

Supplemental Digital Content is available in the text.
Keywords: aging; atherosclerosis; cardiovascular diseases; models, animal; muscle, smooth, vascular; progeria
Abstract
Background:
Progerin, an aberrant protein that accumulates with age, causes the rare genetic disease Hutchinson-Gilford progeria syndrome (HGPS). Patients who have HGPS exhibit ubiquitous progerin expression, accelerated aging and atherosclerosis, and die in their early teens, mainly of myocardial infarction or stroke. The mechanisms underlying progerin-induced atherosclerosis remain unexplored, in part, because of the lack of appropriate animal models.
Methods:
We generated an atherosclerosis-prone model of HGPS by crossing apolipoprotein E–deficient (Apoe–/–) mice with LmnaG609G/G609G mice ubiquitously expressing progerin. To induce progerin expression specifically in macrophages or vascular smooth muscle cells (VSMCs), we crossed Apoe–/–LmnaLCS/LCS mice with LysMCre and SM22αCre mice, respectively. Progerin expression was evaluated by polymerase chain reaction and immunofluorescence. Cardiovascular alterations were determined by immunofluorescence and histology in male mice fed normal chow or a high-fat diet. In vivo low-density lipoprotein retention was assessed by intravenous injection of fluorescently labeled human low-density lipoprotein. Cardiac electric defects were evaluated by electrocardiography.
Results:
Apoe–/–LmnaG609G/G609G mice with ubiquitous progerin expression exhibited a premature aging phenotype that included failure to thrive and shortened survival. In addition, high-fat diet–fed Apoe–/–LmnaG609G/G609G mice developed a severe vascular pathology, including medial VSMC loss and lipid retention, adventitial fibrosis, and accelerated atherosclerosis, thus resembling most aspects of cardiovascular disease observed in patients with HGPS. The same vascular alterations were also observed in Apoe–/–LmnaLCS/LCSSM22αCre mice expressing progerin specifically in VSMCs, but not in Apoe–/–LmnaLCS/LCSLysMCre mice with macrophage-specific progerin expression. Moreover, Apoe–/–LmnaLCS/LCSSM22αCre mice had a shortened lifespan despite the lack of any overt aging phenotype. Aortas of ubiquitously and VSMC-specific progerin-expressing mice exhibited increased retention of fluorescently labeled human low-density lipoprotein, and atheromata in both models showed vulnerable plaque features. Immunohistopathological examination indicated that Apoe–/–LmnaLCS/LCSSM22αCre mice, unlike Apoe–/–LmnaG609G/G609G mice, die of atherosclerosis-related causes.
Conclusions:
We have generated the first mouse model of progerin-induced atherosclerosis acceleration, and demonstrate that restricting progerin expression to VSMCs is sufficient to accelerate atherosclerosis, trigger plaque vulnerability, and reduce lifespan. Our results identify progerin-induced VSMC death as a major factor triggering atherosclerosis and premature death in HGPS.
Editorial, see p 283
Clinical Perspective.
What Is New?
We have generated the first mouse model of progerin-induced acceleration of atherosclerosis, a major medical problem in patients with Hutchinson-Gilford progeria syndrome.
We provide the first direct evidence that progerin expression restricted to vascular smooth muscle cells (VSMCs), but not to macrophages, is sufficient to induce premature atherosclerosis and death.
We demonstrate that progerin-induced loss of VSMCs causes atherosclerotic plaque destabilization that may lead to myocardial infarction.
We show that ubiquitous and VSMC-specific progerin expression increases low-density lipoprotein retention in the aortic media, likely accelerating atherosclerosis.
What Are the Clinical Implications?
Apoe–/–LmnaG609G/G609G mice ubiquitously expressing progerin constitute the first preclinical animal model for the study of accelerated atherosclerosis and premature aging.
Apoe–/–LmnaLCS/LCSSM22αCre mice with VSMC-specific progerin expression constitute the first preclinical animal model for the study of accelerated atherosclerosis isolated from other disease symptoms.
Because atherosclerosis is the main disease symptom leading to death in patients with Hutchinson-Gilford progeria syndrome, our studies point to VSMCs as a potential target for future therapies for this devastating disease.
Cardiovascular disease (CVD) is the leading cause of death worldwide, despite major recent progress in its prevention, diagnosis, and treatment.1,2 Atherosclerosis, defined as the narrowing of the arterial lumen because of atheroma plaque buildup, underlies most manifestations of CVD and can lead to myocardial infarction or stroke.
Accelerated atherosclerosis is a predominant feature of the rare genetic disorder Hutchinson-Gilford progeria syndrome (HGPS). Classical HGPS is caused by a point mutation (c.1824C>T; p.G608G) in the LMNA gene resulting in ubiquitous expression of progerin, a mutant form of the nuclear envelope protein lamin A.3,4 Children with HGPS typically appear normal at birth, but within the first 2 years of life start to display the first signs of the disease, such as growth impairment and skin abnormalities. As childhood progresses, patients develop other symptoms, including alopecia, loss of body fat, joint stiffness, and osteoporosis, resembling many features of physiological aging.5,6 The most clinically relevant feature of HGPS is generalized atherosclerosis that leads to premature death from myocardial infarction or stroke at an average age of 14.6 years.5,7 Atherosclerosis in HGPS is accompanied by pathological changes in the aortic wall involving severe vascular smooth muscle cell (VSMC) depletion in the media, extracellular matrix deposition, calcification, and adventitial thickening.8–10
Atherosclerotic disease in HGPS is particularly intriguing because patients lack the classical CVD risk factors, such as increased low-density lipoprotein (LDL) and C-reactive protein levels in serum.11 HGPS therefore offers a unique opportunity to study CVD in isolation from typical risk factors. Because progerin also accumulates during normal aging,10,12,13 studies of HGPS may enhance our knowledge about physiological aging and CVD.14
As of October 1, 2017, there were 111 identified children living with HGPS worldwide, all of them carrying a progerin-producing mutation in the LMNA gene.15 This extremely low number presents major difficulties for research into disease mechanisms and for clinical trials testing new therapeutic strategies. Several mouse models of HGPS have been generated, including LmnaHG, BAC G608G, LmnaG609G, and Zmpste24–/–.16–22 Although these mice recapitulate many aspects of the disease, none have been reported to develop atherosclerosis, the life-threatening symptom of HGPS. Thus, the aim of this study was to generate a mouse model exhibiting progerin-induced atherosclerosis acceleration and to study the relative contribution to progerin-driven atherosclerosis of macrophages and VSMCs, the key players in atherogenesis.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure (available at the authors’ laboratories).
Study Approval
All experimental and other scientific procedures with animals conformed to EU Directive 2010/63EU and Recommendation 2007/526/EC, enforced in Spanish law under Real Decreto 53/2013. Animal protocols were approved by the local ethics committees and the Animal Protection Area of the Comunidad Autónoma de Madrid (PROEX76/14, PROEX78/14, PROEX167/16). Blood for LDL isolation was obtained from healthy volunteers after informed consent in accordance with local ethics committee guidelines (Comité de Ética de la Investigación del Instituto de Salud Carlos III: CEI PI 12_2016-v2).
Mice
All mice used in this study were males on the C57BL/6J genetic background carrying the following genetic manipulations: Apoe–/– (The Jackson Laboratory, stock no: 002052), LmnaG609G/+,18 LmnaLCS/+,18 SM22αCre (TaglnCre, The Jackson Laboratory, stock no: 017491), and LysMCre.23 These lines were used to generate atherosclerosis-susceptible mouse models with ubiquitous progerin expression (Apoe–/–LmnaG609G/G609G), VSMC-specific progerin expression (Apoe–/–LmnaLCS/LCSSM22αCre), or macrophage (myeloid)-specific progerin expression (Apoe–/–LmnaLCS/LCSLysMCre), together with their corresponding controls expressing normal lamin A/C (Apoe–/–Lmna+/+, used as control for Apoe–/–LmnaG609G/G609G) or lamin C only (Apoe–/–LmnaLCS/LCS used as control for cell type–specific models). Table I in the online-only Data Supplement shows the expression pattern of lamin A, lamin C, and progerin in the above-mentioned models. To compare the atherosclerosis-prone (Apoe–/–) and atherosclerosis-resistant (Apoe+/+) backgrounds, we bred LmnaLCS/LCSSM22αCre, LmnaLCS/LCS, LmnaG609G/G609G, and Lmna+/+ mice.
Longevity Studies
Starting at 4 weeks of age, animals were weighed and inspected for health and survival at least once a week (checks were more frequent for Apoe–/–LmnaG609G/G609G mice). Diseased animals were examined by a specialized veterinarian blinded to genotype. Animals that met humane end point criteria were euthanized and the deaths recorded. Animals euthanized because of hydrocephalus, malocclusion, intermale aggression, or other reasons unconnected to phenotype were excluded from the analysis (normally at a very early stage of the study).
High-Fat Diet Experiments
For diet-induced atherosclerosis experiments, animals were maintained for 8 weeks on a high-fat diet (HFD) (10.7% total fat, 0.75% cholesterol, S9167-E010, Ssniff) starting at 8 weeks of age. Mice were euthanized at 16 weeks of age after overnight fasting.
Hematology and Serum Biochemical Analysis
Animals were fasted overnight for all blood analyses, which were performed by specialized staff from the Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC) Animal Facility. For hematology, blood samples were collected in Microvette 100 EDTA tubes (Sarstedt) and analyzed with a PENTRA 80 hematology analyzer (Horibo). For biochemical analysis, blood samples were collected in plastic tubes, incubated at room temperature for 2 to 3 hours to allow clotting, and centrifuged at 2000g for 5 minutes. Serum was stored at –80°C until samples from all experiments were collected. Because of volume limitations, serum samples were pooled from 2 to 4 animals of the same experimental group. Specimens with overt hemolysis were excluded from testing. Biochemical variables were analyzed using a Dimension RxL Max Integrated Chemistry System (Siemens Healthineers).
Quantification of Atherosclerosis Burden
Mouse aortas were fixed in 4% formaldehyde/phosphate-buffered saline (PBS), cleaned of fatty tissue, and stained with 0.2% Oil Red O (ORO, O0625, Sigma). The thoracic aorta and aortic arch were then cut open longitudinally and pinned out flat, intimal side up, for computer-assisted planimetric analysis. Images were taken with a digital camera (Olympus UC30) mounted on a stereo microscope (Olympus SZX3). The ORO-stained lesion was quantified as a percentage of total area with SigmaScan Pro 5 (Systat Software Inc) by an observer blinded to genotype.
Histology and Immunofluorescence
For all mice with an Apoe–/– background, formaldehyde-fixed aortic arches were cleaned of fatty tissue, incubated in 30% sucrose in PBS overnight at 4°C, and included in Tissue-Tek OCT compound (Sakura) for cryostat sectioning. Serial 8-µm sections were stained with ORO, hematoxylin and eosin, and Masson trichrome. For immunofluorescence, sections were blocked and permeabilized for 1 hour at room temperature (RT) in PBS containing 0.3% Triton X-100 (9002-93-1, Sigma), 5% bovine serum albumin (A7906, Sigma), and 5% normal goat serum (005-000-001, Jackson ImmunoResearch). Sections were then incubated for 2 hours at RT with anti–smooth muscle α-actin-Cy3 (SMA-Cy3, C6198, Sigma, 1:200) antibody and overnight at 4°C with the following antibodies: anti-CD68 (MCA1957, Serotec, 1:200), and anti-progerin/lamin A (sc-20680, Santa Cruz, 1:100). Next, samples were incubated with corresponding secondary antibodies (anti-rat Alexa Fluor-568, A-11077 and anti-rabbit Alexa Fluor-647, A-21245, Invitrogen) and Hoechst 33342 nucleic acid stain (B2261, Sigma) for 2 hours at RT and mounted in Fluoromount-G imaging medium (4958-02, Affymetrix eBioscience).
Aortic arches of LmnaLCS/LCSSM22αCre and LmnaLCS/LCS mice were fixed in 4% formaldehyde in PBS, cleaned of fatty tissue, dehydrated to xylene, and embedded in paraffin. Serial 4-µm sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin and Masson trichrome.
Mouse hearts were fixed in 4% formaldehyde in PBS, divided into 2 parts (the upper portion containing the aortic root and aortic valve was separated for atheroma plaque analysis), dehydrated to xylene, and embedded in paraffin. Heart sections were prepared at least at 6 levels. The aortic root was sectioned throughout the aortic valve. Serial 4-µm sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin, Masson trichrome, von Kossa, and Perl Prussian blue. For immunofluorescence, antigen retrieval was performed using 10 mmol/L sodium citrate buffer (pH 6). Then samples were blocked and permeabilized for 1 hour at RT in PBS containing 0.3% Triton X-100, 5% bovine serum albumin, and 5% normal goat serum, and incubated overnight at 4°C with anti-CD31 (ab28364, Abcam, 1:100) antibody, and for 2 hours at RT with anti-SMA-Cy3 antibody (C6198, Sigma, 1:200). Next, samples were incubated with anti-rabbit Alexa Fluor-647 secondary antibody (111-607-008, Jackson ImmunoResearch) and Hoechst 33342 for 2 hours at RT, and mounted in Fluoromount-G imaging medium.
ORO, hematoxylin and eosin, Masson trichrome, von Kossa, and Perls Prussian blue–stained sections were scanned with a NanoZoomer-RS scanner (Hamamatsu), and images were exported using NDP.view2. Immunofluorescence images were acquired with a Zeiss LSM700 confocal microscope. Images were analyzed using ImageJ Fiji software by an observer blinded to genotype. Aortic media and adventitia thickness and medial lipid and VSMC content were analyzed in ≈3 sections of aortic arch per animal, and mean values were used for statistical analysis. Atherosclerotic plaque area, aortic perimeter affected by atherosclerosis, necrotic core size, collagen content, VSMC content, and plaque calcification were quantified in 3 different zones of the aortic root per animal (at the beginning, in the middle, and at the end of the aortic valve), and mean values were used for statistical analysis. Cardiac alterations were assessed near the aortic valve and at least at 6 different levels of the ventricles by a specialized pathologist blinded to genotype.
mRNA Isolation and Reverse Transcription for PCR Detection of Lamin A and Progerin
Liver, kidney, spleen, and heart were homogenized using a TissueLyser (Qiagen), and total RNA was extracted with QIAzol reagent (Qiagen). The RNA pellet was dissolved in RNase-free water, and concentration was measured in a NanoDrop spectrophotometer (Wilmington). RNA (2 µg) was transcribed to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems).
cDNA (200 ng) was amplified by PCR using DNA polymerase (Biotools). Lamin A and progerin mRNAs were detected according to a protocol adapted from Yang et al.24 PCR products were separated on a 2% agarose gel containing ethidium bromide. Images were taken with a Molecular Imager Gel Doc XR+ System (BioRad) and analyzed with Image Laboratory (BioRad).
LDL Retention in Vivo
Fluorescently labeled human LDL was prepared as described.25 In brief, human blood from healthy donors was collected in K3EDTA-containing tubes (455036, Vacuette). After centrifugation, plasma was collected, mixed with KBr, and layered on a KBr density gradient column. The column was centrifuged at 256 000g for 18 hours in an Optima L-100 ultracentrifuge (Beckman). The 1.063 g/mL density layer containing LDL was collected, purified on a PD10 column (17085101, GE Healthcare), and conjugated to the fluorochrome Atto 565 N-hydroxysuccinimide ester (72464, Sigma-Aldrich). Atto565-LDL was purified on a PD10 column, and purity was tested at the CNIC Proteomics Unit by liquid chromatography-mass spectrometry (High Resolution Orbitrap-Fusion, Thermo Scientific) in an 8% to 27% acetonitrile gradient over 2 hours.
Mice received intravenous injections of Atto565-LDL (15 µg/g body weight). Approximately 20 hours postinjection, animals were anesthetized with pentobarbital (Dolethal, Vetoquinol) and lidocaine (Lidocaine 20 mg/mL, B. Braun Medical S.A.) solution in saline (pentobarbital 1:5, lidocaine 1:50) and perfused with 4% formaldehyde in PBS. Prefixed aortas were extracted, cleaned of fatty tissue, opened longitudinally, and pinned flat on a silicone-covered plate. After overnight fixing in 4% formaldehyde/PBS, aortas were cut into 3 pieces and mounted in glycerol on a microscope slide. Fluorescence images were acquired with a Zeiss LSM780 confocal microscope. Autofluorescence (background) was determined in 2 mice (of each genotype) not injected with Atto565-LDL. ImageJ Fiji software was used to quantify the percentage of the aortic area positive for red fluorescence. Videos were prepared using Imaris software (Bitplane).
Electrocardiography
Mice were anesthetized with 1.5% to 2% isoflurane, and 4 electrocardiography (ECG) electrodes were inserted subcutaneously into the limbs. ECG was performed in the morning using the MP36R system (Biopac Systems). ECG data were analyzed with AcqKnowledge software (Biopac Systems) by specialized staff blinded to genotype.
Statistical Analysis
Experimental data are presented as the mean (error bars indicate SEM) for normally distributed data or median with interquartile range (error bars indicate minimum and maximum) for nonnormally distributed data. Based on all experiments, distribution of a variable (lesion size, nucleus count, etc) was assessed using the Kolmogorov-Smirnov and D’Agostino-Pearson normality tests. If the distribution was normal in most of the experiments, a 2-tailed t test was used. If the distribution of a variable was skewed, the 2-tailed Mann-Whitney test was used. To compare multiple groups, a 1-way ANOVA with the Tukey post hoc test was used for normally distributed data, and a Kruskal-Wallis test with the Dunn post hoc test was used for nonnormally distributed data. A log-rank (Mantel-Cox) test was used to compare Kaplan-Meier survival curves. Differences were considered significant at P<0.05. Statistical analysis was performed with GraphPad Prism 5.
Results
Ubiquitous Progerin Expression in Apoe–/– Mice Accelerates Atherosclerosis
LmnaG609G/G609G knock-in mice ubiquitously expressing progerin recapitulate most clinical manifestations of HGPS, including postnatal growth impairment, lipodystrophy, vascular calcification, and reduced survival (average lifespan: ≈15 weeks).18,26 However, we observed no atherosclerosis in aortas of LmnaG609G/G609G mice even when animals were challenged with a HFD for 8 weeks starting at 8 weeks of age (Figure I in the online-only Data Supplement). This finding is consistent with the observation that mice, unlike humans, are extremely resistant to atherosclerosis development, in part, because of differences in cholesterol and lipoprotein metabolism.27 To circumvent this limitation, we generated an atherosclerosis-prone mouse model of HGPS by crossing LmnaG609G/+ mice with apolipoprotein E–deficient (Apoe–/–) mice, a widely used atherosclerosis model.27 As expected, Apoe–/–LmnaG609G/G609G mice showed reduced body weight and a shortened lifespan (median survival, 18.15 weeks) in comparison with Apoe–/–Lmna+/+ littermates with an intact Lmna gene (Figure 1A through 1C).
Figure 1.

Ubiquitous progerin expression in Apoe–/–LmnaG609G/G609G mice impairs postnatal growth, reduces lifespan, and accelerates atherosclerosis. A, Postnatal body weight curves for Apoe–/–Lmna+/+ mice (n=7) and Apoe–/–LmnaG609G/G609G mice (n=14). B, Kaplan-Meier survival curves. Median survival: 18.15 weeks for Apoe–/–LmnaG609G/G609G mice (n=14) and 117.6 weeks for Apoe–/–Lmna+/+ mice (n=7). C, Representative photograph of 16-week-old males. Scale bar: 1 cm. D, Pre–high-fat diet (HFD) serum levels of total cholesterol, free cholesterol, low-density lipoprotein (LDL), and high-density lipoprotein (HDL) in 8-week-old Apoe–/–LmnaG609G/G609G mice (n=5) and Apoe–/–Lmna+/+ mice (n=7). E, Post-HFD serum levels of total cholesterol, free cholesterol, LDL, and HDL in 16-week-old Apoe–/–LmnaG609G/G609G mice (n=6) and Apoe–/–Lmna+/+ mice (n=8). F, Representative aortic arches (Top) and thoracic aortas (Middle and Bottom) stained with Oil Red O; graphs show quantification of atherosclerosis burden in Apoe–/–LmnaG609G/G609G mice (n=9 aortic arches; n=12 thoracic aortas) and Apoe–/–Lmna+/+ mice (n=6 aortic arches; n=13 thoracic aortas). Scale bar: 2 mm. Studies in A, B, and C were performed with mice fed normal chow, and those in D, E, and F were conducted with mice fed the HFD for 2 months starting at 8 weeks of age. Data are shown as mean±SEM in A, D, and E, and as median with interquartile range and minima and maxima in F. Statistical differences were analyzed by log-rank test in B, by 2-tailed t test in A, D, and E, and by 2-tailed Mann-Whitney test in F. *P<0.05. **P<0.01. ***P<0.001.
To study the influence of progerin on atherosclerosis, 8-week-old mice were challenged for 8 weeks with a HFD and euthanized at 16 weeks of age. Pre-HFD serum lipid levels, including total cholesterol, free cholesterol, LDL, and high-density lipoprotein were indistinguishable between experimental groups (Figure 1D). However, post-HFD levels were significantly lower in 16-week-old Apoe–/–LmnaG609G/G609G mice than in age-matched Apoe–/–Lmna+/+ controls (Figure 1E), probably because of reduced food intake associated with the advanced disease progression in progeroid mice. HFD-fed Apoe–/–LmnaG609G/G609G mice nonetheless had a 1.9-fold higher atherosclerosis burden in the aortic arch and a 2.8-fold higher burden in the thoracic aorta (Figure 1F) as assessed by ORO staining. Although both groups had focal aortic lesions, the aortic surface of progeroid Apoe–/–LmnaG609G/G609G mice was largely covered with lipid deposits that were not seen in control Apoe–/–Lmna+/+ mice (Figure 1F, high magnification in bottom images). Histological examination revealed that this abnormality was attributable to excessive lipid accumulation in the aortic tunica media in atheroma-free zones of Apoe–/–LmnaG609G/G609G aorta (Figure 2A). It is remarkable that Apoe–/–LmnaG609G/G609G mice developed other pathologies present in patients with HGPS, including adventitial thickening (Figure 2A) and massive loss of VSMCs within the media (Figure 2B). Apoe–/–LmnaG609G/G609G mice also showed significant hematologic alterations, including lower pre- and post-HFD lymphocyte and monocyte counts (Figure II in the online-only Data Supplement).
Figure 2.

Apoe–/–LmnaG609G/G609G mice with ubiquitous progerin expression exhibit severe vascular pathology, including lipid retention and vascular smooth muscle cell (VSMC) loss in the media, and adventitial thickening. Mice were fed a high-fat diet for 8 weeks starting at 8 weeks of age. A, Representative staining of aorta sections with Oil Red O (ORO) and hematoxylin and eosin (H&E). Graphs show quantification of lipid content in atheroma-free zones of the media (as % of the ORO-positive area) and adventitia-to-media thickness ratio; n=6 to 8. Scale bar: 50 µm. B, Representative immunofluorescence images of aortas stained with anti–smooth muscle actin (SMA) antibody (red) and Hoechst 33342 (blue). Graphs show quantification of VSMC content in the media as either nucleus count or % of SMA-positive area; n=5 to 8. Scale bar: 50 µm. Box and whisker plots in A show medians, interquartile range, and minima and maxima; data in B are mean±SEM. Statistical analysis was performed by 2-tailed Mann-Whitney test in A, and 2-tailed t test in B. ***P<0.001. a indicates adventitia; and m, media.
VSMC-Specific, But Not Macrophage-Specific, Progerin Expression in Apoe–/– Mice Aggravates Atherosclerosis and Shortens Lifespan
Atherosclerosis is a multifactorial process involving many cell types, including VSMCs and macrophages.28,29 Immunohistopathological characterization of atherosclerotic plaques in patients with HGPS suggests that both cell types contribute to progerin-driven atherogenesis.10 Because Apoe–/–LmnaG609G/G609G mice showed medial VSMC loss and reduced numbers of circulating monocytes (which differentiate to macrophages in atheromata), we generated mice with specific progerin expression in VSMCs or macrophages to study the relative contribution of these cell types to the vascular pathology induced by progerin. We used knock-in LmnaLCS/LCS mice expressing only lamin C, which produce progerin on crossing with transgenic mice expressing Cre recombinase.18 LmnaLCS/LCS mice are apparently normal, but are slightly heavier and longer-lived than wild-type controls.30 We generated Apoe–/–LmnaLCS/LCS mice and analyzed atherosclerosis in 16-week-old mice that received a HFD during the last 8 weeks. In comparison with control Apoe–/–Lmna+/+ littermates, Apoe–/–LmnaLCS/LCS mice showed no significant differences in atherosclerosis burden, serum lipid levels, or hematologic parameters (Figure III in the online-only Data Supplement).
These results demonstrated that Apoe–/–LmnaLCS/LCS mice could serve as controls for the macrophage-specific Apoe–/–LmnaLCS/LCSLysMCre and VSMC-specific Apoe–/–LmnaLCS/LCSSM22αCre models. Progerin expression was examined by immunofluorescence in atheroma-containing aorta and by PCR in other organs. Apoe–/–LmnaLCS/LCS mice did not express progerin in any organ tested (Figures IV and V in the online-only Data Supplement). Apoe–/–LmnaLCS/LCSSM22αCre mice abundantly expressed progerin in VSMCs of the media, to a lesser extent in the adventitia (Figure IV in the online-only Data Supplement), and also in heart (Figure V in the online-only Data Supplement). Progerin expression in Apoe–/–LmnaLCS/LCSLysMCre mice was detected in intimal macrophages (Figure IV in the online-only Data Supplement), but was either absent or negligible in other organs (Figure V in the online-only Data Supplement).
Apoe–/–LmnaLCS/LCSLysMCre mice with macrophage-specific progerin expression showed no significant differences in organ size (Figure VI in the online-only Data Supplement), body weight, or longevity compared with controls (Figure 3A through 3C). Although Apoe–/–LmnaLCS/LCSSM22αCre mice with VSMC-specific progerin expression appeared normal early in life (Figure 3C and Figure VI in the online-only Data Supplement), they stopped gaining weight from ≈20 weeks of age (Figure 3A), and died prematurely at a median age of 34.3 weeks (Figure 3B). Atherosclerosis was studied with the same protocol used for the ubiquitously progerin-expressing Apoe–/–LmnaG609G/G609G mice. The 3 experimental groups showed no intergroup differences in serum lipid levels (Figure 3D and 3E) or hematologic parameters (Figure VII in the online-only Data Supplement), either pre- or post-HFD. Atherosclerosis burden in the thoracic aorta was similar in Apoe–/–LmnaLCS/LCSLysMCre and Apoe–/–LmnaLCS/LCS mice; in contrast, the lesion burden in Apoe–/–LmnaLCS/LCSSM22αCre mice was 4.4-fold higher (Figure 3F). Moreover, histological analysis in Apoe–/–LmnaLCS/LCSSM22αCre mice revealed the same marked aortic phenotype observed in the ubiquitous progeroid model, including massive VSMC loss, lipid accumulation in the media, and adventitial thickening (Figure 4).
Figure 3.

Vascular smooth muscle cell (VSMC)–specific progerin expression in Apoe–/–LmnaLCS/LCSSM22αCre mice reduces lifespan and accelerates atherosclerosis. A, Postnatal body weight curves for Apoe–/–LmnaLCS/LCS mice (n=9), Apoe–/–LmnaLCS/LCSSM22αCre mice (n=12), and Apoe–/–LmnaLCS/LCSLysMCre mice (n=6). The weight curve for Apoe–/–LmnaLCS/LCSSM22αCre mice is shown up to the time when ≈90% of the animals were dead. B, Kaplan-Meier survival curves. Median survival: 34.3 weeks for Apoe–/–LmnaLCS/LCSSM22αCre mice (n=17), 106.3 weeks for Apoe–/–LmnaLCS/LCS mice (n=9), and 116.3 weeks for Apoe–/–LmnaLCS/LCSLysMCre mice (n=6). C, Representative photograph of 16-week-old males. Scale bar: 1 cm. D, Pre–high-fat diet (HFD) serum levels of total cholesterol, free cholesterol, low-density lipoprotein (LDL), and high-density lipoprotein (HDL) in 8-week-old Apoe–/–LmnaLCS/LCS mice (n=5), Apoe–/–LmnaLCS/LCSSM22αCre mice (n=4), and Apoe–/–LmnaLCS/LCSLysMCre mice (n=6). E, Post-HFD serum levels of total cholesterol, free cholesterol, LDL, and HDL in 16-week-old Apoe–/–LmnaLCS/LCS mice (n=10), Apoe–/–LmnaLCS/LCSSM22αCre mice (n=7), and Apoe–/–LmnaLCS/LCSLysMCre mice (n=7). F, Representative examples of thoracic aortas stained with Oil Red O (ORO). The graph shows quantification of atherosclerosis burden in Apoe–/–LmnaLCS/LCS mice (n=24), Apoe–/–LmnaLCS/LCSSM22αCre mice (n=17), and Apoe–/–LmnaLCS/LCSLysMCre mice (n=19). Scale bar: 2 mm. Studies in A, B, and C were performed with mice fed normal chow, and those in D, E, and F were conducted with mice fed the HFD for 2 months starting at 8 weeks of age. Data in A, D, and E are mean±SEM. Box and whisker plots in F show medians, interquartile range, and minima and maxima. Statistical analysis was performed by log-rank test in B, by 1-way ANOVA with the Tukey post hoc test in A, D and E, and by the Kruskal-Wallis test with the Dunn post hoc test in F. ***P<0.001.
Figure 4.

Apoe–/–LmnaLCS/LCSSM22αCre mice with vascular smooth muscle cell (VSMC)–specific progerin expression show severe vascular pathology, including VSMC loss and lipid retention in the media and adventitial thickening. Mice were fed a high-fat diet for 8 weeks starting at 8 weeks of age. A, Representative staining of aorta sections with Oil Red O (ORO) and hematoxylin and eosin (H&E). Graphs show quantification of lipid content in atheroma-free zones of the media (% of ORO-positive area) and adventitia-to-media thickness ratio; n=6 to 8. Scale bar: 50 µm. B, Representative immunofluorescence images of aortas stained with anti–smooth muscle actin (SMA) antibody (red) and Hoechst 33342 (blue). Graphs show quantification of VSMC content in the media as either nucleus count or % of SMA-positive area; n=6 to 8. Scale bar: 50 µm. Box and whisker plots in A show medians, interquartile range, and minima and maxima; data in B are mean±SEM. Statistical analysis was performed by the Kruskal-Wallis test with the Dunn post hoc test in A and by 1-way ANOVA with the Tukey post hoc test in B. *P<0.05. **P<0.01. ***P<0.001. a indicates adventitia; and m, media.
Progerin Expression Accelerates Atherosclerosis in Apoe–/– Mice Fed Normal Chow
To gain insight into the progress of progerin-induced atherogenesis and to assess the role of cholesterol in this process, we performed experiments in 8- and 16-week-old mice fed normal chow. Histological examination of aortas from 8-week-old mice revealed no differences between the progeroid models and their controls, with tissue almost free of atherosclerosis and no obvious structural alterations (Figure VIIIA, VIIIC, and VIIIE in the online-only data supplement). In contrast, the thoracic aortas of 16-week-old Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice presented increased lesion formation than their controls (Figure VIIIB and VIIID in the online-only Data Supplement). Furthermore, aortas of both mutant models contained regions with VSMC loss (Figure VIIIE in the online-only Data Supplement). This vascular phenotype was slightly more pronounced in Apoe–/–LmnaLCS/LCSSM22αCre mice than in Apoe–/–LmnaG609G/G609G mice, with lipid retention in the media and adventitial thickening (data not shown), probably because of normal food intake and a consequent cholesterol level similar to that of control mice.
Aortas of Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre Mice Show Increased LDL Retention
Lipid accumulation in the aortic medial layer is a prominent characteristic of the vascular pathology in both the ubiquitous and the VSMC-specific progeroid mouse models. Because LDL retention in the aortic wall is one of the key processes triggering atherosclerosis initiation and progression, we performed in vivo LDL retention experiments by injecting normal chow–fed 16-week-old mice with human LDL labeled with the fluorochrome Atto565 (Figure 5A). Aortic Atto565-LDL content was quantified 20 hours postinjection, at which time exogenous human LDL is cleared from the circulation.25 In comparison with their respective control littermates, both Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice showed increased aortic LDL retention (Figure 5B and 5C and Figure IXA and IXB in the online-only Data Supplement). Although Atto565-LDL was localized in the intimal lesions in mice of all genotypes, its excessive retention in the medial layer was exclusive to the ubiquitous and VSMC-specific progeria mouse models (Figure 5D and 5E, Figure IXC and IXD in the online-only Data Supplement and Movies I through XVI in the online-only Data Supplement).
Figure 5.

Increased LDL retention in the aortas of Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice. A, Experimental workflow. LDL was isolated from human peripheral blood and labeled with the fluorochrome Atto565. Atto565-LDL was administrated intravenously to 16-week-old mice fed normal chow. At 20 hours postinjection, aortas were extracted and confocal microscope images were acquired of the whole-mount tissue. B and C, Representative tile scan images of aortas from mice of the indicated genotypes; graphs show quantification of Atto565-LDL retention (red florescence as a % of the total area) in the aortic arch and thoracic aorta; n=8 mice in B and n=6 in C. Scale bar: 1 mm. D and E, Still frame images of 3-dimensional reconstruction videos, showing elastin layers (green autofluorescence) and Atto565-LDL retention (red fluorescence) in the aortas of mice of the indicated genotypes. Atto565-LDL retention in the medial layer was principally detected in aortas from Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice (see Movies I through XII in the online-only Data Supplement). Scale bar: 50 µm (Top), 30 µm (Bottom). Data from control mice (not injected with Atto565-LDL) are shown in Figure IX in the online-only Data Supplement and Movies XIII through XVI in the online-only Data Supplement. Data in B and C are presented as mean±SEM. Statistical analysis was performed by 2-tailed t test. *P<0.05. **P<0.01. ***P<0.001.
Progerin Triggers Plaque Vulnerability
Previous studies have shown that VSMC loss in the tunica media can promote atherosclerotic plaque instability31; we therefore characterized atherosclerotic lesions in the aortic root. Consistent with our results in the aorta (Figures 1F and 3F), analysis of atheromata in the aortic root revealed a larger lesion area in HFD-fed Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice than in their respective controls, and this was accompanied by fibrosis and inflammation in the adjacent cardiac tissue (Figure 6A and 6B). Moreover, atherosclerosis affected a higher percentage of the aortic surface in both models (Figure 6A and 6B). Analysis of Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice revealed the presence in atheromata of erythrocytes, indicating healed plaque fissures (Figure 6C and 6D, top). We also found hemorrhage in the aortic valve itself (Figure 6C, bottom), and evidence of thrombus formation (Figure 6D, bottom), which might be related to plaque erosion. Accordingly, atheromata in both models showed features of vulnerable plaques (Table II in the online-only Data Supplement), including larger necrotic cores (Figure 7A and 7B), iron deposits (Figure 7C and 7D), and VSMC loss and disorganization in the fibrous cap (Figure 7E and 7F).
Figure 6.

Enhanced aortic root atherosclerosis in Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice is accompanied by inflammation of adjacent cardiac tissue and plaque destabilization. Mice were fed a high-fat diet for 8 weeks starting at 8 weeks of age. Serial sections of the aortic root were made and 3 different regions were stained with Masson trichrome. A and B, Representative photographs of atheromata in the aortic root of Apoe–/–LmnaG609G/G609G mice (A) and Apoe–/–LmnaLCS/LCSSM22αCre mice (B), and their corresponding controls. Graphs show quantification of plaque area, with each point representing the mean for 3 aortic root regions; n=5. Scale bar: 200 µm. Dashed lines indicate medial perimeter (the last layer of elastin) and dotted lines indicate the luminal surface of the atheroma plaque. C, An example of intraplaque (Top) and intravalve (Bottom) hemorrhage in Apoe–/–LmnaG609G/G6090G mice. Scale bar: 100 µm. D, An example of intraplaque hemorrhage (Top) and thrombus formation (Bottom) in Apoe–/–LmnaLCS/LCSSM22αCre mice. Scale bar: 100 µm. Data are shown as mean±SEM. Statistical analysis was performed by 2-tailed t test. **P<0.01. ***P<0.001.
Figure 7.
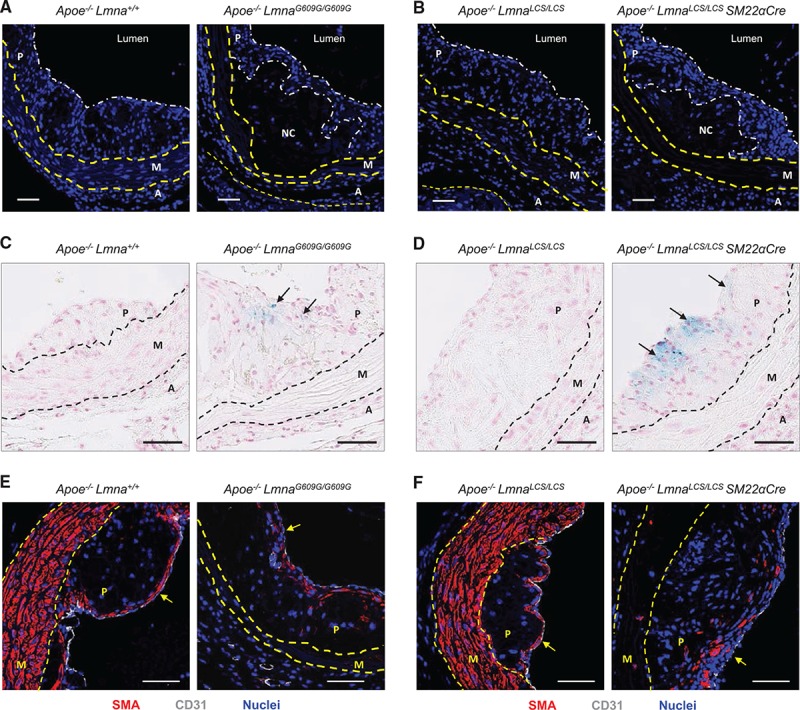
Vulnerable plaque features in Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice. Mice were fed a high-fat diet for 8 weeks starting at 8 weeks of age, and aortas were extracted for histology and immunofluorescence. Representative images are shown. A and B, Hoechst 33342 staining to visualize nuclei (blue). Scale bar: 50 µm. C and D, Perls Prussian blue staining in atherosclerotic plaques to visualize cells with iron deposits (blue; arrows). Scale bar: 50 µm. E and F, Smooth muscle actin (SMA, visualized in red), endothelium (CD31, visualized in white), and nuclei (Hoechst 33342, visualized in blue). Arrows mark the luminal part of the plaque (fibrous cap). Scale bar: 50 µm. A indicates adventitia; M, media; NC, necrotic core; and P, plaque.
Apoe–/–LmnaG609G/G609G Mice Develop Cardiac Electric Defects
Previous studies reported cardiac electric defects in progeroid LmnaG609G/G609G and Zmpste24-deficient (Zmpste24–/–) mice similar to those found in patients with HGPS.6,18,32 We therefore performed ECG in the ubiquitous and VSMC-specific progeria models. As expected, 16-week-old Apoe–/–LmnaG609G/G609G mice exhibited severe bradycardia and prolonged QRS, QT, and QTc intervals (Figure XA in the online-only Data Supplement). Moreover, 2 of 5 mice had arrhythmias (Figure XB in the online-only Data Supplement). In contrast, 16-week-old Apoe–/–LmnaLCS/LCSSM22αCre mice presented no evident ECG alterations (Figure XIA in the online-only Data Supplement). Nevertheless, follow-up revealed a low-voltage QRS complex at 26 weeks of age (Figure IXB in the online-only Data Supplement), consistent with loss of viable myocardial tissue attributable to myocardial infarction. Likewise, many of the 26-week-old Apoe–/–LmnaLCS/LCSSM22αCre mice exhibited T-wave fattening or inversion (Figure IXB in the online-only Data Supplement and data not shown), further supporting the notion that these animals develop age-associated cardiac alterations probably caused by progressive atherosclerosis.
Apoe–/–LmnaLCS/LCSSM22αCre Mice Die of Atherosclerosis-Related Causes
Despite an almost identical vascular phenotype, ubiquitous and VSMC-specific atherosclerosis-prone progeria models differed in their cardiac phenotype and median survival (18.15 weeks versus 34.3 weeks, respectively). Apoe–/–LmnaLCS/LCSSM22αCre mice, which showed no overt aging phenotype, died suddenly between 26 and 66 weeks of age. It is interesting to note that LmnaLCS/LCSSM22αCre mice with an intact Apoe gene also presented VSMC loss and aortic adventitial thickening, but did not develop atherosclerosis and had a normal lifespan (Figure XII in the online-only Data Supplement). These findings point to progerin-driven atherosclerosis as the main cause of death in Apoe–/–LmnaLCS/LCSSM22αCre mice. In contrast, Apoe–/–LmnaG609G/G609G mice progressively lost fat tissue, developed bone problems and muscle weakness, and died between 15 and 24 weeks of age, a median survival very similar to that of atherosclerosis-free LmnaG609G/G609G mice.18 We therefore conclude that the cause of death is likely different in the ubiquitous and VSMC-specific progeria models, at least in the majority of the cases.
To analyze cardiovascular alterations, which could reveal the cause of death, we collected aortas and hearts from chow-fed Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice at an age close to their maximum survival (21–23 weeks and 51 weeks, respectively). Atherosclerosis burden in the aortic arch and thoracic aorta was significantly higher in Apoe–/–LmnaG609G/G609G mice than in Apoe–/–Lmna+/+ controls (Figure 8A). In comparison with Apoe–/–LmnaLCS/LCS controls, Apoe–/–LmnaLCS/LCSSM22αCre mice showed a higher lesion burden in the thoracic aorta but not in the aortic arch (Figure 8B), likely because this athero-prone aortic region is saturated with plaques in 51-week-old (≈1 year) Apoe–/– mice. It is important to note that vascular pathology was much more severe in the aorta of 51-week-old Apoe–/–LmnaLCS/LCSSM22αCre mice than in 21- to 23-week-old Apoe–/–LmnaG609G/G609G mice (Figure 8A through 8C).
Figure 8.

More severe cardiovascular phenotype in Apoe–/–LmnaLCS/LCSSM22αCre mice than in Apoe–/–LmnaG609G/G609G at ages close to their maximum survival. Mice were fed normal chow and euthanized at either 21 to 23 weeks of age (Apoe–/–LmnaG609G/G609G and control Apoe–/–Lmna+/+ mice) or 51 weeks of age (Apoe–/–LmnaLCS/LCSSM22αCre and control Apoe–/–LmnaLCS/LCS mice). A, Representative Oil Red O (ORO) staining in aortas from Apoe–/–LmnaG609G/G609G and Apoe–/–Lmna+/+ mice. Graphs show quantification of atherosclerosis burden in the aortic arch and thoracic aorta; n=6. Scale bar: 2 mm. B, Representative ORO staining in aortas from Apoe–/–LmnaLCS/LCSSM22αCre and Apoe–/–LmnaLCS/LCS mice. Graphs show quantification of atherosclerosis burden in the aortic arch and thoracic aorta; n=5 to 6. Scale bar: 2 mm. C, Higher magnification of ORO-stained thoracic aortas of the indicated genotypes. Scale bar: 1 mm. D, Table summarizing cardiac pathologies assessed in 9 to 11 mice per genotype. Serial heart sections (at least at 6 different levels of the ventricles) were stained with Masson trichrome and analyzed. E, Representative heart sections from Apoe–/–LmnaG609G/G609G and Apoe–/–LmnaLCS/LCSSM22αCre mice. Blue staining indicates fibrosis. Scale bar: 1 mm (whole cross section) and 200 µm (magnified view in insert). Data in A and B are shown as median with interquartile range and minima and maxima. Statistical differences were analyzed by 2-tailed Mann-Whitney test. *P<0.05. **P<0.01. LV indicates left ventricle; RV, right ventricle; and S, septum.
Consistent with the findings in the aorta, pathological findings in the aortic root were more severe in Apoe–/–LmnaLCS/LCSSM22αCre mice than in Apoe–/–LmnaG609G/G609G mice (Figures XIII through XVI in the online-only Data Supplement). Plaques in Apoe–/–LmnaG609G/G609G mice were generally immature, frequently exhibiting intraplaque hemorrhages that were absent in age-matched controls (Figure XIIIA in the online-only Data Supplement). Of the Apoe–/–LmnaG609G/G609G animals, 3 of 9 showed signs of coronary atherosclerosis (Figure XVI, zone 1, in the online-only Data Supplement). In Apoe–/–LmnaLCS/LCSSM22αCre mice, atheromas were mature and severely calcified, and many of them had chondroid metaplasia (Figures XIII and XIV in the online-only Data Supplement). The noncalcified atheromas had thin fibrous caps and large necrotic cores (Figure XIIIA in the online-only Data Supplement). Moreover, 4 of 9 of the analyzed Apoe–/–LmnaLCS/LCSSM22αCre mice developed coronary atherosclerosis (Figure XVI, zone 1, in the online-only Data Supplement). Even though plaques of both mutant models showed features of vulnerable plaques, such as lower collagen and VSMC content (Figure XIIIB in the online-only Data Supplement), Apoe–/–LmnaLCS/LCSSM22αCre mice had more atherosclerosis, fibrosis, and inflammation in the aortic root (Figure XV in the online-only Data Supplement).
We also performed a histopathologic evaluation of the heart ventricles. Apoe–/–LmnaG609G/G609G hearts were characterized by cardiomyocyte vacuolization, which was absent in Apoe–/–Lmna+/+, Apoe–/–LmnaLCS/LCSSM22αCre, and Apoe–/–LmnaLCS/LCS mice (Figure XVII in the online-only Data Supplement and data not shown). Furthermore, 3 of 9 Apoe–/–LmnaG609G/G609G hearts had coronary atherosclerosis (Figure 8D and Figure XVI in the online-only Data Supplement) and 2 had a collagen scar in the septum, indicative of a nonfatal infarct (Figure XVIII in the online-only Data Supplement). Nevertheless, many of the animals analyzed showed no detectable alterations apart from the cardiomyocyte vacuolization (Figure 8D and 8E).
Most Apoe–/–LmnaLCS/LCSSM22αCre hearts presented perivascular and interstitial fibrosis, intimal hyperplasia (without cellular infiltrates) and coronary atherosclerosis (Figure 8D and 8E, Figures XV and XVI in the online-only Data Supplement). Some of the animals analyzed also showed evidence of coronary plaque calcification (Figure 8D). These pathological alterations in the heart suggest that Apoe–/–LmnaLCS/LCSSM22αCre animals are, like patients with HGPS, susceptible to microinfarcts during their lifetime (Figure 8E). Collectively, these histopathologic findings in the aorta and heart indicate that Apoe–/–LmnaLCS/LCSSM22αCre animals die of atherosclerosis complications, whereas Apoe–/–LmnaG609G/G609G mice mostly die of atherosclerosis-independent processes.
Discussion
Several mouse models of HGPS have been generated16–22; however, only a few of these models presented cardiovascular alterations and none showed evidence of atherosclerosis, the pathological process that leads to myocardial infarction– or stroke-related death in most children with HGPS. Based on these models, various molecular mechanisms have been proposed to explain how progerin drives HGPS. Treatments targeting candidate pathways have significantly ameliorated disease progression and extended lifespan in mice; however, these treatments have only had modest therapeutic effects in patients with HGPS.33–35 One possible explanation is that progeric mice, unlike humans, die of atherosclerosis-independent causes. It is important to note that mice are particularly resistant to atherosclerotic plaque formation, in part, because of the murine blood lipid profile, with high atheroprotective high-density lipoprotein and low atherogenic LDL.36 Thus, to study atherosclerosis in mouse models, it is usually necessary to introduce a genetic mutation (such as inactivation of the Apoe or Ldlr genes), to alter their lipid metabolism and make them more prone to developing atherosclerosis.37,38
In this study, we generated Apoe–/–LmnaG609G/G609G mice, the first mouse model of progerin-induced atherosclerosis acceleration. In addition to enhanced atheroma plaque formation, these animals exhibit massive loss of medial VSMCs and prominent adventitial thickening and fibrosis, important hallmarks of vascular disease in HGPS.8–10 These features were previously observed in knock-in LmnaG609G/G609G and transgenic G608G BAC mouse models17–19; however, in earlier models, these features did not manifest as early or as severely as described here and were not accompanied by atherosclerosis development. The Apoe–/–LmnaG609G/G609G model is also the first to show lipid accumulation in atheroma-free zones of the aortic media, which may contribute to enhanced plaque formation. Furthermore, our studies with Apoe–/–LmnaLCS/LCSSM22αCre mice (VSMC-specific progerin expression) and Apoe–/–LmnaLCS/LCSLysMCre mice (macrophage-specific progerin expression) provide the first direct evidence that restricting progerin expression to VSMCs, but not macrophages, is sufficient to accelerate atherosclerosis and reduce lifespan. Although limiting progerin expression to macrophages does not increase atherosclerosis burden or reduce survival, it remains to be determined whether it affects plaque composition. Further studies are also required to ascertain why progerin-expressing Apoe–/– mice have increased lipid retention in the media (eg, increased vascular permeability, changes in the extracellular matrix amount and composition).
Our findings in both the ubiquitous and the VSMC-specific progerin mouse models indicate that VSMCs are particularly sensitive to progerin accumulation. Progerin-induced VSMC death results in increased LDL retention in the media, adventitial thickening, and accelerated atherosclerosis. Similarly to Clarke et al,31 we found that VSMC death triggers features of plaque vulnerability, which may lead to myocardial infarction and premature death. Our results demonstrate the importance of VSMCs in progerin-driven atherosclerosis and identify this cell type as a key target for future treatments.
It is remarkable that we observed that Apoe–/–LmnaG609G/G609G mice have a similar average lifespan to nonatherosclerotic LmnaG609G/G609G mice, suggesting that mice with ubiquitous progerin expression die of atherosclerosis-independent causes, unlike human patients with HGPS. In contrast, Apoe–/–LmnaLCS/LCSSM22αCre mice appear to die of atherosclerosis-related problems, because LmnaLCS/LCSSM22αCre mice, with VSMC-specific progerin expression but an intact Apoe gene, did not develop atherosclerosis and had a normal lifespan. This hypothesis is supported by histopathologic findings in mice at ages close to their maximum survival, which reveal much more severe aortic and coronary atherosclerosis in Apoe–/–LmnaLCS/LCSSM22αCre mice than in Apoe–/–LmnaG609G/G609G mice. It may be that plaque disruption leading to infarction and death occurs in Apoe–/–LmnaG609G/G609G mice, but only in animals surviving to near 26 weeks of age. This is well beyond the median survival in this model (18.15 weeks), but is the age at which Apoe–/–LmnaLCS/LCSSM22αCre mice begin to die. Although Apoe–/–LmnaG609G/G609G mice may die of heart failure, the cause would likely be related to complications such as arrhythmia, rather than atherosclerosis, as supported by ECG studies in this model. Similarly, we previously reported that LmnaG609G/G609G mice have a prolonged QRS-complex duration, indicating defective cardiac conduction.18 Likewise, our studies in Zmpste24-null mice, another model of premature aging caused by defective prelamin A processing, revealed PQ and QRS prolongation and T-wave flattening, which may increase the risk of lethal arrhythmias in the setting of progeria.32 This result is also consistent with the observation that patients with HGPS show repolarization abnormalities resulting in an increased risk of arrhythmias and premature death.32
In summary, atherosclerosis-prone Apoe–/–LmnaG609G/G609G mice faithfully recapitulate most aspects of human HGPS, including growth retardation, shortened lifespan, and early CVD onset, and is therefore a valuable tool for investigation of the general mechanisms underlying HGPS. We have also demonstrated that VSMC-restricted progerin expression in Apoe–/–LmnaLCS/LCSSM22αCre mice is sufficient to induce the vascular features of progeria independently of growth retardation and other disease symptoms, thus providing a unique preclinical model for the study of atherosclerosis and atherosclerosis-associated premature death in isolation from other signs of premature aging. Another advantage of these animals is the lack of overt growth defects, which might permit the testing of diagnostic techniques and interventional strategies that are unavailable in the undersized Apoe–/–LmnaG609G/G609G model.
Acknowledgments
The authors thank F. Osorio and J. Rivera for their support in the initial phases of the study, S. Bartlett for English editing; M. Ricote for providing LysMCre mice; A. de Molina and R. Doohan for support with histology; and V. Zorita, E. Santos, R. Mota, and the CNIC Animal Facility for animal care.
Sources of Funding
Work in Dr Andrés’ laboratory is supported by grants from the Spanish Ministerio de Economía, Industria y Competitividad (MEIC) (SAF2016-79490-R) and the Instituto de Salud Carlos III (AC16/00091, AC17/00067) with cofunding from the Fondo Europeo de Desarrollo Regional (FEDER, “Una manera de hacer Europa”), the Progeria Research Foundation (Established Investigator Award 2014-52), and the Fundació Marató TV3 (122/C/2015). The MEIC supported Dr Hamczyk (“Formación de Personal Investigador” predoctoral contract BES-2011–043938) and Dr Villa-Bellosta (“Juan de la Cierva” JCI-2011–09663 postdoctoral contract). The Instituto Universitario de Oncología is supported by Obra Social Cajastur. The Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC) is supported by the MEIC and the Pro-CNIC Foundation, and is a Severo Ochoa Center of Excellence (award SEV-2015-0505).
Disclosures
None.
Supplementary Material
Footnotes
Sources of Funding, see page 281
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.117.030856/-/DC1.
References
- 1.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 2.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD, 3rd, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Mohd Hanafiah K, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA, 3rd, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez-Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez-Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stöckl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, Memish ZA. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–2260. doi: 10.1016/S0140-6736(12)61766-8. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Lévy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 4.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–2624. doi: 10.1002/ajmg.a.31346. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 6.Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO, 3rd, Gahl WA, Introne WJ. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ullrich NJ, Gordon LB. Hutchinson-Gilford progeria syndrome. Handb Clin Neurol. 2015;132:249–264. doi: 10.1016/B978-0-444-62702-5.00018-4. doi: 10.1016/B978-0-444-62702-5.00018-4. [DOI] [PubMed] [Google Scholar]
- 8.Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol. 1999;8:29–39. doi: 10.1016/s1054-8807(98)00023-4. [DOI] [PubMed] [Google Scholar]
- 9.Stehbens WE, Delahunt B, Shozawa T, Gilbert-Barness E. Smooth muscle cell depletion and collagen types in progeric arteries. Cardiovasc Pathol. 2001;10:133–136. doi: 10.1016/s1054-8807(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 10.Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30:2301–2309. doi: 10.1161/ATVBAHA.110.209460. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon LB, Harten IA, Patti ME, Lichtenstein AH. Reduced adiponectin and HDL cholesterol without elevated C-reactive protein: clues to the biology of premature atherosclerosis in Hutchinson-Gilford Progeria Syndrome. J Pediatr. 2005;146:336–341. doi: 10.1016/j.jpeds.2004.10.064. doi: 10.1016/j.jpeds.2004.10.064. [DOI] [PubMed] [Google Scholar]
- 12.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007;2:e1269. doi: 10.1371/journal.pone.0001269. doi: 10.1371/journal.pone.0001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon LB, Rothman FG, López-Otín C, Misteli T. Progeria: a paradigm for translational medicine. Cell. 2014;156:400–407. doi: 10.1016/j.cell.2013.12.028. doi: 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Progeria Research Foundation. https://www.progeriaresearch.org. Accessed December 7, 2017.
- 16.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG, Collins FS. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103:3250–3255. doi: 10.1073/pnas.0600012103. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osorio FG, Navarro CL, Cadiñanos J, López-Mejía IC, Quirós PM, Bartoli C, Rivera J, Tazi J, Guzmán G, Varela I, Depetris D, de Carlos F, Cobo J, Andrés V, De Sandre-Giovannoli A, Freije JM, Lévy N, López-Otín C. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3:106ra107. doi: 10.1126/scitranslmed.3002847. doi: 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- 19.Lee JM, Nobumori C, Tu Y, Choi C, Yang SH, Jung HJ, Vickers TA, Rigo F, Bennett CF, Young SG, Fong LG. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest. 2016;126:1592–1602. doi: 10.1172/JCI85908. doi: 10.1172/JCI85908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pendás AM, Zhou Z, Cadiñanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F, Tryggvason K, López-Otín C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–99. doi: 10.1038/ng871. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 22.Hamczyk MR, del Campo L, Andrés V. Aging in the cardiovascular system: Lessons from Hutchinson-Gilford Progeria Syndrome. Annu Rev Physiol. 2018;80:27–48. doi: 10.1146/annurev-physiol-021317-121454. doi: 10.1146/annurev-physiol-021317-121454. [DOI] [PubMed] [Google Scholar]
- 23.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 24.Yang SH, Qiao X, Farber E, Chang SY, Fong LG, Young SG. Eliminating the synthesis of mature lamin A reduces disease phenotypes in mice carrying a Hutchinson-Gilford progeria syndrome allele. J Biol Chem. 2008;283:7094–7099. doi: 10.1074/jbc.M708138200. doi: 10.1074/jbc.M708138200. [DOI] [PubMed] [Google Scholar]
- 25.Steffensen LB, Mortensen MB, Kjolby M, Hagensen MK, Oxvig C, Bentzon JF. Disturbed laminar blood flow vastly augments lipoprotein retention in the artery wall: a key mechanism distinguishing susceptible from resistant sites. Arterioscler Thromb Vasc Biol. 2015;35:1928–1935. doi: 10.1161/ATVBAHA.115.305874. doi: 10.1161/ATVBAHA.115.305874. [DOI] [PubMed] [Google Scholar]
- 26.Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acín-Pérez R, Enriquez JA, López-Otín C, Andrés V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013;127:2442–2451. doi: 10.1161/CIRCULATIONAHA.112.000571. doi: 10.1161/CIRCULATIONAHA.112.000571. [DOI] [PubMed] [Google Scholar]
- 27.Fuster JJ, Castillo AI, Zaragoza C, Ibáñez B, Andrés V. Animal models of atherosclerosis. Prog Mol Biol Transl Sci. 2012;105:1–23. doi: 10.1016/B978-0-12-394596-9.00001-9. doi: 10.1016/B978-0-12-394596-9.00001-9. [DOI] [PubMed] [Google Scholar]
- 28.Libby P, Hansson GK. Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res. 2015;116:307–311. doi: 10.1161/CIRCRESAHA.116.301313. doi: 10.1161/CIRCRESAHA.116.301313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrés V, Pello OM, Silvestre-Roig C. Macrophage proliferation and apoptosis in atherosclerosis. Curr Opin Lipidol. 2012;23:429–438. doi: 10.1097/MOL.0b013e328357a379. doi: 10.1097/MOL.0b013e328357a379. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Mejia IC, de Toledo M, Chavey C, Lapasset L, Cavelier P, Lopez-Herrera C, Chebli K, Fort P, Beranger G, Fajas L, Amri EZ, Casas F, Tazi J. Antagonistic functions of LMNA isoforms in energy expenditure and lifespan. EMBO Rep. 2014;15:529–539. doi: 10.1002/embr.201338126. doi: 10.1002/embr.201338126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 32.Rivera-Torres J, Calvo CJ, Llach A, Guzmán-Martínez G, Caballero R, González-Gómez C, Jiménez-Borreguero LJ, Guadix JA, Osorio FG, López-Otín C, Herraiz-Martínez A, Cabello N, Vallmitjana A, Benítez R, Gordon LB, Jalife J, Pérez-Pomares JM, Tamargo J, Delpón E, Hove-Madsen L, Filgueiras-Rama D, Andrés V. Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations. Proc Natl Acad Sci USA. 2016;113:E7250–E7259. doi: 10.1073/pnas.1603754113. doi: 10.1073/pnas.1603754113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109:16666–16671. doi: 10.1073/pnas.1202529109. doi: 10.1073/pnas.1202529109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordon LB, Massaro J, D’Agostino RB, Sr, Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW Progeria Clinical Trials Collaborative. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130:27–34. doi: 10.1161/CIRCULATIONAHA.113.008285. doi: 10.1161/CIRCULATIONAHA.113.008285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordon LB, Kleinman ME, Massaro J, D’Agostino RB, Sr, Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland RH, Nazarian A, Snyder BD, Ullrich NJ, Silvera VM, Liang MG, Quinn N, Miller DT, Huh SY, Dowton AA, Littlefield K, Greer MM, Kieran MW. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford Progeria syndrome. Circulation. 2016;134:114–125. doi: 10.1161/CIRCULATIONAHA.116.022188. doi: 10.1161/CIRCULATIONAHA.116.022188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin W, Carballo-Jane E, McLaren DG, Mendoza VH, Gagen K, Geoghagen NS, McNamara LA, Gorski JN, Eiermann GJ, Petrov A, Wolff M, Tong X, Wilsie LC, Akiyama TE, Chen J, Thankappan A, Xue J, Ping X, Andrews G, Wickham LA, Gai CL, Trinh T, Kulick AA, Donnelly MJ, Voronin GO, Rosa R, Cumiskey AM, Bekkari K, Mitnaul LJ, Puig O, Chen F, Raubertas R, Wong PH, Hansen BC, Koblan KS, Roddy TP, Hubbard BK, Strack AM. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res. 2012;53:51–65. doi: 10.1194/jlr.M019927. doi: 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 38.Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. 1994;93:1885–1893. doi: 10.1172/JCI117179. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]