

Supplemental Digital Content is Available in the Text.
Key Words: raltegravir, once daily, integrase inhibitor, HIV-1 infection, treatment-naive
Abstract
Background:
Raltegravir 1200mg (2×600mg tablets) once daily (QD) demonstrated noninferior efficacy and similar safety to raltegravir 400mg twice daily (BID) at week 48 of the ONCEMRK trial. Here, we report the week 96 results from this study.
Methods:
ONCEMRK is a phase 3, multicenter, double-blind, noninferiority trial comparing raltegravir 1200mg QD with raltegravir 400mg BID in treatment-naive HIV-1–infected adults. Participants were assigned (2:1) to raltegravir 2×600mg QD or 400mg BID, both with emtricitabine and tenofovir disoproxil fumarate (FTC/TDF) for 96 weeks. Randomization was stratified by screening HIV-1 RNA and hepatitis B/C status. Efficacy was assessed as the proportion of participants with HIV-1 RNA <40 copies per milliliter (Food and Drug Administration Snapshot approach); the noninferiority margin was 10 percentage points.
Results:
Of the 797 participants who received study therapy (84.6% were men, 59.3% were white, and mean age was 35.9 years), 694 completed 96 weeks of treatment (87.6% QD; 84.4% BID), with few discontinuations because of lack of efficacy (1.1% for both groups) or adverse events (1.3% QD; 2.3% BID). At week 96, 81.5% (433/531) of QD recipients and 80.1% (213/266) of BID recipients achieved HIV-1 RNA <40 copies per milliliter (difference 1.4%, 95% confidence interval: −4.4 to 7.3). CD4+ T-cell counts increased >260 cells/mm3 from baseline in both groups. Resistance to raltegravir was infrequent, occurring in 0.8% of each treatment group through week 96. Adverse event rates were similar for the 2 regimens.
Conclusions:
In HIV-1–infected treatment-naive adults receiving FTC/TDF, raltegravir 1200mg QD demonstrated noninferior efficacy to raltegravir 400mg BID that was durable to week 96, with a safety profile similar to raltegravir 400mg BID.
INTRODUCTION
Raltegravir 400 mg twice daily (BID), the first integrase strand transfer inhibitor (INSTI) approved for the treatment of HIV-1 infection, has a well-established safety and efficacy profile in treatment-naive and treatment-experienced patients.1–4 A once-daily formulation of raltegravir 1200 mg (2 × 600 mg tablets) has been developed and has the potential to improve treatment adherence and satisfy patient preference for a once-daily regimen.5–8 This new formulation is approved for use in combination with other antiretroviral agents for previously untreated HIV-1 infection in adults and in children weighing ≥40 kg.9 In the ONCEMRK phase 3 trial, raltegravir 1200 mg once daily (QD) showed noninferior efficacy to raltegravir 400 mg BID, with 88.9% and 88.3% of participants, respectively, achieving HIV-1 RNA <40 copies per milliliter at week 48.10 In addition, the immunologic efficacy and overall safety profile of raltegravir QD were similar to those of raltegravir BID at week 48. Here, we report the long-term efficacy and safety of raltegravir 1200 mg QD compared with raltegravir 400 mg BID through week 96 of the ONCEMRK trial.
METHODS
Study Design
ONCEMRK (MK-0518 Protocol 292; ClinicalTrials.gov NCT02131233) was a double-blind, randomized, parallel group, noninferiority study comparing raltegravir 1200 mg QD with raltegravir 400 mg BID, each given with a fixed combination of emtricitabine 200 mg and tenofovir disoproxil fumarate 300 mg (FTC/TDF). The study was conducted according to Good Clinical Practice requirements and applicable statutes and regulations regarding the protection of human participants in biomedical research. An Independent Ethics Committee for each study site reviewed and approved the protocol, and the participants provided written informed consent before any study procedures were performed.
Adults (≥18 years of age) with screening HIV-1 RNA ≥1000 copies per milliliter and no previous antiretroviral therapy for treatment of HIV-1 infection were eligible for the study. Exclusion criteria included documented or known resistance to any of the study regimen components and acute hepatitis or active infections other than chronic hepatitis B or C infection. Participants were randomly assigned (2:1) to raltegravir 1200 mg QD or 400 mg BID, each in combination with open-label FTC/TDF (TRUVADA; Gilead Sciences, Inc., Carrigtohill, County Cork, Ireland) for up to 96 weeks. Randomization was stratified by screening HIV-1 RNA (≤100,000 or >100,000 copies per milliliter) and hepatitis B/C coinfection (yes or no). To conceal the treatment group assignment, participants also received placebo tablets matching the alternate treatment. Participants, study site investigators and staff, and sponsor personnel responsible for monitoring the study remained unaware of the treatment group assignments until the week 96 database lock.
Procedures
Plasma HIV-1 RNA levels were measured by the central laboratory using the Abbott RealTime HIV-1 Assay (lower limit of quantification 40 copies per milliliter). Protocol-defined virologic failure (PDVF) was used to identify participants for viral resistance testing. PDVF consisted of nonresponse (participant did not achieve HIV-1 RNA <40 copies per milliliter by week 24) or rebound (after initial response of HIV-1 RNA <40 copies per millliliter, participant had 2 consecutive measurements of HIV-1 RNA ≥40 copies per milliliter at least 1 week apart). Resistance testing was performed by Monogram Biosciences Inc. using the following assays: PhenoSense GT, PhenoSense Integrase, and either GeneSeq Integrase or GenoSure Integrase; all assays required samples with HIV-1 RNA ≥500 copies per milliliter.
CD4+ T-cell counts were measured at weeks 24, 48, 72, and 96 by the central laboratory according to standard procedures. All clinical adverse events were evaluated for maximum intensity, seriousness, relationship to test drugs, and association with immune reconstitution syndrome. All protocol-required laboratory values were graded according to the Division of AIDS criteria.11
Participants completed a daily medication diary that was reviewed by site personnel at each study visit. A day within the study was counted as “on therapy” if at least 1 tablet of any study medication was taken. Treatment compliance was calculated from the number of days on therapy as recorded in the medication diary (actual/planned × 100). For participants who were compliant with therapy but had HIV-1 RNA >200 copies per milliliter (on 2 consecutive measurements taken at least 1 week apart) on or after week 24 or after previously achieving HIV-1 RNA <40 copies per milliliter, the protocol recommended that discontinuation of study therapy should be considered by the study investigator.
Statistical Analysis
The primary efficacy end point for this trial was assessed at week 48 and has been previously reported.10 This report presents additional data to evaluate the secondary hypothesis that raltegravir 1200 mg QD is noninferior to raltegravir 400 mg BID, each in combination with TDF/FTC, as assessed by the proportion of participants achieving HIV-1 RNA <40 copies per milliliter at week 96. A margin of 10 percentage points was used to assess noninferiority. The week 96 hypothesis was tested sequentially after the week 48 hypothesis was successfully met; therefore, the overall type I error was controlled at one-sided 2.5% alpha level.
The efficacy analyses used the Full Analysis Set population, defined as all randomized participants who received at least 1 dose of study treatment, with participants included in the treatment group to which they were randomized. SAS version 9.3 or 9.4 was used for all analyses.
For the analysis of the proportion of participants with HIV-1 RNA <40 copies per milliliter, all missing data were treated as failures regardless of the reason, including early discontinuation of study therapy (Food and Drug Administration snapshot approach). The difference in proportions between treatment groups and the associated 95% confidence interval (CI) were calculated by the stratum-adjusted Mantel-Haenszel method. A supportive analysis of the proportion of participants with HIV-1 RNA <50 copies per milliliter was also performed using the methods described above.
For the analysis of change from baseline in CD4+ T-cell counts, missing data were handled with the Observed Failure (OF) approach. Baseline values were carried forward for participants who discontinued the assigned therapy because of lack of efficacy and for those who discontinued for non–treatment-related reasons and had HIV-1 RNA ≥40 copies per milliliter; participants with missing data for other reasons were excluded.
To evaluate the consistency of treatment effects, the virologic and immunologic end points were estimated within subgroups based on a variety of prognostic and demographic factors using the OF approach (see above); no hypothesis testing was performed within or between subgroups. The OF approach was used because it focuses on the impact of the antiretroviral treatment, whereas the snapshot approach considers the impact of treatment and non–treatment-related factors.
The safety analyses used the all-participants-as-treated population, which consists of all randomized participants who received at least 1 dose of study treatment and includes participants in the treatment group for the regimen they actually received. Adverse events that occurred after randomization and were reported through the week 96 window were included in the analyses. Adverse event terms from the Medical Dictionary for Regulatory Activities (Version 18.1) were used. For laboratory changes occurring in ≥1% of the participants in either treatment group, between-treatment differences (and 95% CI) were calculated using the Miettinen and Nurminen method.12
RESULTS
Of the 802 randomized participants, 797 received study therapy and were included in the efficacy and safety analyses. Baseline characteristics of the participants were similar between the treatment groups (see Table, Supplemental Digital Content 1, http://links.lww.com/QAI/B166). Mean age of the study population was 35.9 years, and 85% of the participants were men. Mean HIV-1 RNA was 4.6 log10 copies per milliliter, and 28% of participants had >100,000 copies per milliliter; mean CD4+ T-cell count was 415/mm3 and 13% had <200/mm3. A total of 64 participants (12.0%) from the raltegravir QD group and 39 (14.5%) from the raltegravir BID group discontinued early without completing the study. The most common reasons for discontinuation were lost to follow-up and withdrawal by the subject (Fig.1). Treatment compliance was high overall: 97.2% of QD recipients and 95.9% of BID recipients were at least 90% compliant with study therapy.
FIGURE 1.
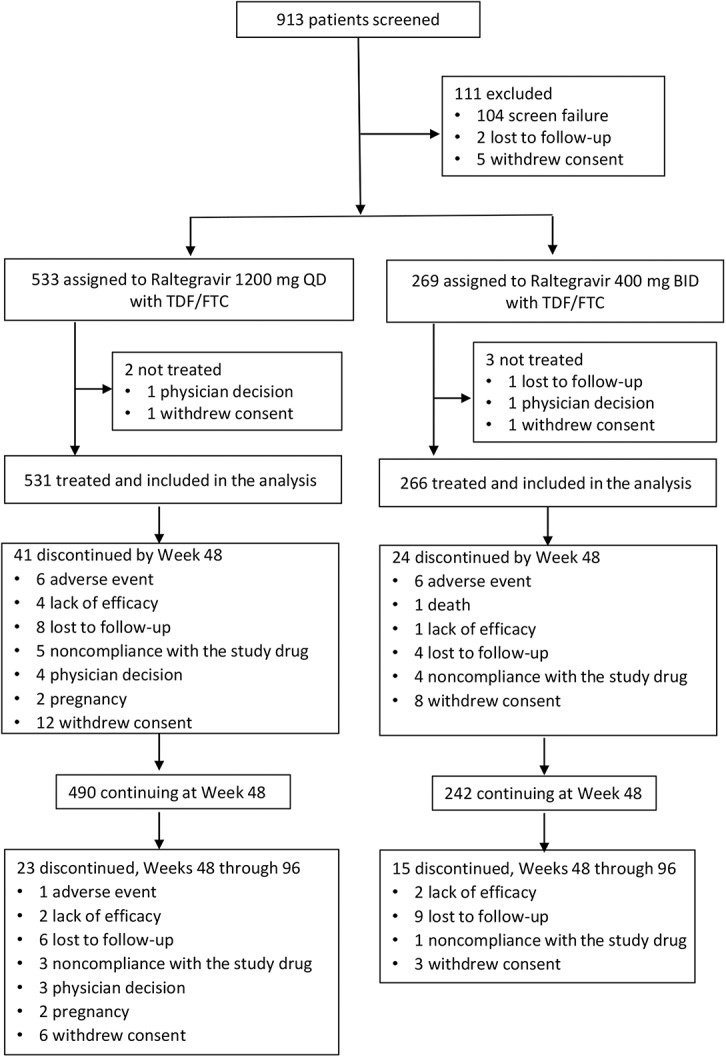
Disposition of study participants.
Efficacy
At week 96, HIV-1 RNA <40 copies per milliliter was achieved by 81.5% (433/531) of the participants who received raltegravir 1200 mg QD and by 80.1% (213/266) of those who received raltegravir 400 mg BID (Fig.2). The treatment difference was 1.4%, with an associated 95% CI of −4.4 to 7.3, demonstrating noninferiority of raltegravir 1200 mg QD to raltegravir 400 mg BID. Other virologic outcomes were comparable between the treatment groups: 9.2% of the QD group and 8.3% of the BID group had HIV-1 RNA ≥40 copies per milliliter, whereas 9.2% and 11.7%, respectively, had no virologic data at week 96 (Table 1).
FIGURE 2.
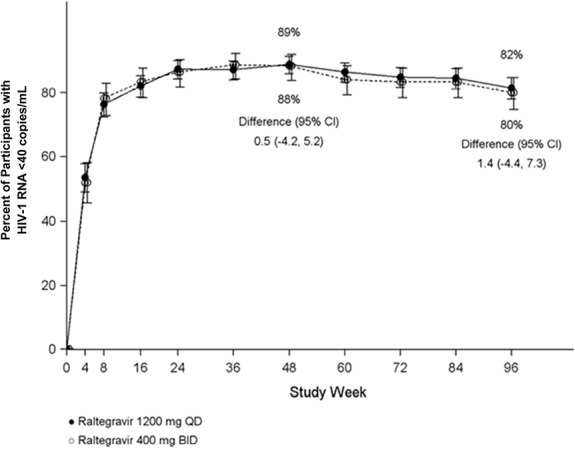
Proportion of participants with HIV-1 RNA <40 copies per milliliterover time, Food and Drug Administration snapshot approach. Raltegravir (RAL) 1200 mg QD and 400 mg BID were given with TDF/FTC.
TABLE 1.
Virologic Outcomes at Week 96
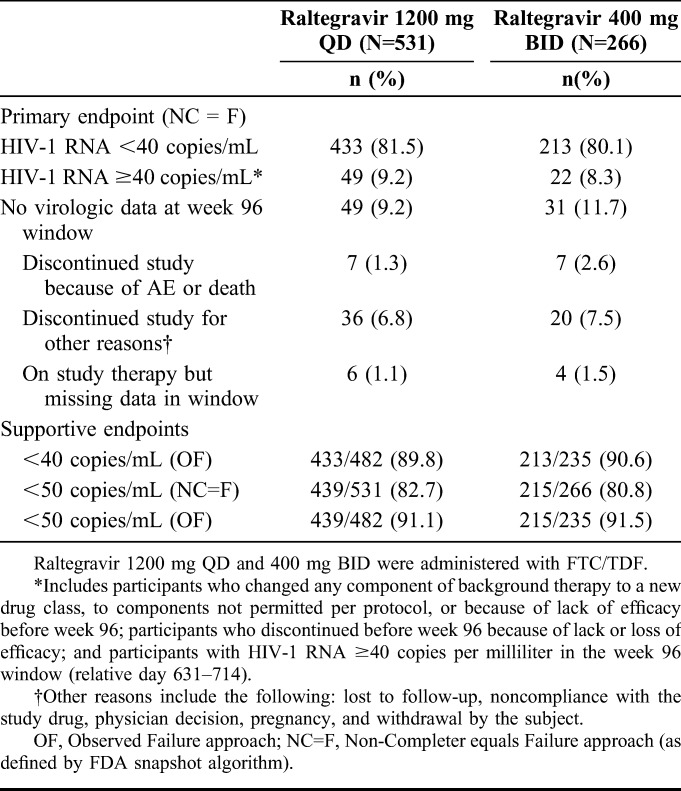
Similar efficacy also was demonstrated between raltegravir QD and raltegravir BID on the supportive endpoint of virologic response defined as HIV-1 RNA <50 copies per milliliter (82.7% vs 80.8%). The mean change from baseline in CD4+ T-cell count was 262 cells/mm3 in both treatment groups, with a treatment difference (95% CI) of −0.6 (−32.8 to 31.6).
In the subgroup analyses, raltegravir 1200 mg QD showed durable antiretroviral and immunologic efficacy comparable to that observed with raltegravir 400 mg BID, regardless of prognostic and demographic factors (Fig.3). Participants with high baseline viral load (>100,000 copies per milliliter) showed similar robust virologic responses at week 96 in both the QD and BID treatment groups (HIV-1 RNA <40 copies per milliliter achieved in 84.7% and 82.9%, respectively), as well as similar increases in CD4+ T-cell counts (297 and 281 cells/mm3, respectively).
FIGURE 3.
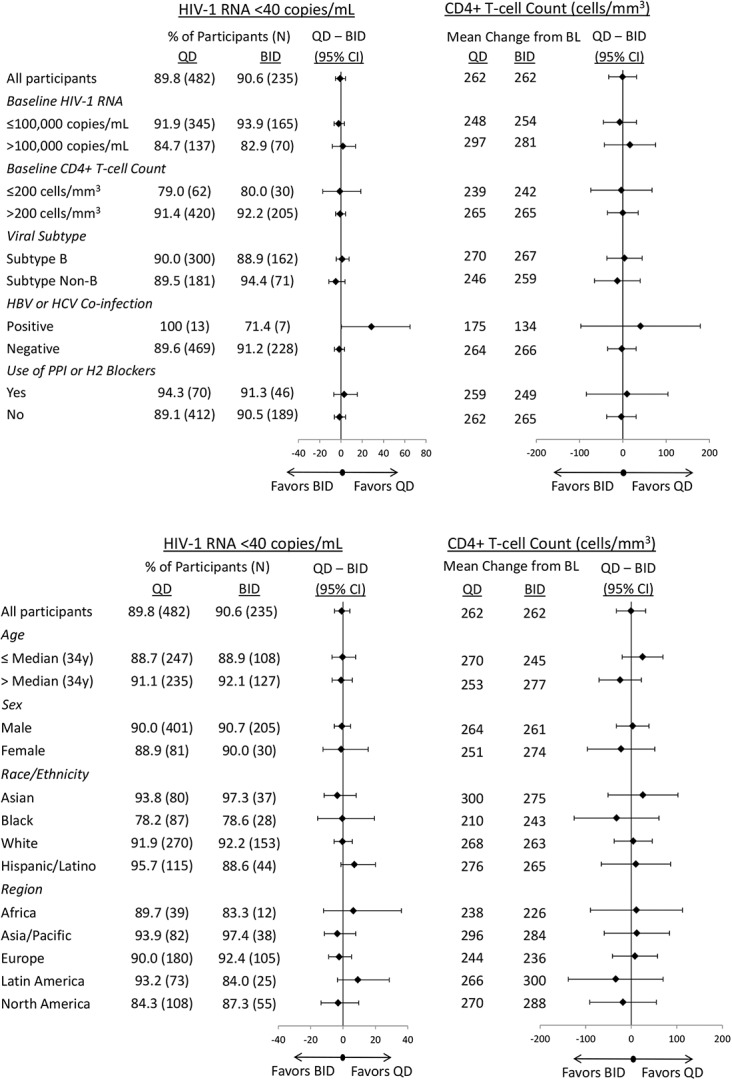
Efficacy of raltegravir 1200 mg QD and raltegravir 400 mg BID by baseline prognostic and demographic factors (Observed Failure approach).
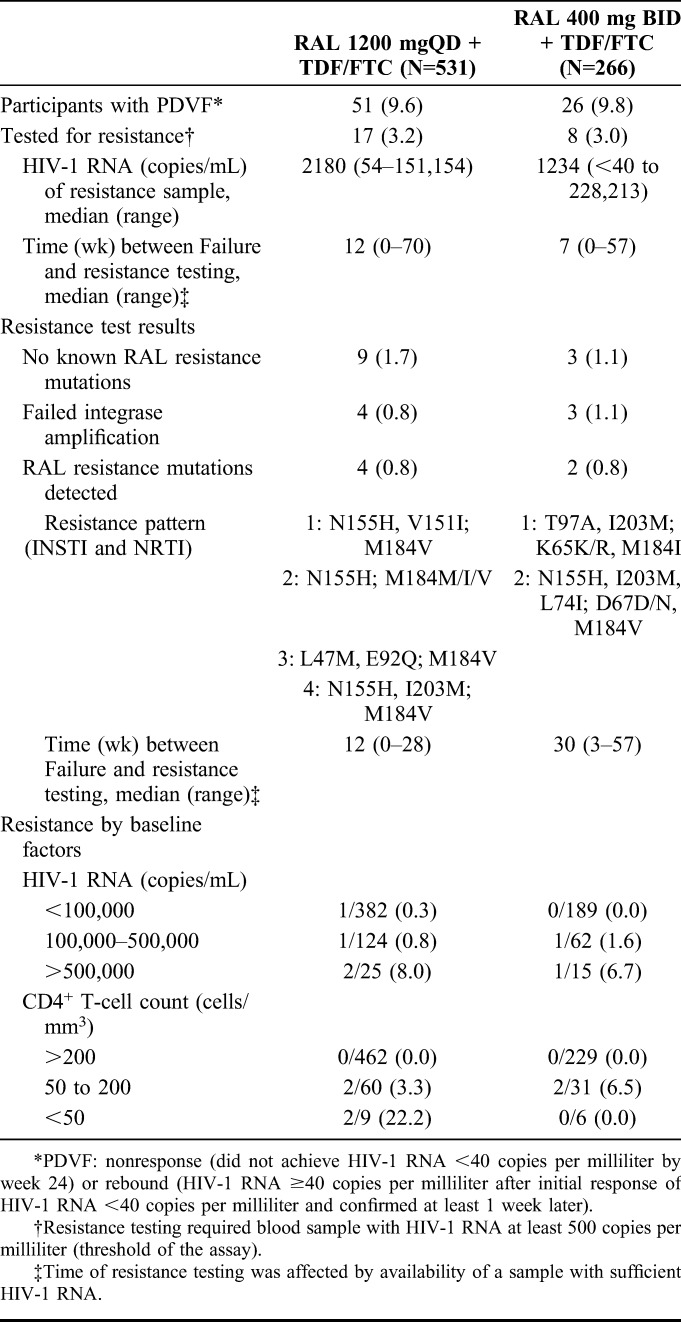
By week 96, a total of 77 participants met the PDVF criteria for nonresponse or rebound, 9.6% (51/531) of the raltegravir QD group and 9.8% (26/266) of the raltegravir BID group, with 23 cases (15 QD; 8 BID) occurring after week 48. Most (92.2%) participants with PDVF were at least 90% compliant with study therapy. A substantial number of participants who met the PDVF criteria remained on study therapy (at the investigator's discretion, as allowed by the protocol) and achieved HIV-1 RNA <40 copies per milliliter at week 96: 45.1% (23/51) in the QD group and 57.7% (15/26) in the BID group.
Most participants with PDVF had low-level viremia, and only 25 (17 QD, 8 BID) could be tested for viral drug resistance because of the assay threshold of HIV-1 RNA ≥500 copies per milliliter (Table 2). Viral resistance to INSTI and nucleoside reverse transcriptase inhibitor (NRTI) occurred in 0.8% of each treatment group (4/531 QD, 2/266 BID), and NRTI resistance alone occurred in 0.4% of each group (2/531 QD; 1/266 BID). The profile of INSTI resistance mutations included integrase N155H, V151I, L74M, E92Q, and I203M. Five of the 6 participants who developed resistance to raltegravir had baseline HIV-1 RNA >100,000 copies per milliliter (3 had >500,000 copies per milliliter), and all 6 had baseline CD4+ T-cell counts <200/mm3 (2 had <50/mm3). Self-reported treatment adherence was high in all 6 of these participants (95%–100% in the QD group; 91%–100% in the BID group).
TABLE 2.
Protocol-Defined Virologic Failure and Resistance Testing
Safety
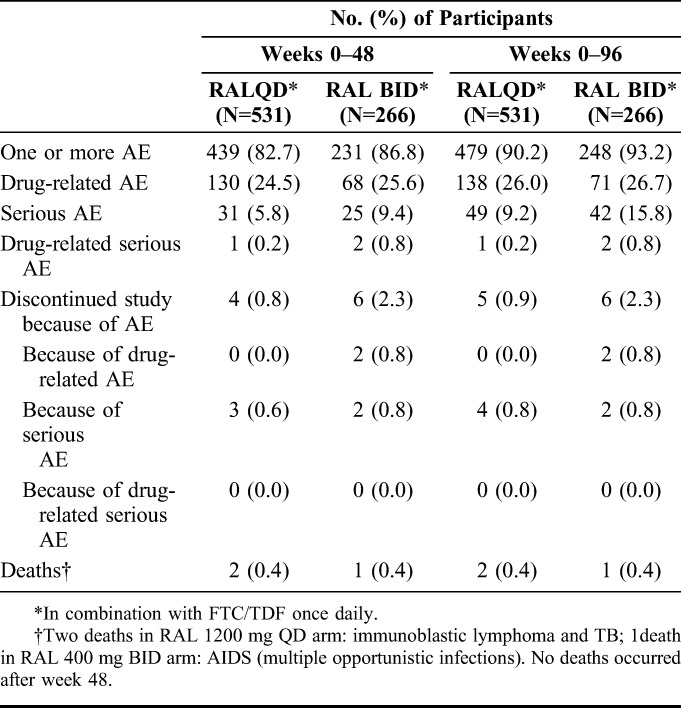
Rates of clinical adverse events were similar between the treatment groups at week 96 and showed minimal change from week 48 to week 96 in both the treatment groups (Table 3). Clinical adverse events led to treatment discontinuation by week 96 in 0.9% of raltegravir QD recipients and 2.3% of raltegravir BID recipients. Only 1 participant (who was in the QD group) discontinued because of a clinical adverse event (facial bone fracture) after week 48; this event was serious and not related to study therapy. No deaths or drug-related serious adverse events occurred after week 48. One participant in the QD group had an adverse event (autoimmune hepatitis) after week 48, which was considered by the investigator to be associated with immune reconstitution inflammatory syndrome; this event was also considered to be related to study therapy. The frequencies of other drug-related adverse events in the QD group were unchanged between week 48 and week 96.
TABLE 3.
Summary of Clinical Adverse Events, Cumulative Rates Through Week 48 and Week 96
Drug-related nervous system adverse events occurred in 37 (7.0%) of raltegravir QD recipients and 26 (9.8%) of raltegravir BID recipients. Specific drug-related nervous system adverse events with >2% incidence were dizziness (2.3% vs 3.4%) and headache (3.0% vs 4.9%). Drug-related psychiatric adverse events occurred in 21 (4.0%) of the QD group and 12 (4.5%) of the BID group; specific drug-related psychiatric AEs occurred in less than 2% of the participants in each treatment group.
Laboratory abnormalities above grade 1 by the Division of AIDS criteria were uncommon and occurred at similar rates in the raltegravir QD and BID groups (Table 4) with 1 exception: grade 2 elevations of alanine aminotransferase (ALT) occurred in 4.2% of the QD group vs 1.5% of the BID group; however, these elevations were generally transient, not associated with clinical adverse events, and not considered clinically significant by the investigators.
TABLE 4.
Laboratory Changes Grade 2 or Higher, Incidence ≥1% in Either Treatment Group, Week 96

Grade 3 or 4 ALT elevations occurred in 12 (2.3%) participants in the QD group and 2 (0.8%) in the BID group by week 96; about half of these events (5 and 1, respectively) occurred after week 48. Most of these events were related to concurrent conditions (hepatitis in 8 participants, hepatocellular injury in 1, hepatic steatosis in 1, and isoniazid treatment in 1). The 3 cases with no obvious etiology resolved on study therapy. Grade 3 or 4 elevations in creatine phosphokinase (CPK) occurred in 35 (6.6%) QD recipients and 12 (4.5%) BID recipients by week 96; of these, 19 and 1, respectively, occurred after week 48. These changes were transient and resolved on therapy in most cases (33 and 11, respectively). Grade 3 or 4 lipase elevations occurred in 17 (3.2%) of the QD group and 4 (1.6%) of the BID group by week 96; of these, 4 and 3, respectively, occurred after week 48. Lipase elevations were not associated with clinical symptoms with the exception of 1 QD recipient who experienced moderate pancreatitis that resolved while continuing study therapy.
Two participants (both in the QD group) discontinued therapy because of laboratory changes: increased ALT and aspartate aminotransferase (AST) (grade 3 and 1, respectively) in 1 and increased CPK (grade 4) in the other; both events occurred before week 48. None of the laboratory events that occurred after week 48 led to treatment discontinuation in either treatment group.
Two participants met the criteria for potential drug-induced liver injury (AST or ALT ≥3xULN [upper limit of the normal range] with bilirubin ≥2xULN and alkaline phosphatase <2xULN): 1 in the QD group before week 48 and 1 in the BID group after week 48. In both cases, there were alternative explanations for the laboratory findings (hepatitis B virus flare and acute hepatitis C virus, respectively).
Among participants with hepatitis coinfection, no drug-related adverse events, serious adverse events, or discontinuations because of adverse events were reported after week 48 in either treatment group. Two of the 15 coinfected participants in the raltegravir QD group had laboratory adverse events after week 48 (grade 4 elevations of lipase and creatine kinase, respectively), but neither event was considered drug-related. No new reports of AST or ALT elevations above grade 2 occurred after week 48 among coinfected participants in either treatment group.
DISCUSSION
The final 96-week results of this pivotal phase 3 trial in treatment-naive adults with HIV-1 infection demonstrate the durable and noninferior efficacy of raltegravir 1200 mg QD compared with raltegravir 400 mg BID, each in combination with FTC/TDF. The noninferior efficacy and favorable safety profile of raltegravir 1200 mg QD were first demonstrated at week 48 and have been previously reported.10 At week 96, the efficacy of raltegravir 1200 mg QD was again shown to be noninferior to that of raltegravir 400 mg BID, as assessed by the proportion of participants with HIV-1 RNA <40 copies per milliliter. Similar results were observed for the supportive virologic endpoint of HIV-1 RNA <50 copies per milliliter, and both treatment groups continued to show substantial increases in CD4+ T-cell counts (over 260 cells/mm3) through week 96. In addition, raltegravir 1200 mg QD showed comparable efficacy to raltegravir 400 mg BID across all subgroups examined, including those with high baseline viral load. Overall, the efficacy of raltegravir 1200 mg QD in this study is comparable to that of dolutegravir and elvitegravir reported at 96 weeks.13–18
PDVF was used to identify participants for drug resistance testing. At week 96, the frequency of PDVF was low and similar between the raltegravir QD and BID groups (9.6% and 9.8%, respectively). The rate of viral drug resistance also remained low, with only 3 additional reports after week 48: 1 in the QD group and 2 in the BID group. By week 96, 0.8% of each group developed resistance to INSTI and resistance to NRTI. Development of resistance was associated with high baseline viral load and low CD4+ T-cell count in 5 of the 6 cases, but did not seem to be related to poor treatment adherence. In previous studies of raltegravir 400 mg BID in treatment-naive adults, rates of resistance to INSTI ranged from 1% to 2% at 96 weeks of follow-up.19,20 The specific integrase mutations observed in this study were consistent with those found in the Phase 3 studies of raltegravir BID in treatment-naive and treatment-experienced adults.20–22 Resistance testing was possible in only a small subset of participants with PDVF; additional resistance may have been selected in participants with confirmed viremia but could not be detected because the HIV-1 RNA was below the assay threshold of 500 copies per milliliter.
The favorable safety profile of raltegravir 400 mg BID was established in clinical trials1–4 and confirmed by postmarketing experience.23–25 At week 96 of the ONCEMRK trial, the QD and BID regimens continued to demonstrate favorable safety and tolerability profiles with small increases in the rates of drug-related adverse events after week 48 (1.5% and 1.1%, respectively), no serious drug-related adverse events after week 48, and only 1 discontinuation because of an adverse event after week 48. The raltegravir 1200 mg QD regimen continued to be well tolerated in participants with chronic hepatitis (B or C) coinfection, an important subpopulation among those with HIV-1 infection.
Treatment-emergent laboratory abnormalities were generally similar for raltegravir 1200 mg QD and raltegravir 400 mg BID. The incidence of grade 3 or 4 elevations in ALT in the QD group (2.2%) was higher than that in the BID group (0.8%) but was similar to rates reported in previous clinical trials of raltegravir 400 mg BID: 1.8% in STARTMRK,20 3.4% in QDMRK,26 3% in NEAT001,27 and 2% in SPRING-2.15 Similarly, the incidence of grade 3 or 4 elevations in AST in the QD group (2.1%) was higher than that in the BID group (0.4%) but was similar to previously reported rates for raltegravir 400 mg BID in STARTMRK (3.2%)20 and QDMRK (3.4%).26 The incidence of grade 3 or 4 CPK elevations (6.6% in the QD group and 4.5% in the BID group) was similar to previously reported rates for raltegravir 400 mg BID in QDMRK (5.4%)26 and NEAT001 (6%).27 The transient nature of the elevations in cases without alternative etiologies (such as viral hepatitis or hepatotoxic concomitant therapies), the small differences between treatment groups, and the finding that most resolved without discontinuation of study therapy suggest that these laboratory changes do not raise new safety concerns for raltegravir QD.
Because the ONCEMRK trial was double-blind, all participants were required to take multiple pills BID; therefore, the potential for improved adherence with the QD regimen could not be evaluated in this trial. The global distribution of the study sites and the racial diversity of the study population support the generalizability of the study results; however, some groups (women, older patients, and those with hepatitis coinfection) were underrepresented. Raltegravir 400 mg BID has shown favorable efficacy and safety in women,28–30 hepatitis C coinfection,31 and older patients25; additional experience with raltegravir 1200 mg QD in these populations will help define further the safety and efficacy profile of the new formulation. The conclusions that can be drawn from the subgroup analyses are limited for several reasons: some of the subgroups were small, no adjustment was made for multiple comparisons, and the study was not powered to detect statistically significant effects within these subgroups.32 Nevertheless, no apparent trends for differences in efficacy within subpopulations were noted in this study.
In summary, the week 96 results of the ONCEMRK trial show that the efficacy of raltegravir 1200 mg QD, previously demonstrated at week 48, was maintained through week 96 and was similar to the efficacy of raltegravir 400 mg BID. Development of resistance to raltegravir remained low (0.8%) in both treatment groups at week 96. In addition, the overall safety profile of raltegravir 1200 mg QD continues to be consistent with the well-established safety profile of raltegravir 400 mg BID.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the study participants and their families and caregivers for participating in the ONCEMRK study. The contributions of the investigators and their staff are likewise gratefully recognized, along with the following employees of Merck & Co., Inc., Kenilworth, NJ: Kim M. Strohmaier for medical writing, Carol Zecca for editorial assistance, and Danielle Mancaruso for graphics support.
APPENDIX 1. The ONCEMRK Study Group
ONCEMRK Primary Investigators (by country):
Argentina: P. E. Cahn, I.Cassetti, M.Losso; Australia: M.T. Bloch, N. Roth, J. McMahon, R. J. Moore, D. Smith; Belgium: N.Clumeck, L.Vanderkerckhove, B.Vandercam, M.Moutschen; Canada: J.Baril, B. Conway, F.Smaill, G. H. R. Smith, A.Rachlis, S. L.Walmsley; Chile: C. Perez, M. Wolff, M. F. Lasso, C. E.Chahin; Colombia: J. D. Velez, O.Sussmann; France: J.Reynes, C.Katlama, Y.Yazdanpanah, S. Ferret, J. Durant, C.Duvivier, I.Poizot-Martin, F.Ajana; Germany: J. K.Rockstroh, G.Faetkanheuer, S.Esser, H. Jaeger, O.Degen, M. Bickel, J.Bogner, K.Arasteh, H.Hartl, A.Stoehr; Guatemala: E. M. Rojas, E.Arathoon, L. D. Gonzalez, C. R. Mejia; Israel: E.Shahar, D. Turner, I. Levy, Z.Sthoeger, H.Elinav; Italy: A.Gori, A.D'ArminioMonforte, G. Di Perri, A.Lazzarin, G.Rizzardini, A.Antinori, B. M.Celesia, F.Maggiolo; Malaysia: T. S. Chow, C. K. C. Lee, R.Iskandar Shah Raja Azwa, M. Mustafa; Peru: M.Oyanguren, R. A. Castillo, L.Hercilla; Phillippines: C.Echiverri; Portugal: F.Maltez, J. G.Saraiva da Cunha, I.Neves, E.Teofilo, R.Serrao; Russia: F.Nagimova, I.Khaertynova, E.Orlova-Morozova, E.Voronin, V.Sotnikov, A. A.Yakovlev, N. G.Zakharova, O. A.Tsybakova; South Africa: M. E.Botes, L.Mohapi, R. Kaplan, M. S.Rassool; Spain: J. R.Arribas, J. M.Gatell, E.Negredo, E. Ortega, J.Troya, J.Berenguer, K.Aguirrebengoa, A.Antela; Switzerland: A.Calmy, M.Cavassini, A. Rauch, M.Stoeckle; Taiwan: W.-H. Sheng, H. -H. Lin, H. -C. Tsai; Thailand: D.Changpradub, A.Avihingsanon, S.Kiertiburanakul, W.R.; United Kingdom: M. R. Nelson, A. Clarke, A.Ustianowski, A. Winston, M. A. Johnson; United States: D. M.Asmuth, J. Cade, J. E. Gallant, P. J.Ruane, P. N. Kumar, A. E.Luque, L. Panther, K. T.Tashima, D. Ward, D. S. Berger, C. A. Dietz, C.Fichtenbaum, S. Gupta, K. M.Mullane, R. M. Novak, D. E. Sweet, G. E.Crofoot, D. P.Hagins, S. T. Lewis, C. K. McDonald, E.DeJesus, L. Sloan, D. J.Prelutsky, J. C.Rondon, S.Henn, A. J.Scarsella, J. O. Morales-Ramirez, L. Santiago, C. D.Zorrilla, M. S.Saag, C. -B. Hsiao.
Footnotes
Supported by Merck & Co., Inc., Kenilworth, NJ.
Presented in part at the 9th IAS Conference on HIV Science;July 23–26, 2017; Paris, France, and the 16th European AIDS Conference;October 25–27, 2017; Milan, Italy.
P.C. is an advisory board member of Merck and ViiV and received research grant support from AbbVie, Merck, and ViiV. P.S. serves as a consultant to or scientific advisory board member of AbbVie, BMS, Gilead, GSK/ViiV, Merck, and Janssen and received research grant support from Bristol Myers Squibb, Gilead, Merck, and GSK/ViiV. K.S. is an advisory board member of Bristol Myers Squibb, Gilead Sciences, Janssen, Merck, and ViiV and received research grant support from Gilead. J.-M.M. is an advisory board member of Merck, Gilead, Bristol Myers Squibb, ViiV, Janssen, and Teva. M.B. received research grants, is an advisory board member, meeting presentations for Amgen, Merck, Gilead Sciences, ViiV Healthcare, and AbbVie. K.S., X.X., Y.Z., B.H., D.H., H.T., G.H., B.-Y.N., and W.G. are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, and may own stock and/or stock options. The remaining authors have no conflicts of interest to disclose.
Registration: ClinicalTrials.govNCT02131233.
Members of the ONCEMRK Study Group are listed in Appendix 1.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
Contributor Information
Collaborators: ONCEMRK Study Group
REFERENCES
- 1.Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: final 5-year results from STARTMRK. J Acquir Immune DeficSyndr. 2013;63:77–85. [DOI] [PubMed] [Google Scholar]
- 2.Eron JJ, Cooper DA, Steigbigel RT, et al. Efficacy and safety of raltegravir for treatment of HIV for 5 years in the BENCHMRK studies: final results of two randomised, placebo-controlled trials. Lancet Infect Dis. 2013;13:587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma M, Walmsley SL. Raltegravir as antiretroviral therapy in HIV/AIDS. Expert OpinPharmacother. 2014;15:395–405. [DOI] [PubMed] [Google Scholar]
- 4.Calcagno A, D'Avolio A, Bonora S. Pharmacokinetic and pharmacodynamic evaluation of raltegravir and experience from clinical trials in HIV-positive patients. Expert Opin Drug MetabToxicol. 2015;11:1167–1176. [DOI] [PubMed] [Google Scholar]
- 5.Parienti JJ, Bangsberg DR, Verdon R, et al. Better adherence with once-daily antiretroviral regimens: a meta-analysis. Clin Infect Dis. 2009;48:484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper V, Moyle GJ, Fisher M, et al. Beliefs about antiretroviral therapy, treatment adherence and quality of life in a 48-week randomised study of continuation of zidovudine/lamivudine or switch to tenofovir DF/emtricitabine, each with efavirenz. AIDS Care. 2011;23:705–713. [DOI] [PubMed] [Google Scholar]
- 7.Cooper V, Horne R, Gellaitry G, et al. The impact of once-nightly versus twice-daily dosing and baseline beliefs about HAART on adherence to efavirenz-based HAART over 48 weeks: the NOCTE study. J Acquir Immune DeficSyndr. 2010;53:369–377. [DOI] [PubMed] [Google Scholar]
- 8.Jayaweera D, Dejesus E, Nguyen KL, et al. Virologic suppression, treatment adherence, and improved quality of life on a once-daily efavirenz-based regimen in treatment-Naive HIV-1-infected patients over 96 weeks. HIV Clin Trials. 2009;10:375–384. [DOI] [PubMed] [Google Scholar]
- 9.ISENTRESS HD (raltegravir) Film-Coated Tablets. Kenilworth, NJ: Merck & Co., Inc; 2017. [Google Scholar]
- 10.Cahn P, Kaplan R, Sax PE, et al. Raltegravir 1200 mg once daily versus raltegravir 400 mg twice daily, with tenofovirdisoproxilfumarate and emtricitabine, for previously untreated HIV-1 infection: a randomised, double-blind, parallel-group, phase 3, non-inferiority trial. Lancet HIV. 2017;4:e486–e494. [DOI] [PubMed] [Google Scholar]
- 11.U.S. Department of Health and Human Services, National Institutes of Health, National Institute of Allergy and Infectious Diseases, Division of AIDS. Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 1.0; 2009. Available at: http://rsc.tech-res.com/docs/default-source/safety/table_for_grading_severity_of_adult_pediatric_adverse_events.pdf. Accessed October 7, 2013. [Google Scholar]
- 12.Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4:213–226. [DOI] [PubMed] [Google Scholar]
- 13.Molina JM, Clotet B, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomised, open-label, phase 3b study. Lancet HIV. 2015;2:e127–36. [DOI] [PubMed] [Google Scholar]
- 14.Walmsley S, Baumgarten A, Berenguer J, et al. Brief report: dolutegravir plus abacavir/lamivudine for the treatment of HIV-1 infection in antiretroviral therapy-naive patients: week 96 and week 144 results from the SINGLE randomized clinical trial. J Acquir Immune DeficSyndr. 2015;70:515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raffi F, Jaeger H, Quiros-Roldan E, et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2013;13:927–935. [DOI] [PubMed] [Google Scholar]
- 16.Wohl D, Oka S, Clumeck N, et al. Brief report: arandomized, double-blind comparison of tenofovir alafenamide versus tenofovir disoproxil fumarate, each coformulated with elvitegravir, cobicistat, and emtricitabine for initial HIV-1 treatment: week 96 results. J Acquir Immune DeficSyndr. 2016;72:58–64. [DOI] [PubMed] [Google Scholar]
- 17.Zolopa A, Sax PE, DeJesus E, et al. A randomized double-blind comparison of coformulate delvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune DeficSyndr. 2013;63:96–100. [DOI] [PubMed] [Google Scholar]
- 18.Rockstroh JK, DeJesus E, Henry K, et al. A randomized, double-blind comparison of coformulate delvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus coformulate demtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune DeficSyndr. 2013;62:483–486. [DOI] [PubMed] [Google Scholar]
- 19.Lennox JL, Landovitz RJ, Ribaudo HJ, et al. Efficacy and tolerability of 3 nonnucleoside reverse transcriptase inhibitor-sparing antiretroviral regimens for treatment-naive volunteers infected with HIV-1: a randomized, controlled equivalence trial. Ann Intern Med. 2014;161:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lennox JL, Dejesus E, Berger DS, et al. Raltegravir versus efavirenz regimens in treatment-naive HIV-1-infected patients: 96-week efficacy, durability, subgroup, safety, and metabolic analyses. J Acquir Immune DeficSyndr. 2010;55:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper DA, Steigbigel RT, Gatell JM, et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008;359:355–365. [DOI] [PubMed] [Google Scholar]
- 22.Steigbigel RT, Cooper DA, Teppler H, et al. Long-term efficacy and safety of Raltegravir combined with optimized background therapy in treatment-experienced patients with drug-resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 Phase III trials. Clin Infect Dis. 2010;50:605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elzi L, Erb S, Furrer H, et al. Adverse events of raltegravir and dolutegravir. AIDS. 2017;31:1853–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Halsema C, Whitfield T, Lin N, et al. Five years' real-life experience with raltegravir in a large HIV centre. Int J STD AIDS. 2016;27:387–393. [DOI] [PubMed] [Google Scholar]
- 25.Naumann U, Moll A, Schleehauf D, et al. Similar efficacy and tolerability of raltegravir-based antiretroviral therapy in HIV-infected patients, irrespective of age group, burden of comorbidities and concomitant medication: real-life analysis of the German 'WIP' cohort. Int J STD AIDS. 2017;28:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eron JJ, Rockstroh JK, Reynes J, et al. Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect Dis. 2011;11:907–915. [DOI] [PubMed] [Google Scholar]
- 27.Raffi F, Babiker AG, Richert L, et al. Ritonavir-boosted darunavir combined with raltegravir or tenofovir–emtricitabine in antiretroviral-naive adults infected with HIV-1: 96 week results from the NEAT001/ANRS143 randomized non-inferiority trial. Lancet. 2014;384:1942–1951. [DOI] [PubMed] [Google Scholar]
- 28.Maliakkal A, Walmsley S, Tseng A. Critical review: review of the efficacy, safety, and pharmacokinetics of raltegravir in pregnancy. J Acquir Immune DeficSyndr. 2016;72:153–161. [DOI] [PubMed] [Google Scholar]
- 29.Squires KE, Bekker LG, Eron JJ, et al. Safety, tolerability, and efficacy of raltegravir in a diverse cohort of HIV-infected patients: 48-week results from the REALMRKstudy. AIDS ResHum Retroviruses. 2013;29:859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Squires K, Bekker LG, Katlama C, et al. Influence of sex/gender and race on responses to raltegravir combined with tenofovir-emtricitabine in treatment-naive human immunodeficiency Virus-1 infected patients: pooled analyses of the STARTMRK and QDMRK studies. Open Forum Infect Dis. 2017;4:ofw047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taramasso L, Cenderello G, Riccardi N, et al. Role of Raltegravir in patients co-infected with HIV and HCV in the era of direct antiviral agents. New Microbiol. 2017;40:227–233. [PubMed] [Google Scholar]
- 32.Wang R, Lagakos SW, Ware JH, et al. Statistics in medicine—reporting of subgroup analyses in clinical trials. N Engl J Med. 2007;357:2189–2194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.