

Abstract
Rationale:
This study aimed to investigate the genetic mutation characteristics of Kallmann syndrome (KS) with CHARGE syndrome through the clinical features and genetic analysis of a pediatric patient with KS in one pedigree.
Patient concerns:
Developmental disorders with olfactory abnormalities, developmental lag, heart malformations, external genital malformations.
Diagnoses:
KS combined with some clinical characteristics of CHARGE syndrome. Molecular genetic analysis found that mutation occurred in the CHD7 gene.
Interventions:
One pediatric patient's clinical data were collected and genomic DNA extracted from the peripheral blood. Nextgeneration gene sequencing technology was used to detect pathogenic genes, and the Sanger method was applied to perform pedigree verification for the detected suspicious pathogenic mutations.
Outcomes:
Gene detection revealed there to be a heterozygous mutation in the CHD7 gene of the patient, which was a missense mutation c.6571G > A (p.E2191K). The father's genotype was wild type, whereas it was the mutant type for the mother and younger brother. The distribution frequency of this mutation was zero in the dbSNP database, Hapmap, 1000 genomes database, and ExAC. Neither the mother nor the younger brother showed any clinical feature of KS or CHARGE syndrome.
Lessons:
This study reports 1 case of KS with some clinical features of CHARGE syndrome as determined via clinical and genetic analysis, and found a new mutation in the CHD7 gene, suggesting that KS has an incomplete penetrance. Meanwhile, data suggested that mutation in the CHD7 gene could be detected in the setting of incomplete clinical manifestations of CHARGE syndrome, or without the usually believed manifestations of combined deafness as well as morphological abnormalities of the ear, providing new evidence for the differential diagnosis of KS with CHARGE syndrome in the future.
Keywords: CHARGE syndrome, CHD7, idiopathic hypogonadotropic hypogonadism, kallmann syndrome
1. Introduction
Kallmann syndrome (KS) is a unique disease phenotype of idiopathic hypogonadotropic hypogonadism (IHH) characterized by developmental disorders and olfactory abnormalities. IHH refers to different degrees of congenital defects in GnRH secretion, resulting in presence of incomplete diseases of pubertal development,[1] belonging to the developmental disorders of hypothalamic gonadotropin. KS accounts for about 50% to 60% of IHH,[2] with the others known as IHH with normal olfaction (nIHH). In l856, Maestre de Sanjuan reported the existence of the disease of sexual dysfunction associated with olfactory dysfunction.[3] In l944, Kallmann et al[4] first reported 9 cases of sexual dysfunction associated with anosmia, proposing that it was a genetic disease, which became known as KS. Since that time, there have been a number of familial and sporadic cases reported, and this disease was found to associate with other congenital malformations.
Typical clinical manifestations of KS are hypogonadism and olfactory dysfunction. Severities of hypoplasia in secondary sex characteristics, sexual dysfunction, as well as hypogenesis of the penis and testicles vary. The development of the olfactory bulb and tract may be affected to varying degrees as well, producing anosmia or selective hyposmia in patients. As olfactory abnormalities of children cannot be easily found in early stages of disease, routine olfactory function tests should be performed on patients with gonadal dysgenesis to detect the condition as early as possible. The morbidity of KS is about 1 in 8000 in men, and 1 in 40000 in women.[5] However, the morbidity of KS in women may be underestimated because some female patients only show mild hypogonadism and patients with primary amenorrhea do not have a clear etiology.[6]
CHARGE syndrome includes ocular tissue defects (C), cardiac abnormalities (H), atresia of the posterior nares (A), growth and/or developmental retardation (R), genital external malformations (G), and ear deformities (E). Ocular tissue defect and atresia of the posterior nares are the major diagnostic criteria, whereas the remaining 4 items are secondary diagnostic criteria. The disease can be confirmed if 4 of the criteria are satisfied (including at least 1 major diagnostic criterion). The genetic model of CHARGE syndrome is mainly one of autosomal dominant inheritance, which is usually because of CHD7 mutation. CHARGE syndrome is similar to some of the clinical features of KS. Patients with CHARGE syndrome suffer from loss of the olfactory bulb and IHH, which are the main characteristics of KS. Jongmans et al[7] suggested that CHD7 gene detection should be performed on patients with deafness, ear deformities, ear hypoplasia or possessing an undeveloped semicircular canal. The majority of patients with CHARGE syndrome are because of deletion mutations in heterozygous CHD7 function. The different mutant genes between CHARGE syndrome and KS contribute to their identification. Molecular genetics is therefore crucial for differential diagnosis when patients present with suspicious features suggesting either of the 2 conditions.
Molecular diagnosis is the best method for studying the diagnosis of KS and its genetic mechanisms. At present, genetic testing is the method of molecular detection used in precise diagnosis, such as Sanger sequencing, amplification refractory mutation system polymerase chain reaction, and differential display of reverse Transcriptional polymerase chain reaction, all of which can detect the sequence of a particular gene. However, these methods are difficult to popularize in clinical diagnosis, especially in relation to diseases with similar clinical features because of the large number of genes, the high cost, as well as long cycles of testing. Next-generation sequencing (NGS) can perform high-throughput detection of multiple genes, as well as all exons of genes related to genetic diseases at once. It cannot only carry out differential diagnosis for diseases associated with known mutation sites, but also reveal new pathogenesis, and is suitable for screening of genetic diseases with similar clinical characteristics as well as the study of genetic diseases.[8] Our research performed sequencing for the exons of genes related to whole blood genetic diseases of the patient by the sequencing method that used a chip to capture high throughput, and successfully found a suspicious pathogenic site. Moreover, this was the first report of this site both nationally and internationally, presenting a great significance for the epidemiological study of KS in China. Meanwhile, it will further supplement the databases of genetic diseases in China, contribute to the diagnosis of diseases, and promote the development of precision medicine, such as preimplantation genetic screening (PGS), preimplantation genetic diagnosis (PGD), prenatal screening, as well as prenatal diagnosis.
2. Materials and methods
2.1. Collection of medical history and related auxiliary examination
This study enrolled 1 patient suspected to be with KS in the Seventh Pediatric Ward and Outpatient Department of Pediatrics of Hunan Provincial People's Hospital in April 2016. The medical history, physical examination data, as well as related auxiliary inspection data were collected after obtaining informed consent from the parents and approval from the Ethics Committee of the hospital. The patient has provided informed consent for publication of the case.
2.2. Detection of pathogenic genes
2.2.1. Specimen collection
Under the premise of informed consent, 4-mL whole blood samples were collected from the patient and the parents as well as the younger brother (ethylenediaminetetraacetic acid anticoagulant), and then genomic DNA of the patient was extracted using BloodGen Midi Kit (Beijing ComWin Biotech Co., Ltd., China).
2.2.2. Capture of target sequence and high-throughput sequencing
Based on the literature and information of OMIM database, Roche NimbleGen capture probe was prepared for exon regions in the genome of >4000 genetic diseases in the OMIM database to perform whole exome capture of the target genes.
2.2.3. Library preparation
Library preparation has been done in following steps:
-
1.
Genome fragmentation: Genes were broken into about 200 bp by Cavoris.
-
2.
Filling-in and repair of the free-end: This was performed for the fragmentized NDA under the role of Klenow Fragment, T4 DNA polymerase, and T4PNK.
-
3.
3‘-terminal adenosine: In the polymerase system, the repaired products obtained by the last step was added with A base at 3’terminal to be prepared for the connection in the next step.
-
4.
Addition of connectors: T4DNA ligase reaction system was configured, whereas adapter and supplementary “A” products were connected after reacting the appropriate temperature for a certain period in Thermo mixer.
-
5.
Amplification: The ligation products went through 4 to 6 rounds of ligation-mediated polymerase chain reaction (LM-PCR) amplification.
-
6.
Hybridization: Library and probe were mixed in a hybridization system at 65°C to be hybridized for 60 to 68 hours.
-
7.
Washing beads and eluting DNA: Elution was performed using eluent after streptomycin beads were incubated with the hybridization samples.
-
8.
Amplification of eluted products: Eluted products were amplified by 10 rounds of LM-PCR.
2.2.4. Sequencing on the Illumina platform
Illumina hiseq2500 platform standardized the operation process of the sequencing. Raw image data were obtained by sequencing, and Illumina official basecall analysis software BclToFastq was used to obtain raw data.
2.3. Data analysis
2.3.1. Analysis of the basic data
Analysis of the basic data was done in following steps:
-
1.
Counting the production of raw data: connector contamination and low quality data were removed.
-
2.
Comparison: Comparative statistics was performed for the data and reference sequence (BWA used), and reference genome was hg19 genome.
-
3.
Single-nucleotide polymorphism (SNP) detection and annotation: Analysis was performed by Samtools software.
-
4.
Indel detection and annotation: Analysis was performed by Pindel software.
-
5.
False positive filter of mutations: Based on the depth of sequencing and quality of mutation, the detected SNP was filtered and screened by Indel, then high-quality and reliable mutations could be obtained.
-
6.
Mutation annotation: SNP and Idel analyzed and obtained the impact of amino acid changes, shear effects, untranslated region, and impact of intron mutation according to the location in genes.
-
7.
Prediction of the effect of screened mutations on protein function: The impact of screened mutations on the protein was predicted by SIFT and the algorithm of homology alignment, as well as conservation of protein structure.
-
8.
The splicing hazard was predicted for the mutations near the splice sites.
2.3.2. In-depth data analysis
In-depth data analysis was done to find mutations in related genes with their genetic patterns and clinical symptoms matching the pediatric patient.
2.4. Validation using first-generation sequencing (Sanger)
Primers were designed according to the sequence of sites validated by the CHD7 gene. Amplification was performed using PCR, and sequencing was conducted with the ABI 3730XL sequencing device, whereas sequencing primers used the original PCR primers. Genetic sequence analysis and alignment were performed using DNASTAR software, and mRNA alignment template was NM_003560. Samples of the pediatric patient, the parents, and the younger brother were validated using first-generation sequencing.
3. Results
3.1. Clinical data
The patient was a 14-year-old boy admitted to hospital because his sexual development lagged 2 years behind, and his right thigh was swollen and painful 1 day after trauma. The pediatric patient was a G3P3 full-term infant with a birth weight of 3.2 kg. Furthermore, he was born without asphyxia at birth and his mother had no special medical history of pregnancy. The pediatric patient started to raise his head at 3 months, sit by himself at 6 months, climb at 9 months, and walk by himself at 12 months, and his growth and development were the same as children of the same age.
3.2. Medical history
In 2005, the pediatric patient underwent splenectomy because of thalassaemia, and recovered well after the operation without complaints of discomfort.
In 2013, the pediatric patient underwent valve repair surgery because of congenital atrial septal defect, and recovered well after the operation without complaints of discomfort to date.
In 2014, his parents complained that his sexual development lagged behind children of the same age after the operation, and that his height was shorter.
On April 11, 2016, the patient received open reduction and internal fixation of the right femur under general anesthesia. The operation was successful and the patient recovered well after the operation. The patient was transferred to our department because his parents wanted to clarify the reasons of the lag in sexual development.
3.3. Family history
His parents were healthy without consanguineous marriage. There was no similar medical history in his family, and no consultable history of special genetic diseases. The patient had a younger brother, who was reported to be healthy.
3.4. Results of examination on admission
-
1.
Physical examination on admission: His height and weight were 148 cm and 32 kg, respectively. The development of the patient lagged behind children of the same age, and his height was shorter, whereas other findings were normal.
-
2.
Postoperative endocrine lab values: Neo-hombreol F was 1.36 nmol/L, and others were normal.
-
3.
Rheumatic labs: Anti-O was 464 IU/mL, immunoglobulin A was 5.14 G/L, immunoglobulin G was 31.7 G/L, and erythrocyte sedimentation rate was 77 mm/h.
-
4.
Specialty examinations:
Olfactory dysfunction;
There was a lack of secondary sexual characteristics, and no growth of armpit hair, Adam's apple development, or beard
Testicular volume was about 3 mL, measured values of bilateral testicules were small, and the length of the penis was 5 cm.
It can be noted from the clinical data that the pediatric patient suffered from developmental disorders with olfactory abnormalities, in line with clinical diagnostic criteria of KS; in addition, the pediatric patient presented with clinical characteristics of CHARGE syndrome, such as developmental lag, heart malformations, as well as external genital malformations. However, the pediatric patient did not meet the diagnostic criteria of CHARGE syndrome, including external ear deformity, deafness, and/or dysplasia or agenesis of the semicircular canal in the inner ear. Therefore, the differential diagnosis of this case required further molecular genetic analysis.
3.5. Genetic detection
Two suspicious mutant sites were found on the CHD7 gene of the pediatric patient through high-throughput sequencing captured by exons, bioinformatics analysis, and clinical database analysis. Validation using first-generation sequencing and pedigree verification (Fig. 1) were performed, respectively. The results are shown as follows.
Figure 1.
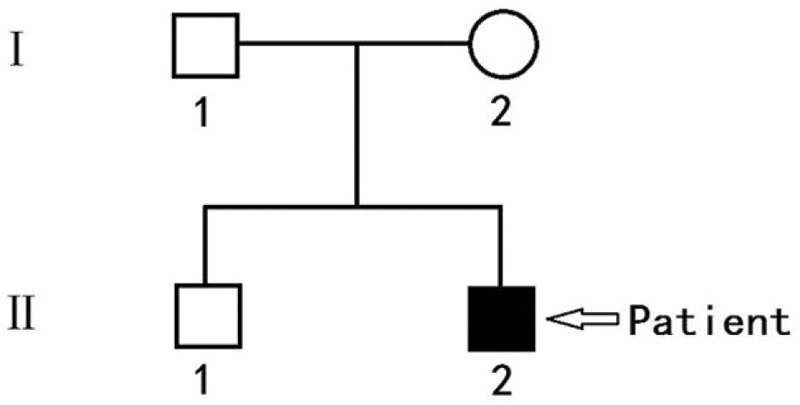
Atlas of the pediatric patient and his pedigree.
Heterozygous missense mutation c.6571G > A (p.E2191K) was found in the CHD7 gene of the pediatric patient (Fig. 2). His mother and younger brother had the same genotype (Figs. 3 and 4), but his father was the wild type (Fig. 5).
Figure 2.
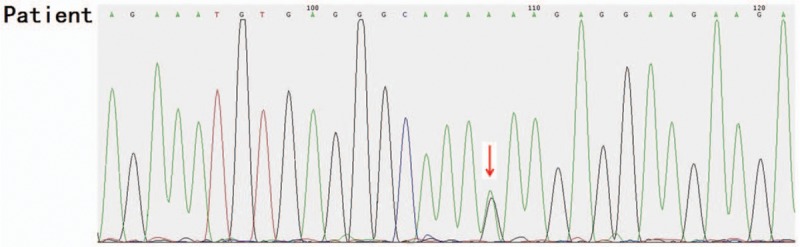
Heterozygous missense mutation c.6571G>A (p.E2191K) was found in the CHD7 gene of the pediatric patient.
Figure 3.
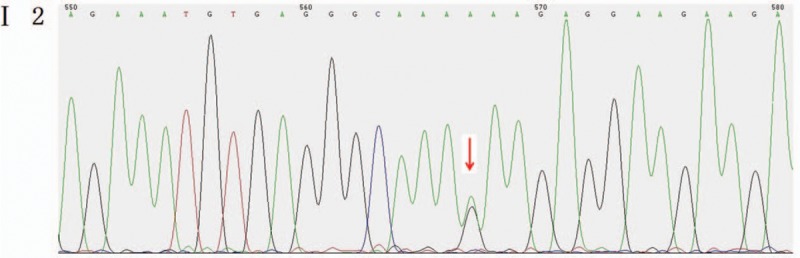
Mutant sites in the CHD7 gene of the pediatric patient's mother were validated to be the mutant type.
Figure 4.
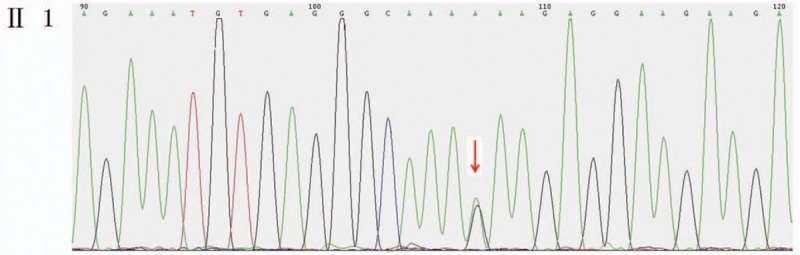
Mutant sites in the CHD7 gene of the pediatric patient's younger brother were validated to be the mutant type.
Figure 5.
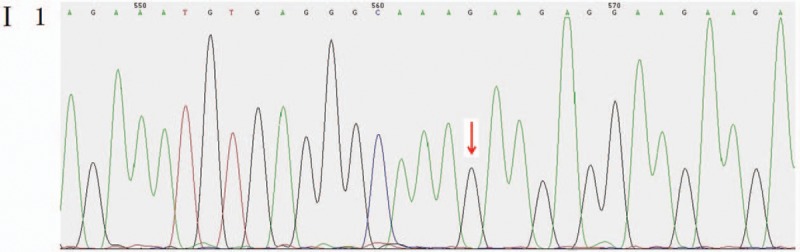
Mutant sites in the CHD7 gene of the pediatric patient's father were validated to be the wild type.
Genetic detection and pedigree validation revealed that the pediatric patient, as well as his mother and younger brother, were mutant types, whereas his father was a wild type, consistent with the dominant inheritance pattern of KS and CHARGE syndrome. The distribution frequency of this mutation was zero in the dbSNP database, Hapmap, 1000 genomes database, and ExAC. This mutation was a new mutation that had not been reported in relevant literature.
In summary, the patient may suffer from KS combined with some clinical characteristics of CHARGE syndrome. Molecular genetic analysis found that mutation occurred in the CHD7 gene, and his mother and younger brother had the same mutant genotype without phenotype, suggesting that the disease may have incomplete penetrance. The molecular mechanism of the disease and the study of the genetic modes of transmission need to be further studied with more cases.
4. Discussion
KS is a unique biological model of IHH characterized by developmental disorders with olfactory abnormalities, caused by congenital defects in GnRH secretion of varying degrees. The pulsatile secretion of GnRH is essential for the hypothalamic-pituitary-gonadal axis function. During embryogenesis, GnRH neurons and olfactory neurons co-originate from the olfactory plate, and migrate to the olfactory bulb through the sieve plate together.[9] It can be considered that GnRH neurons guide migration using the olfactory system. Impaired function of GnRH can be caused by congenital impairment of GnRH neurons in the olfactory plate, abnormal migration of GnRH neurons in embryonic period, abnormal development and maturation of GnRH neurons in hypothalamus, or resistance of the pituitary to GnRH. KS is a unique disease model to study the migration of GnRH neurons and the development of human puberty. Some genes are necessary for the correct differentiation, migration, upstream signal regulation, and function of GnRH neurons in the embryonic period, which can lead to IHH. Some genes that correctly differentiate embryonic GnRH neurons may be correlated with IHH, such as KAL-1, FGFR1, FGF8, PROKR2, PROK2, CHD7, NELF, WDR11, HS6ST1, KISS1R, KISS1, TAC3, TACR3, LEPR, LEP, PCSK1, GNRHR, GNRH1, SEMA3A, and NDN7. Mutations in these genes lead to certain degrees of clinical manifestations. Moreover, KS can be caused by genes such as KAL-1, FGFR1, PROKR2, PROK2, CHD7, and FGF8. Among them, the CHD7 mutation was only found in KS patients with the CHARGE syndrome phenotype, suggesting that if the patient was diagnosed with hypogonadism and anosmia, attention should be paid to investigate the presence of clinical characteristics of CHARGE syndrome. CHD7 should be examined only when combined with deafness, abnormal ear morphology, and/or semicircular canal hypoplasia or aplasia. However, there is no specific method for the investigation of CHARGE syndrome in the various clinical examination methods, including MRI of the auditory organ structure. Moreover, the impact of new genes on the disease is common. Therefore, it is possible to realize molecular diagnosis by full-exome sequencing.
KS is a genetic disease, and its genetic mechanism has become clearer with the development of molecular biology. Currently, genes clearly related to KS include KAL-1, FGFR1, PROKR2, PROK2, CHD7, and FGF8. Genes possibly related to KS include NELF,[10]HS6ST1,[11] and WDR11.[12]KAL-1 gene is considered to only contribute to KS, which is responsible for the X-linked recessive pathogenesis of KS. KAL-1 gene is located on Xp22. 3 with a full length of 210 kb and possesses 14 exons. It encodes the extracellular matrix protein of 680 amino acids, which is also known as Anosmin.[13]KAL-1 gene-encoding protein can regulate neurite outgrowth and recognize target tissues or target cells, as well as participate in the migration of GnRH-secreting neurons and olfactory neurons. Mutation in the KAL-1 gene results in inability to synthesize KAL adhesion protein, affecting the migration of GnRH nerve cells, as well as the formation of the olfactory bulb and olfactory tract. Deficiency in hypothalamic GnRH secretion results in testicular dysfunction and olfactory abnormalities. It is noteworthy that FGFR1, FGF8, PROKR2, and PROK2 lead to not only KS, but also nIHH, whereas CHD7 only appears in patients with CHARGE syndrome.
The etiologies of KS include spontaneous type and hereditary type. Most cases are sporadic, and about one-third of patients have a family history.[14] KS follows the 3 classic Mendelian inheritance patterns of chain recessive inheritance of the X chromosome (OMIM: 308700) (according to KAL-1), autosomal dominant inheritance (OMIM: 147950) (according to FGFR1, FGF8, and CHD7), along with autosomal recessive inheritance (OMIM: 244200) (according to PROKR2, PROK2). Clinical heterogeneity of KS is clear, with incomplete penetrance, mainly manifesting as differences in clinical features as well as genetic mutations. Patients with different genetic mutations often present with some specific nonreproductive system, nonolfactory clinical manifestations. For example, 70% of patients with mutation in KAL-1 present with synkinetic movements, and 30% with unilateral renal hypoplasia. Patients with mutations in FGFR1 or FGF8 often suffer craniofacial deformities (lip/cleft palate, high-arched palate, and ocular signs), as well as dental hypoplasia. PROKR2 or PROK2 mutation is associated with epilepsy, sleep disorders, fibrous dysplasia, and obesity. CHD7 mutation is often accompanied by deafness and hypoplasia of the inner ear's semicircular canal. In addition, pathogenic genes of KS are continuously being discovered, and many other similar cases may have different genetic bases with different patterns in genetic models.
The CHD7 gene is located on chromosome 8q12.1, which encodes DNA-binding protein 7 of helicase in the chromatin region. This protein family has a unique functional domain-binding site, including 2 N-terminal chromatin domains, 1 SWI2/SNF2-like ATP enzyme/solution helix domain, and 1 DNA-binding domain. CHD7 protein complex is expressed in the olfactory epithelium, hypothalamus, as well as the pituitary gland, suggesting that this protein may play an important role in the development of the olfactory bulb and GnRH neuron migration. The genetic pattern of CHD7 gene has not yet been fully understood, and may follow autosomal dominant inheritance, with its mutations accounting for 6% of all IHH patients.[6]
Our research found that the patient had a new mutation inherited from his mother, and his younger brother also carried the same genotype. However, his mother and younger brother did not show clinical features of KS, potentially associated with the clinical heterogeneity of KS.
The clinical heterogeneity of KS mainly manifests in 3 ways: a variety of different phenotypes, ranging from mild to complete loss, exist both in pubertal development and in the degree of olfactory dysfunction, which are not only in sporadic patients, but also in familial patients,[15,16] and even in identical twins; 10% of patients may have different degrees of spontaneous puberty after intermittent exogenous hormone-replacement therapy, which is also known as reversible KS[17–19]; some patients have normal pubertal development, but suffer from KS symptoms in adulthood[20]; which is known as adult-onset KS. This clinical heterogeneity suggests that potential defects do not always prevent the migration of GnRH neurons and/or the secretion of GnRH. The appearance of reversible KS also suggests that the maturation of the GnRH neural network can also occur in the adult period. Therefore, in this study, we should continue to follow the clinical manifestations of the younger brother who may show adult-onset KS.
In addition, clinical heterogeneity suggests that the etiology of KS is far more complex than that of the imagination, and it may not only involve genetic mutation and the 3 genetic models. For example, there are many reports about the “incomplete penetrance” of FGFR1 genetic mutation. Pitteloud et al[17] reported that in a KS pedigree, a R622X mutant showed a reversible gonadal function in propositus, and the mother of the propositus only had history of delayed puberty, whereas the grandfather of the propositus only had anosmia. However, some studies have shown that KS and CHARGE syndrome may have an incomplete penetrance and clinical heterogeneity.[21,22] Thereafter, the patient's mother carried the mutation, but it was likely that she may not be a patient at all, or perhaps present with mild clinical manifestations, and the younger brother may be only temporarily without clinical manifestations.
It was also reported that CHARGE syndrome alone had clinical heterogeneity.[23] In the comparison between the pediatric patient and his parents, clinical manifestations of the parents were only mild symptoms, whereas the pediatric patient showed severe symptoms.[23,24] At present, although KS cannot be cured, and treatment remains limited to hormone-replacement therapy, early diagnosis is the key to treatment. With the development of medicine, the further development of gene knockout technology, as well as gene therapy, genetic disease is now not viewed as an incurable disease. The basis of these treatments is the study of the molecular mechanisms of the disease. Currently, there are many studies of CHD7 gene mutation in KS patients with CHARGE syndrome, and new sites are continuously discovered.[25,26] The newly found site in this study can be used as a further supplement to the molecular genetics library of genetic diseases.
Through clinical and genetic analysis, the patient reported in this study may suffer from KS and some clinical features of CHARGE syndrome. Pedigree verification can be achieved by CHD7 gene mutation c.6571G>A. Through analysis of clinical characteristics, the disease in this pedigree may has incomplete penetrance. There were no relevant reports by searching literature, so this study can be used in molecular diagnosis, genetic counseling, and prenatal screening of KS with CHARGE syndrome.
Meanwhile, this case suggested that the CDH7 gene mutation should be detected when there were no clinical manifestations of complete CHARGE syndrome or there were no usually apparent manifestations of deafness, and morphological abnormalities of the ear, which provided new evidence for the differential diagnosis of KS with CHARGE syndrome.
NGS gene detection technology is becoming an important method in the study at the molecular level such as the pathogenesis of disease and disease classification because of its high throughput and low cost. At present, NGS has been well applied in the fields of precision medicine, such as genetic screening, auxiliary diagnosis of genetic disease, noninvasive prenatal testing, PGS, PGD, individual treatment of tumors, and efficacy monitoring. This case was evaluated with genetic screening using the method of directional acquisition, and the pathogenic site of the pedigree was found precisely, conveniently allowing for the differential diagnosis of multiple pathogenic genes. With the development of sequencing technology, the cost has further been reduced, so that whole exon sequencing will be fully used for disease screening, and even whole-genome application may be possible in the future. Gene sequencing is important to further explore the correlation of genotype–phenotype relationships of disease, to search for therapeutic targets, and to guide clinical work. With the development of science and technology, the possibility of gene therapy for genetic defects is further increased. For example, CRISPR/Cas9 has been used in human embryo knockout experiments, gene therapy for muscular dystrophy has made significant progress, and CAR-T cells were used for the treatment of immunodeficiency. NGS gene detection technology is applied to single cell sequencing, especially in the field of PGS/PGD. Currently, China has been able to screen dozens of single gene diseases, effectively avoiding birth defects. Meanwhile, precise classification and diagnosis of diseases remain the foundations of genetic pathology and form the bases for prenatal screening, prenatal diagnosis, and genetic counseling. We believe that with the accumulation of clinical cases and genetic data in our country, the birth of infants with diseases such as KS can be effectively avoided in the future.
Author contributions
Data curation: Jie WEN.
Formal analysis: Jiang WANG.
Investigation: Li PAN.
Project administration: xuan xu.
Software: Cheng HU.
Footnotes
Abbreviations: GnRH = gonadotropin-releasing hormone, IHH = idiopathic hypogonadotropic hypogonadism, KS = Kallmann syndrome, NGS = next-generation sequencing, PGD = preimplantation genetic diagnosis, PGS = preimplantation genetic screening.
The authors report no conflicts of interest.
References
- [1].Topaloglu AK, Kotan LD. Molecular causes of hypogonadotropic hypogonadism. Curr Opin Obstet Gynecol 2010;22:264–70. [DOI] [PubMed] [Google Scholar]
- [2].Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol 2009;5:569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rugarli EL, Ballabio A. Kallmann syndrome: from genetics to neuro-biology. JAMA 1993;270:2713–6. [DOI] [PubMed] [Google Scholar]
- [4].Kallmann FJ, Schoenfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Ment Defic 1944;48:203–36. [Google Scholar]
- [5].Bonomi M, Libri DV, Guizzardi F, et al. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl 2012;14:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dodé C, Hardelin JP. Clinical genetics of Kallmann syndrome. Ann Endocrinol (Paris) 2010;71:149–57. [DOI] [PubMed] [Google Scholar]
- [7].Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome the clinical overlap with CHARGE syndrome. Clin Genet 2009;75:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011;43:585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature 1989;338:161–4. [DOI] [PubMed] [Google Scholar]
- [10].Xu N, Kim HG, Bhagavath B, et al. Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril 2011;95:1613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tornberg J, Sykiotis GP, Keefe K, et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci U S A 2011;108:11524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kim HG, Layman LC. The role of CHD7 and the newly identified WDR11 gene in patients with idiopathic hypogonadotropic hypogonadism and Kallmann Syndrome. Mol Cell Endocrinol 2011;346:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bhagavath B, Xu N, Ozata M, et al. KAL1 mutations are not a common cause of idiopathic hypogonadotrophic hypogonadism in humans. Mol Hum Reprod 2007;13:165. [DOI] [PubMed] [Google Scholar]
- [14].Pitteloud N, Acierno JS, Jr, Meysing A. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A 2006;103:6281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Santen RJ, Paulsen CA. Hypogonadotropic eunuchoidism. I. Clinical study of the mode of inheritance. J Clin Endocrinol Metab 1973;36:47–54. [DOI] [PubMed] [Google Scholar]
- [16].Lieblich JM, Rogol AD, White BJ, et al. Syndrome of anosmia with hypogonadotropic hypogonadism (Kallmann syndrome): clinical and laboratory studies in 23 cases. Am J Med 1982;73:506–19. [DOI] [PubMed] [Google Scholar]
- [17].Pitteloud N, Acierno JS, Jr, Meysing AU, et al. Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab 2005;90:1317–22. [DOI] [PubMed] [Google Scholar]
- [18].Ribeiro RS, Vieira TC, Abucham J. Reversible Kallmann syndrome: report of the first case with a KAL1 mutation and literature review. Eur J Endocrinol 2007;156:285–90. [DOI] [PubMed] [Google Scholar]
- [19].Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 2007;357:863–73. [DOI] [PubMed] [Google Scholar]
- [20].Mitchell AL, Dwyer A, Pitteloud N, et al. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab 2011;22:249–58. [DOI] [PubMed] [Google Scholar]
- [21].Bosman EA, Penn AC, Ambrose JC, et al. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet 2005;14:3463–76. [DOI] [PubMed] [Google Scholar]
- [22].Kim HG, Kurth I, Lan F, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 2008;83:511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zentner GE, Layman WS, Martin DM, et al. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A 2010;152A:674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bergman JE, Janssen N, Hoefsloot LH, et al. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 2011;48:334–42. [DOI] [PubMed] [Google Scholar]
- [25].Schulz Y, Wehner P, Opitz L, et al. CHD7,the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum Genet 2014;133:997–1009. [DOI] [PubMed] [Google Scholar]
- [26].Wells C, Loundon N, Garabedian N, et al. A case of mild CHARGE syndrome associated with a splice site mutation in CHD7. Eur J Med Genet 2016;59:195–7. [DOI] [PubMed] [Google Scholar]