

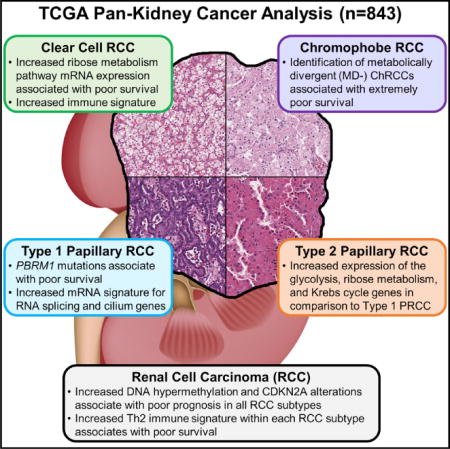
SUMMARY
Renal cell carcinoma (RCC) is not a single disease, but several histologically defined cancers with different genetic drivers, clinical courses, and therapeutic responses. The current study evaluated 843 RCC from the three major histologic subtypes, including 488 clear cell RCC, 274 papillary RCC, and 81 chromophobe RCC. Comprehensive genomic and phenotypic analysis of the RCC subtypes reveals distinctive features of each subtype that provide the foundation for the development of subtype-specific therapeutic and management strategies for patients affected with these cancers. Somatic alteration of BAP1, PBRM1, and PTEN and altered metabolic pathways correlated with subtype-specific decreased survival, while CDKN2A alteration, increased DNA hypermethylation, and increases in the immune-related Th2 gene expression signature correlated with decreased survival within all major histologic subtypes. CIMP-RCC demonstrated an increased immune signature, and a uniform and distinct metabolic expression pattern identified a subset of metabolically divergent (MD) ChRCC that associated with extremely poor survival.
Graphical abstract
In Brief Ricketts et al. find distinctive features of each RCC subtype, providing the foundation for development of subtypespecific therapeutic and management strategies. Somatic alteration of BAP1, PBRM1, and metabolic pathways correlates with subtype-specific decreased survival, while CDKN2A alteration, DNA hypermethylation, and Th2 immune signature correlate with decreased survival within all subtypes.
INTRODUCTION
Renal cell carcinoma (RCC) affects nearly 300,000 individuals worldwide annually and is responsible for more than 100,000 deaths each year. Our understanding of RCC has evolved over the past 40 years, from considering RCC as a single entity to our current understanding that RCC is made up of many different subtypes of renal cancer, each with different histology, distinctive genetic and molecular alterations, different clinical courses, and different responses to therapy (Linehan, 2012; Linehan et al., 2010; Moch et al., 2016). The canonical classification of RCC consists of three major histologic RCC subtypes (Hsieh et al., 2017; Linehan et al., 2006; Moch et al., 2016). Clear cell renal cell carcinoma (ccRCC) is the most common subtype (~75%); papillary renal cell carcinoma (PRCC) accounts for 15%–20% and is subdivided into types 1 and 2; and chromophobe renal cell carcinoma (ChRCC) represents ~5% of RCC.
The Cancer Genome Atlas (TCGA) Research Network has conducted a series of comprehensive molecular characterizations in distinctive histologic types of cancers including ccRCC, ChRCC, and PRCC (Cancer Genome Atlas Research Network, 2013; Cancer Genome Atlas Research Network et al., 2016; Davis et al., 2014). These studies revealed a remodeling of cellular metabolism in ccRCC involving downregulation of Krebs cycle genes, upregulation of pentose phosphate pathway genes, and decreased AMPK in higher-stage, high-grade, and low-survival disease. A distinct PRCC subtype was identified that was characterized by a CpG island methylator phenotype (CIMP-RCC) and associated with early-onset disease, poor survival, and germline or somatic mutation of the fumarate hydratase (FH) gene, and a subset of ChRCC with genomic rearrangements within the TERT promoter region was identified that correlated with highly elevated TERT expression and manifestation of kataegis, uncovering a distinct mechanism of TERT upregulation in cancer. A previous study by Chen et al. (2016) compared all available kidney tumor samples available within TCGA irrelevant of histologic type using cluster analysis of the multi-platform genetic and genomic data to show that the majority of the histologic subtypes could be reconstituted. In addition, this study identified samples that fell outside of the major subtypes and identified several mutation, methylation, and immune expression profiles that correlated with histologic subtypes within the complete TCGA kidney cohort.
The importance of histology cannot be understated in the study of RCC. To highlight the most meaningful somatic alterations in the entire cohort and within each major histologic subtype, we performed an integrated comparative genomic analysis of all available histologically confirmed TCGA samples of ccRCC, PRCC, and ChRCC to identify shared and subtype-specific molecular features that will provide the foundation for the development of disease-specific therapeutic approaches and prognostic biomarkers for RCC.
RESULTS
Evaluation of RCC Histologic Subtypes
In total, 894 samples of kidney cancer were initially submitted to TCGA and were available for analysis, including 537 ccRCC, 291 PRCC, and 66 ChRCC. The initial TCGA analyses of each RCC subtype had excluded several samples due to inconsistent/incorrect histologic classification or therapy prior to sample collection. This included the removal of a small number of samples, such as transitional cell carcinomas, that are kidney cancers that are not classified as RCCs. Additional samples not utilized in previous studies were also re-evaluated by histologic review and removed if considered inappropriate and 15 samples originally submitted as ccRCC were reclassified as ChRCC. This resulted in a final cohort of 843 TCGA-RCC consisting of 488 ccRCC, 274 PRCC, and 81 ChRCC. The 274 PRCC were further divided into four subgroups consisting of 160 type 1 PRCC, 70 type 2 PRCC, 34 unclassified PRCC, and 10 CpG island methylator phenotype-associated (CIMP)-RCC (Table S1).
Comparison of Major RCC Histologic Subtypes
Initial comparison of these RCC was performed using chromosomal copy number profiles, mRNA, miRNA, and lncRNA expression profiles and visualized in a heatmap with the RCCs ordered by histologic subtype, then stage, then vital status (Figure 1A). Clear cell RCC demonstrated significant loss of chromosome 3p and gain of 5q, type 1 PRCC demonstrated gains of chromosomes 7 and 17, and ChRCC demonstrated a pattern of chromosomal losses that included 1, 2, 6, 10, 13, and 17 (Figure 1B). These data confirmed previous observations concerning the copy number patterns within the different RCC histologic subtypes, and somatically gained alterations in chromosomal copy number patterns provide the clearest distinction between subtypes. While specific patterns of copy number alteration were not observed in the CIMP-RCC or the type 2 PRCC, both demonstrated an increased loss of chromosome 22 that encodes NF2 from the HIPPO pathway and SMARCB1, a fundamental component of the SWI/SNF complex, and the CIMP-RCC had loss of chromosome 13q at a similar rate to ChRCC (60% versus 61.3%) that encodes RB1 and BRCA2 (Figure 1B). Analysis of RNA expression across the combined cohort demonstrated distinct mRNA, miRNA, and lncRNA clusters that associated with each histologic RCC type. Two mRNA, three miRNA, and five lncRNA clusters were enriched in ccRCC, while two mRNA, two miRNA, and two lncRNA clusters represented the majority of the PRCC (Figures S1A–S1C). The ChRCC samples demonstrated a distinct uniformity by being present in a single cluster for each RNA type, while the CIMP-RCC had a distinct mRNA cluster and shared a lncRNA cluster with the ChRCC.
Figure 1. Comparison of RCC Histologic Subtypes.
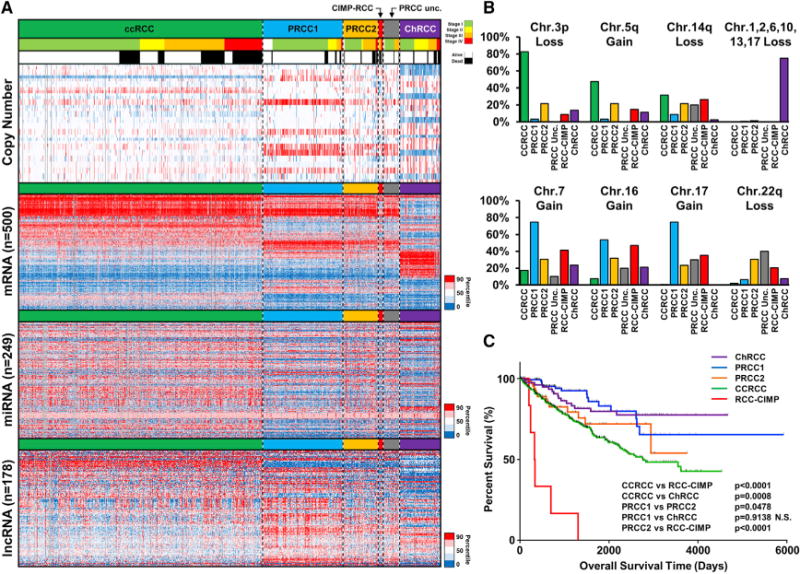
(A) Heatmap representation of chromosomal copy number and RNA expression profiles between the different histologic RCC subtypes. Chromosomal copy number data are ordered by chromosomal arm in descending order (red, gain; blue, loss). The relative RNA expression was assessed for the most variable probes within the complete RCC cohort for either mRNA (n = 500), miRNA (n = 249), or lncRNA (n = 178) (red, increased; blue, decreased). RCC samples were arrayed left to right based on histologic subtype (ccRCC, green; type 1 PRCC, light blue; type 2 PRCC, orange; unclassified [Unc.] PRCC, gray; CIMP-RCC, red; ChRCC, purple), then tumor stage (stage I, light green; stage II, yellow; stage III, orange; stage IV, red), and then vital status (alive, white; deceased, black). (B) Percentage of chromosomal copy number alterations between the different histologic RCC subtypes. (C) Differences in patient overall survival between the different histologic RCC subtypes (log-rank p value).
Survival Differences across the Major RCC Histologic Subtypes
The variation between the RCC histologic subtypes extended to survival outcomes (Figure 1C). Previously, CIMP-RCC was found to have the poorest PRCC survival but now demonstrated the worst survival of all RCC subtypes, including ccRCC (p < 0.0001). Clear cell RCC demonstrated the next poorest survival when compared to all other RCC subtypes, while type 1 PRCC and ChRCC associated with the best survival that was statistically indistinguishable (p = 0.9138). These histologic-specific differences in survival and the uneven representation of each histologic subtype within the cohort produces a potential confounding factor for survival associations evaluated across the entire cohort. With clear distinctions between the histologic subtypes established, survival associations within histologic subtypes are likely to be more relevant than those across the entire cohort.
Gene and Pathway Alteration Associates with Survival in Specific RCC Subtypes
Previous analyses of each histologic RCC subtype had identified a combined total of 16 significantly mutated genes (SMGs) including 9 associated with ccRCC, 11 associated with PRCC, and 2 associated with ChRCC (Figure S2A; Cancer Genome Atlas Research Network, 2013; Cancer Genome Atlas Research Network et al., 2016; Davis et al., 2014). Analysis across RCC types revealed that TP53 and PTEN were the only SMGs shared by ccRCC, PRCC, and ChRCC (2.6% and 4.5%, 1.5% and 3.4%, and 31.1% and 8.1%, respectively). Across the entire cohort, neither TP53 nor PTEN correlated with poor survival, but histologic-specific analysis demonstrated that TP53 mutation correlated with decreased survival in ccRCC (p = 0.0002) and PRCC (p = 0.0049), while PTEN mutation correlated with decreased survival in ChRCC (p = 0.0138) (Figures 2A and S2B). Clear cell RCC and PRCC, but not ChRCC, shared three chromatin remodeling SMGs: PBRM1 (38.0% and 4.5%, respectively), SETD2 (13.2% and 6.4%, respectively), and BAP1 (11.0% and 5.6%, respectively). While BAP1 mutation correlated with decreased survival across the entire cohort (p = 0.0002) and within the ccRCC group (p = 0.0035), BAP1 mutation did not correlate with survival in PRCC or ChRCC. Similarly, PBRM1 mutation, which has been shown to not correlate with survival in ccRCC, was found to correlate with decreased survival in PRCC (p = 0.0008) that was specific to type 1 PRCC (p < 0.0001) (Figures 2A and S2B). CDKN2A mutation, hypermethylation, or deletion was found in 15.8% of tumors, with alterations in each RCC subtype accounting for 16.2% of ccRCC, 5.0% of type 1 PRCC, 18.6% of type 2 PRCC, 100% of CIMP-PRCC, and 19.8% of ChRCC (Figure 2B). Loss of the region of chromosome 9p encoding CDKN2A was the most frequent event across the cohort (11.7%), followed by promoter hypermethylation (4.2%) and mutation (0.7%) (Table S1). CDKN2A alteration provided the sole example of a change that correlated with decreased survival across the entire cohort (p < 0.0001) and in each major histologic subtype, ccRCC (p < 0.0001), type 1 PRCC (p = 0.0067), type 2 PRCC (p = 0.0006), and ChRCC (p = 0.0018) (Figure 2C).
Figure 2. Gene and Pathway Alteration Associates with Survival Predictions in Specific RCC Subtypes.
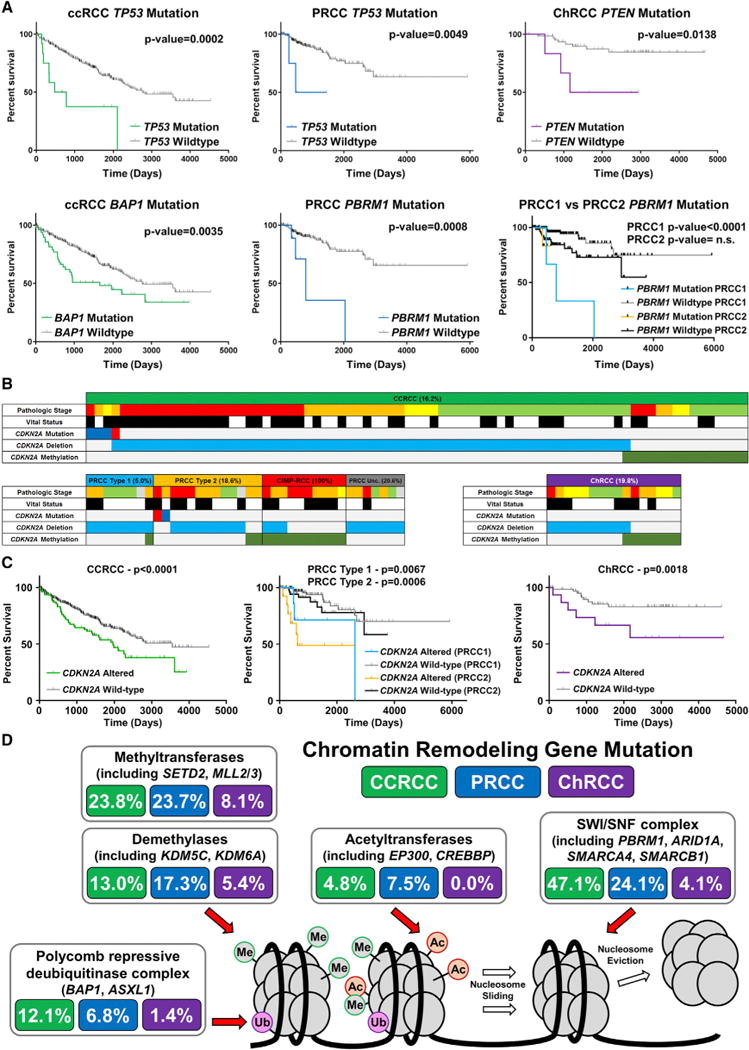
(A) Differences in patient overall survival within histologic RCC subtypes (ccRCC, green; PRCC, blue; ChRCC, purple) dependent upon gene mutation (log-rank p value). (B) Oncoprints for CDKN2A gene deletions, hypermethylation, and mutations for the histologic RCC subtypes (ccRCC, green; type 1 PRCC, light blue; type 2 PRCC, orange; Unc. PRCC, gray; CIMP-RCC, red; ChRCC, purple). Mutations were segregated into nonsense (red) and missense (blue). (C) Differences in patient overall survival within the histologic RCC subtypes (ccRCC, green; type 1 PRCC, light blue; type 2 PRCC, orange; ChRCC, purple) dependent upon CDKN2A alteration (log-rank p value). (D) Chromatin remodeling pathway mutation frequency within histologic RCC subtypes (ccRCC, green; PRCC, blue; ChRCC, purple). Abbreviations: Me, histone methylation; Ac, histone acetylation; Ub, histone ubiquitination.
Eight SMGs were frequently mutated (≥ 2.0%) in more than one RCC subtype. Mutation of at least 1 of the 16 SMGs was found in 81% of ccRCC, 39.1% of PRCC, and 43.2% of ChRCC (Figure S2A). While the overall mutation rate for ChRCC was found to be significantly less than either ccRCC or PRCC (p = 0.0254 and p < 0.0001, respectively), the PRCC mutation rate was higher than ccRCC (p < 0.0001) (Figure S2C). Within PRCC, the most aggressive subtype, CIMP-RCC, was found to have the lowest overall rate of mutation. Pathogenic SMG mutations were not detected in several tumors, particularly PRCC and ChRCC. Several SMGs were members of pathways that contained genes mutated at lower frequencies. In the VHL/HIF pathway, TCEB1 and CUL2 mutations in ccRCC were mutually exclusive with VHL mutation (Figure S2D). HIPPO and NRF2/ARE pathway mutations were present in both PRCC (9.0% and 7.9%, respectively) and ccRCC (3.9% and 3.2%, respectively) (Figure S2D). While chromatin remodeling pathway gene mutations were notably frequent in both ccRCC (69.3%) and PRCC (53.0%), they were less common in ChRCC (14.9%) (Figure S2D and Table S2). Mutations of SWI/SNF complex genes, including PBRM1, ARID1A, and SMARCA4, were the most common chromatin remodeling complex alterations within ccRCC (47.1%), followed by mutation of the histone methyltransferases including SETD2 and MLL3 (23.8%), the histone demethylases including KDM5C (13.0%), the BAP1/ASXL1 histone de-ubiquitinase complex (12.1%), and the histone acetyltransferases (4.8%), compared with frequencies of 24.1%, 23.7%, 17.3%, 6.8%, and 7.5%, respectively, in PRCCs (Figure 2D). Chromatin remodeling gene mutations were more frequent in type 2 PRCC (55.3%) than in type 1 PRCC (40.6%). While mutations of the PI3K/AKT pathway were frequent both across (14.6%) as well as within each RCC subtype (16.2% of ccRCC, 9.8% of PRCC, and 18.9% of ChRCC), they correlated with decreased survival only in ChRCC (p = 0.0018) (Figures S2D and S2E and Table S2).
Mitochondrial (mt) DNA mutation analysis, which was previously performed only in ChRCC (Davis et al., 2014), was conducted in a representative number of tumors from all RCC subtypes. Nonsense or missense mutations in mitochondria-encoded genes with high heteroplasmy (>75%) as well as frameshift mutations with >50% heteroplasmy were considered significant. Mitochondrial DNA mutations were found in 13% of 62 ccRCC, 33% of 99 PRCC (with similar frequencies for type 1 and type 2), and 20% of 65 ChRCC. High-heteroplasmy truncating (nonsense or frameshift) mutations were enriched in ChRCC (14%) compared to PRCC (6%) or ccRCC (2%) (Figure S2F) and mtDNA copy number was increased in ccRCC, PRCC, and ChRCC that carried mtDNA mutations (p = 0.0036, p = 0.0036, and p = 0.0029, respectively) (Figure S2F).
Hypermethylation Correlates with Decreased Survival
Previous analyses of methylation by Chen et al. (2016) had demonstrated that a subset of the DNA methylation probes within the RCC samples highlighted the differences in cell of origin for the major RCC histologic subtypes. This subset of probes was subsequently used to evaluate hypermethylation patterns within the samples but was potentially confounded by the difference in tumor origin. While hypermethylated ccRCC and PRCC samples were identified, no hypermethylated ChRCC samples were observed. An analysis limited to probes that are unmethylated in all normal tissues identified in 1,532 variably hypermethylated markers that identified a cluster of 240 RCCs with increased DNA hypermethylation (methylation cluster 1) that associated with significantly poorer survival (p < 0.0001) (Figure 3A and Table S1). This cluster consisted of the 10 CIMP-RCC, 182 ccRCC (37.3%), 23 type 2 PRCC (32.9%), 16 ChRCC (19.8%), and a small number of type 1 and unclassified PRCC. The remaining two clusters, one containing type 1 and type 2 PRCC (methylation cluster 2) and the other containing ccRCC and ChRCC (methylation cluster 3), had similar survival. In contrast to the distinct CIMP-RCC tumors that had notably high levels of DNA hypermethylation, the remainder of methylation cluster 1 had a less pronounced increase in hypermethylation across the genome. Histologic subtype-specific analysis confirmed decreased survival with the increased hypermethylation pattern in every major RCC histologic subtype (all p < 0.0001) (Figure 3B). Within the PRCC tumors, this correlation remained significant after excluding the CIMP-RCC from the PRCC tumors (p < 0.0001) and when type 1 PRCC (p = 0.0328) and type 2 PRCC (p = 0.0314) were independently evaluated (Figures 3B and S3A). Increased hypermethylation was associated with higher-stage disease in ccRCC, PRCC (with or without CIMP), and ChRCC (all p < 0.0001) and was associated with SETD2 mutation in ccRCC (p < 0.0001), either PBRM1 mutation or SETD2 mutation in type 2 PRCC (p = 0.0053, p = 0.0270, respectively), and TP53 mutation in ChRCC (p = 0.0119) (Figure S3B). Genes represented by the 1,532 probes that characterized the hypermethylated cluster were enriched for genes in the WNT pathway. Previous studies have identified hypermethylation of the WNT pathway regulatory genes, SFRP1 and DKK1, in ccRCC (Ricketts et al., 2014). Increased methylation of probes for these two genes (DKK1, cg07684796; SFRP1, cg15839448) was observed in the methylated cluster 1 samples (Figure S3C), and hypermethylation of either of these genes correlated with poorer survival in ccRCC, PRCC, and ChRCC (p = 0.0015, p < 0.0001, and p = 0.0021, respectively) and in PRCC excluding the CIMP-RCC tumors (p = 0.0035) (Figures 3C and S3D).
Figure 3. Hypermethylation Patterns Associate with Survival Predictions.
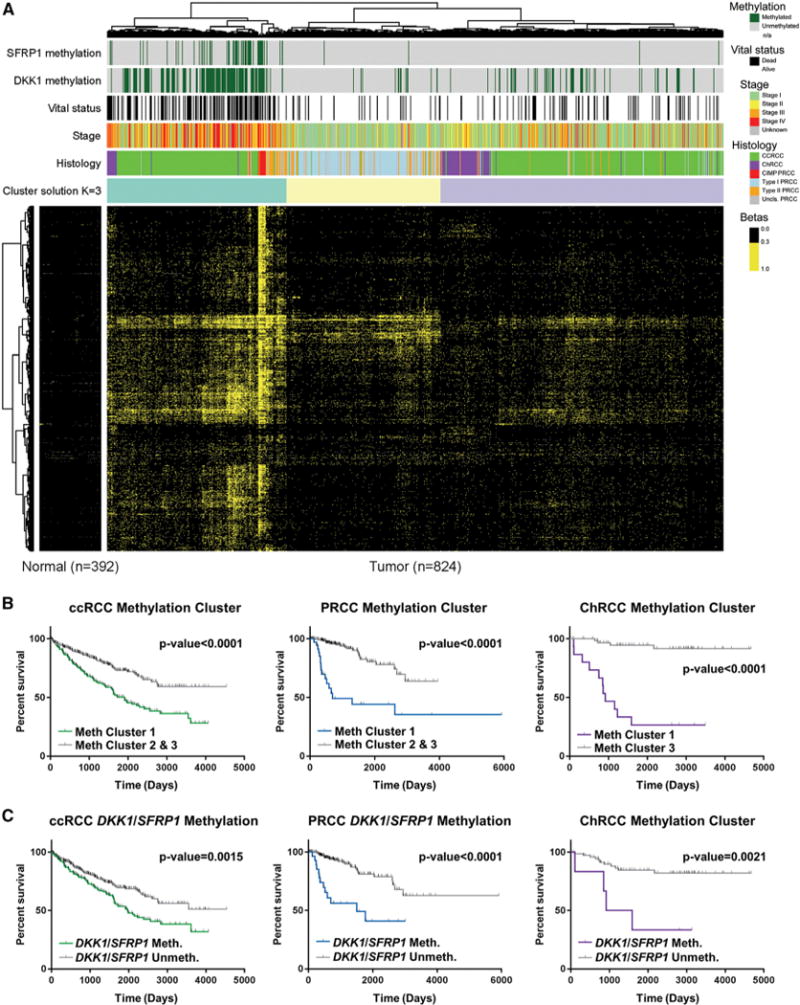
(A) Heatmap representation of the clustering of 1,532 highly variable DNA methylation probes that were unmethylated in the normal tissues. A methylation b-value R 0.3 was considered hypermethylated. Tumors were annotated for histologic RCC subtype (ccRCC, green; type 1 PRCC, light blue; type 2 PRCC, orange; Unc. PRCC, gray; CIMP-RCC, red; ChRCC, purple), tumor stage (stage I, light green; stage II, yellow; stage III, orange; stage IV, red), vital status (alive, white; deceased, black), and DKK1 (cg07684796) and SFRP1 (cg15839448) hypermethylation (hypermethylated, dark green). (B) Differences in patient overall survival within the histologic RCC subtypes (ccRCC, green; PRCC, blue; ChRCC, purple) dependent upon methylation cluster (log-rank p value). (C) Differences in patient overall survival within ccRCC and ChRCC tumors (ccRCC, green; ChRCC, purple) dependent upon hypermethylation of either SFRP1 or DKK1 (log-rank p value).
Specific mRNA Signatures Associate with RCC Histologic Subtypes
A weighted gene co-expression network analysis (WGCNA), performed to identify sets (modules) of highly correlated genes and to assess their relationships to clinical variables and biological functions, revealed several gene modules that differentiated the RCCs by histology, stage, or survival status (Figure 4). Clear cell RCC showed the expected increase in expression of the vasculature development signature, due to activation of the VHL/HIF pathway, and the previously observed increase in immune response signature (p = 4 × 10−86) in comparison to PRCC and ChRCC (Figure 4B). The RNA metabolic process and the mitotic cell cycle signature was specifically increased in ccRCC (p = 5 × 10−26 and p = 5 × 10−25, respectively), while an increased amino acid metabolic process signature (p = 4 × 10−35) and retention of cilium signature (p = 3 × 10−140) was unique to PRCC (Figure 4B). In ChRCC, an increased ion transmembrane transport signature was observed (Figure 4). Subtype analysis of PRCC revealed an increased RNA splicing signature in type 1 PRCC (p = 2 × 10−12) compared to type 2 PRCC, while the cilium signature was significantly higher in the type 1 PRCC (p = 8 × 10−101) than in the type 2 PRCC (p = 4 × 10−7).
Figure 4. RCC Histologic Subtypes Associate with Specific mRNA Signatures.
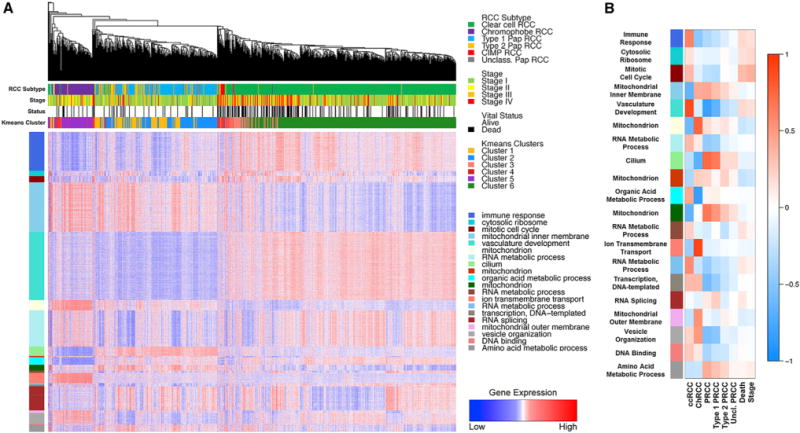
(A) Heatmap representation of the comparison of mRNA expression signatures for major cellular processes between the different histologic RCC subtypes (ccRCC, green; type 1 PRCC, light blue; type 2 PRCC, orange; Unc. PRCC, gray; CIMP-RCC, red; ChRCC, purple). Tumor stage (stage I, light green; stage II, yellow; stage III, orange; stage IV, red) and vital status (alive, white; deceased, black) are indicated above the heatmap. (B) Heatmap representation showing the relationship between gene expression modules and clinical features. Red heatmap shading indicates a positive correlation between a gene module and a clinical feature and blue heatmap shading represents a negative correlation.
Metabolic Gene Expression Associates with Survival
Evaluation of tumor metabolism was performed by comparing the expression profiles for 12 major metabolic processes across all samples (Figure 5A and Table S3). Expression of the Krebs cycle and the electron transport chain (ETC) genes provided a clear distinction between the major histologic subtypes, with low expression in ccRCC and CIMP-RCC, high expression in ChRCC, and intermediate expression in type 1 and type 2 PRCC (Figure 5B). This correlated with increased expression of the pyruvate dehydrogenase complex (PDC) activation genes in ChRCC, that would help fuel the Krebs cycle and oxidative phosphorylation, and the increased expression of PDC suppression genes in ccRCC, which would result in glycolysis-dependent energy production (Figures 5B and S4A). Subtype analysis revealed that glycolytic gene expression was consistently higher in ccRCC and type 2 PRCC, while expression of the Krebs cycle genes was significantly higher in type 2 PRCC compared to type 1 PRCC (p < 0.0001) (Figure S4A). Although expression of PDC activation genes was low in all ccRCC, stage III-IV ccRCC demonstrated significantly lower expression than stage I-II ccRCC (p = 0.0005) and lower PDC activation gene expression in ccRCC was associated with decreased survival (p < 0.0001) (Figures S4A and S4B). Expression of 5′ AMP-activated protein kinase (AMPK), which negatively regulates fatty acid synthesis and positively regulates mitochondria production, was increased in ChRCC and decreased in the CIMP-RCC (Figure 5B). As previously observed in the TCGA ccRCC analysis, AMPK expression was significantly lower in stage III-IV ccRCC compared to stage I-II ccRCC (p = 0.0005), and lower expression correlated with poorer survival (p = 0.0005) (Figures S4A and S4B). Ribose sugar metabolism gene expression was increased in type 2 PRCC compared to type 1 PRCC (p < 0.0001) and greatly increased in CIMP-RCC in comparison to all other RCC subtypes (p < 0.0001) (Figure 5C). The increased ribose sugar metabolism expression previously associated with higher stage and poorer survival prognosis in ccRCC was confirmed in the current study (p = 0.0069), and increased ribose sugar metabolism expression was found to also be associated with decreased survival in PRCC (p = 0.0031) (Figures 5D and S4B).
Figure 5. Metabolic Analysis of RCC Histologic Subtypes.
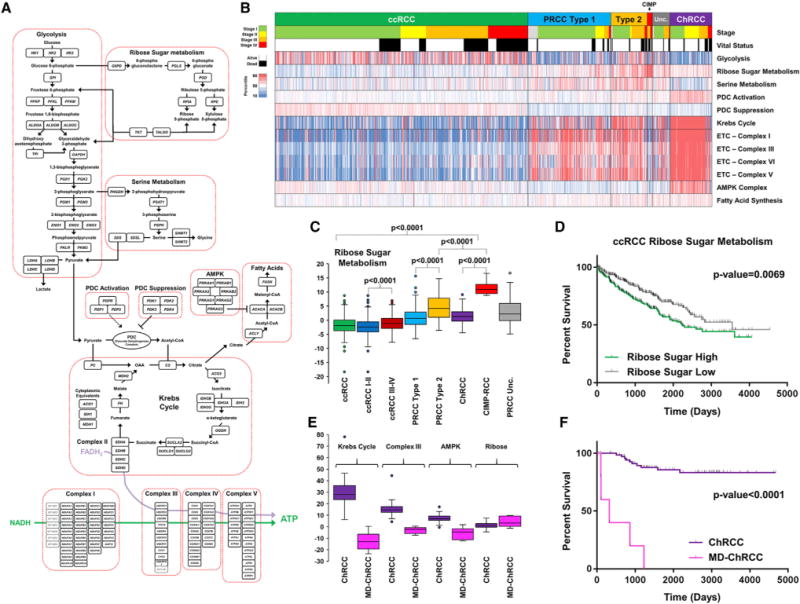
(A) Schematic of metabolic pathway genes selected for metabolic analysis. (B) Heatmap representation of the comparison of mRNA expression signatures for the selected metabolic processes between the different histologic RCC subtypes (ccRCC, green; type 1 PRCC, light blue; type 2 PRCC, orange; Unc. PRCC, gray; CIMP-RCC, red; ChRCC, purple). Tumor stage (stage I, light green; stage II, yellow; stage III, orange; stage IV, red) and vital status (alive, white; deceased, black) are indicated above the heatmap. (C) Comparative expression of the ribose sugar metabolism signature between the different histologic RCC (ccRCC, green; ccRCC stage I/II, dark blue; ccRCC stage III/IV, dark red; type 1 PRCC, light blue; type 2 PRCC, orange; Unc. PRCC, gray; CIMP-RCC, red; ChRCC, purple). (D) Differences in patient overall survival within ccRCC dependent upon expression of the ribose sugar metabolism signature (log-rank p value). (E) Comparative expression of the Krebs cycle, ETC Complex III, AMPK, and ribose sugar metabolism gene signatures between ChRCC and metabolically divergent (MD) ChRCC (ChRCC, purple; MD-ChRCC, pink). (F) Differences in patient overall survival between ChRCC and MD-ChRCC (log-rank p value).
Six ChRCC were identified that presented as distinct metabolic outliers within that histologic subtype (Figure S5A). Compared to the other ChRCC, these samples had low expression of the Krebs cycle and electron transport chain genes, lower expression of the AMPK pathway genes, and increased expression of the genes in the ribose synthesis pathway, and all these features were associated with poorer prognosis in other RCC histologic subtypes (Figure 5E). These metabolically divergent (MD) ChRCC were high stage (stage III or IV), demonstrated the hypermethylation pattern described above, lacked the chromosomal copy number losses normally associated with ChRCC, and were associated with much poorer survival in comparison to other ChRCC (p < 0.0001) (Figures 5F and S5A). Four of the six MD-ChRCC were found to have sarcomatoid de-differentiation (Figure S5B). Several of these MD-ChRCCs were initially misidentified as ccRCC and then re-assigned after a pathology review by urologic pathology experts, reflecting their unusual pathology.
Immune Signature Analysis
An increased immune cell infiltrate gene expression signature in ccRCC in comparison to PRCC and ChRCC has been elucidated by several studies, including importance of single gene markers such as PDCD1 (PD1) and CD247 (PDL1) (Chen et al., 2016; Geissler et al., 2015). Analysis using a refined immune cell gene-specific signatures (Table S4) confirmed that, with the exception of the Th17, IL-8, and CD56bright NK cell gene signatures, there was nearly universal upregulation of these immune signatures in ccRCC compared to the PRCC or ChRCC (Figures 6A and S6A). The T helper 17 cell (Th17) gene signature had increased expression in ChRCC, while the IL-8 and CD56bright NK cell gene signatures had increased expression in PRCC. Separation of the PRCC tumors highlighted distinct differences in the CIMP-RCC compared to the remaining PRCC, including increased expression of the Th2, activated dendritic cell (aDC), plasmacytoid dendritic cell (pDC), and Mast cell gene signatures, that produced a profile more similar to ccRCC (Figures 6B and S6B). T cell receptor (TCR) profiling used to identify TCR clonotype expression within the cohort demonstrated patterns of subtype-specific TCR clonotype expression suggesting variation in T cell response between ccRCC, PRCC, and ChRCC tumors (Figure S6C). In accordance with previous findings, gene signatures correlated with reduced survival, including signatures that represented T cells, B cells, macrophages, dendritic cells, and NK cells (Figure S6D). The T helper 2 cell (Th2) gene signature was increased in most ccRCC, all CIMP-RCC, and in outliers of the ChRCC, with six of the top seven Th2 gene signature scores within the ChRCC tumors representing the aggressive MD-ChRCC tumors (Figure 6B). Notably, an increased Th2 gene signature represented the only biomarker that correlated with poor survival when evaluated within each major histologic subtype, ccRCC (p = 0.0001), PRCC (p = 0.0002), and ChRCC (p = 0.0284) (Figure 6C). Subtype separation of the PRCC demonstrated that this correlation was present only in PRCC type 2 (p = 0.0089) (Figure 6C). Expression of the Th17 gene signature was associated with increased survival in ccRCC (p = 0.0021), with additional positive correlation in ChRCC (p = 0.0362) (Figure S6E).
Figure 6. Immune Signature Analysis.
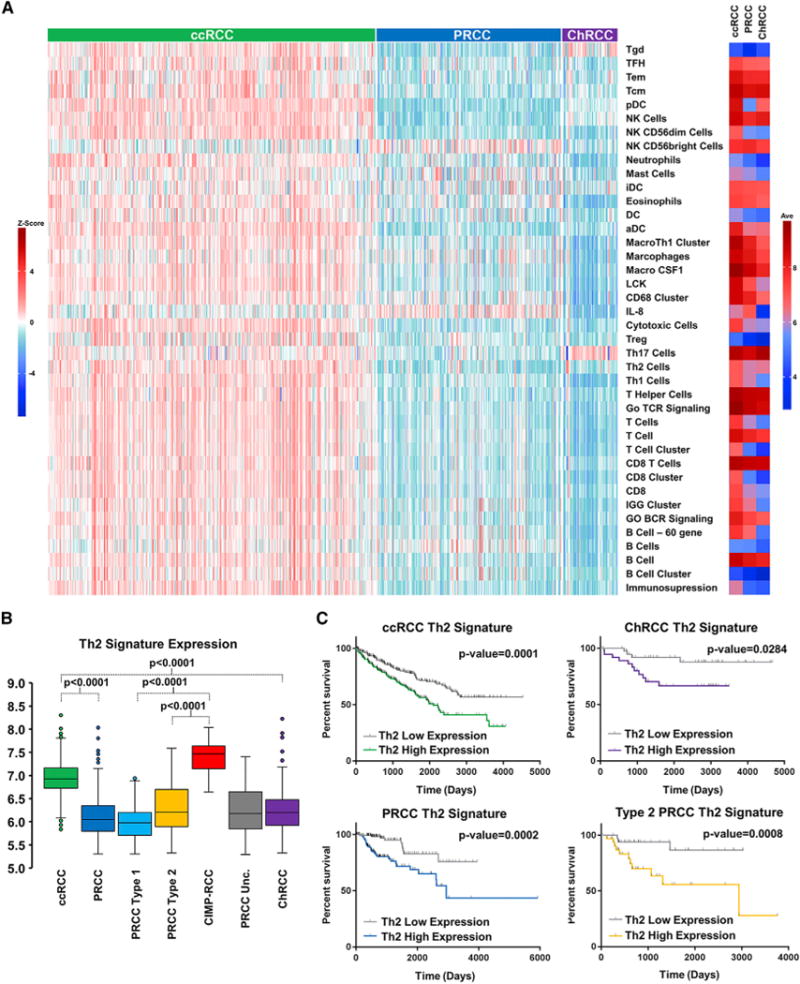
(A) Supervised clustering of immune gene signature (IGS) expression by individual sample (left) or mean IGS expression (right) for the different histologic RCC subtypes (ccRCC, green; PRCC, blue; ChRCC, purple). (B) Comparative expression of the Th2 gene signature between the histologic RCC subtypes (ccRCC, green; PRCC, blue; type 1 PRCC, light blue; type 2 PRCC, orange; CIMP-RCC, red; unclassified PRCC, gray; ChRCC, purple) (t test). (C) Comparative differences in patient overall survival within the histologic RCC subtypes (ccRCC, green; PRCC, blue; type 2 PRCC, orange; ChRCC, purple) dependent upon the Th2 gene signature (log-rank p value).
DISCUSSION
The importance of identifying and differentiating the subtypes and even rare variants of renal cell carcinoma (RCC) is critical for management and treatment of patients affected with this disease. Although histologic subtyping divides tumors into distinct RCC groups, it is limited in its ability to provide in-depth analysis of mechanisms that produce these differences. In the present study, comprehensive genetic and genomic analysis demonstrated that different histologically defined RCC subtypes are characterized by distinctive mutations, chromosomal copy number alterations, and expression patterns of mRNA, miRNA, and lncRNA, and that the combination of histology plus genomics provides unique insight into patient-centered management. These combined differentiating features, obtained via a tumor or liquid biopsy, provide invaluable information and prognostic biomarkers to guide clinical and surgical management.
While this study characterizes the differences between the major RCC histologic subtypes, shared features within the RCC subtypes may also provide more universal prognostic markers and targets for therapy. The loss of CDKN2A, which encodes p16, by either gene deletion, promoter hypermethylation, or mutation, found in 16% of RCC, correlated with poor survival in ccRCC, PRCC, and ChRCC. Loss of CDKN2A is known to correlate with poor outcome in ccRCC, PRCC, and other cancer types, but this demonstrates that it is a universal feature of RCC and is potentially targetable with CDK4/6 inhibitors that target the downstream effects of p16 loss (Hamilton and Infante, 2016). Increased promoter hypermethylation also was found to be associated with decreased survival in ccRCC, PRCC, and ChRCC. Previous studies have shown increased levels of DNA hypermethylation correlating with poorer outcome that was limited to ccRCC and PRCC without identifying potentially impacted pathways (Cancer Genome Atlas Research Network, 2013; Cancer Genome Atlas Research Network et al., 2016; Chen et al., 2016). This study highlighted hypermethylation of WNT pathway regulatory genes and demonstrated that analysis of hypermethylation in two specific WNT regulatory genes, SFRP1 and DKK1, recapitulated the correlation with decreased survival in ccRCC, PRCC, and ChRCC. Increased DNA methylation was associated with SETD2 mutation, which is known to alter DNA methylation patterns (Tiedemann et al., 2016), in ccRCC and PRCC, and increased DNA methylation was similarly associated with PBRM1 mutation in PRCC. Hypermethylation of SFRP1 and DKK1 could provide a prognostic biomarker for RCC and has been previously proposed in ccRCC (Hirata et al., 2011; Ricketts et al., 2014; Urakami et al., 2006). This suggests that treatment with de-methylating agents could be beneficial in patients with increased levels of promoter hypermethylation.
This study also demonstrated features that were shared by some RCC subtypes, but not all, and underlines the importance of evaluating these alterations within each RCC subtype as well as across all subtypes. Previous studies using TCGA data and other cohorts have shown that BAP1 mutation, but not PBRM1 mutation, correlates with poor survival in ccRCC and these correlations were confirmed in a mixed cohort of ccRCC and PRCC TCGA tumors (Chen et al., 2016; Hakimi et al., 2013; Kapur et al., 2013). By analysis of the histologic subtype of RCC, we confirmed these correlations in ccRCC and showed that while BAP1 mutations did not correlate with survival in PRCC, PBRM1 mutations did associate with poor survival in type 1 PRCC.
Assessment of the RCC metabolic states within RCC revealed significant metabolic alterations. High ribose metabolism gene expression was present in both ccRCC and CIMP-RCC, with CIMP-RCC showing the greatest expression, likely due to the increased production of NADPH counteracting the cellular stress induced by the loss of fumarate hydratase in these tumors (Ooi et al., 2011; Patra and Hay, 2014; Sourbier et al., 2014). Type 2 PRCC had increased expression of the glycolysis, ribose metabolism, and Krebs cycle genes in comparison to type 1 PRCC, suggesting a more metabolically active tumor, consistent with its more aggressive nature. Increased expression of the ribose metabolism genes correlated with poor survival in both ccRCC and PRCC. These findings suggest that targeting the ribose metabolism pathway could be a potential therapeutic approach in ccRCC, type 2 PRCC, and CIMP-RCC.
The immune expression signature is an increasingly important feature of ccRCC, given the recent introduction of checkpoint inhibitor therapy (Lee and Motzer, 2016; Motzer et al., 2015), and patterns of immune infiltration in RCC have been observed in several studies (Chen et al., 2016; Geissler et al., 2015). The role of this feature in determining the therapeutic responsiveness of ccRCC will be important in future therapeutic planning. A recent study using TCGA RCC data demonstrated that differences in expression in specific checkpoint-related genes, such as PDCD1 (PD1) and CD247 (PDL1), correlated with patient survival within ccRCC cases (Chen et al., 2016). While we observed the same general pattern as previously seen with PRCC overall demonstrating little expression of the immune signature associated with ccRCC, we found CIMP-RCC to have an increased immune signature expression for select immune gene signatures, including the Th2 gene signature, like that seen in ccRCC. This suggests this most aggressive type of RCC, CIMP-RCC, may benefit from checkpoint inhibitor therapy in a similar manner to ccRCC. Although the Th2 gene signature was considerably higher in ccRCC and CIMP-RCC tumors compared to other tumor subtypes, the relative levels of Th2 gene signature within each major RCC histologic subtype correlated with poor patient survival, as had been previously observed in ccRCC (Şenbabaoǧlu et al., 2016). This suggests that once expression ranges are defined for each subtype, this Th2 gene signature could provide a useful prognostic marker for all RCC subtypes.
While the current study confirmed the previous finding of CIMP-RCC as a specific PRCC subtype, in this analysis we identified a subset of metabolically divergent (MD) ChRCC that also demonstrated a uniform and distinct metabolic expression pattern associated with extremely poor survival. The MD-ChRCC had decreased Krebs cycle, ETC, and AMPK gene expression and increased ribose metabolism gene expression similar to higher-stage ccRCCs. All the MD-ChRCC were high stage and generally lacked the classic ChRCC-associated pattern of chromosomal loss, and most demonstrated sarcomatoid differentiation. A recent study has also shown a correlation between the absence of the classical ChRCC chromosome loss and aggressive, high-grade, metastatic ChRCC (Casuscelli et al., 2017). Many of these MD-ChRCC features are represented in a recently characterized sarcomatoid ChRCC-derived cell line that could provide a model for further investigation of these tumors (Yang et al., 2017). The combination of histopathology and expression analysis may provide a definitive classification for ChRCC and enable the identification of aggressive variants that may require alternative management and therapy, including the potential for adjuvant therapy.
Understanding the molecular and genetic features that characterize the RCC subtypes will provide the foundation for the development of improved methods for both clinical and surgical management and therapies to treat this disease. Besides identifying discrete genomic characteristics that are critical for the understanding of individual RCC subtypes, we have identified unifying features, such as the effect of the Th2 immune gene signature on survival, which cross disease subtypes and which will help provide the foundation for the development of effective forms of therapy for patients with advanced disease.
STAR★METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. W. Marston Linehan (WML@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Biospecimen Acquisition
All biospecimens were acquired by the Cancer Genome Atlas (TCGA) Resource Network. Surgically resected tumor specimens were collected from patients diagnosed with renal cell carcinoma (RCC) that had preferably not received any prior treatment for their disease, such as chemotherapy or radiotherapy. Individual institutional review boards at each tissue source site reviewed the protocols and consent documentation and approved the submission of cases to TCGA. All tumors were staged per the American Joint Committee on Cancer (AJCC) and each primary tumor specimen had a matched normal tissue specimen. The tissue source sites for the Cancer Genome Atlas Research Network are listed in the Cancer Genome Atlas Research Network author list for this project.
The initial 894 samples of kidney cancer that were submitted to TCGA were re-evaluated by a panel of expert pathologists that excluded several samples due to inconsistent or incorrect histologic classification or therapy prior to sample collection. This accounts for the variation in samples compared to the previous Chen et al. study (Chen et al., 2016). The approved 843 tumors were subdivided by histologic subtype into 6 groups consisting of 488 clear cell (cc)RCC, 160 Type 1 papillary (P)RCC, 70 Type 2 PRCC, 34 unclassified PRCC, 10 CpG island methylator phenotype-associated (CIMP-)RCC, and 81 chromophobe (Ch)RCC based on the original pathology reports or re-evaluation by a panel of expert urologic pathologists. Six hundred and ninety-three of the tumors had been analyzed in the three individual TCGA marker papers. The clinical and genetic characteristics of these patients are described in Table S1 in the Supplementary Appendix.
METHOD DETAILS
Somatic Exome Mutation Analysis
Somatic exome sequencing data was available and downloaded for 804 of the 843 pan-kidney tumors representing 463 ccRCC, 266 PRCC, 74 ChRCC. The tumors with sequencing data are designated within Table S1 and all data is accessible via the NCI genome data commons (https://gdc.cancer.gov/).
A combined MAF (Mutation Annotation Format) file for all samples was produced by extracting the relevant sample data from the TCGA unified ensemble “MC3” call set and supplementing this with data from the original three TCGA KIRC, KICH, and KIRP publication for samples not present in the TCGA MC3 dataset. The TCGA unified ensemble “Multi-Center Mutation Calling in Multiple Cancers” (“MC3′”) call set is the public, open-access, dataset of somatic mutation calls (SNVs and indels) produced as part of the capstone project using all available of cases within TCGA using six different algorithms (MuTect, MuSE, Pindel, Somatic Sniper, VarScan2 and Radia) from four centers (Cibulskis et al., 2013; Fan et al., 2016; Koboldt et al., 2012; Larson et al., 2012; Radenbaugh et al., 2014; Ye et al., 2009).
The significantly mutated genes (SMGs) that had been previously identified by the MutSigCV algorithm in the previous TCGA KIRC, KICH, and KIRP publications were used as the reference SMGs when evaluating the entire pan-kidney dataset. Pathway analysis for the HIF pathway, HIPPO pathway, NRF2/ARE pathway, PI3K/AKT pathway and the chromatin remodeling pathways was performed using gene lists described in Table S2. The pathway analysis involving genes known to be activated in cancer, such as MTOR, PIK3CA, and NFE2L2, were limited to missense mutations only.
SNP Array-Based Copy Number Analysis
The gene level copy number data (focal_data_by_genes) generated by Affymetrix SNP 6.0 arrays using protocols at the Genome Analysis Platform of the Broad Institute (McCarroll et al., 2008) was available for 832 of the 843 pan-kidney tumors representing 481 ccRCC, 271 PRCC, and 80 ChRCC. Tumors with copy number data are designated within Table S1 and all data is accessible via the NCI genome data commons (https://gdc.cancer.gov/). Estimates for gross chromosomal arm gain or loss were produced by averaging the copy number values for all genes within each region. Average values greater than 0.3 were considered chromosomal gain and average values less than −0.3 were considered chromosomal loss. For individual gene copy number analysis, such as CDKN2A loss, copy number values of less than −0.4 were considered to represent deletion.
RNA Expression Data Analysis
The level 3 RNA-Seq upper quartile normalized RSEM data was available for 839 of the 843 pan-kidney tumors representing 485 ccRCC, 273 PRCC, and 81 ChRCC. Tumors with RNA-seq data are designated within Table S1 and all data is accessible via the NCI genome data commons and the Gene Expression Omnibus (https://gdc.cancer.gov/ and https://www.ncbi.nlm.nih.gov/geo/). Analysis of the RNA data was split into miRNA analysis, lncRNA analysis, mRNA signature analysis, and immune gene signature analysis.
mRNA Signature Analysis
Raw count data for each sample included was obtained from Gene Expression Omnibus (GSE62944) (Rahman et al., 2015). All subsequent analyses were performed in R open source programming language. For differential expression analysis, RPKM values were calculated from RNaseq raw counts and upper quantile normalized. For hierarchical clustering and WGCNA, raw count data were processed and normalized using the variance stabilizing transformation (VST) algorithm implemented by the DESeq2 package (Love et al., 2014).
Scale-free weighted signed gene co-expression networks were constructed by the WGCNA package (Langfelder and Horvath, 2008). Using the top 11000 varying genes according to their standard deviation, WGCNA was restricted to the 9000 most connected genes. First, a pairwise gene correlation matrix was calculated with a Pearson correlation analysis, which was transformed into a weighted matrix to produce an adjacency matrix after raising values by an exponent beta (β = 16). Then the adjacency was transformed into a topological overlap matrix (TOM). The dynamic tree cut method was used for module identification from the hierarchical clustering of genes using 1-TOM as the distance measure with a deepSplit value of 2 and a minimum size cutoff of 50 genes. Highly similar modules were identified by clustering and then merged together with a height cut-off of 0.2. Finally, modules and their relationship to clinical traits were identified using Pearson correlation analysis between the modules and external traits. Functional annotation of identified modules was performed using tools provided by the WGCNA package.
Kmeans consensus clustering was performed using ConsensusClusterPlus package (Wilkerson and Hayes, 2010). The K-value of 6 was selected according to the consensus cumulative distribution function, where K > 6 did not produce any appreciable increase in consensus (Monti et al., 2003; Wilkerson and Hayes, 2010). Hierarchical unsupervised cluster analysis was performed using 7738 genes pertaining to selected WGCNA modules (see Figure 4 for modules). Hierarchical clustering was performed using average linkage of Euclidean distance.
Non-coding RNA (lncRNA and miRNA) Sequencing and Analysis
mRNA sequence reads were aligned to the human reference genome (hg38) and transcriptome (Ensembl v82, September 2015) using STAR 2.4.2a (Dobin et al., 2013). STAR was run with the following parameters: minimum/maximum intron sizes were set to 30 and 500,000, respectively; noncanonical, unannotated junctions were removed; maximum tolerated mismatches was set to 10; and the outSAMstrandField intron motif option was enabled. The Cuffquant command included with Cufflinks 2.2.1 (Trapnell et al., 2013) was used to quantify the read abundances per sample, with fragment bias correction and multiread correction enabled, and all other options set to default. To calculate normalized abundance as fragments per kilobase of exon per million fragments mapped (FPKM), the Cuffnorm command was used with default parameters. From the FPKM matrix for the 80 tumor samples, we extracted 8167 genes with “lincRNA” and “processed_transcript” Ensembl biotypes.
From the matrix of 8167 lncRNAs (above), we extracted FPKM profiles for 499 lncRNAs that were robustly expressed (mean FPKM ≥ 1) and highly variable (≥ 92.5th FPKM variance percentile) across the n = 833 primary tumor cohort. We identified groups of samples with similar expression profiles by unsupervised consensus clustering with ConsensusClusterPlus (CCP) 1.20.0 (Wilkerson and Hayes, 2010). Calculations were performed using Pearson correlations, partitioning around medoids (PAM), a gene fraction of 0.95, and 200 iterations. It was anticipated that a hierarchically-related series of finer-grained and coarser-grained sets of subtypes may be available from a clustering analysis, that a particular clustering solution (i.e., number of subtypes) from such a series may be a more informative choice for a particular question and context, and that results from multiple data types may need to be considered in order to identify a clustering solution to report on because it is effective in contributing to the overall insights (Aine et al., 2015; Ronan et al., 2016). A consensus clustering solution for lncRNAs was selected by initially considering information for different numbers of clusters and for a range of clustering approaches. The reported clustering solution considered four main factors: a) the consensus membership heatmaps and dendrograms; b) the ‘delta’ plot showing how the area under the cumulative distribution function of consensus membership values increases as the numbers of clusters increases; c) the profile of silhouette width calculated from the consensus memberships, which we take as a measure of typical versus atypical cluster membership; and d) how KIRC, KIRP Type 1 and 2, and KICH samples were separated and subdivided by the clusters. Thus, we selected an 8-cluster solution after assessing consensus membership heatmaps, dendrograms, and CCP clustering metrics for up to 10 clusters. To visualize typical versus atypical cluster members, we used the R cluster package to calculate a profile of silhouette widths (Wcm) from the consensus membership matrix. To generate an abundance heatmap for the 8-cluster result, used the pheatmap R package (v1.0.2). We ordered the columns to correspond to the above consensus clustering result. We manually transferred the upper dendrogram graphic from the consensus result to the heatmap graphic that we were generating. For the rows, we identified a subset of lncRNAs that had a mean FPKM ≥ 10 and a SAM multiclass (samr 2.0) (Li and Tibshirani, 2013) q value of 0.0 across the clusters (see differential abundance, below), transformed the FPKM matrix by log10(FPKM + 1), then, in pheatmap, scaled the rows and clustered them with a Pearson distance metric and Ward clustering.
We compared unsupervised clusters to clinical and molecular covariates by calculating contingency table association p values using R, with a Chi-square or Fisher exact test for categorical data, and a Kruskal-Wallis test for real-valued data.
We generated miRNA sequencing (miRNA-seq) data from messenger RNA-depleted RNA, as describe in (Chu et al., 2016). Briefly, we aligned ~22-nt reads to the GRCh37/hg19 reference human genome, assigned read count abundances to miRBase v16 stem-loops and 5p and 3p mature strands, and assigned miRBase v20 mature strand names to MIMAT accession IDs. Note that while we used only reads with exact-match alignments in calculating miRNA abundances, BAM files available from the Genomics Data Commons (https://gdc.cancer.gov/) include all sequence reads.
For miRNA, mature strand (miR) sequencing data for n = 811 primary tumors, we extracted normalized abundance (RPM) data matrices for ccRCC (n = 457), PRCC (n = 274), and ChRCC (n = 80, which included n = 65 KICH and n = 15 that were originally part of the KIRC cohort). From RPM data matrices for the 457, 274 and 65 original samples respectively, we identified the 304 miRs that were the most-variant 25% (of 1214 miRBase v16 strands) for each cohort. Combining the three lists gave 369 unique miR names. In a batch-corrected data matrix containing 743 miRs and 9,555 primary tumor samples (of 10,825 total samples), 367 of the 369 miRs were available, and we generated a batch-corrected data matrix with 367 miR and 811 primary tumor samples that was the input to unsupervised clustering.
Using ConsensusClusterPlus v1.40.0 we assessed consensus membership heatmaps and other metrics for six approaches, using Pearson or Spearman correlations as distance metrics, and hierarchical, partitioning around meoids (PAM) or k-means clustering. For each approach, we assessed solutions with between two and nine clusters. We report on a 6-cluster solution for Spearman correlations, PAM clustering, and 1000 iterations with a random mature-strand fraction of 0.85 for each iteration. We used a similar selection methodology for the 6-cluster solution as was described above for the lncRNAs.
We used an approach similar to that described above for lncRNAs to generate a clustering heatmap for miRNAs. We first identified miRNAs that were differentially abundant between the unsupervised miRNA clusters using a SAM multiclass analysis (samr 2.0) (Li and Tibshirani, 2013) in R, with the 367-×-811 RPM input data matrix, 1000 permutations, no array centering, a Wilcoxon test statistic, and an FDR threshold of 0.05. For the heatmap we used miRNAs that had larger SAMseq scores and q-values of 0.0. We ordered the data matrix columns to match the clustering result, manually transferred over the upper dendrogram from the consensus clustering graphic, then transformed each row of the matrix by log10(RPM+1) and used the pheatmap R package (v1.0.2) to scale and cluster only the rows.
We generated a Kaplan-Meier plot for the miRNA clusters using the R survival package v2-41.3. We compared unsupervised clusters to clinical and molecular covariates by calculating contingency table association p values using R, with a Chi-square or Fisher exact test for categorical data, and a Kruskal-Wallis test for real-valued data.
Immune Gene Signature Analysis
Immune gene signatures were derived from previously published works (Beck et al., 2009; Bindea et al., 2013; Fan et al., 2011; Iglesia et al., 2014; Kardos et al., 2016; Palmer et al., 2006; Rody et al., 2009; Rody et al., 2011; Schmidt et al., 2008; Teschendorff et al., 2007). RSEM upper quartile normalized, log-2 transformed, and mean centered RNA-seq data was matched to predefined immune gene signature clusters via Entrez IDs. Each gene signature was calculated as the average value of all genes included in the signature (Table S4). Differential expression for each gene signature was analyzed between kidney cancer types and subtypes via one-way ANOVA. These p values were adjusted for multiple testing using the Benjamini-Hochberg procedure. For hazard ratio forest plots, univariate Cox proportional hazards (CoxPH) model was used with signature/clinical variable as a continuous variable compared to patient overall survival. T cell receptor repertoire analysis was performed using MiXCR v1.7.1 on default alignment and assemble settings (Bolotin et al., 2013). Diversity measurements were analyzed between kidney cancer types and subtypes via Mann-Whitney U-test.
DNA Methylation Analysis
Two generations of Illumina Infinium DNA Methylation BeadArrays, including the HumanMethylation27 (HM27) and HumanMethylation450 (HM450) arrays, were used to assay 824 pan-kidney tumors (65 KICH, 485 KIRC and 274 KIRP) and 392 normal kidney samples in total (Table S1). All data is available from the NCI genome data commons (https://gdc.cancer.gov/).
Data from HM27 and HM450 were combined and further normalized using a probe-by-probe proportional rescaling method to yield a common set of 22,601 probes with comparative methylation levels between the two platforms, as described in details on Synapse (Syn7073804). Briefly, we rescaled data on HM27 based on between-platform difference measured by technical replicates. Probes were further filtered based on 34 technical replicates measured together with the KIRC samples by removing those showing a standard deviation of 0.05 or above. Unsupervised clustering was performed based on cancer-specific autosomal loci, which were defined as unmethylated probes in all normal tissue types as well as sorted blood populations (mean beta value < 0.2), but methylated (beta value > 0.3) in more than 5% samples within any of the kidney tumor type (for tumor type with less than 100 samples, we require the portion of methylated samples to be greater than 10% instead). To minimize the influence of tumor purity, we dichotomize the methylation data into 0’s and 1’s with a beta value cut off of 0.3, and used Ward’s method to cluster the distance matrix computed with the Jaccard Index. Heatmaps were generated based on row and column orders calculated as above and colored by dichotomized beta values.
The DNA methylation level as interrogated by cg07684796, cg15839448 was used for DKK1, and SFRP1, respectively, with a beta value of 0.3 or more considered evidence for epigenetic silencing.
Survival Analysis
The Kaplan-Meier method was used to generate curves for overall survival and the Log-rank test was used to assess the univariate survival differences with no correction for multiple testing, unless otherwise stated in specific analyses. Overall survival was defined as the time from the nephrectomy to death of any cause.
mtDNA Sequence and Copy Number Analysis
Whole exome sequencing (WXS) BAM files, sequenced at BCM Sequencing Center, were obtained for 66 ChRCC, 153 ccRCC, and 128 PRCC tumor samples and corresponding blood or normal tissue DNA. BAM files were used as input of the MToolBox pipeline, that includes GSNAP, MUSCLE, and SAMtools, to align reads to the Revised Cambridge Reference Sequence (rCRS) for human mitochondrial DNA, extract variant alleles, quantify their heteroplasmy levels and related confidence intervals, and obtain functional annotation of the identified variants.(Calabrese et al., 2014; Edgar, 2004; Li et al., 2009; Wu and Watanabe, 2005) Samples with > 75% mtDNA sequence coverage in Tumor and Normal DNA and variants with > 5% mutation load were considered for further analysis (61 ChRCC, 66 ccRCC, and 99 PRCC). Variant tables from tumor and corresponding normal DNA were compared to determine somatic mutations, which were then classified according to criteria outlined in Figure S2F.
The mtDNA copy number (m) was calculated for samples with mtDNA sequence data as the ratio of the number of sequencing reads aligning to the mitochondrial genome (rm) and the nuclear genome (rn) according to the following formula: m = rm/rn × R. Correction for tumor ploidy and purity (R) was calculated as RTumor = (Purity × Ploidy+(1 Purity) × 2)/2. Allele-specific copy number and estimates of tumor ploidy and purity were calculated with ASCAT (Reznik et al., 2016; Reznik et al., 2017; Van Loo et al., 2010) using matched Affymetrix SNP6 array data from tumor and normal tissue. Batch effect on exome enrichment was corrected for by applying a linear model that accounted for plate and center IDs as well as tissue type.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all analyses, significance was determined as a p value < 0.05 and corrected for multiple testing where specified. Univariate analysis was performed unless otherwise specified. Survival analyses were performed using GraphPad Prismâ (GraphPad Software, Inc.) or by individually specified methodologies. In all cases the “n” represents individual patients from which a single tumor was evaluated.
DATA AND SOFTWARE AVAILABILITY
Raw and processed clinical, array and sequence data are all available via the Genomic Data Commons download portal (https://portal.gdc.cancer.gov) or Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/-GSE62944) and the digital pathology images are all available from the Cancer Digital Slide Archive (http://cancer.digitalslidearchive.net/)
Supplementary Material
Highlights.
BAP1, PBRM1, and metabolic pathway changes correlate with RCC subtype-specific survival
DNA hypermethylation/CDKN2A alterations associate with poor survival in all RCC subtypes
Immune gene signatures increased in ccRCC and CIMP-RCC
Increased Th2 gene signature within each RCC subtype associates with poorer survival
Acknowledgments
This study was performed using the following grants: NIH Genome sequencing center grants U54 HG003273 for R.A.G., U54 HG003067 for S. Gabriel and E.S. Lander, and U54 HG003079 for R.K. Wilson; NIH Genome data analysis center and genome characterization center grants U24 CA143799 for T.P. Speed and P.T.S., U24 CA143835 for I. Shmulevich, U24 CA143840 for M. Ladanyi and C. Sander, U24 CA143843 for R.A.G. and D.A.W., U24 CA143845 for G. Getz and L. Chin, U24 CA143848 for D.N. Hayes and C.M. Perou, U24 CA143858 for J. Stuart, C. Benz, and D.H. Haussler, U24 CA143866 for M.A. Marra, U24 CA143867 for S. Gabriel and M.L. Meyerson, U24 CA143882 for S.B. Baylin and P.W. Laird, U24 CA143883 for G.B.M., R.A., W.K.A. Yung, and J.N. Weinstein, and U24 CA144025 for R.S. Kucherlapati; and NIH PCC grant P30 CA016672 for G.B.M. This project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research, NIH, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.075.
AUTHOR CONTRIBUTIONS
Conceptualization, C.J.R., P.T.S., W.K.R., and W.M.L.; Methodology, C.J.R., A.A.D., H.F., C.C.S., M.L., E.R., R. Bowlby, E.A.G., and A.G.R.; Investigation, C.J.R., A.A.D., H.F., C.C.S., M.L., E.R., R. Bowlby, E.A.G., S.H., R.S.K., B.K., W.L., H.S., B.G.V., A.G.R., W.K.R., and W.M.L.; Resources, A.A.D., R.A., R. Beroukhim, D.P.B., T.K.C., R.A.G., A.K.G., A.A.H., E.P.H., J.J.H., T.H.H., D.J.K., W.L., M.J.M., G.B.M., J.M., M.L.N., V.E.R., L.S.S., C.S.S., B.S., S.S., R.S., P.T., G.T., C.D.V., D.A.W., L.Y., W.T.K., A.G.R., P.T.S., W.K.R., and W.M.L.; Data Curation, C.J.R., A.A.D., and A.G.R.; Writing - Original Draft, C.J.R., P.T.S., W.K.R., and W.M.L.; Writing - Review & Editing, C.J.R., A.A.D., H.F., C.C.S., M.L., E.R., R. Bowlby, E.A.G., R.A., R. Beroukhim, D.P.B., T.K.C., R.A.G., A.K.G., S.H., A.A.H., E.P.H., J.J.H., T.H.H., R.S.K., B.K., D.J.K., W.L., M.J.M., G.B.M., J.M., M.L.N., V.E.R., L.S.S., C.S.S., H.S., B.S., S.S., R.S., P.T., G.T., B.G.V., C.D.V., D.A.W., L.Y., W.T.K., A.G.R., P.T.S., W.K.R., and W.M.L.; Visualization, C.J.R., A.A.D., H.F., C.C.S., M.L., E.R., R. Bowlby, E.A.G., S.H., R.S.K., B.K., W.L., H.S., B.G.V., W.T.K., A.G.R., W.K.R., and W.M.L.; Supervision, P.T.S., W.K.R., and W.M.L. P.T.S., W.K.R., and W.M.L. are joint chairs of TCGA Pan-Kidney Project.
DECLARATION OF INTERESTS
Michael Seiler, Peter G. Smith, Ping Zhu, Silvia Buonamici, and Lihua Yu are employees of H3 Biomedicine, Inc. Parts of this work are the subject of a patent application: WO2017040526 titled “Splice variants associated with neomorphic sf3b1 mutants.” Shouyoung Peng, Anant A. Agrawal, James Palacino, and Teng Teng are employees of H3 Biomedicine, Inc. Andrew D. Cherniack, Ashton C. Berger, and Galen F. Gao receive research support from Bayer Pharmaceuticals. Gordon B. Mills serves on the External Scientific Review Board of Astrazeneca. Anil Sood is on the Scientific Advisory Board for Kiyatec and is a shareholder in BioPath. Jonathan S. Serody receives funding from Merck, Inc. Kyle R. Covington is an employee of Castle Biosciences, Inc. Preethi H. Gunaratne is founder, CSO, and shareholder of NextmiRNA Therapeutics. Christina Yau is a part-time employee/consultant at NantOmics. Franz X. Schaub is an employee and shareholder of SEngine Precision Medicine, Inc. Carla Grandori is an employee, founder, and shareholder of SEngine Precision Medicine, Inc. Robert N. Eisenman is a member of the Scientific Advisory Boards and shareholder of Shenogen Pharma and Kronos Bio. Daniel J. Weisenberger is a consultant for Zymo Research Corporation. Joshua M. Stuart is the founder of Five3 Genomics and shareholder of NantOmics. Marc T. Goodman receives research support from Merck, Inc. Andrew J. Gentles is a consultant for Cibermed. Charles M. Perou is an equity stock holder, consultant, and Board of Directors member of BioClassifier and GeneCentric Diagnostics and is also listed as an inventor on patent applications on the Breast PAM50 and Lung Cancer Subtyping assays. Matthew Meyerson receives research support from Bayer Pharmaceuticals; is an equity holder in, consultant for, and Scientific Advisory Board chair for OrigiMed; and is an inventor of a patent for EGFR mutation diagnosis in lung cancer, licensed to LabCorp. Eduard Porta-Pardo is an inventor of a patent for domainXplorer. Han Liang is a shareholder and scientific advisor of Precision Scientific and Eagle Nebula. Da Yang is an inventor on a pending patent application describing the use of antisense oligonucleotides against specific lncRNA sequence as diagnostic and therapeutic tools. Yonghong Xiao was an employee and shareholder of TESARO, Inc. Bin Feng is an employee and shareholder of TESARO, Inc. Carter Van Waes received research funding for the study of IAP inhibitor ASTX660 through a Cooperative Agreement between NIDCD, NIH, and Astex Pharmaceuticals. Raunaq Malhotra is an employee and shareholder of Seven Bridges, Inc. Peter W. Laird serves on the Scientific Advisory Board for AnchorDx. Joel Tepper is a consultant at EMD Serono. Kenneth Wang serves on the Advisory Board for Boston Scientific, Microtech, and Olympus. Andrea Califano is a founder, shareholder, and advisory board member of DarwinHealth, Inc. and a shareholder and advisory board member of Tempus, Inc. Toni K. Choueiri serves as needed on advisory boards for Bristol-Myers Squibb, Merck, and Roche. Lawrence Kwong receives research support from Array BioPharma. Sharon E. Plon is a member of the Scientific Advisory Board for Baylor Genetics Laboratory. Beth Y. Karlan serves on the Advisory Board of Invitae.
References
- Aine M, Eriksson P, Liedberg F, Sjödahl G, Höglund M. Biological determinants of bladder cancer gene expression subtypes. Sci Rep. 2015;5:10957. doi: 10.1038/srep10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, Varma S, Marinelli RJ, van de Rijn M, West RB. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin Cancer Res. 2009;15:778–787. doi: 10.1158/1078-0432.CCR-08-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Bolotin DA, Shugay M, Mamedov IZ, Putintseva EV, Turchaninova MA, Zvyagin IV, Britanova OV, Chudakov DM. MiTCR: software for T-cell receptor sequencing data analysis. Nat Methods. 2013;10:813–814. doi: 10.1038/nmeth.2555. [DOI] [PubMed] [Google Scholar]
- Calabrese C, Simone D, Diroma MA, Santorsola M, Guttà C, Gasparre G, Picardi E, Pesole G, Attimonelli M. MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics. 2014;30:3115–3117. doi: 10.1093/bioinformatics/btu483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, Wheeler DA, Murray BA, Schmidt L, Vocke CD, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–145. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casuscelli J, Weinhold N, Gundem G, Wang L, Zabor EC, Drill E, Wang PI, Nanjangud GJ, Redzematovic A, Nargund AM, et al. Genomic landscape and evolution of metastatic chromophobe renal cell carcinoma. JCI Insight. 2017;2:2. doi: 10.1172/jci.insight.92688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang Y, Şenbabaoglu Y, Ciriello G, Yang L, Reznik E, Shuch B, Micevic G, De Velasco G, Shinbrot E, et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016;14:2476–2489. doi: 10.1016/j.celrep.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A, Robertson G, Brooks D, Mungall AJ, Birol I, Coope R, Ma Y, Jones S, Marra MA. Large-scale profiling of microRNAs for The Cancer Genome Atlas. Nucleic Acids Res. 2016;44:e3. doi: 10.1093/nar/gkv808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. The Cancer Genome Atlas Research Network The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Prat A, Parker JS, Liu Y, Carey LA, Troester MA, Perou CM. Building prognostic models for breast cancer patients using clinical variables and hundreds of gene expression signatures. BMC Med Genomics. 2011;4:3. doi: 10.1186/1755-8794-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Xi L, Hughes DS, Zhang J, Zhang J, Futreal PA, Wheeler DA, Wang W. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17:178. doi: 10.1186/s13059-016-1029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler K, Fornara P, Lautenschlager C, Holzhausen HJ, Seliger B, Riemann D. Immune signature of tumor infiltrating immune cells in renal cancer. OncoImmunology. 2015;4:e985082. doi: 10.4161/2162402X.2014.985082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, et al. ccRCC Cancer Genome Atlas (KIRC TCGA) Research Network investigators Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res. 2013;19:3259–3267. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–138. doi: 10.1016/j.ctrv.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Hirata H, Hinoda Y, Nakajima K, Kawamoto K, Kikuno N, Ueno K, Yamamura S, Zaman MS, Khatri G, Chen Y, et al. Wnt antagonist DKK1 acts as a tumor suppressor gene that induces apoptosis and inhibits proliferation in human renal cell carcinoma. Int J Cancer. 2011;128:1793–1803. doi: 10.1002/ijc.25507. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009. doi: 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesia MD, Vincent BG, Parker JS, Hoadley KA, Carey LA, Perou CM, Serody JS. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin Cancer Res. 2014;20:3818–3829. doi: 10.1158/1078-0432.CCR-13-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur P, Peña-Llopis S, Christie A, Zhrebker L, Pavía-Jiménez A, Rathmell WK, Xie XJ, Brugarolas J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol. 2013;14:159–167. doi: 10.1016/S1470-2045(12)70584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, Iglesia MD, Milowsky MI, Parker JS, Kim WY, Vincent BG. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight. 2016;1:e85902. doi: 10.1172/jci.insight.85902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Motzer RJ. Immune checkpoint therapy in renal cell carcinoma. Cancer J. 2016;22:92–95. doi: 10.1097/PPO.0000000000000177. [DOI] [PubMed] [Google Scholar]
- Li J, Tibshirani R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-Seq data. Stat Methods Med Res. 2013;22:519–536. doi: 10.1177/0962280211428386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan WM. Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome Res. 2012;22:2089–2100. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan WM, Coleman JA, Walther MM, Zbar B. Genetic basis of kidney cancer. In: Vogelzang NJ, Scardino PT, Shipley WU, DeBruyne FMJ, Linehan WM, editors. Comprehensive Textbook of Genitourinary Oncology. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 701–709. [Google Scholar]
- Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol. 2016;70:93–105. doi: 10.1016/j.eururo.2016.02.029. [DOI] [PubMed] [Google Scholar]
- Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn. 2003;52:91–118. [Google Scholar]
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. CheckMate 025 Investigators Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BWH, Tan MH, Zhang Z, Yang XJ, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. 2006;7:115. doi: 10.1186/1471-2164-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radenbaugh AJ, Ma S, Ewing A, Stuart JM, Collisson EA, Zhu J, Haussler D. RADIA: RNA and DNA integrated analysis for somatic mutation detection. PLoS ONE. 2014;9:e111516. doi: 10.1371/journal.pone.0111516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M, Jackson LK, Johnson WE, Li DY, Bild AH, Piccolo SR. Alternative preprocessing of RNA-sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics. 2015;31:3666–3672. doi: 10.1093/bioinformatics/btv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznik E, Miller ML, Şenbabaoǧlu Y, Riaz N, Sarungbam J, Tickoo SK, Al-Ahmadie HA, Lee W, Seshan VE, Hakimi AA, Sander C. Mitochondrial DNA copy number variation across human cancers. eLife. 2016;5:5. doi: 10.7554/eLife.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznik E, Wang Q, La K, Schultz N, Sander C. Mitochondrial respiratory gene expression is suppressed in many cancers. eLife. 2017;6:6. doi: 10.7554/eLife.21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts CJ, Hill VK, Linehan WM. Tumor-specific hypermethylation of epigenetic biomarkers, including SFRP1, predicts for poorer survival in patients from the TCGA Kidney Renal Clear Cell Carcinoma (KIRC) project. PLoS ONE. 2014;9:e85621. doi: 10.1371/journal.pone.0085621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rody A, Holtrich U, Pusztai L, Liedtke C, Gaetje R, Ruckhaeberle E, Solbach C, Hanker L, Ahr A, Metzler D, et al. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res. 2009;11:R15. doi: 10.1186/bcr2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, Gaetje R, Solbach C, Ahr A, Metzler D, et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011;13:R97. doi: 10.1186/bcr3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan T, Qi Z, Naegle KM. Avoiding common pitfalls when clustering biological data. Sci Signal. 2016;9:re6. doi: 10.1126/scisignal.aad1932. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Böhm D, von Törne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kölbl H, Gehrmann M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405–5413. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- Şenbabaoǧlu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, Miao D, Ostrovnaya I, Drill E, Luna A, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17:231. doi: 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourbier C, Ricketts CJ, Matsumoto S, Crooks DR, Liao PJ, Mannes PZ, Yang Y, Wei MH, Srivastava G, Ghosh S, et al. Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell. 2014;26:840–850. doi: 10.1016/j.ccell.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007;8:R157. doi: 10.1186/gb-2007-8-8-r157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann RL, Hlady RA, Hanavan PD, Lake DF, Tibes R, Lee JH, Choi JH, Ho TH, Robertson KD. Dynamic reprogramming of DNA methylation in SETD2-deregulated renal cell carcinoma. Oncotarget. 2016;7:1927–1946. doi: 10.18632/oncotarget.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakami S, Shiina H, Enokida H, Hirata H, Kawamoto K, Kawakami T, Kikuno N, Tanaka Y, Majid S, Nakagawa M, et al. Wnt antagonist family genes as biomarkers for diagnosis, staging, and prognosis of renal cell carcinoma using tumor and serum DNA. Clin Cancer Res. 2006;12:6989–6997. doi: 10.1158/1078-0432.CCR-06-1194. [DOI] [PubMed] [Google Scholar]
- Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- Yang Y, Vocke CD, Ricketts CJ, Wei D, Padilla-Nash HM, Lang M, Sourbier C, Killian JK, Boyle SL, Worrell R, et al. Genomic and metabolic characterization of a chromophobe renal cell carcinoma cell line model (UOK276) Genes Chromosomes Cancer. 2017;56:719–729. doi: 10.1002/gcc.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.