

Abstract
Local drug presentation made possible by drug-eluting depots has demonstrated benefits in a vast array of diseases, including in cancer, microbial infection and in wound healing. However, locally-eluting depots are single-use systems that cannot be refilled or reused after implantation at inaccessible sites, limiting their clinical utility. New strategies to noninvasively refill drug-eluting depots could dramatically enhance their clinical use. In this report we present a refillable hydrogel depot system based on bioorthogonal click chemistry. The click-modified hydrogel depots capture prodrug refills from the blood and subsequently release active drugs locally in a sustained manner. Capture of the systemically-administered refills serves as an efficient and non-toxic method to repeatedly refill depots. Refillable depots in combination with prodrug refills achieve sustained release at precancerous tumor sites to improve cancer therapy while eliminating systemic side effects. The ability to target tissues without enhanced permeability could allow the use of refillable depots in cancer and many other medical applications.
Keywords: Biomaterials, Drug Delivery, Cancer Therapy, Drug Depot, Sustained Release
Graphical abstract
Introduction
Local drug presentation made possible by drug-eluting depots has provided benefits in a vast array of diseases, including in cancer[1–4], microbial infection[5–7] and in wound healing[8–11]. Drug-eluting depots provide a number of advantages to systemic drug administration (e.g., oral or intravenous [i.v.] dosing) that make them promising therapeutics for cancer. First, drug-eluting depots provide high doses of drugs locally at disease sites. Second, sustained drug release prevents peaks and troughs in drug presentation, obviating the need for long i.v. infusions. Third, drug-eluting depots provide continuous drug presence, improving disease outcomes [12,13] and patient compliance[14]. Finally, local drug presentation prevents systemic side effects often seen with systemic drug dosing[15]. These advantages make depots particularly promising in cancer therapy for the prevention of tumor recurrence at dirty surgical margins following surgical resection[16], where sustained drug presentation can affect missed cancer cells without appreciable systemic side effects. One clinically-approved therapy, Gliadel[3], affords sustained release of carmustine into the extracellular fluids of the brain, eliminating the need for the drug to cross the blood-brain barrier.
However, the utility of locally-eluting hydrogel depots is limited because these are single-use systems that cannot be refilled or reused after implantation at sites accessible only through invasive surgical procedures. One limitation of depot technologies is the relatively short period of drug release available with many systems. For example, Gliadel releases the majority of its cargo in 5–10 days and demonstrates a burst release in the first 12 hours. Because the initial burst release translates to excessive local or systemic drug concentrations, the burst release may limit the total amount of drug loaded into the depots. Increasing elution time by a single half-life necessitates doubling the initial dose, while increasing elution times by two half-lives requires 4× the dose. Refillable depots provide an opportunity to extend release by multi half-lives without reaching the toxicity limit. Additionally, the finite release periods for these systems leads to a requirement for physical replacement to continue the dosing regimen. In applications where the depot is implanted using an invasive procedure such as during a tumor resection, the invasive procedure would need to be repeated for depot replacement. Refilling of the depots also provides an opportunity for temporally separating different doses, or refilling and releasing one therapeutic before refilling and releasing a second one at a later time. Finally, both the identity and the dose of the therapeutic is set at depot implantation with conventional systems, making it impossible to change drug dose or drug identity with disease progression or recurrence. Thus, new strategies to noninvasively refill local depots could have dramatic impact on their clinical use.
Recently, a new paradigm in local drug delivery has been introduced, the replenishment of drug depots in vivo in a minimally-invasive manner (Fig. 1). In early versions of this technology, injectable alginate hydrogels were conjugated to chemically-stabilized oligonucleotides which captured drug-carrying nanoparticles from the circulation, refilling the depots inside tumors. These refillable depots increased nanoparticle targeting of tumors and improved treatment efficacy compared to controls in which passive targeting of nanoparticles was utilized[17]. However, due to the size of the refills, enhanced vascular permeability was required for the refills to be able to target the injected gel, thus limiting the scope of the approach.
Figure 1.
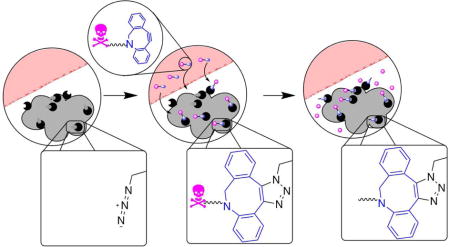
Refillable drug depots allow non-invasive targeting of tissues. Schematic for noninvasive refilling of therapeutic depots. Therapeutic depots (gray) carrying azide groups are implanted at tumor sites during surgery (left). Non-toxic prodrug consisting of cytotoxic drug conjugated to a targeting motif through a cleavable linker is administered systemically and is covalently captured at the depot (middle). Cleavage of the linker over a period of weeks allows release of drug (right).
Bioorthogonal click chemistry constitutes an alternative approach to oligonucleotide targeting for refilling drug depots. Highly specific click reactions are compatible with the biological milieu[18–24]. The click reaction between azides and cyclooctynes is particularly promising for in vivo use due to a good balance between efficient kinetics and in vivo stability. The reactive partners in bioorthogonal click chemistry are much smaller than oligonucleotides, allowing them to extravasate better out of blood vessels and to penetrate further into tissues. Moreover, conjugation of therapeutic compounds to click chemical groups provides an opportunity to reduce systemic toxicity through creation of prodrugs. The click group can be conjugated to drug through a traceless cleavable linker whose cleavage kinetics would be far slower than the clearance time of prodrug from the circulation. Thus, only prodrugs that efficiently conjugate to implanted hydrogels will have the opportunity and the time to cleave off the payload, releasing active therapeutic directly into the vicinity of the gel. We, therefore, reasoned that in addition to targeting established tumors[25,26], click-based targeting of drugs to resident gels would improve both the efficiency and the safety of refillable depots at site where only a few tumor cells were present.
In contrast to previous methods that used in vivo click chemistry to target drugs to tumor tissues for cancer treatment, our system carries several advantages. Previous studies have used metabolic incorporation of azide groups[25,27] for subsequent drug targeting. The reliance on cell-driven enzymatic processes is hampered by tumor heterogeneity and the need for metabolic processes that are truly unique to cancer cells. Additionally, our strategy has the advantage of working at sites of tumor embolization or next to dirty surgical margins with few remaining tumor cells. Additionally, the small molecule refills used in this report do not rely on the enhanced permeability and retention (EPR) effect for tumor penetration, allowing for efficient capture and drug release at sites lacking EPR and poorly vascularized sites. In contrast to click-to-release systems[28,29] our technology decouples the click-mediated conjugation of prodrugs and the hydrolysis-mediated release chemistry, allowing the two reactions kinetics to be independently optimized and creating application-specific refilling and release. Finally, in contrast to systems reliant on tetrazine-TCO chemistry[22,26,30] the chemical stability of the azide groups under physiological conditions, in sharp contrast to tetrazine[23], allows our system to remain active months and potentially years after initial placement. Taken together this report presents an innovative drug delivery system in which a refillable azide-conjugated hydrogel depot serves as a target for nontoxic prodrug refills, allowing multiple refillings and sustained release of drugs at sites of tumor resection, in the absence of enhanced vascular permeability.[31]
Methods
Azide conjugation to alginate
Medical grade, high guluronic acid content, high molecular weight (Mw) alginate (MVG) was purchased from FMC Biopolymers (Princeton, NJ). 200mg of alginate (0.8 micromoles, 1 eq.) was dissolved overnight in 200mL MES buffer (100mM MES, 300mM NaCl, pH=5.5). 11-Azido-3,6,9-trioxaundecan-1-amine (349 mg, 1.6mmole, 2000 eq, Sigma-Aldrich 17758) was added to the solution and the mixture was stirred for an additional 1 hour at room temperature. A mixture of 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (306.7mg, 1.6 mmol, 2000eq, Sigma-Aldrich E7750) and sulfo-N-hydroxysuccinimide (173 mg, 800 umoles, 1000 eq, Thermo Fisher 24510) was added in three equal doses eight hours apart and the solution was stirred for an additional eight hours. The solution was dialyzed against 4L of water with successively lower salt content, changing solution 2–3 times per day. Dialyzed solutions were frozen and lyophilized under high vacuum. Final weight: 150–160 mg. Yield: 75–80%.
Subcutaneous hydrogel targeting
All animal experiments were performed according to established animal protocols. 8-week old, female CD-1 background outbred strains were purchased from Charles River Laboratories. Animals were anesthetized with isoflurane (2 wt%). 20mg/mL alginate conjugated to azide was crosslinked with 4% wt/v CaSO4 (1.22M) in PBS and thus obtained alginate-azide hydrogel (50 μL) was injected through a 25 gauge needle subcutaneously into the right dorsal flank of the animal (n=3 each group). As a control, unconjugated alginate gel was similarly crosslinked with 4% wt/v CaSO4 (1.22M) in PBS and a 50 μL amount injected into the left dorsal flank of the same animal. 24 hours after gel injection, mice were injected using a retro-orbital route with 100 μL of 20 μg/mL Cy7-DBCO (Click Chemistry Tools). Mice were imaged at 24 and 48 hours after i.v.-injection on an IVIS Spectrum in vivo imager (excitation: 745 nm, emission: 820nm). Mice did not show adverse effects from these administrations and no mouse had to be euthanized prior to completion of experiment. No mice were excluded from the analysis.
Intratumoral hydrogel targeting
Tumors were created by injecting 106 MDA-MB-231 cells (American Type Culture Collection, VA) in a 1:1 solution of Matrigel (BD Biosciences, 100 μl) and calcium-crosslinked alginate (100 μl, 2 wt%, 40mM CaSO4), in a total volume of 200μL, intradermally into the dorsal flank of 8 week old J:Nu (Foxn1nu/Foxn1nu) mice (Jackson Labs, ME). 40, 90 and 140 days after gel/cell implantation, mice were injected intravascularly using the retro-orbital route 100 μL 20 μg/mL Cy7-DBCO (Click Chemistry Tools). Mice were imaged 24 hours after i.v.-injection on an IVIS Spectrum in vivo imager (excitation: 745 nm, emission: 820nm). No mice were excluded from the analysis.
Oral dosing of small molecule
8-week old, female CD-1 background outbred strains were purchased from Charles River Laboratories. Animals were anesthetized by intraperitoneal injections of ketamine (80mg/kg) and xylazine (5 mg/kg). Hindlimb ischemia was induced by unilateral external iliac artery and vein ligation. At the time of surgery, mice were randomized to one of two groups (n = 3 per group). 20mg/mL alginate conjugated to azide or unconjugated alginate control were crosslinked with 4% wt/v CaSO4 (1.22M) in PBS and 50 μL of each alginate hydrogel was injected through a 25 gauge needle near the distal end of the ligation site (n=3 each group). In control animals, following surgery, unconjugated alginate was injected intramuscularly. Incisions were closed by 5–0 Ethilon sutures (Johnson & Johnson, NJ). Ischemia in the hindlimb was confirmed by laser Doppler perfusion imaging (LDPI) system (Perimed AB, Sweden).
24 hours after gel injection, mice were administered 100 μL 1mg/mL Cy7-DBCO (Click Chemistry Tools) through oral gavage. Mouse cages were changed daily to prevent coprophagia. One week after oral gavage administration, mice were imaged on an IVIS Spectrum in vivo imager (excitation: 745 nm, emission: 820nm). Mice did not show adverse effects from these administrations and no mouse had to be euthanized prior to completion of experiment. No mice were excluded from the analysis.
Synthesis of prodrug
To form prodrug 4, doxorubicin was conjugated to DBCO-containing moiety through L-cysteine. DBCO-NHS ester 2(25 mg, Click Chemistry Tools, A133) was dissolved in dimethylformamide to a resulting concentration of 30 mg/mL. To this solution was added cysteine 1 (Sigma Aldrich C-7755, 7.51mg in 750 μL sodium borate buffer). The reaction solution was vortexed for 10 seconds and allowed to sit for 5 minutes. Doxorubicin-EMCH 3 (50 mg MedKoo Biosciences 201550) was dissolved in 2 mL DMSO and added to the DMF solution of cysteine-DBCO reaction mixture prepared in the previous step. The reaction was vortexed to dissolve the sudden precipitant and allowed to sit for 5 minutes. Prodrug 4 was purified on HPLC using with a gradient of 5% acetonitrile/95% TEAA to 75% acetonitrile/25% TEAA over 10 minutes on a zorbax C18 5 μm HPLC column on an Agilent 1100 Series Purification HPLC. HPLC fractions containing purified, desired product were lyophilized to dryness to yield prodrug 4 in 60% yield. Resulting dry, red powder was dissolved in 40% hydroxypropyl cyclodextrin (Abcam, ab145002). Concentration was confirmed through measuring the absorbance of prodrug solutions at 498 nm and comparing to the extinction coefficient of doxorubicin. The material was diluted to standard concentrations, sterile filtered, aliquoted and frozen for animal experiments. 1H NMR spectra were recorded on a Varian Inova-500 (500 MHz) at ambient temperature. High Res MS [M+H]+: 1159.3975 Da.
Hydrolysis kinetics of prodrug
Kinetics of hydrolysis of prodrug into active doxorubicin were measured by dissolving Doxorubicin prodrug 4 at 172 nM in phosphate buffered saline of different pH (pH = 6.0, 6.5, 7.0, 7.5). Cleavage of prodrug was monitored by liquid chromatography/mass spectrometry on an Agilent 1290/6140 Ultra High Performance Liquid Chromatography/Mass Spectrometer (UHPLC/MS) consisting of a 1290 Infinity LC binary pump interfaced with an Agilent 1290 Diode Array Detector, an Agilent 1290 Infinity Autosampler, an Agilent Thermostat Column Compartment, and an Agilent 6140 Quadrupole MSD system. Integration of the LCMS peak corresponding to the doxorubicin was measured and confirmed with loss of the prodrug peak.
Click chemistry kinetics of DBCO-Cy7 and DBCO-prodrug
Azide-conjugated alginate was dissolved in 1× PBS at 1 wt%. 20 μL of the alginate solution (~100 azides/strand) and 100 mole equivalence of DBCO-Cy7 or DBCO-prodrug were combined in a total volume of 1mL and added to a quartz cuvette. The reaction of DBCO with azides was quantitated spectrophotometrically based on the Δε308 = 11,800 M−1*cm−1 and a 1 cm path-length cuvette on a spectrophotometer.
Doxorubicin release from click-conjugated prodrug loaded on azide-alginate
A 2% w/v alginate with bound azides solution was prepared in PBS with the addition of 5×10−9 moles DBCO-conjugated prodrug (~10% of the administered dose). The solution was mixed overnight. Calcium-crosslinked gels were prepared from this material and placed at 37C in a solution of PBS (with .1M CaCl2). Gels were incubated and samples taken off at various time points for analysis.
Cell toxicity of doxorubicin prodrug
Tumor cells were plated in 96-well plates 24 hours prior to the start of toxicity experiments. Doxorubicin and prodrug 4 were dissolved in phosphate buffered saline at different concentrations (from .02 uM to 50uM). These solutions were added, in triplicate, to cancer cells and incubated for 1 hour. After incubation, the cells were washed twice with DMEM and cells were cultured for 24 hours in DMEM. Cells were tested for metabolic activity through addition of 10 μL alamar blue solution (Thermo Scientific 88951) for 1 hour and fluorescence was measured on a fluorescence plate reader (excitation: 530 nm, emission: 580 nm).
Animal toxicity of doxorubicin prodrug
15 five-week old CD1 mice were divided into three groups. Mice received 100 μL intraperitoneal administration of doxorubicin prodrug (14.5 mg/kg, 12.5 micromoles/kg in PBs containing 40% hydroxypropyl beta cyclodextrin), doxorubicin (12.5 micromoles/kg, 6.8 mg/kg in PBS containing 40% hydroxypropyl beta cyclodextrin) or vehicle (PBS containing 40% hydroxypropyl beta cyclodextrin) twice weekly for five weeks. Mice were weighed twice weekly and euthanized at the end of administration period. After euthanasia, lung, heart, liver, kidneys, spleen and tissue from the injection site were collected from each mouse into 10% neutral buffered formalin for fixation, then routinely processed into paraffin blocks. Sections were stained with hematoxylin and eosin for light microscopic evaluation by a board-certified veterinary pathologist. Findings were subjectively graded for the degree of pathology by a professional pathologist blinded to the group identity using a scale of 1–5: 1 (minimal), 2 (mild), 3 (moderate), 4 (marked) or 5 (severe). Tissues in which no findings were recognized were scored as Within Normal Limits (+). No mice were excluded from the analysis.
Animal toxicity of implanted gel
15 five-week old CD1 mice were divided among three groups. Each mouse received 100 μL injection intramuscular into the thigh of right hind limb of 2 wt% calcium-crosslinked azide-conjugated alginate gel, 2 wt% calcium-crosslinked unconjugated control gel or vehicle (PBS) (n=5/group). After 3 weeks, mice were euthanized. After euthanasia, lung, heart, liver, kidneys, spleen and tissue from the injection site were collected from each mouse into 10% neutral buffered formalin for fixation, then routinely processed into paraffin blocks. Sections were stained with hematoxylin and eosin for light microscopic evaluation by a board-certified veterinary pathologist. Findings were subjectively graded for the degree of pathology by a professional pathologist blinded to the group identity using a scale of 1–5: 1 (minimal), 2 (mild), 3 (moderate), 4 (marked) or 5 (severe). Tissues in which no findings were recognized were scored as Within Normal Limits (+). No mice were excluded from the analysis.
Animal experiment: tumor growth with prodrug
Tumors were created by administration of calcium-crosslinked alginate (2 wt%, 40mM CaSO4) with 105 MDA-MB-231 cells (American Type Culture Collection, VA) in Matrigel (BD Biosciences, CA) to a total volume of 200μL (100 μl alginate and 100 μl Matrigel) subcutaneously to 8 week old J:Nu (Foxn1nu/Foxn1nu) mice (Jackson Labs, ME). Mice received 100 μL intraperitoneal administration of doxorubicin prodrug (14.5mg/kg, 12.5 micromoles/kg in PBS with hydroxypropyl beta cyclodextrin) or vehicle (PBS with hydroxypropyl beta cyclodextrin) twice weekly for 10 weeks. Throughout the study, tumor volume was measured twice per week with digital calipers in two dimensions and tumor volume was approximated by the equation volume = (length * width * width)/2[32]. Mice were sacrificed when tumors grew beyond 20mm in any dimension or became ulcerated. No mice were excluded from the analysis.
Statistical analysis
Statistical analysis was performed in conjunction with statisticians at the Biostatistics Shared Resource at the Lineberger Comprehensive Research Center at the University of North Carolina.
Results
Selective click chemistry-mediated targeting to alginate hydrogels at subcutaneous and intratumoral sites
We recently reported that azide-modified alginate hydrogels efficiently capture circulating small molecule payloads at intramuscular sites[33]. It was now investigated whether subcutaneous hydrogels could similarly capture small molecules from the blood. Calcium-crosslinked azide-conjugated alginate hydrogels and unconjugated controls were injected subcutaneously on the dorsal backs of CD-1 mice. Twenty four hours after hydrogel injection, cyclooctyne-conjugated far-red fluorophore (DBCO-Cy7) was administered i.v. to the animals. Azide-conjugated gels subsequently showed a significant increase in fluorescence as compared to unmodified control hydrogels (Fig. 2A,B).
Figure 2. Click chemistry-mediated targeting of a drug surrogate to subcutaneous space.
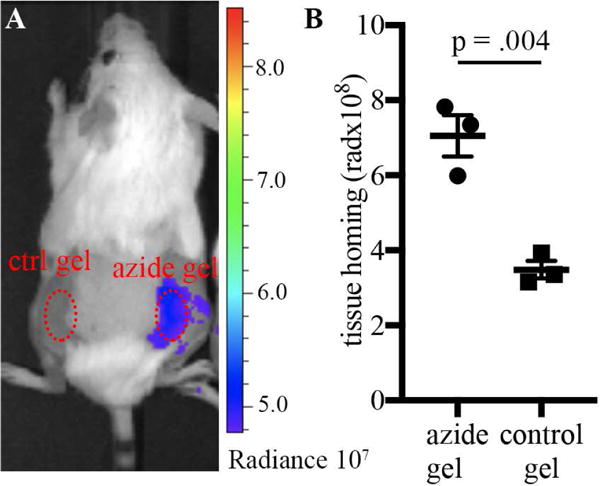
Mice were injected with unconjugated control alginate hydrogels (left flank) or azide-conjugated hydrogels (right flank). Targeting to this gel was tested through i.v. injection of a fluorescently labeled DBCO. A) representative IVIS images, B) quantification of fluorescence at the gel site after 48 hours. Values represent the mean +/− SEM, n=3. p-value from Student’s two-tailed t-test (homoscedastic).
To confirm that the azide-conjugated hydrogels would provide a stable target over a long period of time, the ability of gels to capture circulating molecules in a tumor environment was investigated over a period of 4.5 months. In previous studies[33] tetrazine-conjugated alginates were found to be unstable to in vivo incubation over a period of one week while azide-conjugated alginates were stable for as long as 30 days. Calcium-crosslinked alginate gels (100 μL, 2 wt% in 40mM CaCl2) were combined with triple-negative breast cancer tumor cells (MDA-MB-231, 1×105 cells) and injected subcutaneously into the dorsal flank of nude mice, so that tumors grew around the gel. At days 40, 90 and 140 after gel implantation, the ability of implanted gels to capture cyclooctyne-conjugated molecules was tested. At all time points, azide-conjugated gels efficiently captured circulating cyclooctyne-conjugated molecules (Fig. 3).
Figure 3. Click chemistry-mediated targeting to intratumoral hydrogels.
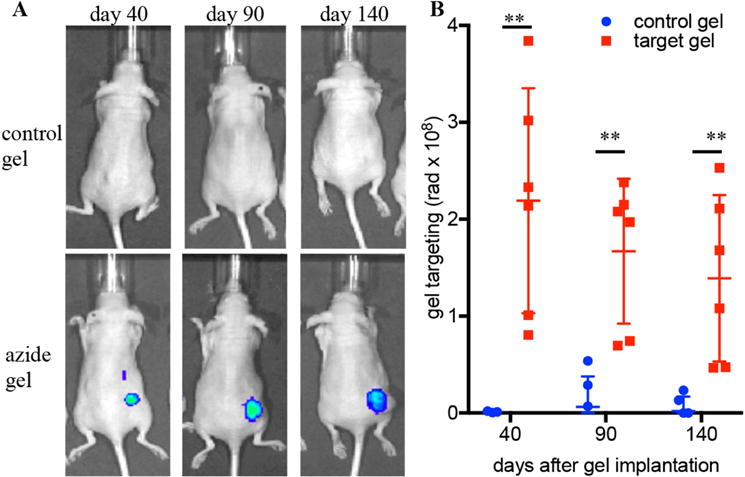
Mice were injected with MDA-MB-231 tumor cells combined with hydrogels (azide-conjugated or control). Targeting to this gel was tested through i.v. injection of a fluorescently labeled DBCO at different time points. A) representative I VIS images, B: quantification of fluorescence at the gel site 24 hours after i.v. injection. Values represent the mean +/− SEM, n=6. ** = p <0.005 by Student’s two-tailed t-test.
It was hypothesized that this approach to depot refiling, which exploits small molecules, would lend itself to oral refilling. To test whether oral administration of small molecule would lead to their concentration at injected gels, azide-conjugated alginate gels were evaluated for their ability to capture cyclooctyne-conjugated fluorophores given by oral gavage. Intramuscular gels efficiently captured cyclooctyne-conjugated fluorophores after oral administration (Supplemental Fig. 1).
The biocompatibility and immunogenicity of azide-conjugated alginate gels were investigated in vivo. Outbred CD-1 mice were injected intramuscularly with 50 μL calcium-crosslinked azide-conjugated gels or unconjugated control gels. Mice receiving vehicle were used as a second control. Three weeks after IM injection, major organs (heart, lungs, liver, kidneys, spleen) as well as the injected muscles were isolated and analyzed by histology and pathology. No differences were observed in the organs of mice in the three groups. Very minor localized physical disruption was observed at the injection site, which did not result in significant inflammation or any evidence of toxic injury. Additionally, no inflammation or toxic injury was observed in response to either the azide-conjugated or the control gels (see supplemental information for full pathology report).
Taken together, these experiments demonstrate that biocompatible azide-conjugated alginate gels placed at subcutaneous and intratumoral sites can noninvasively capture small molecules from the circulation. In the case of intratumor refilling, high vascularization likely aids in gel refilling, however, targeting at subcutaneous location demonstrated that gel replenishment does not require the presence of permeable blood vessels.
Synthesis, kinetics and toxicity of click-conjugated doxorubicin prodrug
Efficient click chemistry-based targeting of azide-conjugated hydrogels requires that drugs are modified with the click partner, such as a cyclooctyne, and this serves as an opportunity to modify drug properties and reduce toxicity. We chose to connect the chemotherapeutic doxorubicin to the cyclooctyne through a hydrazone linker, previously reported to be cleavable at low pH. In this case, however, rather than rapid cleavage of the molecule at low pH, a sustained hydrolysis over a long period of time at more neutral pH was desired.
Synthesis of doxorubicin prodrug is shown in Fig. 4A. Cysteine 1 was conjugated to NHS ester-activated cyclooctyne 2. This solution was directly reacted with aldoxorubicin 3, a maleimide-containing doxorubicin hydrazide currently undergoing clinical trials. The resultant conjugate incorporated a carboxylic acid (red) in order to increase solubility. Prodrug 4 was purified by HPLC in 60% yield.
Figure 4. A slow-cleaving, DBCO-conjugated doxorubicin prodrug.
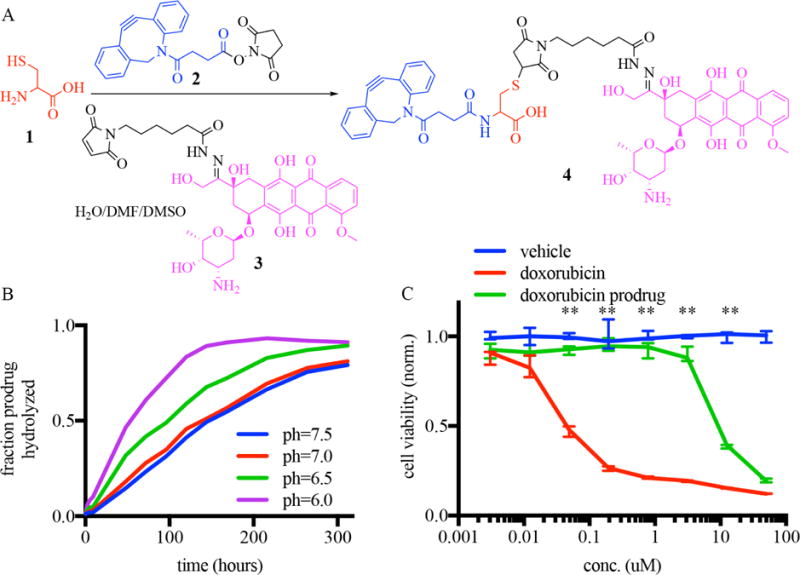
A) Synthesis of a chemotherapeutic prodrug linking doxorubicin (purple) to cyclooctyne (blue) through a hydrolyzable linker. B) LCMS quantitation of the hydrolysis of doxorubicin prodrug over time at different pH. C) In vitro toxicity of doxorubicin, doxorubicin prodrug and vehicle incubated with MDA-MB-231 mammalian cells. ** = p<01 determined by Holm-Sidak method for multiple comparison testing. n=3.
The kinetics of reaction of the DBCO-conjugated prodrug (84.4 M−1s−1) was measured to be somewhat lower than the kinetics for DBCO-Cy7 (179 M−1s−1). This reduction in binding efficiency was attributed at least partly to a lower solubility for the prodrug in PBS as compared to the DBCO-Cy7.
Since sustained drug presentation at the gel site requires sustained release kinetics, both the solution-state cleavage of doxorubicin prodrug 4 to give doxorubicin and the release of doxorubicin after prodruging conjugation to azide-gels was evaluated over time. We observed sustained release of doxorubicin from the alginate gel after conjugation of the prodrug to the azide-alginate gels (Supplemental Fig. 2). In addition, while the prodrug was relatively unstable at low pH with a half-life of 50 hours at pH=6.0. At neutral pH, the cleavage was significantly slower, with a half-life of 144 hours at pH=7.5 (Fig. 4B). A neutral pH is expected at resection sites, which are free of tumor burden.
In our strategy, it was desired that doxorubicin prodrug 4 would be less toxic than the parent doxorubicin molecule, and doxorubicin prodrug 4 showed significantly less toxicity in vitro compared to free doxorubicin. While doxorubicin demonstrated toxicity against tumor cells at concentration above 10nM, the prodrug did not cause toxicity at 300-fold this concentration (Fig. 4C).
To confirm that the low toxicity of prodrug was not limited to in vitro application, administration of doxorubicin prodrug 4 was compared to doxorubicin and vehicle in vivo. Doxorubicin, doxorubicin prodrug 4 and vehicle were administered to mice at 12.5 micromoles/kg twice-weekly over 5 weeks. Repeated administration of doxorubicin caused significant weight loss in mice (Fig. 5), as expected, while equimolar administrations of doxorubicin prodrug did not exhibit this effect. In addition to weight loss, mice receiving doxorubicin also exhibited alopecia, lethargy, and signs of dehydration. Mice receiving doxorubicin intraperitoneally showed significant peritonitis.
Figure 5. In vivo safety of click-enabled doxorubicin prodrugs.
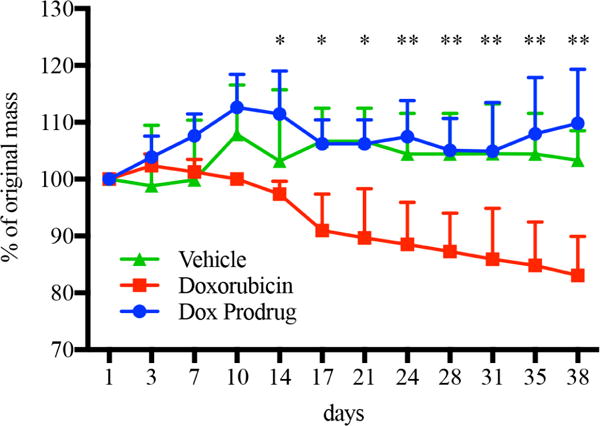
Mouse weight over time with administrations of doxorubicin (red), its cyclooctyne-linked prodrug (blue), or vehicle (green). Twice-weekly intraperitoneal administrations were evaluated for five weeks with doxorubicin or molar equivalent of prodrug. * = p<05 and ** = p<01 doxorubicin vs. dox prodrud for multiple comparison t-test with Holm-Sidak correction. n=5.
Histological examination of the major organs from mice receiving doxorubicin revealed cardiomyopathy, evident by hypereosinophilic myofibers, clumped and fragmented sarcoplasm, and enlarged (reactive) nuclei; splenic atrophy, including both the lymphopoietic and the hematopoietic (leukoid and erythroid) elements; moderate atrophy of skin adnexa (follicles and glands), mild degenerative changes in the subcutaneous muscles, panniculus carnosus, and marked serous atrophy of hypodermal fat. Two out of three mice receiving doxorubicin had atrophic changes in the local mammary tissue. In stark contrast to the doxorubicin-administered animals, mice receiving doxorubicin prodrug 4 showed no deviation from those receiving the vehicle (see supplemental information for full pathology report).
Taken together, these data demonstrate that prudent linkage of cyclooctyne to doxorubicin through a slowly hydrolyzing linker effectively masks the toxicity of doxorubicin. Because the hydrolysis of the hydrazone bond at neutral pH happens over a period of weeks, it is expected that circulating prodrug will be cleared from the circulation without hydrolysis while the prodrug captured by azide gel will have the opportunity to slowly release active drug into surroundings tissues.
Refillable depots prevent tumor growth in rodent cancer models
It was next investigated whether refillable alginate-azide gels in combination with doxorubicin prodrug 4 could prevent tumor growth in a “dirty margins” model of tumor growth. MDA-MB-231 triple-negative breast cancer cells were injected subcutaneously into nude mice in concert with azide-conjugated gels. Unconjugated gels were used as controls. Cyclooctyne-conjugated doxorubicin prodrug or vehicle control were administered twice weekly. The combination of azide-conjugated gels and doxorubicin prodrugs was effective in preventing tumor growth (Fig. 5A). Additionally, the refillable depot combination also prevented side effects from the treatment and could be continued throughout the entire length of the study (Fig. 5B). Importantly, delivering the doxorubicin prodrug to control gels that were incapable of their capture did not slow tumor growth, as the growth kinetics were similar to mice administered vehicle instead of chemotherapeutic (Fig. 5A).
Discussion
This report describes a drug delivery technology based on the use of bioorthogonal click chemistry to refill intratumoral depots with chemotherapeutic prodrug. This strategy was effective in combating tumor growth while eliminating systemic side effects. Azide-modified alginate gels were demonstrated to capture small molecules from the circulation in a subcutaneous setting, where both low vascularization and lack of EPR effects typically diminish drug delivery. The gels were shown to be active over a period of at least 4 months. Therapeutic refills were formulated as chemotherapeutic drug conjugated to cyclooctyne - virtually eliminating systemic toxicity from the chemotherapy. The combination of refillable azide-linked gel depots with a cyclooctyne-linked prodrug significantly inhibited tumor growth while minimizing side effects of the cancer chemotherapy.
Azide-conjugated, calcium-crosslinked alginate gels were used in this study as refillable gel depots. The azides provide a selective binding site for circulating cyclooctyne-linked therapeutics. The selectivity of drug targeting is due largely to the innate selectivity of the click reaction between azide and cyclooctyne moieties[34–36]. We demonstrate that the azide motifs at these gels show good stability after months of implantation. In contrast, we and others have shown that tetrazine conjugates, although stable in buffer, lose function in a matter of hour in vivo[23,33]. The magnitude of capture was similar at days 40, 90 and 140, suggesting that the depot largely retains its capture efficiency over this time. Calcium-crosslinked alginate gels that have not been rendered biodegradable through oxidation[37] or irradiation[38] are expected to be very stable in the body, and have even been considered a “permanent” implant in clinical trials.[39]
Alginate gels have been used as implants in a variety of clinical and preclinical applications[40–44], demonstrating good general biocompatibility and minimal foreign body response. These biocompatible traits were confirmed in this report through analyzing the major organs and the injection site three weeks after gel injection, which revealed minimal physiological response from immunocompetent CD-1 mice. Although the animal model used in this report showed efficacy with ~105 injected cells, the functionality of our system at subcutaneous sites suggests to use that it may also prove useful when only a few cells are left at resection margins. We suggest that these gels could be injected into the void left after a tumor resection, tumor debulking or following tumor ablation. In addition, refillable gel depots would also allow the injection of empty gels (no initial drug load) during tumor biopsy, with the drug therapy chosen after analysis of the biopsied material and subsequently i.v. injected to load the depot for sustained release.
The conjugation of cyclooctyne to drug provides an opportunity to connect the drug to the cyclooctyne through a cleavable linker and thereby create a non-toxic prodrug. Choosing a linker with a slow cleavage profile prevents systemic side effects as prodrug is cleared from the circulation before appreciable amounts of drug are released. The linker is expected to cleave and release drugs into the local microenvironment only at the gel site, where prodrugs were previously captured through click chemistry. Current chemotherapy relies on i.v. infusions because these provide sustained concentrations of drugs within a relatively narrow therapeutic window. The low systemic toxicity and local action of the prodrug provides an opportunity for alternative administration methods, including i.p. (as done in this study), subcutaneous or even oral administrations. Indeed, our data show that the developed system can concentrate orally bioavailable molecules.
The low toxicity of prodrug creates an opportunity for the administration of prodrug at concentrations far higher than those of parent drug, potentially providing weeks or months of chemotherapy in a single dose. In a previous report, it was found that the click-conjugated hydrogels capture about 7.5% of the administered dose of DBCO molecule[33]. Using a conservative estimate of 5% administrative dose captured, the maximum theoretical number of refills possible before saturation of the azide groups was calculated to be in excess of 100 (see Supplemental Information), suggesting that capture of higher doses of prodrug is feasible. In addition, improved azide conjugation methods would allow significant increases in the maximum numbers of refills.
The lack of toxicity from the doxorubicin prodrug is hypothesized to stem from the inability of this compound to cross the cell membrane. Alternative methods for conjugating different types of prodrug would involve sterically blocking an active functionality and would be applicable to peptides[45,46], oligonucleotides[47] and large biological proteins[48]. The hydrazone-linked doxorubicin is currently undergoing clinical trials, suggesting that it is a promising and non-toxic linker for this application[49].
Recent reports have highlighted tumor-associated stromal desmoplasia as a significant barrier to drug targeting[50,51]. One main goal of this technology is to target drug refills to areas of tumor resection, where little stromal desmoplasia is expected. However, the low toxicity of the reported prodrugs provide the opportunity to significantly increase dosing as compared to standard chemotherapy, increasing the amount of chemotherapy at the disease site in absolute terms, even if relative amounts are unchanged.
The refillable hydrogel depot present an alternative to intratumoral[52] or systemic infusion of drugs through a catheter. Intratumoral and systemic infusions mediated by catheter are being actively explored in clinical application. However, the catheters carry a risk of infection[53] or displacement [54], The technology is also complementary to antibody-directed drug delivery such as ADEPT and PDEPT, in which an antibody or nanoparticle is “pretargeted” to a specific site in the body[55–57], and the drug given secondarily. These systems rely on the presence of specific cellular markers of disease, which may be absent in many clinical application[58].
Refillable depots in combination with therapeutic, prodrug refills was shown to improved outcomes in a tumor model. The tumor model chosen was meant to mimic the injection of a hydrogel depot into a resection cavity with dirty surgical margins. In this tumor model, a slow-growing cell line was combined with a relatively small number of cells (2×105). Although early refilling events occur in a relatively avascular environment, we expect that the tumors to grow next to and potentially around, the gels, potentially increased refilling efficiency at later time points. It is hypothesized that refillable depots implanted during tumor resection could be highly beneficial to preventing local recurrence, such as that seen in glioblastoma[59], liposarcoma[60], breast[61] and ovarian cancer[62,63]. The small molecule refills are able to find and refill hydrogels even in the absence of increased permeability because small molecules have higher vessel extravasation, tissue diffusion, and gel permeation as compared to nanoparticle systems. Thus, the refillable depots have potential to be used at site of resection which lack many of the hallmarks of the tumor microenvironment, including tortuous, permeable blood vessels and lowered pH.
Conclusion
Bioorthogonal click chemistry can be used to repeatedly refill drug-releasing depots at a tumor site, allowing repeated release of a drug at a site of tumor resection. A major benefit of refillable depots is the potential for repeated, easily controlled and long-term local release of materials without systemic side effects. The ability to target tissues without enhanced permeability could allow the use of refillable depots in many medical applications, including in antimicrobial therapy for local infections (osteomyelitis, implant associated infections), administration of immunosuppressive drugs to prevent organ rejection, or for refilling blood-contacting devices such as drug-eluting stents, vascular grafts and catheters.
Supplementary Material
Figure 6. Efficacy of doxorubicin prodrugs in combination with refillable depots.
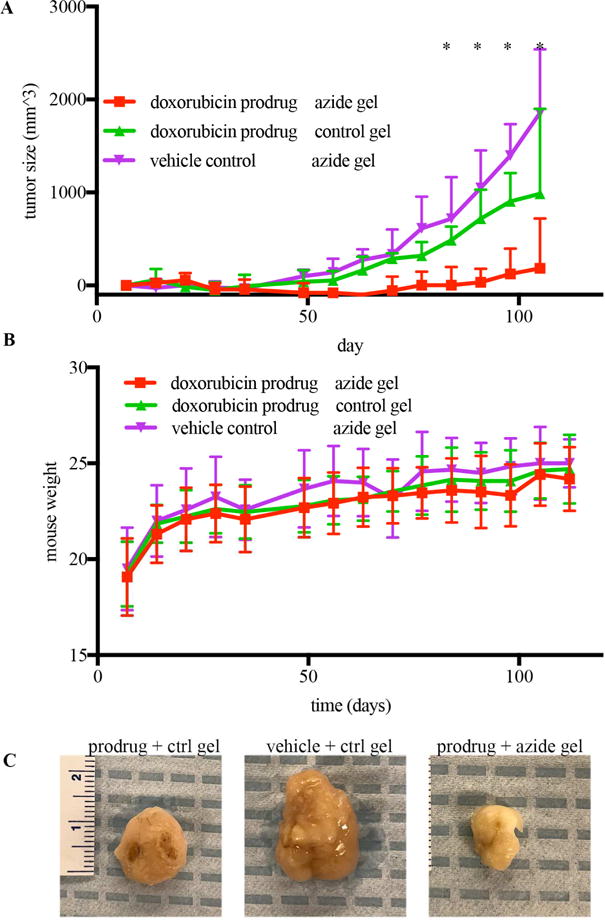
A) Average tumor size over time in MDA-MB-231 bearing mice treated with azide-conjugated or control gels and twice-weekly adminsitration doxorubicin prodrug or vehicle. B) Mouse weight over time for the three experimental groups. * p<.05 by Wicoxon’s post-hoc comparisons testing. n=12. C) Repe- sentative tumor images.
Acknowledgments
We are grateful to the assistance of Chris Johnson and Michael Lewandowski at the Materials Characterization Core at the Wyss Institute for help with LCMS measurements and HPLC purification. We are grateful to Dr. Kathleen Pritchett-Corning at the Harvard Office of Animal Welfare for help with designing animal studies. We are grateful to Dr. Xianming Tan at the Lineberger Comprehensive Cancer Center Biostatistics Resource for consulting on proper statistical analytical methods. This work was supported by the Wyss Institute for Biologically Inspired Engineering and NIH (R01 CA223255). YB gratefully acknowledges funding support from the Wyss Technology Development Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
The raw data required to reproduce these findings will be made available to download from a dedicated web address after publication. The processed data required to reproduce these findings are available to download from a dedicated web address after publication.
References
- 1.Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers. J Control Release. 2012;159:14–26. doi: 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manabe T, Okino H, Maeyama R, Mizumoto K, Nagai E, Tanaka M, Matsuda T. Novel strategic therapeutic approaches for prevention of local recurrence of pancreatic cancer after resection: trans-tissue, sustained local drug-delivery systems. J Control Release. 2004;100:317–330. doi: 10.1016/j.jconrel.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, Whittle IR, Jääskeläinen J, Ram Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Indolfi L, Ligorio M, Ting DT, Xega K, Tzafriri AR, Bersani F, Aceto N, Thapar V, Fuchs BC, Deshpande V, Baker AB, Ferrone CR, Haber DA, Langer R, Clark JW, Edelman ER. A tunable delivery platform to provide local chemotherapy for pancreatic ductal adenocarcinoma. Biomaterials. 2016;93:71–82. doi: 10.1016/j.biomaterials.2016.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nandi SK, Bandyopadhyay S, Das P, Samanta I, Mukherjee P, Roy S, Kundu B. Understanding osteomyelitis and its treatment through local drug delivery system. Biotechnol Adv. 2016;34:1305–1317. doi: 10.1016/j.biotechadv.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Zalavras CG, Patzakis MJ, Holtom P. Local antibiotic therapy in the treatment of open fractures and osteomyelitis. Clin Orthop Relat Res. 2004:86–93. doi: 10.1097/01.blo.0000143571.18892.8d. [DOI] [PubMed] [Google Scholar]
- 7.Hanssen AD. Local antibiotic delivery vehicles in the treatment of musculoskeletal infection. Clin Orthop Relat Res. 2005:91–96. doi: 10.1097/01.blo.0000175713.30506.77. [DOI] [PubMed] [Google Scholar]
- 8.Attanasio S, Snell J. Therapeutic angiogenesis in the management of critical limb ischemia: current concepts and review. Cardiology in Review. 2009;17:115–120. doi: 10.1097/CRD.0b013e318199e9b7. [DOI] [PubMed] [Google Scholar]
- 9.Kearney CJ, Mooney DJ. Macroscale delivery systems for molecular and cellular payloads. Nat Mater. 2013;12:1004–1017. doi: 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- 10.Hubbell JA. Hydrogel systems for barriers and local drug delivery in the control of wound healing. J Control Release. 1996;39:305–313. [Google Scholar]
- 11.Mayet N, Choonara YE, Kumar P, Tomar LK, Tyagi C, Du Toit LC, Pillay V. A comprehensive review of advanced biopolymeric wound healing systems. J Pharm Sci. 2014;103:2211–2230. doi: 10.1002/jps.24068. [DOI] [PubMed] [Google Scholar]
- 12.Eisenberg MJ, Konnyu KJ. Review of randomized clinical trials of drug-eluting stents for the prevention of in-stent restenosis. Am J Cardiol. 2006;98:375–382. doi: 10.1016/j.amjcard.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 13.Hill RA, Dündar Y, Bakhai A, Dickson R, Walley T. Drug-eluting stents: an early systematic review to inform policy. Eur Heart J. 2004;25:902–919. doi: 10.1016/j.ehj.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 14.Cramer MP, Saks SR. Translating safety, efficacy and compliance into economic value for controlled release dosage forms. Pharmacoeconomics. 1994;5:482–504. doi: 10.2165/00019053-199405060-00005. [DOI] [PubMed] [Google Scholar]
- 15.Brem H, Piantadosi S, Burger PC, Walker M, Selker R, Vick NA, Black K, Sisti M, Brem S, Mohr G. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995;345:1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 16.Fakhari A, Anand Subramony J. Engineered in-situ depot-forming hydrogels for intratumoral drug delivery. J Control Release. 2015;220:465–475. doi: 10.1016/j.jconrel.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Brudno Y, Silva EA, Kearney CJ, Lewin SA, Miller A, Martinick KD, Aizenberg M, Mooney DJ. Refilling drug delivery depots through the blood. Proc National Acad Sci. 2014;111:12722–12727. doi: 10.1073/pnas.1413027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR. Copper-free click chemistry in living animals. Proceedings of the National Academy of Sciences. 2010;107:1821–1826. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prescher JA, Dube DH, Bertozzi CR. Chemical remodelling of cell surfaces in living animals. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 20.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun W, Chu T. In vivo click reaction between Tc-99m-labeled azadibenzocyclooctyne-MAMA and 2-nitroimidazole-azide for tumor hypoxia targeting. Bioorg Med Chem Lett. 2015;25:4453–4456. doi: 10.1016/j.bmcl.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Rossin R, Läppchen T, van den Bosch SM, Laforest R, Robillard MS. Diels–Alder Reaction for Tumor Pretargeting: In Vivo Chemistry Can Boost Tumor Radiation Dose Compared with Directly Labeled Antibody. J Nucl Med. 2013;54:1989–1995. doi: 10.2967/jnumed.113.123745. [DOI] [PubMed] [Google Scholar]
- 23.Rossin R, Verkerk PR, van den Bosch SM, Vulders RCM, Verel I, Lub J, Robillard MS. In vivo chemistry for pretargeted tumor imaging in live mice. Angew Chem Int Ed Engl. 2010;49:3375–3378. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- 24.Devaraj NK, Thurber GM, Keliher EJ, Marinelli B, Weissleder R. Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc Natl Acad Sci U S A. 2012;109:4762–4767. doi: 10.1073/pnas.1113466109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Wang R, Cai K, He H, Liu Y, Yen J, Wang Z, Xu M, Sun Y, Zhou X, Yin Q, Tang L, Dobrucki IT, Dobrucki LW, Chaney EJ, Boppart SA, Fan TM, Lezmi S, Chen X, Yin L, Cheng J. Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat Chem Biol. 2017;13:415–424. doi: 10.1038/nchembio.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oneto JM, Khan I, Seebald L, Royzen M. In Vivo Bioorthogonal Chemistry Enables Local Hydrogel and Systemic Pro-Drug To Treat Soft Tissue Sarcoma. Acs Central Sci. 2016;2:476–482. doi: 10.1021/acscentsci.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S, Koo H, Na JH, Han SJ, Min HS, Lee SJ, Kim SH, Yun SH, Jeong SY, Kwon IC, Choi K, Kim K. Chemical tumor-targeting of nanoparticles based on metabolic glycoengineering and click chemistry. ACS Nano. 2014;8:2048–2063. doi: 10.1021/nn406584y. [DOI] [PubMed] [Google Scholar]
- 28.Versteegen RM, Rossin R, ten Hoeve W, Janssen HM, Robillard MS. Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew Chem Int Ed. 2013;52:14112–14116. doi: 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- 29.Rossin R, van Duijnhoven SMJ, ten Hoeve W, Janssen HM, Kleijn LHJ, Hoeben FJM, Versteegen RM, Robillard MS. Triggered Drug Release from an Antibody–Drug Conjugate Using Fast “Click-to-Release” Chemistry in Mice. Bioconjug Chem. 2016;27:1697–1706. doi: 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- 30.Oneto MJM, Gupta M, Leach KJ, Lee M. Implantable Biomaterial Based on Click Chemistry for Targeting Small Molecules. Acta Biomater. 2014 doi: 10.1016/j.actbio.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 31.Koukourakis GV, Kouloulias V, Zacharias G, Papadimitriou C, Pantelakos P, Maravelis G, Fotineas A, Beli I, Chaldeopoulos D, Kouvaris J. Temozolomide with radiation therapy in high grade brain gliomas: pharmaceuticals considerations and efficacy; a review article. Molecules. 2009;14:1561–1577. doi: 10.3390/molecules14041561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 33.Brudno Y, Desai RM, Kwee BJ, Joshi NS, Aizenberg M, Mooney DJ. In Vivo Targeting through Click Chemistry. Chem Med Chem. 2015;10:617–620. doi: 10.1002/cmdc.201402527. [DOI] [PubMed] [Google Scholar]
- 34.McKay CS, Finn MG. Click chemistry in complex mixtures: bioorthogonal bioconjugation. Chem Biol. 2014;21:1075–1101. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Best MD. Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochemistry. 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]
- 36.Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3+ 2] azide- alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 37.Bouhadir KH, Lee KY, Alsberg E, Damm KL, Anderson KW, Mooney DJ. Degradation of partially oxidized alginate and its potential application for tissue engineering. Biotechnol Prog. 2001;17:945–950. doi: 10.1021/bp010070p. [DOI] [PubMed] [Google Scholar]
- 38.King K. Changes in the functional properties and molecular weight of sodium alginate following γ irradiation. Food Hydrocoll. 1994;8:83–96. [Google Scholar]
- 39.Anker SD, Coats AJS, Cristian G, Dragomir D, Pusineri E, Piredda M, Bettari L, Dowling R, Volterrani M, Kirwan BA, Filippatos G, Mas JL, Danchin N, Solomon SD, Lee RJ, Ahmann F, Hinson A, Sabbah HN, Mann DL. A prospective comparison of alginate-hydrogel with standard medical therapy to determine impact on functional capacity and clinical outcomes in patients with advanced heart failure (AUGMENT-HF trial) Eur Heart J. 2015;36:2297–2309. doi: 10.1093/eurheartj/ehv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Augst AD, Kong HJ, Mooney DJ. Alginate hydrogels as biomaterials. Macromol Biosci. 2006;6:623–633. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 41.Liew CV, Chan LW, Ching AL, Heng PW. Evaluation of sodium alginate as drug release modifier in matrix tablets. International Journal of Pharmaceutics. 2006;309:25–37. doi: 10.1016/j.ijpharm.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 42.Matthew IR, Browne RM, Frame JW, Millar BG. Subperiosteal behaviour of alginate and cellulose wound dressing materials. Biomaterials. 1995;16:275–278. doi: 10.1016/0142-9612(95)93254-b. [DOI] [PubMed] [Google Scholar]
- 43.Halberstadt C, Austin C, Rowley J, Culberson C, Loebsack A, Wyatt S, Coleman S, Blacksten L, Burg K, Mooney D, Holder W. A hydrogel material for plastic and reconstructive applications injected into the subcutaneous space of a sheep. Tissue Engineering. 2002;8:309–319. doi: 10.1089/107632702753725067. [DOI] [PubMed] [Google Scholar]
- 44.Silva EA, Kim ES, Kong HJ, Mooney DJ. Material-based deployment enhances efficacy of endothelial progenitor cells. Proc Natl Acad Sci U S A. 2008;105:14347–14352. doi: 10.1073/pnas.0803873105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorganic & Medicinal Chemistry Letters. 2006;16:5350–5355. doi: 10.1016/j.bmcl.2006.07.076. [DOI] [PubMed] [Google Scholar]
- 46.Ezra A, Hoffman A, Breuer E, Alferiev IS, Mönkkönen J, Hanany-Rozen NEl, Weiss G, Stepensky D, Gati I, Cohen H, Törmälehto S, Amidon GL, Golomb G. A peptide prodrug approach for improving bisphosphonate oral absorption. J Med Chem. 2000;43:3641–3652. doi: 10.1021/jm980645y. [DOI] [PubMed] [Google Scholar]
- 47.Iyer RP, Yu D, Devlin T, Ho NH, Agrawal S. Acyloxyaryl prodrugs of oligonucleoside phosphorothioates. Bioorg Med Chem Lett. 1996;6:1917–1922. [Google Scholar]
- 48.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EEM, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ, Sagert JG, Hostetter DR, Han F, Gee J, Flandez J, Markham K, Nguyen M, Krimm M, Wong KR, Liu S, Daugherty PS, West JW, Lowman HB. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med. 2013;5:207ra144. doi: 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- 49.Sachdev E, Sachdev D, Mita M. Aldoxorubicin for the treatment of soft tissue sarcoma. Expert Opin Investig Drugs. 2017;26:1175–1179. doi: 10.1080/13543784.2017.1371134. [DOI] [PubMed] [Google Scholar]
- 50.Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, Babykutty S, Huang Y, Jung K, Rahbari NN, Han X, Chauhan VP, Martin JD, Kahn J, Huang P, Desphande V, Michaelson J, Michelakos TP, Ferrone CR, Soares R, Boucher Y, Fukumura D, Jain RK. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016;6:852–869. doi: 10.1158/2159-8290.CD-15-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crispino P, De Toma G, Ciardi A, Bella A, Rivera M, Cavallaro G, Polistena A, Fornari F, Unim H, Pica R, Cassieri C, Mingazzini PL, Paoluzi P. Role of desmoplasia in recurrence of stage II colorectal cancer within five years after surgery and therapeutic implication. Cancer Invest. 2008;26:419–425. doi: 10.1080/07357900701788155. [DOI] [PubMed] [Google Scholar]
- 52.Walter KA, Tamargo RJ, Olivi A, Burger PC, Brem H. Intratumoral chemotherapy. Neurosurgery. 1995;37:1128–1145. [PubMed] [Google Scholar]
- 53.Pinilla JC, Ross DF, Martin T, Crump H. Study of the incidence of intravascular catheter infection and associated septicemia in critically ill patients. Crit Care Med. 1983;11:21–25. doi: 10.1097/00003246-198301000-00007. [DOI] [PubMed] [Google Scholar]
- 54.Kaur R, Samra T, Chaudhary L, Jain A. Intratumoral migration of central venous catheter in a patient with malignant bronchial carcinoid. J Anaesthesiol Clin Pharmacol. 2015;31:415. doi: 10.4103/0970-9185.161706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satchi R, Connors TA, Duncan R. PDEPT: polymer-directed enzyme prodrug therapy. I. HPMA copolymer-cathepsin B and PK1 as a model combination. Br J Cancer. 2001;85:1070–1076. doi: 10.1054/bjoc.2001.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Syrigos KN, Epenetos AA. Antibody directed enzyme prodrug therapy (ADEPT): a review of the experimental and clinical considerations. Anticancer Res. 1999;19:605–613. [PubMed] [Google Scholar]
- 57.Rossin R, van Duijnhoven SMJ, Läppchen T, van den Bosch SM, Robillard MS. Trans-cyclooctene tag with improved properties for tumor pretargeting with the diels-alder reaction. Mol Pharm. 2014;11:3090–3096. doi: 10.1021/mp500275a. [DOI] [PubMed] [Google Scholar]
- 58.Au JLS, Yeung BZ, Wientjes MG, Lu Z, Wientjes MG. Delivery of cancer therapeutics to extracellular and intracellular targets: Determinants, barriers, challenges and opportunities. Adv Drug Deliv Rev. 2016;97:280–301. doi: 10.1016/j.addr.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Durmaz R, Erken S, Arslantaş A, Atasoy MA, Bal C, Tel E. Management of glioblastoma multiforme: with special reference to recurrence. Clin Neurol Neurosurg. 1997;99:117–123. doi: 10.1016/s0303-8467(97)00014-0. [DOI] [PubMed] [Google Scholar]
- 60.Singer S, Antonescu CR, Riedel E, Brennan MF. Histologic subtype and margin of resection predict pattern of recurrence and survival for retroperitoneal liposarcoma. Ann Surg. 2003;238:358. doi: 10.1097/01.sla.0000086542.11899.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schnitt SJ, Abner A, Gelman R, Connolly JL, Recht A, Duda RB, Eberlein TJ, Mayzel K, Silver B, Harris JR. The relationship between microscopic margins of resection and the risk of local recurrence in patients with breast cancer treated with breast-conserving surgery and radiation therapy. Cancer. 1994;74:1746–1751. doi: 10.1002/1097-0142(19940915)74:6<1746::aid-cncr2820740617>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 62.Alwan L, Goff B, Liao J. Altered recurrence patterns after extended treatment with bevacizumab for ovarian, fallopian tube, and primary peritoneal cancer. Gynecol Oncol. 2013;130:e118–e119. doi: 10.1016/j.ygyno.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tandar A, Abraham G, Gurka J, Wendel M, Stolbach L. Recurrent peritoneal mesothelioma with long-delayed recurrence. J Clin Gastroenterol. 2001;33:247–250. doi: 10.1097/00004836-200109000-00018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.