J-P, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, Beer DG, Cope L, Creighton CJ, Danilova L, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33:2938–2940. doi: 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JC, Stolovitzky G. Seeking the Wisdom of Crowds Through Challenge-Based Competitions in Biomedical Research. Clin. Pharmacol. Ther. 2013;93:396–398. doi: 10.1038/clpt.2013.36. [DOI] [PubMed] [Google Scholar]
- Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41:e67–e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis ER, et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012;22:1589–1598. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Houlahan KE, Hu Y, Ellrott K, Caloian C, Yamaguchi TN, Bare JC, P’ng C, Waggott D, Sabelnykova VY, et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods. 2015;12:623–630. doi: 10.1038/nmeth.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Xi L, Hughes DST, Zhang J, Zhang J, Futreal PA, Wheeler DA, Wang W. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17:178. doi: 10.1186/s13059-016-1029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode DL, Hunter SM, Doyle MA, Ma T, Rowley SM, Choong D, Ryland GL, Campbell IG. A simple consensus approach improves somatic mutation prediction accuracy. Genome Med. 2013;5:90. doi: 10.1186/gm494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, Stojanov P, McKenna A, Lander ES, Gabriel S, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmaier RJ, Albacker LA, Chmielecki J, Bailey M, He J, Goldberg ME, Ramkissoon S, Suh J, Elvin JA, Chiacchia S, et al. High-Throughput Genomic Profiling of Adult Solid Tumors Reveals Novel Insights into Cancer Pathogenesis. Cancer Res. 2017;77:2464–2475. doi: 10.1158/0008-5472.CAN-16-2479. [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Speed TP. Comparing somatic mutation-callers: beyond Venn diagrams. BMC Bioinformatics. 2013;14:189. doi: 10.1186/1471-2105-14-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radenbaugh AJ, Ma S, Ewing A, Stuart JM, Collisson EA, Zhu J, Haussler D. RADIA: RNA and DNA Integrated Analysis for Somatic Mutation Detection. PLoS One. 2014;9:e111516. doi: 10.1371/journal.pone.0111516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, Robertson AG, Cherniack AD, Gupta M, Getz G, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32:185–203. e13. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD. The case for cloud computing in genome informatics. Genome Biol. 2010;11:207. doi: 10.1186/gb-2010-11-5-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull C. Introducing Whole Genome Sequencing into routine cancer care: The Genomics England 100,000 Genomes project. Ann. Oncol. 2018 doi: 10.1093/annonc/mdy054. [DOI] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Wang J, Jayasinghe R, Lameijer E-W, McMichael JF, Ning J, McLellan MD, Xie M, Cao S, Yellapantula V, et al. Systematic discovery of complex insertions and deletions in human cancers. Nat. Med. 2015;22:97–104. doi: 10.1038/nm.4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Project GENIE Goes Public. Cancer Discov. 2017;7:118.2–118. doi: 10.1158/2159-8290.CD-NB2017-002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S2: Overlap of mutation calls with analysis working group MAFs at the time of Pancan12, 2013. Related to STAR Methods. Top) Proportion bar plots indicate the percentage intersection of previous efforts with MC3 variants (teal) and the percentage of variants that are unique to previous variant calling efforts (red). Bottom) Proportion bar plot indicates the percentage of MC3 variants that overlap with variants produced by previous efforts (teal) and the percentage of variants that are unique to the MC3 MAFs (blue).
Figure S3: Tumor purity scores calculated by ABSOLUTE, are shown in box-plot format. Related to STAR Methods. Cancer types on the y-axis are sorted by increasing median purity estimates.
Figure S4: Composition of validation data. Related to Figure 3. (A) Composition of the Variant Allele Fraction (VAF) of mutations in the validation set, the full mutation call set and the filtered open-access data set. Validation data has a clear bias toward lower VAF mutations, selected for validation because they were harder to call. (B) The composition of the validation data by cancer type. Most of the calls come from UCEC, COAD, and LUAD.
Figure S5: The effects of filtering on mutation counts by gene. Related to Figure 2. Mutation count analysis was performed for the pre- and post-filtering mutations using the “PASS” filter flag. Variants used for this analysis were restricted to the exonic regions only. (A) The height of each bar represents the total number of called mutations for each gene and is split by “PASS” calls and not “PASS” calls. The top genes 50 genes with the largest difference (‘Not passed’ minus ‘passed’) are plotted in order according to increasing gene length. (B) This panel is identical to panel (A) but is subset to 50 cancer genes identified by Kandoth et. al 2013.