

Abstract
Purpose
We evaluated the combination of talimogene laherparepvec plus ipilimumab versus ipilimumab alone in patients with advanced melanoma in a phase II study. To our knowledge, this was the first randomized trial to evaluate addition of an oncolytic virus to a checkpoint inhibitor.
Methods
Patients with unresectable stages IIIB to IV melanoma, with no more than one prior therapy if BRAF wild-type, no more than two prior therapies if BRAF mutant, measurable/injectable disease, and without symptomatic autoimmunity or clinically significant immunosuppression were randomly assigned 1:1 to receive talimogene laherparepvec plus ipilimumab or ipilimumab alone. Talimogene laherparepvec treatment began in week 1 (first dose, ≤ 4 mL × 106 plaque-forming units/mL; after 3 weeks, ≤ 4 mL × 108 plaque-forming units/mL every 2 weeks). Ipilimumab (3 mg/kg every 3 weeks; up to four doses) began week 1 in the ipilimumab alone arm and week 6 in the combination arm. The primary end point was objective response rate evaluated by investigators per immune-related response criteria.
Results
One hundred ninety-eight patients were randomly assigned to talimogene laherparepvec plus ipilimumab (n = 98), or ipilimumab alone (n = 100). Thirty-eight patients (39%) in the combination arm and 18 patients (18%) in the ipilimumab arm had an objective response (odds ratio, 2.9; 95% CI, 1.5 to 5.5; P = .002). Responses were not limited to injected lesions; visceral lesion decreases were observed in 52% of patients in the combination arm and 23% of patients in the ipilimumab arm. Frequently occurring adverse events (AEs) included fatigue (combination, 59%; ipilimumab alone, 42%), chills (combination, 53%; ipilimumab alone, 3%), and diarrhea (combination, 42%; ipilimumab alone, 35%). Incidence of grade ≥ 3 AEs was 45% and 35%, respectively. Three patients in the combination arm had fatal AEs; none were treatment related.
Conclusion
The study met its primary end point; the objective response rate was significantly higher with talimogene laherparepvec plus ipilimumab versus ipilimumab alone. These data indicate that the combination has greater antitumor activity without additional safety concerns versus ipilimumab.
INTRODUCTION
New therapies, such as immune checkpoint inhibitors and oncolytic viruses, have transformed patient care in advanced melanoma. Potentially, combination therapy strategies targeting complementary cancer immunity pathways could improve antitumor responses versus single-agent therapy.1 Consistent with this hypothesis, combination therapy with ipilimumab plus nivolumab improved response rates in untreated metastatic melanoma versus ipilimumab monotherapy, albeit with increased toxicity.2,3 These results advocate for evaluation of novel combinations with lower toxicity.
Ipilimumab is an anti–cytotoxic T-lymphocyte antigen 4 (CTLA-4) antibody.4 In previously treated advanced melanoma, ipilimumab improved overall response versus gp100 (10.9% v 1.5%; P < .001; complete response [CR], 1.5% v 0%).5 In treatment-naive advanced melanoma, ipilimumab treatment resulted in an objective response rate (ORR) of 19.0% (CR, 2.2%) and median time to objective response of 2.8 months (range, 2.5 to 12.4 months).2 Improvement in overall survival (OS) with ipilimumab monotherapy relative to gp1005 and in combination with dacarbazine versus dacarbazine alone has been reported.6 Talimogene laherparepvec is a genetically modified herpes simplex virus type 1 that expresses the immunostimulatory cytokine granulocyte-macrophage colony-stimulating factor.7 In the phase III single-agent registration study OPTiM talimogene laherparepvec improved durable response rate (primary end point) versus subcutaneous granulocyte-macrophage colony-stimulating factor (16% v 2%; P < .001).8
Talimogene laherparepvec and ipilimumab enhance T-cell activation through different mechanisms. Talimogene laherparepvec is designed to increase tumor-specific immune activation by improving antigen presentation and T-cell priming,9 whereas CTLA-4 blockade with ipilimumab promotes T-cell expansion.10 Combining these therapies may enhance antitumor immune responses, and thereby provide greater antitumor activity than either agent alone. Specifically, we hypothesized that ipilimumab would augment systemic antitumor responses triggered by talimogene laherparepvec, yielding improved outcomes.
To test this hypothesis, we evaluated talimogene laherparepvec in combination with ipilimumab in patients with unresected advanced melanoma in a phase Ib/II study. Phase Ib results have been reported previously.11 Herein, we report the primary analysis of the randomized phase II portion, which compared talimogene laherparepvec plus ipilimumab with ipilimumab alone in patients with stages IIIB to IV melanoma.
METHODS
Patients
Eligible patients (≥ 18 years of age) had histologically confirmed stages IIIB to IVM1c malignant melanoma not suitable for surgical resection, but suitable for injection (≥ 1 cutaneous/subcutaneous/nodal lesion ≥ 5 mm in longest diameter); measurable disease per contrast-enhanced or spiral computed tomography (CT; visceral lesions) or calipers (cutaneous/subcutaneous lesions); Eastern Cooperative Oncology Group performance status ≤ 1; and adequate hematologic, hepatic, and renal function. Patients were initially required to be treatment naive; however, a protocol amendment that was intended to account for the availability of new melanoma therapies allowed the enrollment of patients who had received one line of systemic anticancer therapy if BRAF wild-type or ≤ 2 lines if BRAF mutant (one must have been a BRAF inhibitor). Patients with primary uveal or mucosal melanoma, history of melanoma in an immunodeficient state, clinically active cerebral metastases, active herpetic lesions that require systemic treatment with antiherpetic drugs, evidence of clinically significant immunosuppression, history of inflammatory bowel disease and/or other symptomatic autoimmune disease, or prior exposure to talimogene laherparepvec and/or other oncolytic immunotherapy were excluded. Prior ipilimumab therapy was permitted if patients had previously had a partial response (PR), CR, or stable disease for ≥ 6 months. Prior anti–programmed death-1 or anti–CTLA-4 antibody therapy was permitted if discontinuation did not result from treatment-related adverse events (AEs).
Patients provided written informed consent. Institutional review boards/ethics committees at each site approved study procedures.
Study Design
The phase II part of the study was an open-label, multicenter, randomized trial evaluating talimogene laherparepvec in combination with ipilimumab (Appendix Fig A1, online only). Patients were randomly assigned (1:1) to receive talimogene laherparepvec plus ipilimumab or ipilimumab alone. Before the protocol amendment, random assignment was stratified by disease stage (IIIB/IIIC/IVM1a/IVM1b v IVM1c) and BRAFV600E status (mutant v wild-type); after the amendment, random assignment was stratified by disease stage (stage IIIB/IIIC/IVM1a v IVM1b/IVM1c) and prior therapy (treatment naive v previous systemic anticancer immunotherapy v systemic anticancer treatment other than immunotherapy) to better distinguish between patients whose disease had or had not spread beyond the skin and/or lymph nodes.
In the combination arm, talimogene laherparepvec was injected intralesionally on day 1 of week 1 at a dose of 106 plaque-forming units/mL (≤ 4.0 mL total injection volume; new and larger lesions were prioritized) followed by administration on day 1 of week 4, and every 2 weeks thereafter at 108 plaque-forming units/mL (≤ 4.0 mL)12; ipilimumab (3 mg/kg) was administered intravenously every 3 weeks beginning on day 1 of week 6 for up to four infusions. Talimogene laherparepvec acts in the earliest stages of the cancer immunity cycle by initiating antigen release/presentation.9,13 Ipilimumab was initiated after two doses of talimogene laherparepvec to potentially maximize the agents’ complementary mechanisms of action. In the ipilimumab arm, ipilimumab (3 mg/kg) was administered intravenously every 3 weeks beginning on day 1 of week 1 for up to four infusions. Talimogene laherparepvec treatment continued until CR, all injectable tumors had disappeared, confirmed disease progression per modified immune-related response criteria (irRC),14 or intolerance. Ipilimumab treatment continued for four infusions, until disease progression per irRC or unacceptable ipilimumab-related toxicity. Talimogene laherparepvec dose reductions were not permitted, other than a reduction in injection volume as a result of a reduction in lesion dimensions. Treatment could be delayed and/or withheld as a result of toxicity; delays > 4 weeks as a result of AEs resulted in permanent discontinuation. Ipilimumab dose reductions were not permitted; dosing could be withheld and/or discontinued as a result of toxicity. If talimogene laherparepvec was delayed and/or discontinued, ipilimumab treatment could continue. The study protocol includes complete dose modification rules.
End Points
The primary end point was investigator-assessed ORR per modified irRC (Study Protocol).14 Secondary end points included OS, best overall response, disease control rate, time to response, duration of response, progression-free survival (PFS), and safety.
Assessments
Radiographic imaging was done at baseline, every 12 weeks thereafter (independent of treatment cycle) until clinically relevant disease progression, and at the safety follow-up. Tumor assessments included measurement of cutaneous/subcutaneous/nodal lesions by calipers. Radiographic assessments of the chest, abdomen, and pelvis were performed by CT, positron emission tomography/CT, or magnetic resonance imaging. Response assessments were per irRC14 and required response/disease progression to be confirmed by a second consecutive clinical/radiographic assessment ≥ 4 weeks after the first documented response/disease progression. Confirmation of disease progression was only required in the absence of rapid clinical deterioration. AEs occurring during the study were recorded and graded using Common Terminology Criteria for Adverse Events version 3.0.
Statistical Analysis
The planned sample size of approximately 200 patients was estimated to provide 90% power to detect a significant difference in investigator-assessed ORR with an overall two-sided 5% significance level for testing the null hypothesis of no treatment effect, assuming a range of potential ORRs in the ipilimumab arm. Assuming an ORR for ipilimumab of 15.0%, 90% power was achieved if the ORR in the combination arm was at least 35.7%. The intent-to-treat analysis set included all randomly assigned patients analyzed according to the treatment to which they were randomly assigned. The safety analysis set included all patients who received ≥ 1 dose of talimogene laherparepvec or ipilimumab, analyzed according to the treatment received. The between-arm treatment comparison in ORR was evaluated using a χ2 test with continuity correction with a two-sided 5% significance level.15 Exact binomial two-sided 95% CIs were calculated for each arm for ORR and disease control rate.16 Logistic regression was used to evaluate treatment effect adjusted for covariates. An unstratified log-rank test was used to compare PFS (defined as time from random assignment to the earlier of disease progression or death), time to response, and duration of response between arms. Treatment effects on PFS, time to response, and duration of response were estimated using unstratified Cox proportional hazards models; P values for these end points were descriptive. The primary analysis of OS (using an unstratified log-rank test; two-sided α = 0.05 conditional on a statistically significant ORR increase) was planned for approximately 3 years after the last patient was randomly assigned.
RESULTS
Patients
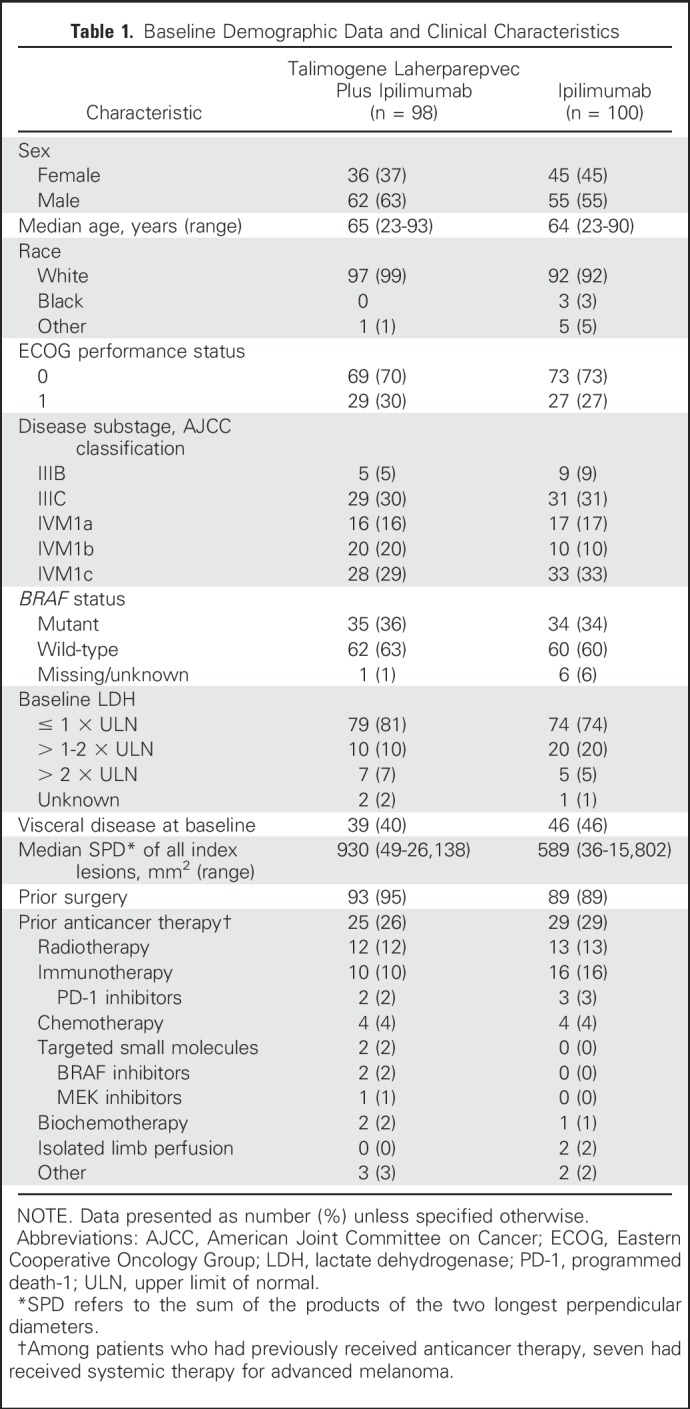
Between August 2013 and February 2016, 198 patients from 45 sites in the United States, France, and Germany were randomly assigned to talimogene laherparepvec plus ipilimumab (n = 98), or ipilimumab (n = 100). At the time of the primary analysis (August 23, 2016), five patients were continuing to receive talimogene laherparepvec and three patients (all in the combination arm) were continuing to receive ipilimumab (Fig 1). Baseline characteristics were generally balanced between arms (Table 1).
Fig 1.

Disposition of patients in the study. (*) Of the 76 patients who were not randomly assigned, 57 did not meet eligibility criteria. (†) Ninety-one patients in the talimogene laherparepvec plus ipilimumab arm received at least one dose of both study treatments. (‡) Eight patients were enrolled in the study but did not receive any study medication: talimogene laherparepvec plus ipilimumab (n = 3); ipilimumab alone (n = 5). All but one patient withdrew consent before treatment began; the remaining patient withdrew for insurance reasons. All were included in the intent-to-treat analysis of efficacy.
Table 1.
Baseline Demographic Data and Clinical Characteristics
Exposure
The median duration of talimogene laherparepvec treatment was 21.1 weeks; the median duration of ipilimumab treatment was 9.1 weeks in both arms. The median number of talimogene laherparepvec doses was 10 (range, 1 to 45 doses). Of the 95 patients in each arm who received treatment, 63 patients in the combination arm and 66 patients in the ipilimumab arm received four ipilimumab doses. The mean (± standard deviation) number of ipilimumab doses administered was 3.3 (± 1.1) in the combination arm and 3.5 (± 0.9) in the ipilimumab arm. At the time of analysis, 29% of patients in the combination arm and 27% of patients in the ipilimumab arm were being followed for tumor response. The median follow-up time was 68 weeks (range, 0 to 156 weeks) in the combination arm and 58 weeks (range, 0 to 152 weeks) in the ipilimumab arm.
Response Rate
The ORR was significantly improved in the combination arm versus the ipilimumab arm. Among randomly assigned patients, 38 patients (39% [CR, 13%; PR, 26%]) in the combination arm and 18 patients (18% [CR, 7%; PR, 11%]) in the ipilimumab arm had an objective response (see footnote in Table 2). Some patients were unevaluable for response (Table 2). The between-arm difference in ORR was 20.8% (odds ratio [OR], 2.9; 95% CI, 1.5 to 5.5; χ2, P = .002). Seven patients in the combination arm and one patient in the ipilimumab arm had disease progression per irRC before response.
Table 2.
Objective Response Rate (intent-to-treat analysis)

The ORR for patients with stage IIIB/IIIC/IVM1a disease was 44% in the combination arm and 19% in the ipilimumab arm (OR, 3.3; 95% CI, 1.4 to 7.8; P = .007); for patients with stage IVM1b/IVM1c disease, the ORR was 33% and 16%, respectively (OR, 2.6; 95% CI, 0.9 to 7.0; P = .09; Fig 2A). Among BRAF wild-type patients, the ORR was 42% (26 of 62 patients) in the combination arm versus 10% (six of 60 patients) in the ipilimumab arm (OR, 6.5; 95% CI, 2.4 to 17.4; P < .001). Among patients with BRAF-mutant tumors, the ORR was 34% (12 of 35 patients) in the combination arm versus 32% (11 of 34 patients) in the ipilimumab arm (OR, 1.1; 95% CI, 0.4 to 3.0; P = 1.0; Fig 2A). The estimated OR for treatment effect in a stepwise logistic regression model was 2.73 (P = .004; Appendix Table A1, online only).
Fig 2.

Subgroup analysis of objective response rate (A) and disease control rate (B) per immune-related response criteria. LDH, lactate dehydrogenase; OR, odds ratio; SPD, sum of the products of the two longest perpendicular diameters; ULN, upper limit of normal.
The median time to response was 5.8 months (95% CI, 5.4 to 10.9 months) in the combination arm (n = 38) and not estimable (95% CI, not estimable) in the ipilimumab arm (n = 18; HR, 1.41; 95% CI, 0.8 to 2.5; Table 2). The median duration of response was not reached in either arm (Table 2). At the time of analysis, 89% of patients in the combination arm and 83% of patients in the ipilimumab arm with a response remained in response. The disease control rate was 58% in the combination arm and 42% in the ipilimumab arm (OR, 1.9; 95% CI, 1.1 to 3.4; P = .033; Table 2). The disease control rate in all subgroups, with the exception of patients with BRAF mutations, favored the combination arm (Fig 2B).
Most patients in both arms had reductions in tumor burden from baseline (Fig 3A). Responses occurred in both injected and uninjected lesions (including visceral lesions). Injected lesion burden was calculated as the sum of tumor areas from all injected lesions. Among 89 patients in the combination arm with evaluable injected lesion burdens (defined as having baseline and at least one postbaseline assessment), 62 (70%) had a reduction in injected lesion burden from baseline (Fig 3B). Among 37 patients in the combination arm and 80 in the ipilimumab arm with evaluable uninjected, nonvisceral lesion burdens, 21 (57%) and 45 (56%), respectively, had a decrease in their corresponding lesion burden (Fig 3C). Among 31 patients in the combination arm and 22 patients in the ipilimumab arm with evaluable visceral metastasis disease burdens, 16 (52%) and 5 (23%), respectively, had a decrease in visceral lesion burden (Fig 3D).
Fig 3.

Change in lesion burden from baseline (A) for all lesions, (B) injected lesions, (C) uninjected nonvisceral lesions, and (D) visceral lesions. Per protocol, visceral lesions were not injected. Evaluable patients had baseline and at least one postbaseline evaluation of lesion burden. In panel D, two patients identified as having stage IVM1a disease at baseline had visceral index lesions (one with an adrenal lesion and one with a lung lesion; indicated with (*) and (†), respectively). The remaining patients with stages IIIC and IVM1a disease developed visceral lesions during the study.
Progression-Free Survival
PFS analysis was descriptive (Fig 4). Fifty-two of 98 patients (53%) in the combination arm and 51 of 100 patients (51%) in the ipilimumab arm had PFS events. The median PFS was 8.2 months (95% CI, 4.2 to 21.5 months) in the combination arm and 6.4 months (95% CI, 3.2 to 16.5 months) in the ipilimumab arm (HR, 0.83; 95% CI, 0.56 to 1.23; P = .35). PFS was defined as time from random assignment to the earlier of disease progression or death; consequently, patients in the combination arm initiated ipilimumab therapy 5 weeks later than patients in the ipilimumab arm and had received only two ipilimumab doses at the first response assessment (12 weeks). At data cutoff, 20 of 98 patients (20%) in the combination arm and 23 of 100 patients (23%) in the ipilimumab arm had died. The HR for OS was 0.80 (95% CI, 0.44 to 1.46).
Fig 4.

Kaplan-Meier estimate of progression-free survival (PFS). (*) P value is descriptive.
Safety
Ninety-three patients (98%) in the combination arm and 90 patients (95%) in the ipilimumab arm had ≥ 1 AE (Table 3). Incidences of grade ≥ 3 AEs were 45% and 35%, respectively. The most frequently occurring AEs and events with greatest between-arm differences in incidence were similar to those seen in other talimogene laherparepvec8 and ipilimumab studies2,5 and included fatigue (59%, 42%), chills (53%, 3%), diarrhea (42%, 35%), pruritus (40%, 36%), rash (39%, 28%), and nausea (38%, 24%, respectively; Table 3). The incidence of grade ≥ 3 ipilimumab-related AEs was 19% in the combination arm and 18% in the ipilimumab arm; the most frequently occurring were GI disorders (colitis: 5%, 4%; diarrhea: 3%, 3%; autoimmune colitis: 2%, 3%, respectively). The incidence of grade ≥ 3 talimogene laherparepvec–related AEs was 15%; the most frequently occurring were influenza-like illness (4%) and lymphopenia (4%). Two patients (2%) in the combination arm discontinued talimogene laherparepvec as a result of AEs; 12 patients (13%) in the combination arm and 16 patients (17%) in the ipilimumab arm discontinued ipilimumab as a result of AEs. Three patients in the combination arm had fatal AEs (myocardial infarction [n = 1], and disease progression [n = 2]); none were considered treatment related.
Table 3.
Incidence of Treatment-Emergent Adverse Events

DISCUSSION
This study, which to our knowledge is the first to evaluate an oncolytic virus in combination with a checkpoint inhibitor, met its primary end point: talimogene laherparepvec plus ipilimumab resulted in a significantly higher ORR versus ipilimumab alone (OR, 2.9; 95% CI, 1.5 to 5.5; P = .002). Responses occurred in patients with and without visceral disease and in uninjected lesions after combination treatment. The confirmed ORR (39%) in the combination arm is higher than historical controls of either agent administered as monotherapy (ipilimumab, 11% to 19%2,5,17,18; talimogene laherparepvec, 26%8). In OPTiM, talimogene laherparepvec monotherapy was associated with a ≥ 50% size decrease in 64% of injected lesions and 15% of visceral lesions,19 suggesting that additional responses might be achievable through the addition of T-cell checkpoint inhibition. In the previously reported phase Ib portion, we observed changes in T-cell subsets consistent with this hypothesis,11 and, in this phase II portion, we found evidence of enhanced systemic antitumor response. Individual lesion-type analysis indicated that responses occurred in both injected and uninjected tumor burden. The higher rate of decrease (combination arm, 52%; ipilimumab arm, 23%) and higher rate of complete reduction in visceral tumor burden (combination arm, 23%; ipilimumab arm, 0%) is consistent with a systemic effect of the combination because talimogene laherparepvec was not injected into these lesions. The nonvisceral uninjected lesion tumor burden response was similar across arms. However, the number of patients in this category in the combination arm was smaller than in the control arm because patients with injected nonvisceral lesions were excluded. These results indicate that the combination elicited a greater systemic antitumor response than could be achieved with either agent alone.
Response rate favored the combination arm for both disease-stage subsets (IIIB/IIIC/IVM1a, 44% v 19%; IVM1b/IVM1c, 33% v 16%). Although response rates were high in patients with both BRAF wild-type and BRAF-mutant tumors, the effect size favoring combination treatment was greater in the BRAF wild-type subgroup (42% v 10%). The response rate was also greater in the combination arm for patients with BRAF wild-type versus BRAF-mutant tumors (42% v 34%). Although small patient numbers in these subgroups may have contributed to this finding, similar results were observed in a previous phase III study of ipilimumab plus nivolumab.3 Treatment effect differences between BRAF-mutant and BRAF wild-type subgroups might reflect differences in biologic response to the combination; however, we cannot exclude the possibility that they were driven by other patient and/or tumor characteristics.
Despite ipilimumab treatment starting 5 weeks later in the combination arm, PFS was 8.2 months in the combination arm versus 6.4 months in the ipilimumab arm (HR, 0.83; 95% CI, 0.56 to 1.23). The median PFS in the combination arm was also greater than that previously observed in ipilimumab monotherapy studies (2.8 to 2.9 months)5,17,18 and similar to the median time to treatment failure in the OPTiM study (8.2 months).8 The analysis of OS was immature; patients are still being followed for response and OS.
Toxicity observed with the combination was consistent with expectations from the phase Ib study.11 The incidence of grade ≥ 3 ipilimumab-related toxicities was similar between arms; no unexpected grade ≥ 3 treatment-related AEs occurred. AEs with the greatest between-arm differences were consistent with monotherapy studies that have found flu-like symptoms to be characteristic of treatment with talimogene laherparepvec8; these events were typically grades 1 and 2. There was no evidence that the combination was associated with cardiotoxicity, as has been reported with ipilimumab plus nivolumab,20 and there was no meaningful increase in grade ≥ 3 AEs with combination therapy. Overall, combination treatment was not associated with unexpected AEs or increase in incidence or severity of AEs, suggesting that combination therapy is tolerable for patients with advanced melanoma.
Many combination strategies are under investigation to augment efficacy of single-agent therapies. Ipilimumab plus nivolumab improves patient outcomes, but is associated with significant toxicity. In trials evaluating ipilimumab plus nivolumab, increases in clinically significant grade ≥ 3 AEs were observed in the combination arm versus the monotherapy arm(s)2,3; the National Comprehensive Cancer Network recommends careful patient selection and monitoring for this regimen.21 Combination regimens with lower toxicity may allow for their use in a broader range of patients. Since this study was initiated, other immune checkpoint inhibitors have been introduced for melanoma. The combination of talimogene laherparepvec with an anti–programmed death-1 antibody is being evaluated in MASTERKEY-265, a phase Ib/III trial of talimogene laherparepvec plus pembrolizumab in patients with previously untreated stages IIIB to IV melanoma.22,23 Results from the phase Ib portion of MASTERKEY-265 showed a response rate > 50% with no dose-limiting toxicities; the phase III portion is enrolling patients.22,23
Although the outcome of this trial was positive, as a phase II study it had certain limitations. First, because of the population size of 198 patients, there were relatively small patient numbers in certain subgroups. Second, this was an open-label study without central review of investigator-assessed response. Third, the duration of follow-up at the time of this analysis limited the interpretation of time-to-event end points. Fourth, delayed ipilimumab initiation may have limited the interpretation of benefit from combination therapy. Fifth, there were between-arm differences in certain baseline characteristics (eg, disease stage/substage, visceral disease, prior therapy) that may have influenced treatment-effect estimates. However, improvement in ORR in the combination arm remained significant in a final logistic regression analysis after adjustment for potentially imbalanced covariates. Finally, differences in eligibility criteria (eg, injectable disease requirement) may confound between-study comparisons.
This randomized trial of an oncolytic immunotherapy in combination with a checkpoint inhibitor, which is the first such study to our knowledge, met its primary end point: talimogene laherparepvec plus ipilimumab resulted in a significantly higher ORR versus ipilimumab alone. The combination seems to have greater efficacy (including in uninjected and visceral lesions), without additional safety concerns versus ipilimumab alone in patients with unresected stages IIIB to IV melanoma. The combination of talimogene laherparepvec and a checkpoint inhibitor may have significant clinical utility in treatment of advanced melanoma.
ACKNOWLEDGMENT
We thank Meghan Johnson and Ali Hassan at Complete Healthcare Communications (West Chester, PA), a CHC Group company, whose work was funded by Amgen, for medical writing assistance in the preparation of this article. We also thank the patients who participated in this study; the clinical trial personnel including Stacy Baum, Cynthia Swartz, and Katie Golladay at the University of Louisville; and the clinical protocol office staff at the University of North Carolina at Chapel Hill, especially Diana Wallack.
Appendix
Fig A1.

Study schema for phase II. ECOG, Eastern Cooperative Oncology Group; irRC, immune-related response criteria; IV, intravenous; ORR, objective response rate; OS, overall survival; PD-1, programmed death-1; PFS, progression-free survival; PFU, plaque-forming unit; R, randomization. (*) irRC described in Wolchok et al.14
Table A1.
Stepwise Regression Model of Objective Response Rate per Modified Immune-Related Response Criteria

Footnotes
Funded by Amgen.
Presented at the European Society for Medical Oncology 2016 Congress, Copenhagen, Denmark, October 7-11, 2016; and at the American Society of Clinical Oncology 2017 Annual Meeting, Chicago, Illinois, June 2-6, 2017.
Clinical trial information: NCT01740297.
AUTHOR CONTRIBUTIONS
Conception and design: Jason Chesney, Igor Puzanov, Merrick Ross, Jennifer Gansert, Robert H.I. Andtbacka, Howard L. Kaufman
Provision of study materials or patients: Igor Puzanov, Frances Collichio, Howard L. Kaufman
Collection and assembly of data: Jason Chesney, Igor Puzanov, Frances Collichio, Parminder Singh, John Glaspy, Omid Hamid, Philip Friedlander, Claus Garbe, Theodore F. Logan, Axel Hauschild, Celeste Lebbé, Jenny J. Kim, Jennifer Gansert, Howard L. Kaufman
Data analysis and interpretation: Jason Chesney, Igor Puzanov, Frances Collichio, Mohammed M. Milhem, John Glaspy, Omid Hamid, Theodore F. Logan, Axel Hauschild, Celeste Lebbé, Lisa Chen, Jenny J. Kim, Jennifer Gansert, Robert H.I. Andtbacka, Howard L. Kaufman
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Jason Chesney
Consulting or Advisory Role: Amgen
Research Funding: Amgen
Travel, Accommodations, Expenses: Amgen
Igor Puzanov
Consulting or Advisory Role: Amgen, Genentech, Bristol-Myers Squibb
Travel, Accommodations, Expenses: Amgen, Merck
Frances Collichio
Consulting or Advisory Role: Amgen (Inst)
Research Funding: Amgen (Inst), Novartis (Inst), Mayo Phase II (Inst), Vascular Biogenics (Inst), Morphotek (Inst)
Parminder Singh
Consulting or Advisory Role: Genentech
Speakers’ Bureau: Genentech
Mohammed M. Milhem
Consulting or Advisory Role: Genentech, EMD Serono, Novartis, Eisai, Bristol-Myers Squibb, Amgen
John Glaspy
Research Funding: Amgen
Omid Hamid
Consulting or Advisory Role: Amgen, Novartis, Roche, Bristol-Myers Squibb, Merck
Speakers’ Bureau: Bristol-Myers Squibb, Genentech, Novartis, Amgen
Research Funding: AstraZeneca (Inst), Bristol-Myers Squibb (Inst), Celldex (Inst), Genentech (Inst), Immunocore (Inst), Incyte (Inst), Merck (Inst), Merck Serono (Inst), MedImmune (Inst), Novartis (Inst), Pfizer (Inst), Rinat (Inst), Roche (Inst)
Merrick Ross
Honoraria: Merck, Amgen, Castle Biosciences
Consulting or Advisory Role: Merck, Amgen
Speakers’ Bureau: Amgen
Research Funding: Amgen (Inst)
Travel, Accommodations, Expenses: Merck, Amgen, Castle Biosciences, Provectus
Philip Friedlander
Stock or Other Ownership: Allergan, Incyte, Merrimack, Bristol-Myers Squibb
Consulting or Advisory Role: Novartis, Castle Biosciences, Genentech, EMD Serono, Pfizer
Research Funding: Celgene, Amgen, Merck, Genentech, Oncovir
Travel, Accommodations, Expenses: Castle Biosciences, Novartis
Claus Garbe
Honoraria: Amgen, Bristol-Myers Squibb, MSD, Novartis, Philogen, Roche, Incyte
Consulting or Advisory Role: Amgen, Bristol-Myers Squibb, MSD, Novartis, Philogen, Roche, Incyte
Research Funding: Bristol-Myers Squibb (Inst), Novartis (Inst), Roche (Inst)
Travel, Accommodations, Expenses: Amgen, Bristol-Myers Squibb, Novartis, Philogen, Roche, MSD
Theodore F. Logan
Consulting or Advisory Role: Prometheus
Research Funding: Abbott Laboratories (Inst), Abraxis BioScience (Inst), Amgen (Inst), Argos Therapeutics (Inst), AstraZeneca (Inst), AVEO (Inst), BioVex (Inst), Bristol-Myers Squibb (Inst), Eisai (Inst), Eli Lilly (Inst), GlaxoSmithKline (Inst), Roche (Inst), Immatics (Inst), Merck (Inst), Novartis (Inst), Pfizer (Inst), Prometheus (Inst), Roche (Inst), Synta (Inst), Threshold Pharmaceuticals (Inst), Millennium (Inst), TRACON Pharma (Inst), Cerulean Pharma (Inst), EMD Serono (Inst), Acceleron Pharma (Inst)
Axel Hauschild
Honoraria: Amgen, Bristol-Myers Squibb, MedImmune, Merck Serono, Merck Sharp & Dohme, Novartis, OncoSec, Roche, Nektar, Philogen, Provectus, Regeneron
Consulting or Advisory Role: Amgen, Bristol-Myers Squibb, MedImmune, Merck Serono, Merck Sharp & Dohme, Novartis, OncoSec, Roche, Nektar Therapeutics, Philogen, Provectus, Regeneron
Research Funding: Amgen, Bristol-Myers Squibb, Celgene, Eisai, GlaxoSmithKline, Merck Serono, Merck Sharp & Dohme, Novartis, Roche
Travel, Accommodations, Expenses: Amgen, Bristol-Myers Squibb, MedImmune, Merck Serono, Merck Sharp & Dohme, Novartis, OncoSec, Roche, Nektar Therapeutics, Philogen, Provectus, Regeneron
Celeste Lebbé
Honoraria: Roche, Bristol-Myers Squibb, Novartis, MSD, Amgen
Consulting or Advisory Role: Roche, Bristol-Myers Squibb, Novartis, Amgen, MSD
Speakers’ Bureau: Roche, Bristol-Myers Squibb, Amgen, Novartis
Research Funding: Roche (Inst), Bristol-Myers Squibb (Inst)
Travel, Accommodations, Expenses: Roche, Bristol-Myers Squibb, Novartis, Amgen
Lisa Chen
Employment: Amgen
Stock or Other Ownership: Amgen
Jenny J. Kim
Employment: Amgen
Stock or Other Ownership: Amgen
Jennifer Gansert
Employment: Amgen
Stock or Other Ownership: Amgen
Patents, Royalties, Other Intellectual Property: Amgen
Robert H.I. Andtbacka
Honoraria: Amgen, Merck/Schering Plough
Consulting or Advisory Role: Amgen, Merck/Schering Plough
Research Funding: Amgen (Inst), Viralytics (Inst), Takara Bio (Inst), OncoSec (Inst)
Howard L. Kaufman
Employment: Compass Therapeutics
Leadership: Compass Therapeutics
Honoraria: Amgen, EMD Serono, Merck, Celldex, Prometheus, Turnstone Bio, Compass Therapeutics
Consulting or Advisory Role: Amgen, Merck, Merck Serono, Paometheus, Celldex, Turnstone Bio, Bristol-Myers Squibb, Compass Therapeutics
Speakers’ Bureau: Merck
Research Funding: Amgen (Inst), Merck (Inst)
Travel, Accommodations, Expenses: EMD Serono, Turnstone Bio
REFERENCES
- 1.Antonia SJ, Larkin J, Ascierto PA: Immuno-oncology combinations: A review of clinical experience and future prospects. Clin Cancer Res 20:6258-6268, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. : Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373:23-34, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Postow MA, Chesney J, Pavlick AC, et al. : Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372:2006-2017, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.YERVOY, ipilimumab. Bristol-Myers Squibb, Princeton, NJ, 2015 [Google Scholar]
- 5.Hodi FS, O’Day SJ, McDermott DF, et al. : Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711-723, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert C, Thomas L, Bondarenko I, et al. : Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364:2517-2526, 2011 [DOI] [PubMed] [Google Scholar]
- 7.IMLYGIC, talimogene laherparepvec. Amgen, Thousand Oaks, CA, 2015 [Google Scholar]
- 8.Andtbacka RH, Kaufman HL, Collichio F, et al. : Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 33:2780-2788, 2015 [DOI] [PubMed] [Google Scholar]
- 9.Kohlhapp FJ, Kaufman HL: Molecular pathways: Mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res 22:1048-1054, 2016 [DOI] [PubMed] [Google Scholar]
- 10.Subudhi SK, Aparicio A, Gao J, et al. : Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc Natl Acad Sci USA 113:11919-11924, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puzanov I, Milhem MM, Minor D, et al. : Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol 34:2619-2626, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffner B, Iodice GM, Gasal E: Administration and handling of talimogene laherparepvec: An intralesional oncolytic immunotherapy for melanoma. Oncol Nurs Forum 43:219-226, 2016 [DOI] [PubMed] [Google Scholar]
- 13.Chen DS, Mellman I: Oncology meets immunology: The cancer-immunity cycle. Immunity 39:1-10, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Wolchok JD, Hoos A, O’Day S, et al. : Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res 15:7412-7420, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Fleiss JL, Tytun A, Ury HK: A simple approximation for calculating sample sizes for comparing independent proportions. Biometrics 36:343-346, 1980 [PubMed] [Google Scholar]
- 16.Clopper CJ, Pearson ES: The use of confidence and fiducial limits illustrated in the case of the binomial. Biometrika 26:404-413, 1934 [Google Scholar]
- 17.Robert C, Schachter J, Long GV, et al. : Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 372:2521-2532, 2015 [DOI] [PubMed] [Google Scholar]
- 18. Wolchok J, Chiarion-Sileni V, Gonzalez R, et al: Updated results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naive patients (pts) with advanced melanoma (MEL) (CheckMate 067). J Clin Oncol 34, 2016 (suppl; abstr 9505) [Google Scholar]
- 19.Andtbacka RH, Ross M, Puzanov I, et al. : Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann Surg Oncol 23:4169-4177, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson DB, Balko JM, Compton ML, et al. : Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med 375:1749-1755, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.National Comprehensive Cancer Network . 2017. : NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Melanoma version 1.2017. Fort Washington, PA, [Google Scholar]
- 22. Long GV, Dummer R, Ribas A, et al: Efficacy analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec (T-VEC) and pembrolizumab (pembro) for unresectable stage IIIB-IV melanoma. J Clin Oncol 34, 2016 (suppl; abstr 9568) [Google Scholar]
- 23.Long GV, Dummer R, Ribas A, et al. : Primary analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec and pembrolizumab for unresectable stage IIIB-IV melanoma, 12th International Congress of the Society for Melanoma Research. San Francisco, CA, November 18-21, 2015 [Google Scholar]