

Abstract
Purpose
No standard treatment exists for patients with cholangiocarcinoma for whom first-line gemcitabine-based therapy fails. Fibroblast growth factor receptor 2 (FGFR2) fusions/translocations are present in 13% to 17% of intrahepatic cholangiocarcinomas. BGJ398, an orally bioavailable, selective pan-FGFR kinase inhibitor, has shown preliminary clinical activity against tumors with FGFR alterations.
Methods
A multicenter, open-label, phase II study (ClinicalTrials.gov identifier: NCT02150967) evaluated BGJ398 antitumor activity in patients age ≥ 18 years with advanced or metastatic cholangiocarcinoma containing FGFR2 fusions or other FGFR alterations whose disease had progressed while receiving prior therapy. Patients received BGJ398 125 mg once daily for 21 days, then 7 days off (28-day cycles). The primary end point was investigator-assessed overall response rate.
Results
Sixty-one patients (35 women; median age, 57 years) with FGFR2 fusion (n = 48), mutation (n = 8), or amplification (n = 3) participated. At the prespecified data cutoff (June 30, 2016), 50 patients had discontinued treatment. All responsive tumors contained FGFR2 fusions. The overall response rate was 14.8% (18.8% FGFR2 fusions only), disease control rate was 75.4% (83.3% FGFR2 fusions only), and estimated median progression-free survival was 5.8 months (95% CI, 4.3 to 7.6 months). Adverse events included hyperphosphatemia (72.1% all grade), fatigue (36.1%), stomatitis (29.5%), and alopecia (26.2%). Grade 3 or 4 treatment-related adverse events occurred in 25 patients (41%) and included hyperphosphatemia (16.4%), stomatitis (6.6%), and palmar-plantar erythrodysesthesia (4.9%).
Conclusion
BGJ398 is a first-in-class FGFR kinase inhibitor with manageable toxicities that shows meaningful clinical activity against chemotherapy-refractory cholangiocarcinoma containing FGFR2 fusions. This promising antitumor activity supports continued development of BGJ398 in this highly selected patient population.
INTRODUCTION
Cholangiocarcinoma, the second most common primary liver cancer, develops when bile duct cholangiocytes undergo neoplastic transformation into intrahepatic, perihilar, or distal extrahepatic tumors.1 Global prevalence is highest in Southeast Asia; in Korea, the incidence is approximately 10-fold higher than in the United States or Europe.2-4 Cholangiocarcinomas are often diagnosed at an advanced unresectable stage, with few treatment options available after disease progression while receiving gemcitabine and cisplatin first-line chemotherapy, resulting in a poor patient prognosis.
Heterogeneous genetic changes within cholangiocarcinomas and our increasing understanding of the functional consequences of these genetic modifications form the foundation for targeted therapeutics.5 Alterations in genes encoding fibroblast growth factor receptors (FGFRs), which regulate cell proliferation, survival, migration, and angiogenesis, can promote aberrant FGF pathway activation and tumorigenesis.6 FGFR translocations (ie, fusion events) represent driver mutations in cholangiocarcinoma. They are present in 13% to 17% of intrahepatic cholangiocarcinomas and may predict tumor sensitivity to FGFR inhibitors.7-9 In one study, FGFR pathway alterations were identified in seven (13%) of 55 patients with intrahepatic cholangiocarcinoma compared with one (5%) of 20 extrahepatic cases, with most alterations being mutations.10
BGJ398 is an orally bioavailable, selective, ATP-competitive pan-FGFR kinase inhibitor with activity against tumor models harboring FGFR alterations.11 In early-phase clinical evaluation, BGJ398 showed a tolerable safety profile and single-agent activity. Common adverse events included hyperphosphatemia, constipation, decreased appetite, and stomatitis.12,13 There is no established treatment approach for patients with cholangiocarcinoma and disease progression while receiving standard gemcitabine-based therapy. We herein report the results of a phase II study of BGJ398 in patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or other FGFR genetic alterations who have progressed on or were intolerant of cytotoxic therapy.
METHODS
Study Design and Participants
On the basis of BGJ398 activity against various cancer model systems and single-agent clinical activity against tumors with FGFR genetic alterations, a multicenter, open-label, phase II study was conducted to evaluate BGJ398 antitumor activity in patients with advanced or metastatic cholangiocarcinoma harboring FGFR genetic alterations whose disease progressed despite prior systemic therapy (Appendix Fig A1, online only).
Patients had histologically or cytologically confirmed advanced or metastatic intrahepatic or extrahepatic cholangiocarcinoma with FGFR2 fusions or other FGFR genetic alterations identified by local Clinical Laboratory Improvement Amendments–certified testing or at a central facility (Appendix Fig A1). All patients provided consent for genetic prescreening unless tumor molecular status was already established, and FGFR alterations were confirmed either locally or centrally by the sponsor. Patients provided written informed consent for study participation after molecular eligibility (prescreening) and before study-specific screening (performed < 21 days before treatment initiation). Approximately 55 patients were planned to be treated in this trial, of whom, approximately 40 patients were expected to harbor FGFR2 gene fusions and up to 15 patients were expected to have other FGFR genetic alterations
Patients were required to have evidence of measurable or evaluable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1,14 an Eastern Cooperative Oncology Group performance status of 0 or 1,15 and evidence of disease progression after one or more prior regimens of gemcitabine-based combination therapy or gemcitabine monotherapy. If prior treatment was discontinued because of toxicity, the patient was required to have persistent evidence of measurable or evaluable disease. Patients with hepatitis B or C virus were eligible. Exclusion criteria included cancer of the gallbladder or ampulla of Vater, prior or current treatment with an MEK inhibitor or selective FGFR inhibitor, current significant corneal or retinal disorder/keratopathy, history and/or current extensive tissue calcification or alterations in calcium or phosphate homeostasis, GI impairment or disease that could alter BGJ398 absorption, concurrent treatment with strong cytochrome P450 3A4 inducers/inhibitors, intake of foods with potential CYP3A4 interaction, or drugs or supplements that increase phosphorous or calcium levels, and participation in concurrent investigational drug or device studies.
Procedures
In this single-arm study, all patients received BGJ398 125 mg once daily for 21 days followed by 7 days off in 28-day cycles on the basis of phase I trial experience.12 To manage hyperphosphatemia, prophylactic use of sevelamer, a phosphate-binding agent, was recommended on days of BGJ398 administration per the product packaging information and institutional guidelines. Patients were also instructed to adhere to a low-phosphate diet.
Patients continued BGJ398 treatment until unacceptable toxicity, disease progression, and/or investigator discretion, or consent withdrawal. Dose modifications were based on the worst preceding toxicity. Treatment was resumed after resolution or reduction to grade 1 toxicity, with each patient allowed two dose reductions (100 mg, 75 mg) before BGJ398 discontinuation.
Outcomes
Tumor response to BGJ398 was assessed per RECIST version 1.114 using computed tomography or magnetic resonance imaging. The primary study end point was overall response rate (ORR; complete response plus partial response) as assessed by local site radiographic review. Secondary efficacy end points included progression-free survival (PFS) and disease control rate (DCR; complete response plus partial response plus stable disease). Adverse events (AEs) were assessed according to the Common Terminology Criteria for Adverse Events, version 4.03,16 during treatment and until 30 days after the last dose was administered.
FGFR genetic alteration was required to confirm patient eligibility. These and other concurrent genetic alterations were correlated with clinical outcome. CA 19-9 values were measured at baseline and while receiving treatment to explore the effects of BGJ398 treatment. Investigators were given central laboratory–supplied kits for sample collection, including archival or newly obtained tumor tissue, and shipping for next-generation sequencing mutational analysis at Foundation Medicine (Cambridge, MA).
Statistical Analysis
Data were combined from all participating study sites for the analyses. Summary data are reported for patient baseline demographics and efficacy and safety data. Categorical data are presented as frequencies and percentages, and continuous data are presented as the median and range, unless otherwise stated. The Kaplan-Meier method was used to analyze PFS. Efficacy analyses included all patients who received at least one BGJ398 dose (full analysis set). Safety analyses included all patients who received at least one dose of study treatment and had at least one postbaseline safety assessment (safety set). Additional statistical analysis details and sample size justification are provided as online supplementary content.
Study Oversight
The study protocol was approved by the appropriate ethics committee or institutional review board at each study site. The study was conducted in accordance with Guidelines for Good Clinical Practice, following applicable local regulations and the ethical principles of the Declaration of Helsinki. All patients provided written informed consent before enrollment. Participating study sites and Novartis personnel promptly reviewed safety data and continually monitored for emerging safety signals.
RESULTS
Sixty-one patients (35 women, 26 men; median age, 57 years) participated in this study (Table 1). The majority of patients were white. All patients had received prior antineoplastic therapy (67.2% had received at least two prior regimens), and the majority (82%) initiated study treatment within 6 months of the end of their last antineoplastic medication. All patients had measurable disease.
Table 1.
Baseline Patient Demographics and Clinical Characteristics (N = 61)
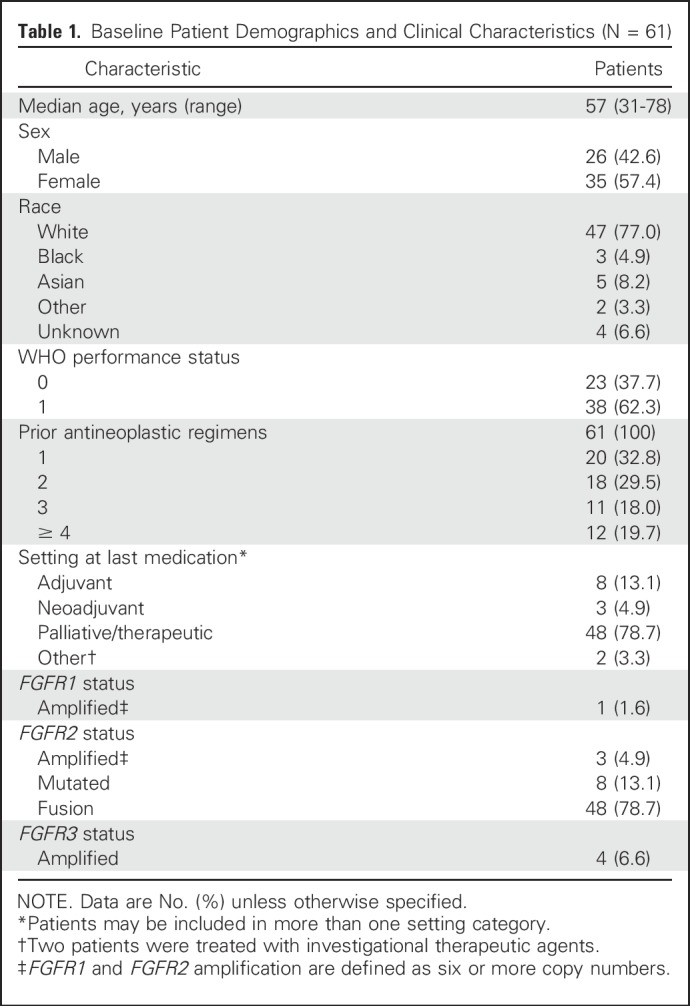
Most patient tumors harbored FGFR2 genetic alterations, including FGFR2 fusions (n = 48), mutations (n = 8), and amplifications (n = 3; Table 1). FGFR1 and FGFR3 amplifications were detected in one patient and four patients, respectively. Among the 14 tumors with sufficient tissue available for repeat testing, FGFR alterations identified at local sites were confirmed centrally by next-generation sequencing in 11 patients and partially confirmed in one patient with locally identified FGFR2 amplification and FGFR2 mutation. Central analysis confirmed the FGFR2 amplification and newly identified an FGFR2 fusion but did not detect an FGFR2 mutation.
The median duration of exposure to BGJ398 was 4.7 months, with a range of exposure from 1 day to 13.6 months (Table 2). Most patients (62.3%) required a dose reduction while receiving therapy (Appendix Table A1, online only) to achieve an extended duration of exposure (Fig 1A). Patients required a median of two (range, one to three) dose reductions, with the majority of reductions made due to an AE (37.7%) or per protocol (26.2%). A total of 47 patients (77%) required a BGJ398 dose interruption during the study, with patients experiencing a median of three (range, one to 12) interruptions, mostly because of an AE (70.5%; Appendix Table A1).
Table 2.
Response to Treatment (full analysis set; N = 61)
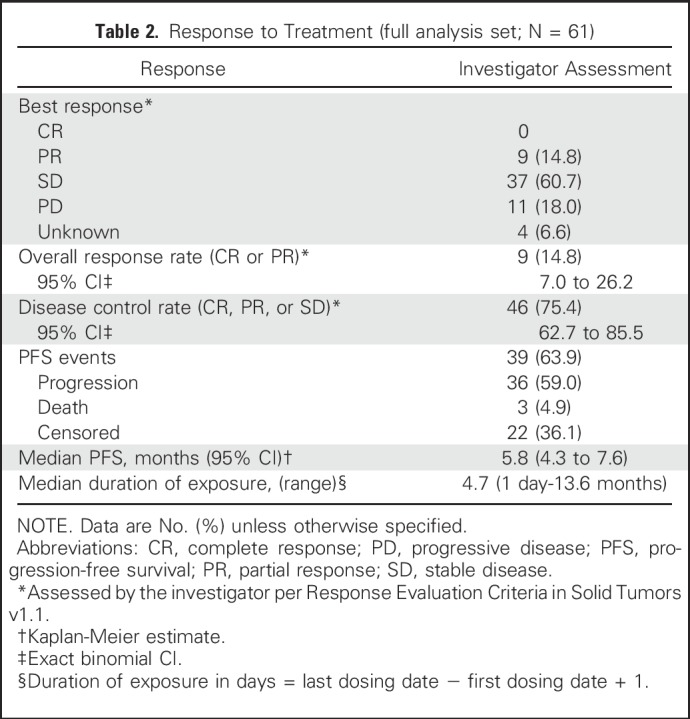
Fig 1.

(A) Duration of exposure. Swimplot presenting duration of exposure, dosing history, and efficacy assessments. (B) Best percentage change in sum of longest tumor diameters from baseline. Only patients with baseline and at least one postbaseline assessment are included. N, number of patients who received at least one study treatment; n, number of patients with baseline and at least one postbaseline assessment.
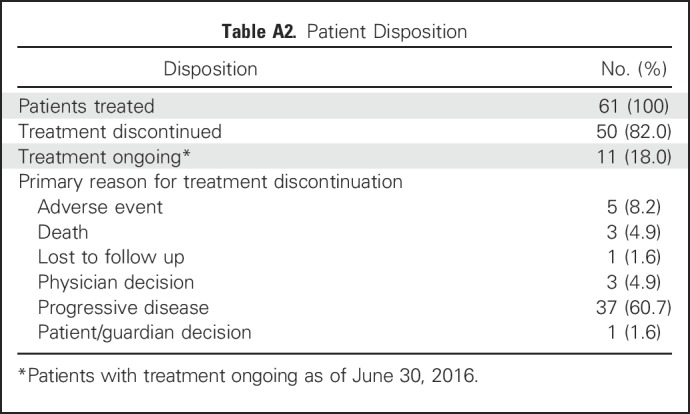
At the time of this analysis (June 30, 2016), 50 patients (82%) had discontinued BGJ398 treatment (Appendix Table A2, online only), most commonly due to progressive disease (60.7%). ORR, the primary efficacy end point, was 14.8% (95% CI, 7.0% to 26.2%; Table 2). BGJ398 achieved a DCR of 75.4%, with 39 PFS events. The median duration of disease control (among the 46 patients who achieved a best overall response of stable disease, partial response, or complete response) was 7.5 months (95% CI, 5.6 to 7.6 months), as estimated by the Kaplan-Meier method (24 disease progressions, one death, and 21 censored, either because they were still receiving treatment without disease progression at the time of data cutoff, or initiated new antineoplastic therapy). The degree of disease control is reflected in the best change in tumor size (Fig 1B), for which the majority of patients showed some tumor regression. As of the cutoff date, the median PFS was estimated at 5.8 months (95% CI, 4.3 to 7.6 months; Table 2 and Fig 2). Thirty-nine of the 61 patients (63.9%) had a PFS event (three deaths and 36 patients whose disease progressed).
Fig 2.

Progression-free survival (full analysis set). Kaplan-Meier plot of progression-free survival as per investigator assessment according to Response Evaluation Criteria in Solid Tumors v1.1.
The response rate among patients with tumors bearing FGFR2 fusions was 18.8% (nine of 48 patients with a partial response), and the DCR was 83.3% (40 of 48 patients). Thirty-six (75%) of the 48 patients with tumors bearing FGFR2 fusions showed reduced target lesion size in at least one disease evaluation. None of the four patients with tumors bearing FGFR3 amplification responded to BGJ398. One tumor with an FGFR2 mutation showed a 23% reduction in size, and another tumor with FGFR2 amplification was reduced by 27%.
All (> 5% frequency) and grade 3 and 4 AEs suspected to be study drug–related are reported in Table 3. AEs were mostly predictable on the basis of prior clinical experience with BGJ39812 and were largely reversible. Treatment-emergent hyperphosphatemia reflected BGJ398 on-target treatment effects and was the most commonly reported AE suspected as potentially treatment related (72.1% all grade; 16.4% grade 3 or 4). Hyperphosphatemia also prompted study drug adjustment or temporary interruption in 26 patients (42.6%). Other frequent all-grade treatment-related AEs included fatigue (36.1%), stomatitis (29.5%), and alopecia (26.2%). Nail and eye toxicities were also reported, which included dry eye (21.3%), blurred vision (14.8%), and onychomadesis (18%). Twenty-five patients (41%) experienced a grade 3 or 4 AE suspected to be related to treatment (Table 3). Among the safety set, 23 patients (37.7%) experienced at least one serious AE. The most frequently reported serious AEs included abdominal pain (4.9%) and anemia, ascites, cellulitis, cholangitis, constipation, hepatic failure, hypercalcemia, hyponatremia, hypophosphatemia, and sepsis (all 3.3%).
Table 3.
All (> 5%) and Grade 3 and 4 AEs Suspected to be Study Related (N = 61)
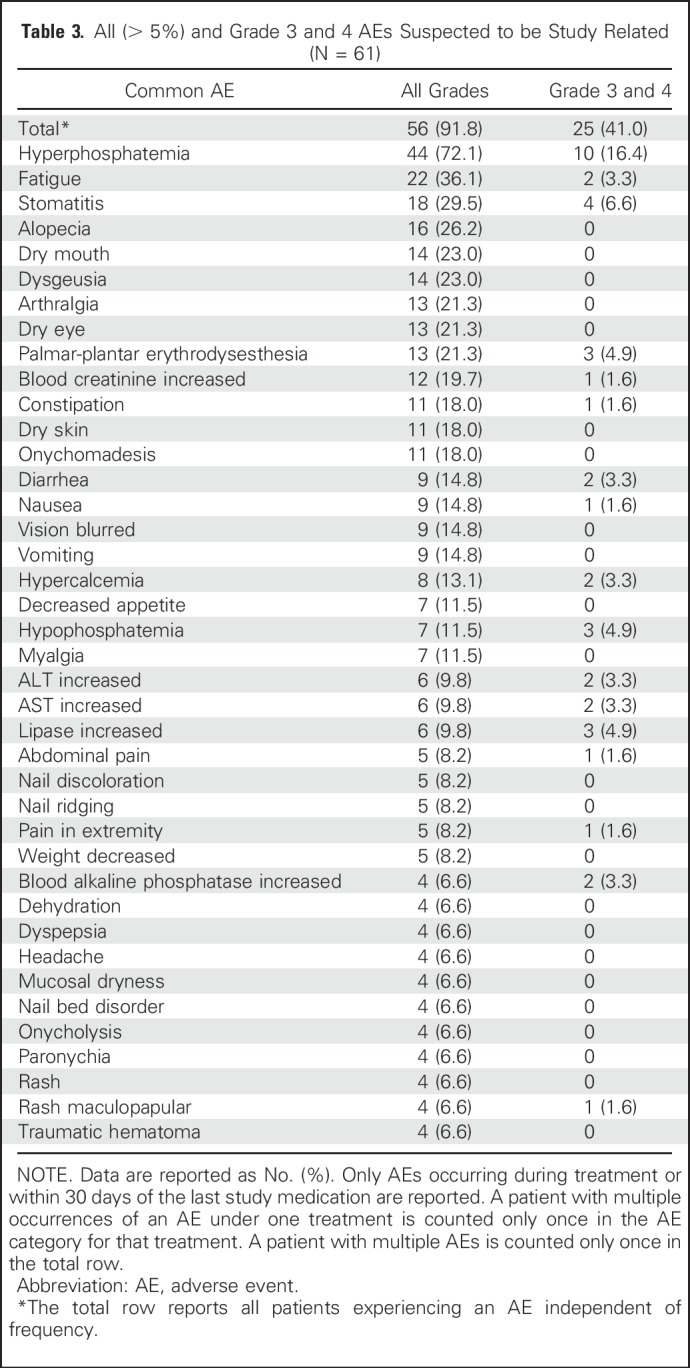
Frequent grade 1 laboratory abnormalities included elevations in ALT (51.2%), AST (56.7%), creatinine (93.9%), and magnesium (40.0%). New or worsened grade 1 hematology abnormalities included elevated hemoglobin (66.7%), leukocytes (18.2%), absolute neutrophils (6.5%), and platelets (41%). A total of five patients (8.2%) discontinued BGJ398 treatment because of AEs. Five patients died on study (four as a result of disease progression and one as a result of an ischemic bowel).
Tumor genomic alterations were assessed (Appendix Fig A2A, online only) and compared with historical and Cancer Genome Atlas data on cholangiocarcinoma (Appendix Fig A2B). Consistent with previous reports suggesting that IDH and FGFR mutations are mutually exclusive,8,10,13,17 the one patient with an IDH1 mutation was noted to have an FGFR2 W290C variant but no FGFR2 fusion. Potential association, positive or negative, between treatment efficacy and frequent gene alterations (excluding FGFR alterations) was also analyzed. No statistically significant association was detected between antitumor activity (best overall response or PFS) and gene alterations.
Serum CA19-9 concentration was also measured sequentially to assess the relationship between BGJ398 antitumor activity and changes in CA19-9 biomarker expression. Among the 30 patients with postbaseline data for both CA19-9 and lesion measurements, 80% had a best change in tumor size that was directionally similar to the best change in serum CA19-9 concentration with treatment (Appendix Fig A3, online only).
DISCUSSION
Cholangiocarcinoma is a rare disease that generally presents at an advanced stage. Treatment options after disease progression while receiving first-line gemcitabine with or without cisplatin chemotherapy are limited,18 because of tumor heterogeneity and resistance to currently available therapies.7,19 Therefore, advanced cholangiocarcinoma has poor prognosis with limited treatment options. Nontargeted therapeutics typically show dismal efficacy in these patients, with response rates in the single digits and an approximate 3-month median PFS.20,21
This phase II study evaluated the efficacy of BGJ398, a first-in-class pan-FGFR kinase inhibitor, in patients with advanced or metastatic cholangiocarcinoma and FGFR2 fusions or other FGFR alterations whose disease progressed after cytotoxic therapy or who were intolerant to treatment. At the time of data cutoff (June 30, 2016), BGJ398 administered at the recommended phase II dose of 125 mg once daily on a 3-weeks-on/1-week-off schedule demonstrated an ORR of 14.8% and a DCR of 75.4%, with a predictable and manageable safety profile. This study provides the largest clinical database of patients with cholangiocarcinoma and FGFR alterations.
Commonly reported AEs included hyperphosphatemia, fatigue, eye and fingernail toxicities, and stomatitis. A similar safety profile was observed in the BGJ398 phase 1 trial12 and with JNJ-42756493, a pan-FGFR inhibitor.22 Hyperphosphatemia was the most commonly observed AE at 72.1%, which was similar to the 74.2% rate observed previously.12 Hyperphosphatemia is believed to represent an on-target treatment effect on the basis of the importance of FGFR pathway signaling in FGF23-mediated phosphate homeostasis.23,24 Intermittent dosing (3-weeks-on/1-week-off), prophylactic use of a phosphate-lowering agent, active monitoring, and early intervention effectively mitigated the effects of hyperphosphatemia and helped patients continue to receive therapy. Although the majority of patients required some level of dose modification, dose reduction, and/or dose delay, particularly for managing nail toxicities, effective mitigation strategies helped achieve a median 4.7 months of therapy.
These promising preliminary results and reports of targetable tumor FGFR genetic alterations25,26 support FGFR inhibitor therapy as a viable therapeutic option in advanced biliary tract malignancies. Although the duration of disease control is encouraging, all patients ultimately will experience disease progression, which suggests the emergence of acquired resistance to FGFR inhibition. Indeed, serial analysis of cell-free circulating DNA in a subset of patients from this trial demonstrated the emergence of recurrent secondary resistance mutations in the FGFR2 kinase domain at disease progression.25 These results demonstrate the FGFR-dependent state of the tumors and the on-target activity of BGJ398. Collectively, these data support the further clinical evaluation of BGJ398 for the treatment of patients with intrahepatic cholangiocarcinoma and in the presence of FGFR2 alterations.
Comprehensive genomic profiling of cholangiocarcinoma has revealed extensive and complex genetic heterogeneity, which provides a strong rationale for developing targeted therapies in this disease.5,8,10,27,28 A variety of FGFR genetic alterations, including fusions, mutations, and amplifications, can alter FGFR kinase activity and result in constitutively active FGFR signaling that promotes cell proliferation and inhibits apoptosis. From genomic sequencing of tumor DNA, FGFR2 fusion events were identified in 13% of intrahepatic cholangiocarcinomas.9 The majority of the population treated in the current study (78.7%) had a documented FGFR2 fusion. Although other FGFR changes and other genetic alterations were also detected, enrollment of only a few patients with FGFR alterations other than FGFR2 fusions limited the accurate estimate of BGJ398 activity in these patients. Detection of multiple different FGFR2 alterations likely reflects tumor clonal heterogeneity and confirms FGFR2 as an important oncogenic driver.
Certain FGFR alterations, such as FGFR2 fusions in cholangiocarcinoma and FGFR3 mutations and/or fusions in bladder/urothelial carcinoma, are regarded as dominant oncogenic drivers that enable malignant transformation.9 In particular, gene fusions can induce malignancy and alter cell homeostasis in tumors.7,26 These genetic changes also confer sensitivity to BGJ398-mediated FGFR inhibition, are altered more frequently in cholangiocarcinomas than other genetic tumor drivers such as ERBB2 amplification and ROS1 fusions,7 and occur almost mutually exclusive of KRAS, IDH1, and BRAF mutations. These factors suggest that certain FGFR alterations may serve as biomarkers for personalized cancer therapy.7,26 Serum CA19-9 concentration was also associated with BGJ398 antitumor activity, in agreement with a previously reported correlation between serum CA19-9 and clinical outcome in patients with cholangiocarcinoma.29,30 However, additional or alternative biomarkers may better predict response to BGJ398. A larger patient population is required to determine if other genetic changes can serve as markers predictive of response to BGJ398 therapy.
This study demonstrates that a prospective trial evaluating targeted treatment efficacy in a genetic subset of patients with cholangiocarcinoma is feasible and can produce promising clinical findings. A median PFS of 5.8 months in this patient population is encouraging. Single-agent BGJ398 antitumor activity was observed in patients with cholangiocarcinoma harboring different types of FGFR2 genetic alterations, strongly supporting its further biologic and clinical investigation.
ACKNOWLEDGMENT
We thank all patients, their families, the study investigators, and personnel from each participating center. Medical writing assistance was provided by Christopher Barnes of ArticulateScience and funded by Novartis Pharmaceuticals Corporation. We thank Reham Abdel-Wahab, MD, for assistance with the manuscript.
This study was designed and conducted and data were analyzed by the sponsor (Novartis) or their designee. The funder provided study drugs and participated in regulatory and ethics approval, safety monitoring, data collection, and statistical analyses. All authors, including those employed by the sponsor, had full access to study data for interpretation and analysis, participated in manuscript preparation, and approved the final manuscript for submission for publication.
Appendix
Methods
Analysis Sets.
The full analysis set consisted of all patients who received one or more intended/planned doses of BGJ398 study drug. The full analysis set was used as the default analysis set for all analyses. The safety set included all patients who received one or more doses of BGJ398 and had one or more valid postbaseline safety assessments. Summaries of safety data include only those assessments collected no later than 28 days after study treatment discontinuation.
Efficacy.
The primary end point of overall response rate was analyzed when all patients had completed at least six cycles of treatment or discontinued from the trial. Any complete response or partial response occurring up to the data cutoff date was considered as a responder for overall response rate, irrespective of when it occurred. Best overall response was summarized based on the overall response rate and the disease control rate.
Progression-free survival (PFS) was defined as the date of the start of treatment until the date of the event, defined as the first documented disease progression or death as a result of any cause. If a patient did not have an event, PFS was censored at the date of the last adequate tumor assessment. The Kaplan-Meier method was used for the estimation of PFS.
Safety.
Safety analyses were performed on the safety set before treatment initiation (from the day of patient’s informed consent to the day before first dose of study medication), while receiving treatment (from the day of first dose of study medication to 30 days after last dose of study medication), and after treatment was discontinued (starting at day 31 after last dose of study medication). Tolerability was assessed by summarizing the number of dose interruptions, dose reductions, and relative intensity of exposure for each patient.
Biomarkers.
A correlative analysis to evaluate the association between genetic alterations (fusion, mutation, amplification, or deletion) and antitumor activity was performed. The efficacy end points considered were response (best overall response of complete response or partial response), disease control (best overall response of complete response, partial response, or stable disease), and progression-free survival. Genes altered in at least 10% of patients were considered in this analysis. Fisher’s exact test was used to evaluate the association between genetic alteration and binary efficacy end points (response and disease control). Cox proportional hazards model was used to evaluate the association between genetic alteration and progression-free survival with Bonferroni adjustment.
Sample Size Justification.
Approximately 55 patients were planned to be treated in this trial, out of whom approximately 40 patients were expected to harbor FGFR2 gene fusions and up to 15 patients with other FGFR genetic alterations. If the true underlying overall response rate (ORR) for the FGFR2 fusion group is 35%, then, with 40 patients, the probability of success (success defined as observed ORR > 25%) is 87.9%, and the chances of falsely claiming success (when the true ORR is 10%) is 0.1%. A Bayesian statistical model (binomial likelihood and beta distribution for ORR) was used for designing the study and evaluating the operating characteristics as mentioned above.
Fig A1.

Study design. (*) Safety follow-up was conducted for all patients. (†) Disease progression follow-up was conducted for patients who discontinued treatment for any reason other than disease progression. Follow-up continued until the first incidence of disease progression, initiation of a subsequent cancer therapy, or death. (‡) Survival follow-up will continue until all patients have discontinued study drug treatment and at least 80% of patients have died, withdrawn consent, or been lost to follow-up. CT, computed tomography; MRI, magnetic resonance imaging; PO, orally; RECIST, Response Evaluation Criteria in Solid Tumors.
Fig A2.

Genetic alterations in cholangiocarcinoma. (A) Clustered alteration profiles. (B) Most frequent gene alterations in cholangiocarcinoma.
Fig A3.

CA19-9 data: postbaseline data versus tumor regression. Best percentage change from baseline in sum of longest diameters and CA19-9 concentration (full analysis set). Analysis excluded any patient with no postbaseline assessment.
Table A1.
Dose Reductions and Interruptions (N = 61)
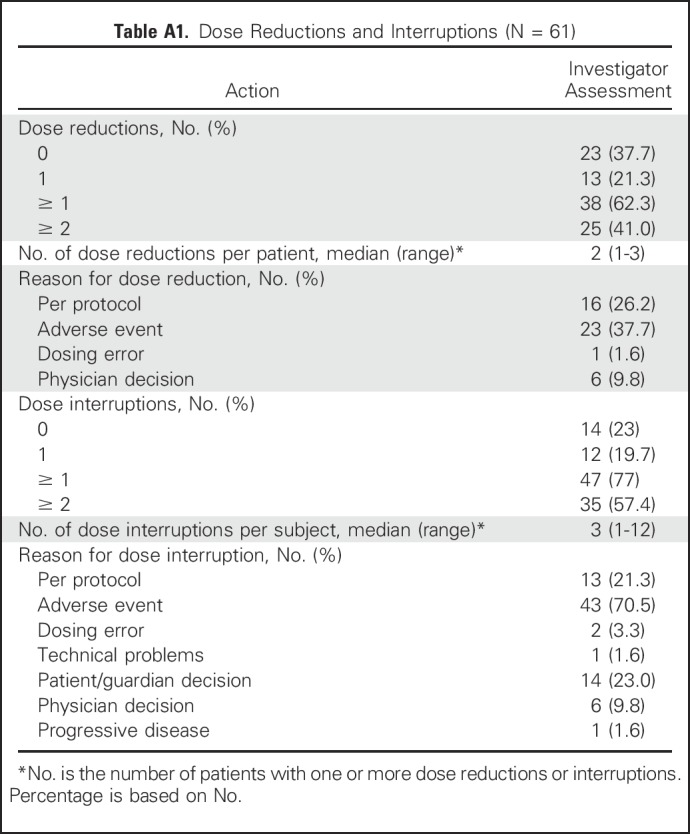
Table A2.
Patient Disposition
Footnotes
Supported by Novartis Pharmaceuticals.
Presented in part at the ASCO Gastrointestinal Cancers Symposium, San Francisco, CA, January 21-23, 2016.
Clinical trial information: NCT02150967.
AUTHOR CONTRIBUTIONS
Conception and design: Milind Javle, Anuradha Patel, Suman Sen, Randi Isaacs, Ghassan K. Abou-Alfa, Tanios Bekaii-Saab
Administrative support: Philip A. Philip
Provision of study materials or patients: Ramesh K. Ramanathan, Su Pin Choo, Philip A. Philip, Richard S. Finn, Andrew X. Zhu, Ghassan K. Abou-Alfa, Tanios Bekaii-Saab
Collection and assembly of data: Milind Javle, Maeve Lowery, Rachna T. Shroff, Karl Heinz Weiss, Christoph Springfeld, Mitesh J. Borad, Ramesh K. Ramanathan, Lipika Goyal, Saeed Sadeghi, Teresa Macarulla, Robin Kate Kelley, Ivan Borbath, Su Pin Choo, Do-Youn Oh, Philip A. Philip, Li-Tzong Chen, Thanyanan Reungwetwattana, Eric Van Cutsem, Kun-Huei Yeh, Kristen Ciombor, Richard S. Finn, Anuradha Patel, Suman Sen, Randi Isaacs, Andrew X. Zhu, Tanios Bekaii-Saab
Data analysis and interpretation: Milind Javle, Lipika Goyal, Saeed Sadeghi, Teresa Macarulla, Anthony El-Khoueiry, Su Pin Choo, Kristen Ciombor, Richard S. Finn, Anuradha Patel, Suman Sen, Dale Porter, Randi Isaacs, Andrew X. Zhu, Ghassan K. Abou-Alfa, Tanios Bekaii-Saab
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Milind Javle
No relationship to disclose
Maeve Lowery
Consulting or Advisory Role: Agios Pharmaceuticals, Celgene
Rachna T. Shroff
Consulting or Advisory Role: Celgene, Codiak Biosciences, Amgen, Agios Pharmaceuticals, Halozyme Therapeutics
Research Funding: Eli Lilly, Celgene, Agios Pharmaceuticals, Halozyme Therapeutics
Travel, Accommodations, Expenses: Celgene, Codiak Biosciences, Amgen, Agios Pharmaceuticals, Halozyme Therapeutics
Karl Heinz Weiss
Consulting or Advisory Role: Bayer AG, Bristol-Myers Squibb, Alexion Pharmaceuticals, Wilson Therapeutics (Inst), Univar (Inst)
Research Funding: Novartis (Inst), Bayer AG (Inst), Alexion Pharmaceuticals (Inst), Univar (Inst), Wilson Pharmaceuticals (Inst)
Christoph Springfeld
Consulting or Advisory Role: SERVIER, Baxalta, Celgene
Mitesh J. Borad
Stock or Other Ownership: GlaxoSmithKline Pharmaceuticals, Gilead Sciences
Consulting or Advisory Role: G1 Therapeutics, TD2, Fujifilm (Inst), Agios Pharmaceuticals (Inst), Insys Therapeutics (Inst), Novartis (Inst), ArQule (Inst), Celgene (Inst), Inspyr Therapeutics, Halozyme Pharmaceuticals (Inst)
Research Funding: Boston Biomedical (Inst), Mirna Therapeutics (Inst), Senhwa Biosciences (Inst), MedImmune (Inst), Bioline Pharmaceutical (Inst), Agios Pharmaceuticals (Inst), Halozyme Pharmaceuticals (Inst), Celgene (Inst), Threshold Pharmaceuticals (Inst), Toray Pharmaceuticals (Inst), Dicerna Pharmaceuticals (Inst), Sillajen (Inst), Eisai (Inst), Taiho Pharmaceutical (Inst), EMD Serono (Inst), Isis Pharmaceuticals (Inst), Incyte (Inst), Sun BioPharma (Inst), ARIAD Pharmaceuticals (Inst), ImClone Systems (Inst)
Travel, Accommodations, Expenses: ArQule, Celgene, AstraZeneca
Ramesh K. Ramanathan
Honoraria: Taiho Pharmaceutical, Cerulean Pharma, Pharmacyclics, Insys Therapeutics, Novocure
Consulting or Advisory Role: Cerulean Pharma, Novocure, Insys Therapeutics, Pharmacyclics
Research Funding: AbbVie (Inst), Celgene (Inst), Merck/Schering Plough (Inst), Merrimack Pharmaceuticals (Inst), Boston Biomedical (Inst), Berg Pharma (Inst), Superlab Far East (Inst)
Lipika Goyal
Honoraria: Ribon Therapeutics
Travel, Accommodations, Expenses: Debiopharm Group, Taiho Pharmaceutical
Saeed Sadeghi
No relationship to disclose
Teresa Macarulla
No relationship to disclose
Anthony El-Khoueiry
Honoraria: Bayer AG, Bristol-Myers Squibb, Novartis, Genentech, AstraZeneca, Celgene, CytomX Therapeutics
Consulting or Advisory Role: Bristol-Myers Squibb Brazil, Bayer AG, AstraZeneca, CytomX Therapeutics
Speakers' Bureau: Merrimack Pharmaceuticals
Robin Kate Kelley
Consulting or Advisory Role: Bayer AG (Inst), Debiopharm Group (Inst), Agios Pharmaceuticals (Inst), Astra Zeneca (Inst), Bristol-Myers Squibb (Inst)
Research Funding: Eli Lilly (Inst), Exelixis (Inst), Regeneron (Inst), Celgene (Inst), Tekmira (Inst), Sanofi (Inst), Novartis (Inst), Bristol-Myers Squibb (Inst), MedImmune (Inst), Merck Sharp & Dohme (Inst), Agios Pharmaceuticals (Inst), AstraZeneca (Inst), Adaptimmune Therapeutics (Inst)
Ivan Borbath
Consulting or Advisory Role: Novartis (Inst)
Research Funding: Ipsen (Inst), Celgene (Inst), Novartis (Inst)
Su Pin Choo
Honoraria: Bristol-Myers Squibb, Bayer AG
Consulting or Advisory Role: Bristol-Myers Squibb, Novartis, Celgene, Shire
Research Funding: Bristol-Myers Squibb
Travel, Accommodations, Expenses: Merck, Amgen
Do-Youn Oh
Research Funding: AstraZeneca
Philip A. Philip
Honoraria: Celgene, Amgen, Roche, Sanofi, Bayer AG, Bristol-Myers Squibb, Novartis, Ipsen, Halozyme Therapeutics, Merrimack Pharmaceuticals
Consulting or Advisory Role: Celgene, Novartis, Halozyme Therapeutics, Merrimack Pharmaceuticals, Gilead Sciences, Ipsen
Speakers' Bureau: Celgene, Bayer AG, Amgen, Roche, Sanofi
Research Funding: Bayer AG (Inst), Incyte (Inst), Karyopharm Therapeutics (Inst), Merck (Inst), Taiho Pharmaceutical (Inst), Momenta Pharmaceuticals (Inst), Novartis (Inst), Plexxikon (Inst), Immunomedics (Inst), Regeneron (Inst), Genentech (Inst)
Other Relationship: Celgene, Halozyme Therapeutics, Sanofi, Roche, EMD Serono
Li-Tzong Chen
Consulting or Advisory Role: Ono Pharmaceutical, Bristol-Myers Squibb, Eli Lilly, MSD, PharmaEngine, Merrimack Pharmaceuticals, TTY Biopharm, Syncore, Five Prime Therapeutics, Novartis
Research Funding: Novartis, TTY Biopharm, Syncore, GlaxoSmithKline, Merck Serono, Polaris
Patents, Royalties, Other Intellectual Property: Anti-alpha enolase (ENO-1) monoclonal antibody to HuniLife Biotechnology
Thanyanan Reungwetwattana
Honoraria: AstraZeneca, Novartis, Roche, Bristol-Myers Squibb, MSD
Consulting or Advisory Role: AstraZeneca, Novartis, Roche, Bristol-Myers Squibb, MSD
Research Funding: AstraZeneca, Roche, Novartis, MSD
Eric Van Cutsem
Consulting or Advisory Role: Bayer AG, Eli Lilly, Roche, SERVIER
Research Funding: Amgen (Inst), Bayer AG (Inst), Boehringer Ingelheim (Inst), Eli Lilly (Inst), Novartis (Inst), Roche (Inst), Sanofi (Inst), Celgene (Inst), Ipsen (Inst), Merck (Inst), Merck KGaA (Inst), SERVIER (Inst)
Kun-Huei Yeh
Honoraria: Bayer AG, MSD, Merck Serono, Bristol-Myers Squibb, Ono Pharmaceutical, Amgen, Novartis, Eli Lilly, Takeda Pharmaceuticals, Boehringer Ingelheim
Consulting or Advisory Role: Bayer AG, MSD, Merck Serono, Bristol-Myers Squibb, Ono Pharmaceutical, Amgen, Novartis, Eli Lilly, Takeda Pharmaceuticals, Boehringer Ingelheim
Kristen Ciombor
Research Funding: Pfizer, Boston Biomedical, MedImmune, Onyx Pharmaceuticals, Bayer AG, Boehringer Ingelheim, Bristol-Myers Squibb, Merck, Novartis, Incyte, Amgen, Sanofi
Richard S. Finn
Consulting or Advisory Role: Pfizer, Bayer AG, Novartis, Bristol-Myers Squibb, Merck, Eisai
Research Funding: Pfizer (Inst), Novartis (Inst), Bristol-Myers Squibb (Inst), Bayer AG (Inst), Eisai (Inst)
Anuradha Patel
Employment: Novartis
Stock or Other Ownership: Novartis
Suman Sen
Employment: Novartis
Stock or Other Ownership: Novartis
Dale Porter
Employment: Novartis
Stock or Other Ownership: Novartis
Randi Isaacs
Employment: Novartis
Stock or Other Ownership: Novartis
Patents, Royalties, Other Intellectual Property: Novartis
Andrew X. Zhu
Consulting or Advisory Role: Bayer AG, Bristol-Myers Squibb Brazil, Novartis, Sanofi, Merck, Eisai
Research Funding: Bayer AG, Eli Lilly, Novartis
Ghassan K. Abou-Alfa
Consulting or Advisory Role: Astellas Pharma, Celsion, IntegraGen, Vicus Therapeutics, Aduro Biotech (I), Celgene (I), Sanofi (I), Silenseed (I), Celgene, Sillajen, Boston Scientific, CASI Pharmaceuticals, Merrimack Pharmaceuticals, Merrimack Pharmaceuticals (I), Onxeo, Array BioPharma (I), Roche, Bristol-Myers Squibb (I), EMD Serono (I), Gilead Sciences (I), Momenta Pharmaceuticals (I), Vicus Therapeutics (I), SERVIER, Agios Pharmaceuticals, ASLAN Pharmaceuticals, Bayer AG, Blueprint Medicines, Delcath Systems, Eisai, Halozyme Therapeutics (I), Ipsen, Janssen (I), VAXIMM (I), Merck Serono, New B Innovation, Newlink Genetics (I), Opsona Therapeutics (I), Pfizer (I), Sirtex Medical, Westhaven, AstraZeneca, Medimmune
Research Funding: Amgen (Inst), Bayer AG (Inst), Exelixis (Inst), Genentech (Inst), Polaris (Inst), Vicus Therapeutics (Inst), Roche (Inst), Chugai Pharmaceutical (Inst), CASI Pharmaceuticals (Inst), MedImmune (Inst), AstraZeneca (Inst), Bristol-Myers Squibb (Inst), Incyte (Inst), MabVax Therapeutics (Inst), Momenta Pharmaceuticals (Inst), OncoMed (Inst)
Travel, Accommodations, Expenses: Polaris
Tanios Bekaii-Saab
Consulting or Advisory Role: Amgen, Celgene, National Comprehensive Cancer Network, Genentech, Bayer AG, Boehringer Ingelheim, Research to Practice, Merrimack Pharmaceuticals, Glenmark Pharmaceuticals, Ipsen (Inst), Eli Lilly, Bristol-Myers Squibb (Inst)
Research Funding: Boston Biomedical (Inst), AbGenomics International (Inst), Celgene (Inst), Eli Lilly (Inst), Merck (Inst), Bayer AG (Inst), Boehringer Ingelheim (Inst), Glenmark Pharmaceuticals (Inst)
Other Relationship: Exelixis, Merck, Sillajen, ARMO BioSciences
REFERENCES
- 1.Alpini G, Prall R, LaRusso NFThe pathobiology of biliary epithelia, in Arias IM, Boyer JL, Chisari FV, et al.: The Liver: Biology and Pathobiology (ed 4). Philadelphia, PA, Lippincott Williams & Wilkins, 2001, pp 421-435 [Google Scholar]
- 2.American Cancer Society : Bile duct cancer (cholangiocarcinoma). https://www.cancer.org/cancer/bile-duct-cancer.html
- 3.Banales JM, Cardinale V, Carpino G, et al. : Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 13:261-280, 2016 [DOI] [PubMed] [Google Scholar]
- 4.Oh CM, Won YJ, Jung KW, et al. : Cancer statistics in Korea: Incidence, mortality, survival, and prevalence in 2013. Cancer Res Treat 48:436-450, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brandi G, Farioli A, Astolfi A, et al. : Genetic heterogeneity in cholangiocarcinoma: A major challenge for targeted therapies. Oncotarget 6:14744-14753, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner N, Grose R: Fibroblast growth factor signalling: From development to cancer. Nat Rev Cancer 10:116-129, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Graham RP, Barr Fritcher EG, Pestova E, et al. : Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 45:1630-1638, 2014 [DOI] [PubMed] [Google Scholar]
- 8.Ross JS, Wang K, Gay L, et al. : New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 19:235-242, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farshidfar F, , Zheng S, Gingras MC, et al. : Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep 18:2780-2794, 2017. [Erratum: Cell Rep 19:2878-2880, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Churi CR, Shroff R, Wang Y, et al. : Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS One 9:e115383, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guagnano V, Kauffmann A, Wöhrle S, et al. : FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov 2:1118-1133, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Nogova L, Sequist LV, Perez Garcia JM, et al. : Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol 35:157-165, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Javle MM, Shroff RT, Zhu A, et al. : A phase 2 study of BGJ398 in patients (pts) with advanced or metastatic FGFR-altered cholangiocarcinoma (CCA) who failed or are intolerant to platinum-based chemotherapy. J Clin Oncol 34, 2016. suppl 4S; abstr 335 [Google Scholar]
- 14.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Oken MM, Creech RH, Tormey DC, et al. : Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649-655, 1982 [PubMed] [Google Scholar]
- 16.National Cancer Institute : Common Terminology Criteria for Adverse Events (CTCAE) v4.03. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf
- 17.Jiao Y, Pawlik TM, Anders RA, et al. : Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 45:1470-1473, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valle JW, Furuse J, Jitlal M, et al. : Cisplatin and gemcitabine for advanced biliary tract cancer: A meta-analysis of two randomised trials. Ann Oncol 25:391-398, 2014 [DOI] [PubMed] [Google Scholar]
- 19.National Comprehensive Cancer Network : NCCN Clinical Practice Guidelines in Oncology: Kidney Cancer. V2. http://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf [DOI] [PubMed]
- 20.Rogers JE, Law L, Nguyen VD, et al. : Second-line systemic treatment for advanced cholangiocarcinoma. J Gastrointest Oncol 5:408-413, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brieau B, Dahan L, De Rycke Y, et al. : Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 121:3290-3297, 2015 [DOI] [PubMed] [Google Scholar]
- 22.Tabernero J, Bahleda R, Dienstmann R, et al. : Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 33:3401-3408, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Perwad F, Zhang MY, Tenenhouse HS, et al. : Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol 293:F1577-F1583, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Gattineni J, Bates C, Twombley K, et al. : FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 297:F282-F291, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goyal L, Saha SK, Liu LY, et al. : Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov 7:252-263, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu YM, Su F, Kalyana-Sundaram S, et al. : Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 3:636-647, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H, Wang K, Johnson A, et al. : Comprehensive genomic profiling of extrahepatic cholangiocarcinoma reveals a long tail of therapeutic targets. J Clin Pathol 69:403-408, 2016 [DOI] [PubMed] [Google Scholar]
- 28.Xie D, Ren Z, Fan J, et al. : Genetic profiling of intrahepatic cholangiocarcinoma and its clinical implication in targeted therapy. Am J Cancer Res 6:577-586, 2016 [PMC free article] [PubMed] [Google Scholar]
- 29.Liu SL, Song ZF, Hu QG, et al. : Serum carbohydrate antigen (CA) 19-9 as a prognostic factor in cholangiocarcinoma: A meta-analysis. Front Med China 4:457-462, 2010 [DOI] [PubMed] [Google Scholar]
- 30.Yoo T, Park SJ, Han SS, et al. : Postoperative CA19-9 change is a useful predictor of intrahepatic cholangiocarcinoma survival following liver resection. Dis Markers 2015:298985, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]