

Short abstract
This article estimates potential future savings from biosimilars, summarizes the experience to date with the first marketed biosimilar, and discusses key policy issues surrounding biosimilars in the United States.
Keywords: Health Care Costs, Health Economics, Health Insurance, Pharmaceutical Drugs
Abstract
The Biologics Price Competition and Innovation Act (BPCIA), enacted as part of the 2010 Patient Protection and Affordable Care Act (ACA), authorized the U.S. Food and Drug Administration (FDA) to create a new regulatory approval pathway for biosimilars, which are biologic drugs that are very similar to already approved “reference” biologics in terms of potency, safety, and efficacy, but are manufactured by different companies. In the seven years since the ACA, many drug manufacturers worked to push new biosimilars through development and FDA review. As of July 2017, there were three marketed biosimilars and two more that were approved by the FDA but not yet marketed. BPCIA's shorter, lower-cost biosimilar approval pathway was designed to introduce competition among biologic manufacturers.
This article estimates potential future savings from biosimilars in the United States, summarizes the experience to date with the first marketed biosimilar in the United States, and discusses key policy issues surrounding biosimilars. We estimate that biosimilars will reduce direct spending on biologic drugs by $54 billion from 2017 to 2026, or about 3 percent of total estimated biologic spending over the same period, with a range of $24 to $150 billion. While our estimate uses recent data and transparent assumptions, we caution that actual savings will hinge on industry and regulatory decisions as well as potential policy changes to strengthen the biosimilar market.
The Biologics Price Competition and Innovation Act (BPCIA), enacted as part of the 2010 Patient Protection and Affordable Care Act (ACA), authorized the U.S. Food and Drug Administration (FDA) to create a new regulatory approval pathway for biosimilars, which are biologic drugs that are very similar to already approved “reference” biologics in terms of potency, safety, and efficacy, but manufactured by different companies. In the seven years since the ACA, many drug manufacturers worked to push new biosimilars through development and FDA review. As of July 2017, there were three marketed biosimilars and two more that were approved by the FDA but not yet marketed. BPCIA's shorter, lower-cost biosimilar approval pathway was designed to introduce competition among biologic manufacturers, leading to savings in spending on biologics. This article estimates potential future savings from biosimilars in the United States, summarizes the experience to date with the first marketed biosimilar in the United States, and discusses key policy issues surrounding biosimilars. We estimate that biosimilars will lead to a reduction of $54 billion in direct spending on biologic drugs from 2017 to 2026, or about 3 percent of total estimated biologic spending over the same period, with a range of $24 to $150 billion. While our estimate uses recent data and transparent assumptions, we caution that actual savings will hinge on industry, regulatory, prescriber, and insurer decisions, as well as potential future policy changes to strengthen the biosimilar market.
Background
U.S. spending on prescription drugs increased by 4.8 percent to $323 billion from 2015 to 2016.1 The increasing use of specialty drugs—including biologics—is one of the main drivers of spending growth. Biologics are complex, protein-based drugs manufactured in living systems and include insulin; monoclonal antibodies to block inflammation in rheumatoid arthritis; and a range of drugs to treat cancer, multiple sclerosis, and other serious diseases. Biologics are a primary treatment option for several cancers and other serious conditions. While only 1–2 percent of the U.S. population is treated with a specialty drug each year—a category that includes biologics and other complex, often expensive drugs,2 biologics alone accounted for 38 percent of U.S. prescription drug spending in 2015 due to their high cost per dose,3 and for 70 percent of drug spending growth between 2010 and 2015.4
For over 30 years, the United States has implemented successful policies and regulations to promote competition between manufacturers of simpler, “small molecule” drugs after key patents expire or are successfully challenged in court. However, the provisions of the Hatch-Waxman Act of 1984 that created the U.S. generic drug industry do not apply to biologics, and until recently it did not make economic sense in most cases for manufacturers to bring competing biologics to market, even after the key patents and FDA-granted exclusivity periods protecting originator biologics expired.5 The BPCIA, enacted as part of the ACA, authorized the FDA to create a regulatory approval pathway for biologics that would be shorter and less expensive than a full new drug application. These biosimilar drugs are very similar to approved “reference” biologics in potency, safety, and efficacy, but with minor differences because they are derived from living organisms.
In the years following passage of the ACA, the FDA began releasing guidance to industry outlining its approach to regulating biosimilars.6 Many drug manufacturers intensified or initiated biosimilar development programs. The first applications under the FDA's biosimilar pathway were submitted in 2014. In March 2015, a biosimilar of filgrastim—a drug used to treat low white blood cell counts due to chemotherapy, among other causes—was the first to be approved. As of July 2017, there are three marketed biosimilars (one to filgrastim and two to infliximab), two biosimilars that are FDA-approved but not yet marketed (one each to adalimumab and etanercept),7 and over 60 biosimilar molecules in development for more than 20 reference biologics.8
Roadmap
In this article, we build on an earlier RAND analysis to describe how the developing U.S. biosimilar market could reduce spending on biologics.9 Specifically, we expand our prior literature review on estimates of the cost savings potential of biosimilars to include studies conducted through March 2017. Drawing on assumptions from the literature review and our subject-matter experience, we then estimate the potential cost savings from biosimilars, using recent data on all biologic drugs as a baseline. Finally, we discuss sources of uncertainty and evolving policy issues in the U.S. biosimilar market that could affect the cost savings.
The Cost Savings Potential of Biosimilars
The rationale for a biosimilar approval pathway is to promote competition among manufacturers to lower prices and potentially increase access to medications. Figure 1 illustrates the relationships between manufacturers, providers, insurers, pharmacy benefit managers (PBMs),10 and patients that drive both competition and potential savings. Biosimilars and their respective reference biologics are expected to compete on price to gain market share. Both insurers and providers are, in a way, “buyers” of biologics and can steer patients toward one product or another. Providers buy biologics from manufacturers or wholesalers and administer biologics to patients. Insurers influence prescribers by setting their own payment rates and through utilization management tools, such as prior authorization, that require prescribers to provide justification and documentation to support the insurer paying for a drug at all. Patients are also “buyers” of biologics to the extent that they pay for part of the cost of drugs through cost-sharing. The manufacturer offering the best price to providers (including hospitals, physician practices, and pharmacies) and the largest rebates to insurers should expect to gain market share and revenue. Over time, patients could benefit from price competition through lower insurance premiums, lower out-of-pocket costs, and increased access to medications.
Figure 1.
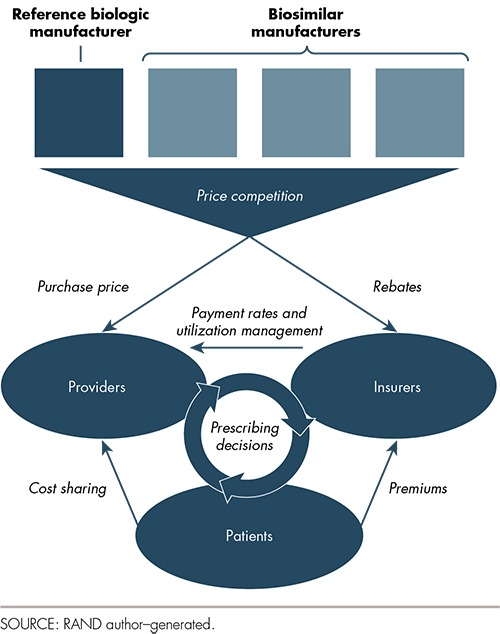
Biologic Market Relationships
Price competition will result in savings if utilization remains constant. Lower prices, however, may increase utilization of biologics. For example, some patients who choose not to take biologics due to high out-of-pocket cost might change their minds if biosimilars offer lower prices. Providers relying on margins that scale with the acquisition price of biologics for revenue (as we describe later in this article) may write more prescriptions or increase the volume of other services to compensate for the reduction in price. The net change in spending due to biosimilars may be positive or negative depending both on the magnitude of the price reduction and on how insurers, providers, and patients respond to lower prices in terms of volume.
A previous RAND Perspective described a framework based on economic theory to link not only competition from biosimilars, but also other drivers to changes in spending on biologic drugs.11 The other drivers—including the safety, efficacy, and real-world effectiveness of biosimilars; payment; and the acceptability of biosimilars to prescribers and patients—work with and through competition to determine biosimilars' market share, prices, and impact on spending. While these other drivers are important, we identified competition as the most important factor in determining impact on spending.
An Evolving Market
While biosimilar approvals and launches are major milestones, the U.S. biosimilar market is still in its infancy. The ultimate features of the market, such as the number of biosimilar manufacturers; the intensity of competition; the reception of biosimilars by prescribers, payers, and patients; and the potential savings from biosimilars, will continue to evolve over the next few years. Some of the most important evolving features of the U.S. biosimilar market include
uncertainty surrounding intellectual property (IP): The BPCIA introduced new procedures for resolving patent disputes between biosimilar and reference biologic manufacturers prior to their launch. Some early biosimilar manufacturers have launched their products without going through BPCIA's “patent dance” provisions, which involve notification and sharing of patent information between biosimilar and reference biologic manufacturers. Other manufacturers with FDA-approved biosimilars have delayed entering the market to avoid patent litigation risk. A recent Supreme Court decision clarified some of the uncertainty about when the “patent dance” provisions apply.12 This decision and other early cases will start to set the tone for how patents and litigation will influence biosimilar entry into the market.
interchangeability: Biosimilar regulations allow the FDA to designate some biosimilars as interchangeable with their reference biologic; in other words, pharmacies could dispense one or the other without needing a prescriber to authorize the change. None of the biosimilars approved to date have sought the interchangeable designation, and the FDA only recently released a draft guidance document outlining the requirements for the interchangeable designation.13 Without interchangeability, prescribers must choose a specific biosimilar or reference biologic by name, which limits the potential for biosimilars to gain market share and compete on price. Relatedly, many states have enacted laws to define pharmacists' ability to substitute biosimilars for reference biologics.14 State laws differ in their specific features, but many include provisions that could affect biosimilar uptake: for example, requiring patients to be notified of biosimilar substitution and allowing prescribers to prevent substitution by noting “dispense as written” on the prescription.
payment rates: Medicare pays for drugs administered in physicians' offices or hospital outpatient departments (such as many biologics) based on the average sales price, net rebates, and other discounts reported to the government, plus a fixed percentage. Under this payment approach, providers would usually be penalized for choosing a lower-cost drug because the markup on a lower price is smaller in dollar terms. To prevent providers from facing this financial disincentive to prescribe lower-cost biosimilars, BPCIA requires Medicare payment for biosimilars to include a fixed percentage based on the more-expensive reference biologic. Subsequently, Medicare implemented a new payment policy for biosimilars that pays a blended average sales price for all biosimilars that share a common reference biologic drug, plus the fixed percentage of the more-expensive reference biologic, as required by BPCIA. Medicare's payment approach could shift over time and it is not yet known whether private insurers are more aggressively incentivizing biosimilars through payment.
Prior Cost Savings Estimates
For over a decade, academics and policymakers have debated just how much savings biosimilars might create in the United States, drawing on European experience with generic drugs and with drugs that resemble biosimilars in the United States.15 In an earlier Perspective, we identified peer-reviewed papers and other literature and extracted key assumptions and results from these studies.16 We found considerable variation in assumptions, time frames, and perspectives across these estimates. Based on our findings from this review, we used 2016 sales data for a wide range of biologic drugs to estimate the potential savings from biosimilars over a ten-year period under a “base case” set of assumptions and other reasonable assumptions that yielded higher and lower savings. We estimated that biosimilars would lead to a $44 billion reduction in direct spending on biologic drugs from 2014 through 2024, or about 4 percent of total biologic spending over the same period, with a range of $13 to $66 billion.
The Need for Updated Cost Savings Estimates
There are several reasons to revisit the question of the likely magnitude of savings from biosimilars. First, biologics have become an increasingly important share of the overall U.S. prescription drug market. Several important biologics will face the end of their regulatory exclusivity period over the next ten years. Second, we have data to analyze the actual experience with at least one biosimilar—Sandoz's filgrastim-sndz (Zarxio®), a biosimilar to filgrastim (Amgen's Neupogen®). Because filgrastim-sndz is the only U.S.-marketed biosimilar product with a significant sales history and because of the unique characteristics of the filgrastim market, including a competing next-generation reference biologic, we cannot use the experience to date to estimate cost savings in other markets. Still, it is useful to compare how the price and market share of filgrastim-sndz changed over time with the assumptions that feed into cost savings estimates. Finally, we now have a better—but still incomplete—understanding of the timing of biosimilar development, regulatory review, and market entry that can be used to produce more-realistic savings estimates.
Recent Literature on Biosimilar Cost Savings
We updated our earlier review of the literature to include recent (2014 through March 2017) peer-reviewed and other literature on the potential direct cost savings from biosimilars. Our overall literature review covers the period 2006 through March 2017. Box 1 describes our search methods and the number of publications that we identified.
Box 1.
Literature Review Methodology
Search terms: Search terms included “biosimilar,” “follow-on biologic,” and “biogeneric” combined with terms focusing on economic and financial impacts, such as “cost,” “price,” and “savings.” Searches for the term “biosimilar” produced the most results, while searches for “follow-on biologic” and “biogeneric” returned relatively few results. In addition to the search terms, we also mined citations and performed forward searches to identify other articles that cited key publications. Each study abstract or summary was reviewed by one of the authors. We reviewed in full studies reporting the actual or predicted impacts of multisource biologics (i.e., similar biologics produced by different manufacturers) on (a) prices, (b) utilization, or (c) health outcomes and we catalogued key outcomes using an abstraction sheet. We did not review editorial, opinion, or perspectives articles without empirical analyses.
Databases: To create the library of peer-reviewed literature, we searched databases including PubMed, Web of Science, and Google Scholar. We searched for non–peer-reviewed “grey” literature using Google and reference mining.
Abstracting: With studies that satisfied our search criteria, we coded articles based on their methodology to estimate the spending impact of biosimilars, i.e., those that dealt only with changes in unit price and those that accounted for potential utilization changes due to price reductions. In addition, we collected information on biosimilar market share predictions (either empirical or implied) and categorized reports by the type of cost savings reported (as a percentage of biologic spending, in absolute numbers on a societal level, or in savings relative to a specific indication or therapeutic area).
We reviewed 150 sources and identified 15 with model-based estimates of biosimilar cost savings. We found that assumptions on biosimilar price relative to originator price ranged from 10 to 51 percent (mean 27 percent). Biosimilar market share assumptions ranged from 5 to 60 percent (mean 28 percent). These two important assumptions—biosimilar price relative to reference biologic price and biosimilar market share—help to determine the magnitude of cost savings estimates. In terms of results, cost savings estimates as a share of total biologic spending ranged from 0.2 to 10.5 percent (mean 3.1 percent, or $3.3 billion of total 2016 biologic spending).
The studies that we reviewed varied in scope in terms of all biologics versus select therapeutic classes and in terms of the entire U.S. health care system versus individual payers, such as Medicare, employers, and others. Studies also employed different reporting metrics, either as cost savings in absolute or relative terms. While we used midpoint estimations for our review, all reviewed studies highlighted relative uncertainty regarding potential cost savings due to possible variation in key assumptions.
The key drivers of biosimilar cost savings that were discussed but rarely modeled in cost savings estimates were the number and timing of biosimilar entrants; patient and prescriber acceptability; biosimilar development cost; life-cycle management strategies by the innovator manufacturers, such as the launch of second- or third-generation biologics in a class and efforts to switch patients to these new biologics without biosimilar competitors; changes in market size, share, or prices over time; payer coverage and payment policies; cost-sharing; and regulatory policies, including interchangeability.
Our findings from the updated literature review echo those from our earlier Perspective. Because of limited U.S. experience with biosimilars, the key assumptions on market share and biosimilar prices are “best guesses” based on anecdotes or professional opinion. In some cases, these assumptions are informed by Europe's experience with biosimilars or historical data from U.S. small-molecule generic or multisource biologic markets. There are well-founded concerns that the U.S. biosimilars market is different than these other markets.
We also separately analyzed sales data on biologic drugs to understand how market share and prices have changed over time in the filgrastim market. Our data17 include quarterly volume and sales and distinguish between the filgrastim reference biologic (Amgen's Neupogen®), a second filgrastim product approved through the full regulatory approval pathway (Teva's Granix®; tbo-filgrastim),18 and a biosimilar to filgrastim (Sandoz's Zarxio®; filgrastim-sndz) (see Box 2). We found that market share for Zarxio® and Granix® increased gradually over time to capture a combined 30-percent market share by sales19 and a 40-percent share in terms of volume by the end of 2016. The prices calculated from these data do not reflect rebates paid by manufacturers to insurers and therefore do not truly reflect the cost per unit. Keeping this limitation in mind, we found that Zarxio® and Granix® had pre-rebate prices that were 30-percent and 45-percent below the reference biologic's price. Overall, our empirical analysis of filgrastim data aligns with the estimates from our literature review. The average market share of 28 percent from the literature review maps to our empirical finding of 30 percent. The average price reduction of 24 percent from the literature review is consistent with the larger filgrastim pre-rebate price differences, especially if Amgen started offering larger rebates to insurers to compete with Zarxio® and Granix® (which would decrease the 30-percent and 45-percent price reductions).
Box 2.
The U.S. Experience with Filgrastim Biosimilars Through 2016
In March 2015, the FDA approved Zarxio® (filgrastim-sndz) to the reference biologic Neupogen® (filgrastim) as the first biosimilar product in the United States.20 The new drug, marketed by Sandoz (part of Novartis), became available on the U.S. market in September 2015. Earlier, in August 2012, the FDA approved Granix® (tbo-filgrastim) under the full regulatory approval pathway (a biosimilar pathway did not exist then) for one of Neupogen's® five indications, severe neutropenia in patients with non-myeloid malignancies. Granix® entered the market in late 2013. In quarter 4 of 2016, Granix® and Zarxio® together shared 30 percent of the total filgrastim market (see Figure 2), and are currently offered at a 30-percent and 45-percent discount to Neupogen®, respectively.21 Yet all three filgrastims (Neuopgen®, Granix®, and Zarxio®) represented just 15 percent of the market share at the end of 2016 if peg-filgrastim (a longer-acting version of filgrastim marketed by Amgen as Neulasta®) is included. Neulasta® was approved in early 2002 and has gradually increased in market share relative to the other filgrastim products combined.
Figure 2.
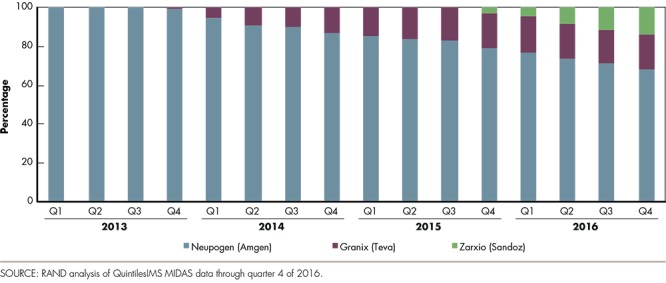
Market Share by Filgrastim Brand (As a Percentage of Sales Before Rebates)
While the market share of Zarxio® and Granix® reached a significant level by 2016, total spending on all three filgrastims has been declining: Net sales for all three dropped from $931 million in 2013 to $766 million in 2016 at an average annual rate of 6.3 percent, while the number of units sold dropped by 2.3 percent per year during this period, suggesting that biosimilar competition has had a nontrivial impact on the (average) cost per unit. However, the net price of Amgen's Neupogen® remained virtually constant between 2013 and 2016, while both Granix® and Zarxio® experienced large reductions in price per unit between their respective launches and quarter 4 of 2016.22
Calculating an Updated Estimate of Biosimilar Cost Savings
Our updated cost savings estimate focuses on the potential reduction in spending due to lower unit prices for biologics. We do not estimate how utilization and spending increase as price declines for two reasons. First, very few of the estimates of biosimilar cost savings that we reviewed address volume effects at all. Second, the change in demand for biologics, access to biologics, and prescriber decisions associated with lower prices will likely vary significantly across different components of the fragmented U.S. health care system.
We used 2016 U.S. sales data on over 100 biologics, including all blockbuster biologics with sales over $1 billion and many products with smaller markets, to estimate the potential direct cost savings from biosimilars.23 We excluded filgrastim ($766 million in total 2016 U.S. sales) from our estimate because this market is already experiencing competition from biosimilars. The remaining products had combined 2016 sales of $104.4 billion across all distribution channels. We expect the biosimilar market for insulins and human growth hormones—where there are already multiple competing products—to look different than the market for other biologics. We divided the $104.4 billion total into two parts, leaving $29.9 billion in 2016 sales for the “established” insulin and human growth hormone markets and $74.5 billion for all other biologic markets.
Our estimate of savings is an aggregate estimate summed over ten calendar years (2017 through 2026). Using our baseline assumptions, we modeled 2.5-percent quarter-on-quarter market growth (or about 7-percent year-on-year growth), a 30-percent biosimilar market share, constant reference biologic prices, and biosimilar prices that are 70 percent of the reference biologic price.24 We reviewed biosimilar development pipelines and FDA regulatory exclusivity expiration dates to assign each biologic drug market to one of seven categories, which describe how close a biosimilar is to launch in the market (see Box 3). We developed a set of probabilities to describe the likelihood of biosimilar entry in each year and then extrapolated quarterly entry probabilities. We assumed that established insulin and human growth hormone markets have one-half the biosimilar penetration and price discounts of other markets.
Box 3.
Biosimilar Entry Probabilities Over Time
As part of our modeling, we needed to estimate when biosimilars would enter individual biologic drug markets throughout the ten-year time horizon. We reviewed biosimilar development pipelines by searching for clinical trials, company announcements, and other sources for all large molecules (biologics) currently on the market. We also collected FDA regulatory exclusivity expiration dates to assign each biologic drug market to one of seven categories:
At least one already marketed biosimilar (e.g., filgrastim)
At least one already approved—but not yet marketed—biosimilar (i.e., biosimilar manufacturers have decided not to launch due to IP or other considerations)
At least one biosimilar application under FDA review
At least one biosimilar in late-stage development
At least one biosimilar in early-stage development
Biosimilars not in development now, but with exclusivity expiration several years away
Biosimilars not in development now, and exclusivity has already expired.
Our estimates of potential cost savings should reflect the fact that biosimilar entry is much more likely in the second category (already approved but not yet marketed) than in the last category (where biosimilar entry in the near future is very unlikely). There is no single data source that we could use to project biosimilar launch probabilities over time. Analogies from small-molecule drugs or outside the United States do not feel appropriate to apply to our context. As a result, we developed our own set of entry probabilities for categories 2 through 7 (see Figure 3). For example, for each market with at least one FDA-approved-but-not-yet-launched biosimilar (the yellow line in the figure), we assume that biosimilar entry by year one in our model (2018) will happen with 50-percent probability, entry by year two will happen with 80-percent probability, entry by year three will happen with 90-percent probability, and so on. Entry probabilities for categories 6 and 7 are 0 percent in the first two years and they increase very slowly, reflecting our assumption that some markets are not likely to face biosimilar competition within a ten-year time horizon. Our main cost savings estimates use the entry probabilities in Figure 3. We also report results from a sensitivity analysis where the entry probabilities are reduced by 25 percent and increased by 25 percent. The lower-entry probability estimates may be more realistic if patents—which we did not consider in determining the earliest entry date for each biologic market—delay entry beyond the expiration of regulatory exclusivity.
Figure 3.
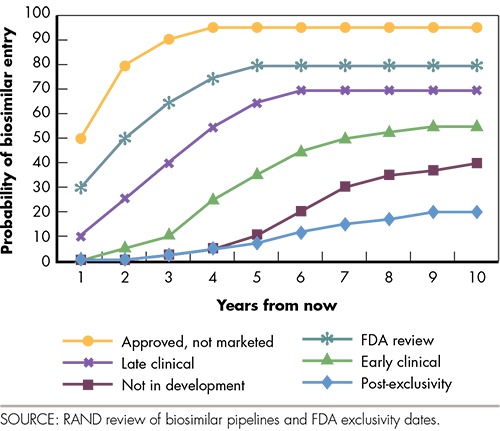
Probability of Biosimilar Entry Over Time
These baseline assumptions—while informed by previously published studies, subject-matter expertise, and our empirical analysis of the filgrastim market through 2016—are informed guesses, and as a result, we vary many of the assumptions with sensitivity analyses. We calculated the estimated cost savings under an “upper-bound” set of assumptions where biosimilar market share is 50 percent and biosimilar prices are 50 percent of reference biologic prices. We also calculated estimated cost savings under a “lower-bound” set of assumptions where biosimilar market share is 20 percent and biosimilar prices are 20-percent lower than reference biologic prices.
Under our baseline assumptions, we calculate potential direct cost savings of $54.0 billion over ten years25 or about 2.8 percent of total biologic sales over the same period.26 The lower- and upper-bound scenarios yield ten-year cost saving estimates of $24 and $150 billion, respectively. Table 1 reports results for a range of other combinations of market share and price assumptions. Using the baseline assumptions but reducing entry probabilities by 25 percent decreases ten-year cost savings to $40.5 billion, while 25 percent–higher entry probabilities increases ten-year cost savings to $67.5 billion.
Table 1.
Sensitivity Analysis Results: Ten-Year Biosimilar Cost Savings
| Biosimilar Price Relative to Reference Biologic | Biosimilar Market Share Assumption | ||||||
|---|---|---|---|---|---|---|---|
| 20% | 25% | 30% | 35% | 40% | 45% | 50% | |
| 50% | $60 | $75 | $90 | $105 | $120 | $135 | $150 |
| 55% | $54 | $67 | $81 | $94 | $108 | $121 | $135 |
| 60% | $48 | $60 | $72 | $84 | $96 | $108 | $120 |
| 65% | $42 | $52 | $63 | $73 | $84 | $94 | $105 |
| 70% | $36 | $45 | $54 | $63 | $72 | $81 | $90 |
| 75% | $30 | 37 | $45 | $52 | $60 | $67 | $75 |
| 80% | $24 | $30 | $36 | $42 | $48 | $54 | $60 |
SOURCE: RAND authors' calculations.
NOTE: All dollar amounts are in billions.
The potential for cost savings will vary across biologic classes based on sales, the degree of competition, and the timing of biosimilar entry. We used the same assumptions to generate estimates of potential savings for specific classes of biologics (see Figure 4). Anti–tumor necrosis factor (TNF) products, monoclonal antibody antineoplastics, and immunostimulants excluding interferons (including filgrastim) alone account for 87 percent of estimated savings. The anti-TNF category includes some of the largest biologic products by sales and biosimilars that are already marketed or close to market entry.27
Figure 4.
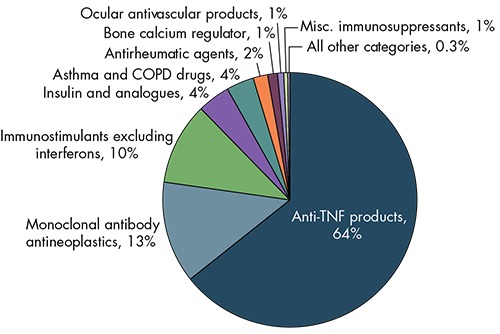
Estimated Cost Savings, by Biologic Class
Limitations of Our Cost Savings Estimate
Our estimate, like any other, is based on assumptions. While we use recent data and are transparent in our assumptions, we know that the actual cost savings from biosimilars over a ten-year period will differ from our estimate. We did not account for growth in demand for biologics due to lower prices. While additional utilization will narrowly increase spending on drugs, improving access to appropriate biologics may reduce health care spending on other services, such as hospitalizations. We did not model interactions between different biologics, i.e., we did not capture spillover effects when the entry of a biosimilar changes the pricing strategy of a reference biologic manufacturer, or when the entry of a biosimilar to one reference biologic might affect prices in other markets. We also did not explicitly include prescriber and patient decisionmaking; insurer efforts to steer decisions toward biosimilars through utilization management, payment, and cost-sharing; or other drivers of biosimilar cost-sharing in our model. In general, we did not separately model scenarios with and without some biosimilars obtaining the interchangeable designation from the FDA. However, these other drivers of cost savings are related to our market share and price assumptions.
Who Will Benefit from Biosimilar Cost Savings?
Biologics include self-administered drugs obtained at retail pharmacies, drugs administered in inpatient settings, and drugs administered in physician office and hospital outpatient department settings. The balance of cost savings to health care payers, providers, and patients is different for each setting due to differences in payment and cost-sharing arrangements.
Insurers benefit from lower biologic prices across all delivery settings in the short term through lower payment rates, and, over time, commercial insurers may transfer savings to patients in the form of lower insurance premiums. Lower spending on biologics in public systems, such as Medicare, will ultimately benefit taxpayers.
Providers, such as physicians, purchase the biologics that they administer to patients in their offices and are reimbursed retroactively. Depending on how they are paid, they may benefit from lower prices for the biologics that they administer in their offices. In Medicare, physician-administered drugs are reimbursed at “average sales price” (ASP), plus a fixed percentage. Biosimilars are paid at a blended, biosimilar-specific ASP of all biosimilars that share a common reference biologic drug, plus the fixed percentage of the more-expensive reference biologic. The reference biologic drug is reimbursed based on its own ASP. As a result, providers could be reimbursed at a lower rate for the biosimilar than their purchase price, depending on the specific biosimilars that they stock. This could serve as a disincentive to administer biosimilars over reference biologics. Hospitals and other facilities purchase the biologics that are administered in the inpatient and outpatient facility settings and could also benefit from lower prices, depending on the biosimilars that they stock.
Whether and for how long physicians and facilities benefit from cost savings hinges on insurers. If insurers aggressively lower payment levels for biologics to those of biosimilars or incentivize biosimilars directly, the savings will accrue to insurers, rather than providers. Insurers may, however, be slow to reduce payment levels. In some cases (e.g., prospective payment for inpatient healthcare), insurers make a single payment for a bundle of services. It may be challenging to adjust payment rates to reflect lower biologic prices in those cases.
Patients are often subject to cost-sharing for biologics. Many biologics are “specialty drugs” that are on separate specialty formulary tiers with co-insurance rates of 20 to 35 percent. Cost-sharing often applies to self-administered biologics, and can also apply to physician-administered biologics, depending on the patient's insurance coverage. Deductibles and co-pays also factor into total patient out-of-pocket spending. In most cases, and especially when co-insurance plays a major role in patient out-of-pocket spending, lower biologic prices will benefit patients.29
Since biosimilars first entered the market, insurance plans and PBMs have leveraged new competition to negotiate lower prices on biologics in the same class and have, in some cases, restricted reimbursement for original reference biologics altogether. For example, one major PBM, Express Scripts, recently listed Zarxio® and Granix®, but not Neupogen® (the reference biologic for Zarxio®), on its 2018 National Preferred Formulary,30 while another large PBM, CVS Caremark, restricted coverage of Neupogen® in July 2017.31
The Future of the U.S. Biosimilars Market
Whether actual cost savings end up above or below our baseline estimate hinges in large part on whether manufacturers continue to have a business case to invest in developing and marketing biosimilars. Several key challenges and sources of uncertainty remain, which we discuss in this section.
IP: Manufacturers with FDA-approved biosimilars have already delayed market entry to avoid patent litigation. While some litigation (including the suit brought by Amgen against Sandoz that was recently decided by the U.S. Supreme Court32) focused more on procedural issues, other ongoing litigation (for example, between Novartis and Amgen related to patents protecting Amgen's Enbrel® and between Amgen and AbbVie related to patents protecting Humira®) will deal with potential infringement by the biosimilar manufacturer of one or more patents held by the reference biologic manufacturer. Evolving litigation strategies on both sides—biosimilar and reference biologic—will affect the timing of future biosimilar launches.
Payment: Payment arrangements for biologics in general depend on whether the drug is covered under a plan's medical or pharmacy benefit, how the drug is administered, and other factors (see Box 4). To date, Medicare and most commercial payers are evaluating and applying variations on these established payment approaches to biosimilars. While Medicare pays for Part B biosimilars on a fee-for-service basis as it does for other physician-administered drugs, its approach of consolidating biosimilars to the same reference biologic in a single Part B blended payment rate is a new, biosimilar-specific policy. There are some concerns that Medicare's treatment of biosimilars covered under Part D could actually incentivize the use of reference biologics over less-expensive biosimilars.33 More broadly, payers are experimenting with new payment approaches, including value-based purchasing and bundled payment, both of which have been discussed in the context of Medicare payment of self-administered and physician-administered biologics.34 Changes in broader payment models may have profound impacts on physician and facility incentives to shift to biosimilars, leading to changes in utilization and spending. In many cases, the availability of lower-cost biosimilars may help providers respond to cost-control incentives put in place by payment policies and programs. For example, by including physician-administered drugs in the range of services covered by Medicare's Bundled Payments for Care Improvement (BPCI) initiative,35 participating providers might have more of an incentive to use lower-cost biosimilars to keep costs down.36
Box 4.
Common Payment Arrangements for Biologics
Self-administered, pharmacy-dispensed biologics: Most self-administered outpatient drugs are paid for on a fee-for-service basis and the final amount paid by insurers reflects several transactions, including a confidential rebate payment. First, wholesalers and pharmacy chains purchase biologics from manufacturers at market rates. Patients then obtain biologics from pharmacies, often with cost-sharing in the form of a co-pay or co-insurance. Next, pharmacies bill insurance companies for the net market rate of cost-sharing, plus a dispensing fee. As a final step, manufacturers often deliver a rebate payment to insurers in exchange for favorable placement on the insurers' formulary.
Biologics used in the inpatient facility setting: Most inpatient procedures are paid on a prospective, bundled basis (through, e.g., diagnosis-related group payments). The costs associated with biologics administered in inpatient settings are incorporated into these prospective payments. Facilities purchase and stock biologics directly from manufacturers and wholesalers or through Group Purchasing Organizations (GPOs). Health care professionals may bill separately for administration of the drug and related services.
Biologics used in the outpatient facility setting: As in the inpatient hospital setting, in most cases facilities purchase drugs for use in an outpatient setting from manufacturers and wholesalers or through GPOs. Some facilities are eligible to use the 340B Drug Discount Program28 to obtain biologics for outpatient use at reduced prices. Unlike the inpatient setting, many insurers pay for biologic drugs separately under fee-for-service arrangements. Some low-cost biologics are “packaged” into Medicare outpatient hospital payments for other services and are not separately reimbursed. Health care professionals may bill separately for administration of the drug and related services.
Biologics administered in the physician office setting: Physician offices purchase drugs directly from wholesalers and manufacturers or through GPOs. Medicare pays physicians a fixed-fee-schedule–based price (ASP, which is reported to the Centers for Medicare and Medicaid Services [CMS] by manufacturers), plus a margin to cover acquisition and stocking costs. Currently, Medicare reimburses the cost of all biosimilars that share a common reference biologic drug based on their blended ASP, plus a fixed percentage of the reference biologic's ASP. Commercial insurers also use a cost-plus-margin payment approach, although the base and margin can differ from the Medicare rates. In most cases, physicians bill separately for administration of the drug and related services.
Price competition and switching: The prices paid for drugs by most insurers hinge on negotiated rebates between the insurer (or a separate PBM) and drug manufacturers. In general, manufacturers are willing to give larger discounts if insurers can successfully steer significant prescription volume to their products. Insurers' willingness and ability to shift utilization to a specific drug may be different for biosimilars compared with other drugs. There are potential concerns that switching from one drug (such as a reference biologic) to a similar drug (such as a biosimilar) for a nonclinical reason may have safety and other consequences for patients.37 On the other hand, insurers routinely change benefit design and preferred drug designations to reduce spending on prescription drugs, which can benefit patients. Because none of the biosimilars on the market now are interchangeable with their reference biologics, the extent of price reductions depends as much on insurers' willingness to use utilization management and other tools to shift volume as it does on manufacturers' willingness to provide discounts. The fact that large PBMs recently changed their formularies, making Zarxio® preferred over Neupogen®, suggests that there are both cost savings and effective tools to promote biosimilar uptake.38
Non-price competition from reference biologic manufacturers: Biologic manufacturers in some markets are developing next-generation biologics that offer improvements over their older reference biologics facing the potential of biosimilar competition. These next-generation biologics will compete with biosimilars and older reference biologics for market share. Whether payers, patients, and prescribers will switch to these next-generation biologics rather than to biosimilars depends on their relative safety, efficacy, convenience, and cost. In addition to the development of new products, manufacturers of originators and biosimilar drugs may differentiate their products by offering value-added services, such as patient support and medication therapy management.
Naming conventions: A vigorous debate has been taking place regarding the sharing of a nonproprietary name between reference and biosimilar products (which could lead to inadvertent substitution when a biosimilar is not designated as interchangeable) and the requirement to include the suffix in biosimilar products only (which could incorrectly signal lower efficacy and safety standards and lead to a lower uptake by patients and providers).39 According to an FDA guidance,40 biological drugs licensed under the Public Health Service Act—including biosimilars and other biologics—will bear a name that is “a combination of the core name and a distinguishing suffix that is devoid of meaning and composed of four lowercase letters.”41 It is not yet known with certainty how the new naming convention will affect perceptions and acceptance of biosimilars, although prescribers and patients could perceive lower or different efficacy and safety for biosimilars with a suffix until suffixes are also added to reference biologic names.42
Interchangeability: The potential for cost savings from biosimilars could increase when at least one biosimilar is determined by the FDA to be interchangeable with the reference biologic. Patients and prescribers could view an interchangeable biosimilar as a closer substitute to a reference biologic even though all FDA-approved biosimilars (interchangeable or not) are highly similar to reference biologics without clinically meaningful differences in safety, purity, and potency. None of the currently approved or marketed biosimilars has this designation, and the FDA has only recently released draft guidance to industry outlining what would be required to receive the interchangeable designation.43 The eventual advent of interchangeable biosimilars may signal changes in competition, payment, and delivery. Then again, insurers in particular may already have adequate incentives and tools to shift patients to biosimilars, even without the interchangeability designation.
Is Policy Change Necessary?
The pervasive uncertainty in the U.S. biosimilar market—including questions as to whether the market will be sustainable and lead to cost savings, as intended—presents two choices for policymakers. One strategy is to let the market continue to develop under current policies. Increasing FDA and industry experience with approval requirements, precedent through early legal decisions, and evolving pricing and market-share trends will eventually provide clarity on the stability of the U.S. biosimilar market and the significance of biosimilars to the health care system.
As an alternative, policymakers could choose to intervene to help steer the U.S. biosimilar market more quickly to a sustainable, competitive state. For example, regulators at the FDA could experiment with new approaches to provide stronger, earlier signals through guidance documents or other mechanisms on expectations surrounding interchangeability and other topics. The current FDA commissioner, Scott Gottlieb, commented on the need for timely FDA biosimilar regulatory decisions prior to his nomination.44 The FDA only recently released a draft guidance focusing on interchangeability, and it is not clear when a final guidance will be released.45 This gradual approach to policymaking offers some advantages, including the benefit of “learning by doing” through early biosimilar applications and other FDA action, but on the other hand, it makes it difficult for manufacturers to anticipate the FDA's requirements.
In terms of payment, Medicare's drug payment policies could be reworked to more actively incentivize biosimilar uptake. A recent proposal for Medicare Part B physician-administered biologics from the Medicare Payment Advisory Commission (MedPAC), an independent government entity that advises CMS on payment issues and policy, suggests switching to a single consolidated payment rate based on a blend of reference biologic and biosimilar prices.46 While this policy and Medicare's current policy of blending only biosimilar payment could catalyze price competition, there is a risk that a “race to the bottom” in terms of price could shrink or eliminate incentives for manufacturers to invest in developing new biosimilars. An alternative would pay for each biosimilar based on its individual ASP, plus the fixed add-on percentage of the reference biologic, which could dampen price competition comparatively in the short term but attract more biosimilar manufacturers to the market and potentially generate greater savings in the long term. Another option would be to pay providers under current policy but with a bonus payment (i.e., an additional percentage or flat fee on top of the fixed percentage calculated from the reference biologic's ASP) to incentivize biosimilar uptake. Any change to payment policy would require careful set-up and implementation to ensure that the right balance of incentives is in place.
Policymakers could also target several Medicare Part D policies to ensure that patients and providers have the right incentives to use lower-cost biosimilars, including lowering or eliminating cost-sharing for biosimilars47 and applying the “gap discount” to Part D biosimilars. While changes to Medicare's handling of Part B and Part D biosimilars may target just one segment of the broader U.S. health care system, in many cases the policies implemented by Medicare spill over and are implemented by other public and private insurers. Commercial insurers have other tools at their disposal—including formulary design and utilization management tools—to introduce price competition and incentivize biosimilar uptake, leading to savings.
Beyond FDA regulation, payment, and coverage, both government and industry could play a role in educating patients and providers about the potential cost savings from biosimilars, much like both groups have done for generic drugs. Investments in education and outreach will complement “under-the-hood” changes in payment and FDA regulation and will help the current administration achieve its goal of lowering drug spending.48 While our study does not address whether policy action is needed now, it is likely that the answer will become clearer over the next one to three years as the market continues to develop.
Conclusion
We estimated the cost savings potential of biosimilars to be $54 billion over ten years using recent baseline data and transparent assumptions, with a lower- to upper-bound range of $25 billion to $150 billion. Actual savings will hinge on an evolving biosimilar regulatory and competitive landscape. Payment arrangements, regulatory policies and guidance, patient and prescriber acceptance of biosimilars, and other issues will also influence the magnitude of potential savings. Savings will accrue to a range of stakeholders in the short term, although patients and taxpayers will benefit in the long term. Future research as additional biosimilars are marketed in the United States will help us to assess whether the BPCIA achieved competition and cost savings through the creation of a biosimilar regulatory approval pathway.
Notes
The research described in this article was sponsored by Sandoz, a Novartis Company, and conducted within RAND Health.
QuintilesIMS, “QuintilesIMS Institute Study: U.S. Drug Spending Growth of 4.8 percent in 2016,” press release, May 5, 2017. As of September 27, 2017: https://www.quintilesims.com/press-releases/quintilesims-institute-study-us-drug-spending-growth-of-48-percent-in-2016
Steve Miller, “The $250 Billion Potential of Biosimilars,” Express Scripts, April 23, 2013. As of September 27, 2017: http://lab.express-scripts.com/lab/insights/industry-updates/the-$250-billion-potential-of-biosimilars
Allan Coukell, “Specialty Drugs and Health Care Costs,” Pew Charitable Trusts, Drug Spending Research Initiative, November 16, 2015. As of September 27, 2017: http://www.pewtrusts.org/en/research-and-analysis/fact-sheets/2015/11/specialty-drugs-and-health-care-costs
National Pharmaceutical Services, “Specialty Drug Spending,” white paper, undated. As of September 27, 2017: https://www.pti-nps.com/nps/wp-content/uploads/2017/04/NPS_Specialty-Medication-White-Paper-Web.pdf
Public Law 98–417, The Drug Price Competition and Patent Term Restoration Act, 1984. This act is more commonly known as the Hatch-Waxman Act.
FDA, “Guidances (Drugs): Biosimilars,” web page, September 21, 2017. As of September 27, 2017: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm290967.htm
Center for Drug Evaluation and Research, “List of Licensed Biological Products with (1) Reference Product Exclusivity and (2) Biosimilarity or Interchangeability Evaluations to Date,” undated. As of September 27, 2017: https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM560162.pdf
Janet Woodcock, Peter Marks, and Jeffrey Shuren, “Prescription Drug User Fee Act Reauthorization (PDUFA VI), Medical Device User Fee Act Reauthorization (MDUFA IV), Generic Drug User Fee Act Reauthorization (GDUFA II), and Biosimilar User Fee Act Reauthorization (BsUFA II),” testimony before the U.S. Senate Committee on Health, Education, Labor, and Pensions, Washington, D.C., March 21, 2017. As of September 27, 2017: https://www.fda.gov/NewsEvents/Testimony/ucm547898.htm
Andrew W. Mulcahy, Zachary Predmore, and Soeren Mattke, The Cost Savings Potential of Biosimilar Drugs in the United States, Santa Monica, Calif.: RAND Corporation, PE-127-SANI, 2014. As of September 27, 2017: https://www.rand.org/pubs/perspectives/PE127.html
PBMs design and administer prescription drug programs under contract for some commercial and public health insurers. Other insurers perform the functions of a PBM in-house. We use the term insurer to refer both to those who perform PBM functions internally and to those who contract with a separate PBM.
Mulcahy, Predmore, and Mattke, 2014.
U.S. Supreme Court, Sandoz Inc. v Amgen Inc., No. 15-1039, June 12, 2015.
FDA, “Considerations in Demonstrating Interchangeability with a Reference Product: Guidance for Industry,” Washington, D.C.: U.S. Department of Health and Human Services, draft guidance, January 2017a. As of September 27, 2017: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf
Richard Cauchi, “State Laws and Legislation Related to Biologic Medications and Substitution of Biosimilars,” web page, National Conference of State Legislatures, August 1, 2017. As of September 27, 2017: http://www.ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx
This experience includes situations in which multiple, similar drugs were approved in the United States through the full New Drug Application (NDA) or Biologics License Application, or via the 505(b)(2) “paper” NDA pathway.
Mulcahy, Predmore, and Mattke, 2014.
The IMS MIDAS extract available for this analysis reports ex-manufacturer volume and sales (excluding rebates and discounts) across all distribution channels for individual biologic products. IMS collects these data through audits and projects both volume and sales to the national level. Note that IMS was recently acquired by Quintiles and is now referred to as QuintilesIMS.
While Granix® is not a biosimilar (i.e., it was not approved through the biosimilar regulatory approval pathway), it is the second entrant in the filgrastim market and competes directly with the first brand-name filgrastim, Neupogen®, and with the biosimilar, Zarxio®.
Sales reported in IMS MIDAS do not reflect rebates or discounts. If rebates and discounts are similar in relative terms for biosimilar and reference biologics, then the actual share in terms of sales post-rebates and discounts will be similar to the result reported here.
FDA, “FDA Approves First Biosimilar Product Zarxio,” press release, March 6, 2015. As of September 28, 2017: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm436648.htm
RAND analysis of QuintilesIMS data.
These results could be different if the manufacturers of Neupogen®, Granix®, or Zarxio® introduce significantly different levels of rebates or other discounts.
We used 2016 QuintilesIMS MIDAS data for 101 individual biologic drugs. These drugs included all products in major biologic Anatomical Therapeutic Classification classes (e.g., insulins, growth hormones, interferons, anti-tumor necrosis factor alpha products, and monoclonal antibody neoplastics) as well as select biologics in other classes (e.g., other antineoplastics). We excluded vaccines and blood products due to unique manufacturing and market considerations for these products.
Note that the magnitudes of these baseline assumptions are based on our literature review and expert opinion. They happen to be relatively close to what we observe in the filgrastim market.
The ten-year period is from 2017 to 2026. We chose 2016 as a base period because it was the last year for which complete status quo sales data were available.
Our earlier baseline estimate (from Mulcahy, Predmore, and Mattke, 2014) was smaller in magnitude ($44.2 billion from 2014 through 2024), but it accounted for a larger share of biologic spending (4.0 percent). The increase on one hand but decrease on the other between the prior and current estimates is due to a growing overall biologic market. Several changes to our methodology—including more-granular assumptions on biosimilar entry timing and probabilities—led to lower savings in relative terms in the current estimate. The wider range between lower- and upper-bound assumptions in the current estimate compared with the prior estimate reflects ongoing uncertainty in how biosimilar markets will evolve over time.
A biosimilar to infliximab is now marketed and biosimilars to adalimumab and etanercept are FDA approved but not yet marketed.
The 340B Drug Discount Program creates a ceiling price for outpatient drugs that is based on Medicaid's payment rate for drugs. 340B pricing is available to a variety of safety-net provider organizations, including federally qualified health centers and disproportionate share hospitals.
Biosimilars covered by Medicare Part D (which covers primarily patient-administered biologics, such as insulins) are a potential exception because they are not eligible for a “gap discount” when patients are in the coverage gap (also known as the “donut hole”). The gap discount helps patients reach the catastrophic coverage threshold more quickly. As a result, patients may have to pay more out of pocket before reaching catastrophic coverage. For beneficiaries eligible for low-income subsidies, the cost-sharing amount is the same for Part D biosimilars and reference biologics and patients therefore do not have a financial incentive to switch to a biosimilar.
Express Scripts, “2018 National Preferred Formulary,” undated. As of September 28, 2017: https://www.express-scripts.com/art/pdf/npf2018.pdf
CVS Caremark, “Formulary Drug Removals,” July 2017. As of September 28, 2017: https://www.caremark.com/portal/asset/Formulary_Exclusion_Drug_List.pdf
Sandoz Inc. v. Amgen Inc., 2015.
More specifically, biosimilars are treated like small-molecule generic drugs and are therefore ineligible for a discount when patients are in the coverage gap. As a result, Medicare Part D plans face lower spending when the patient uses a reference biologic.
Medicare Payment Advisory Commission, “Chapter 5: Medicare Payment Systems and Follow-on Biologics,” in Report to the Congress: Improving Incentives in the Medicare Program, 2009.
CMS, “Bundled Payments for Care Improvement (BPCI) Initiative: General Information,” September 25, 2017. As of September 28, 2017: https://innovation.cms.gov/initiatives/bundled-payments
However, Medicare Part D drugs, including some patient-administered biologics and potential biosimilars, are out of scope for BPCI.
Elaine Nguyen, Erin R. Weeda, Diana M. Sobieraj, Brahim K. Bookhart, Catherine Pak Piech, and Craig I. Coleman, “Impact of Non-Medical Switching on Clinical and Economic Outcomes, Resource Utilization and Medication-Taking Behavior: A Systematic Literature Review,” Current Medical Research and Opinion, Vol. 32, No. 7, March 8, 2016, pp. 1281–1290. As of September 28, 2017: http://dx.doi.org/10.1185/03007995.2016.1170673
PBMs also made Teva's Granix® preferred over Neupogen®, although Granix® is not a biosimilar.
Lauren Santye, “Debate Continues Around Biosimilar Naming Conventions,” Specialty Pharmacy Times, August 24, 2016. As of September 28, 2017: http://www.amcp.org/WorkArea/DownloadAsset.aspx?id=21373
FDA, “Nonproprietary Naming of Biological Products: Guidance for Industry,” Washington, D.C.: U.S. Department of Health and Human Services, January 2017b. As of September 28, 2017: https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf
For example, Pfizer's Inflectra®, a biosimilar to Remicade®, is named infliximab-dyyb. Note that filgrastim-sndz was assigned before this guidance was finalized.
Following the FDA's final guidance (FDA, 2017b), approved biologics (including nonbiosimilar biologics) will be assigned suffixes, although as of July 2017 suffixes had been added to previously approved nonbiosimilar biologics.
FDA, 2017a.
Scott Gottlieb and Gillian Woollett, “Helping Patients Benefit from Biotech Drugs,” Forbes, September 30, 2013. As of September 28, 2017: https://www.forbes.com/sites/scottgottlieb/2013/09/30/helping-patients-benefit-from-biotech-drugs/#4e46ad2a4335
FDA, 2017a.
Kim Neuman, Nancy Ray, and Brian O'Donnell, “Medicare Part B Drug Payment Policy Issues,” MedPAC briefing slides, April 6, 2017. As of September 28, 2017: http://www.medpac.gov/docs/default-source/default-document-library/part-b-drug-presentation-april-2017-for-public.pdf?sfvrsn=0; and Allan Coukell and Chuck Shih, “Can Biosimilar Drugs Lower Medicare Part B Drug Spending?” Pew Charitable Trusts, Drug Spending Research Initiative, January 3, 2017. As of September 28, 2017: http://www.pewtrusts.org/en/research-and-analysis/issue-briefs/2017/01/can-biosimilar-drugs-lower-medicare-part-b-drug-spending?utm_campaign=LM+-+GP+-+SDRI+-+Biosimilars+Medicare+-+12+28+16&utm_medium=email&utm_source=Pew
MedPAC, “Improving Medicare Part D,” in Report to the Congress: Medicare and Health Care Delivery System, Washington, D.C., June 2016, pp. 155–200. As of September 28, 2017: http://www.medpac.gov/docs/default-source/reports/chapter-6-improving-medicare-part-d-june-2016-report-.pdf
Scott Gottlieb, “First Remarks to FDA Staff,” speech given at FDA All Hands Meeting, Silver Spring, Md., May 15, 2017. As of September 28, 2017: https://www.fda.gov/NewsEvents/Speeches/ucm558566.htm; and Carolyn Y. Johnson, “Trump Administration Signals Initial Steps to Deal with Drug Prices,” Washington Post, June 21, 2017. As of September 28, 2017: https://www.washingtonpost.com/news/wonk/wp/2017/06/21/trump-administration-signals-initial-steps-to-deal-with-drug-prices/?utm_term=.a910be6ac0d1